Improved methods and resources for paramecium genomics: transcription units, gene annotation and gene expression
Texte intégral
Figure
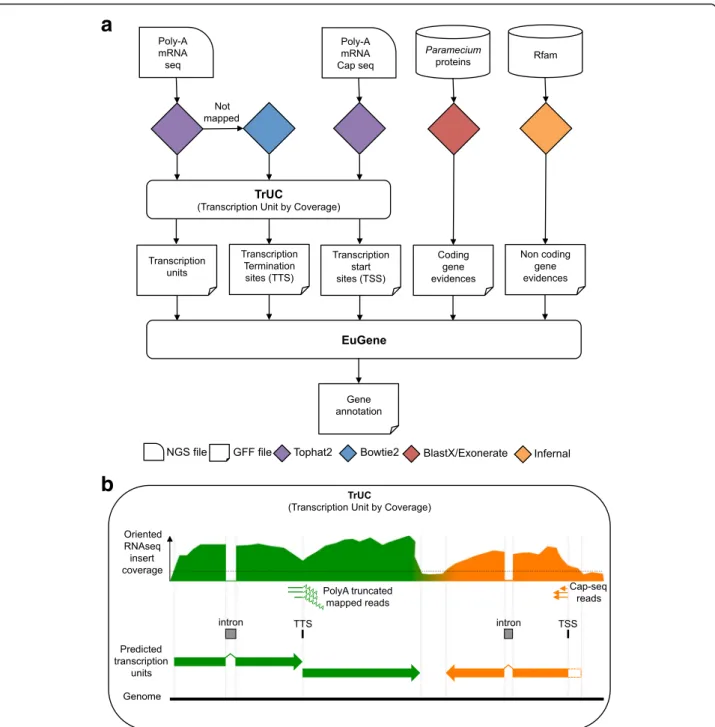
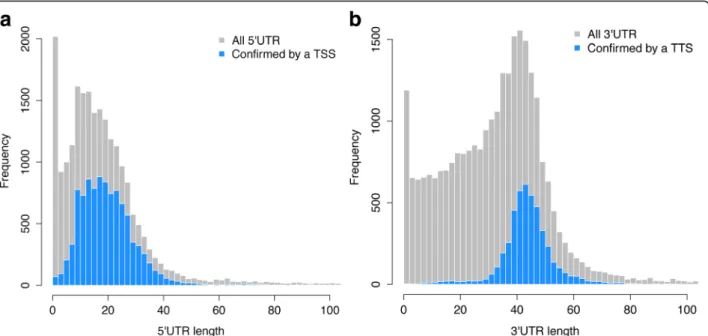
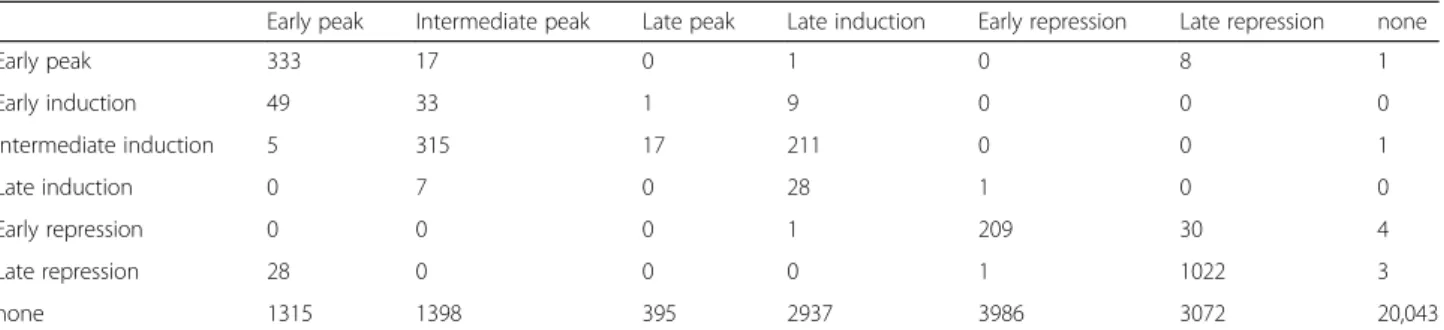
Documents relatifs
Keywords: respiratory-related evoked potentials, respiratory afferents, upper airway, respiratory sensations, obstructive sleep apnea syndrome, event-related
S'ils se prenaient pour des Rambo c'était leur problème, mais certainement pas le sien et elle, elle n'était pas sur terre pour se faire chier, surtout dans ce quartier de
Heckel G, Burri R, Fink S, Desmet J-F, Excoffier L (2005) Genetic structure and colonization processes in European populations of the common vole Microtus arvalis.
Même si les résultats obtenus par la mesure de confiance issue du SRAP sont performants pour détecter les documents mal indexables, l’index de compacité sémantique permet
Capitaux permanents Immobilisations Dettes à court Et Moyen terme Stocks Créances clients Y compris effets TRESORERIE Emplois Ressources ACTIF PASSIF...
En 1957, Bickerstaff décrivit huit patients avec un tableau de dysfonction aiguë du tronc cérébral survenant dans les suites d’une infection. La rhombencéphalite
Notre étude permet d’avoir un aperçu sur les principaux symptômes physiques accompagnant les troubles anxieux et dépressifs chez les patients consultants en
By using several combinations of serum and cycloheximide, a protein synthesis inhibitor, we showed that: i) addition of cycloheximide to resting cell elicits an increase in c-fos
