Development of mass spectrometry based technologies for quantitative cell signaling phosphoproteomics : the epidermal growth factor receptor family as a model system
Texte intégral
Figure
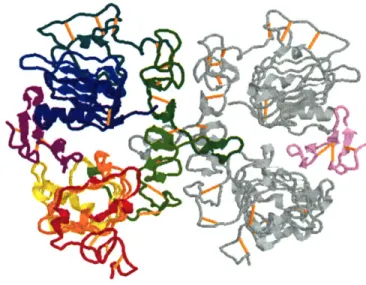
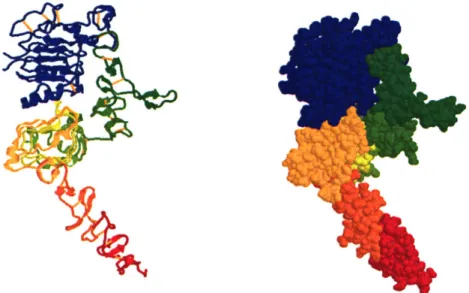
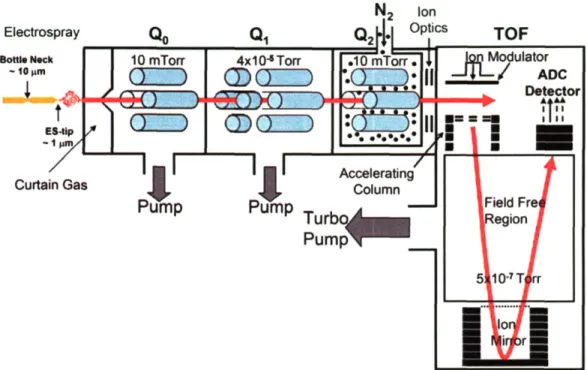
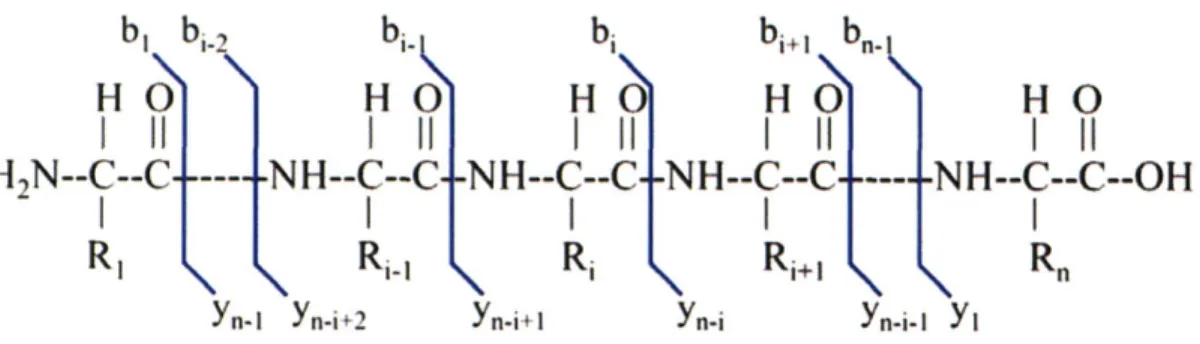
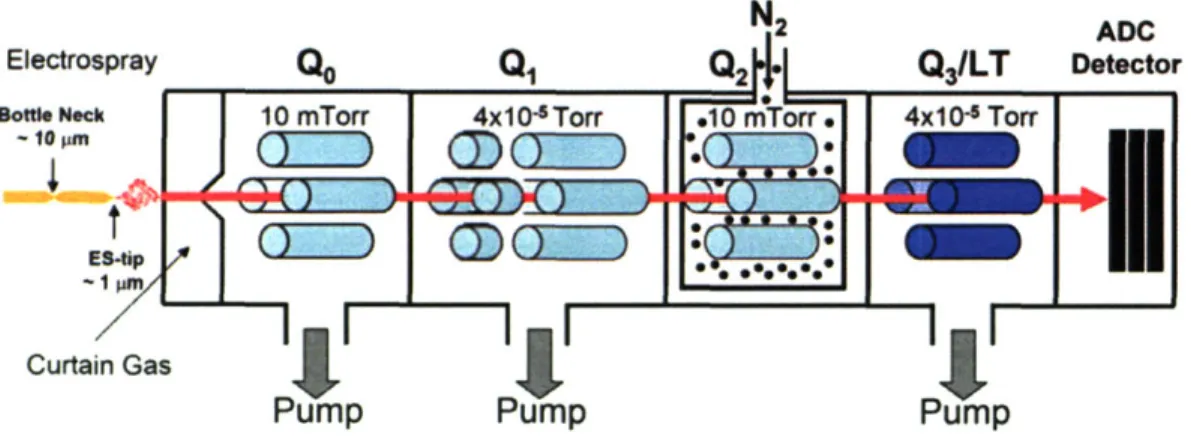
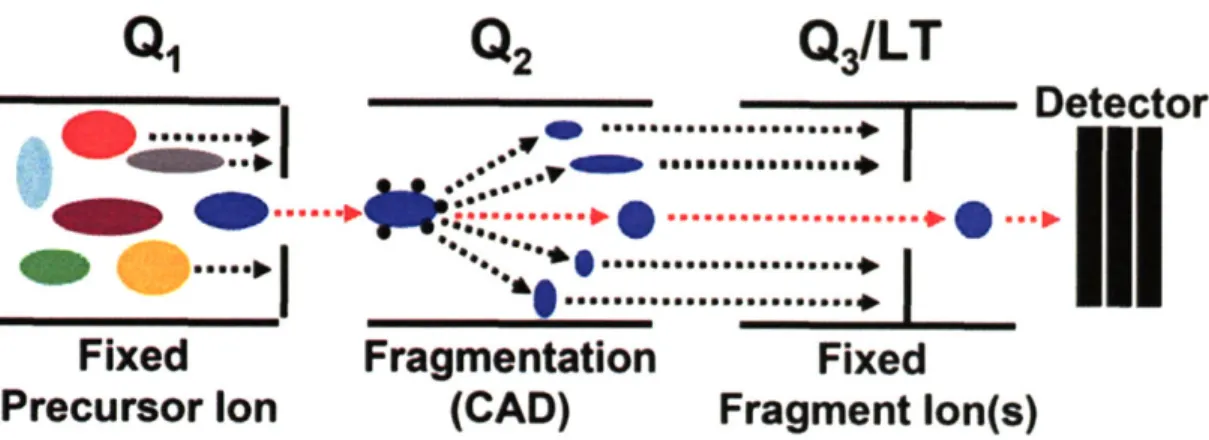
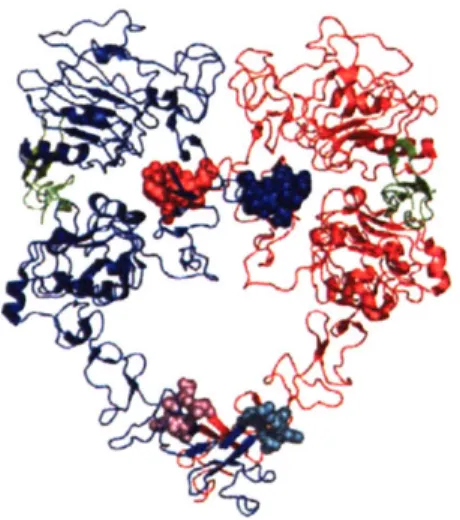
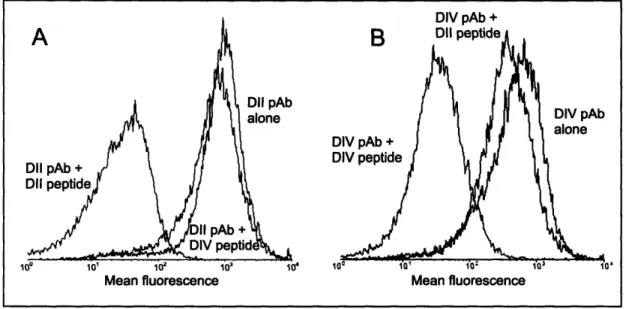
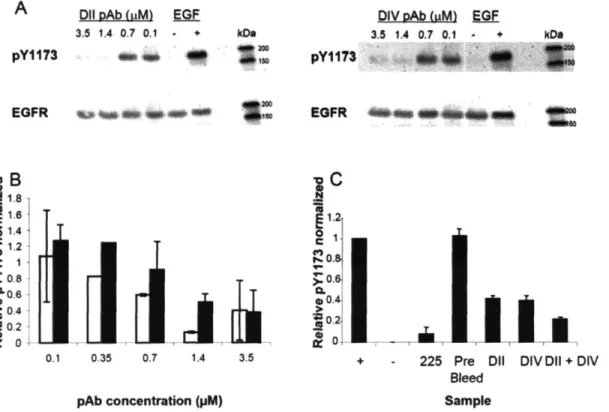
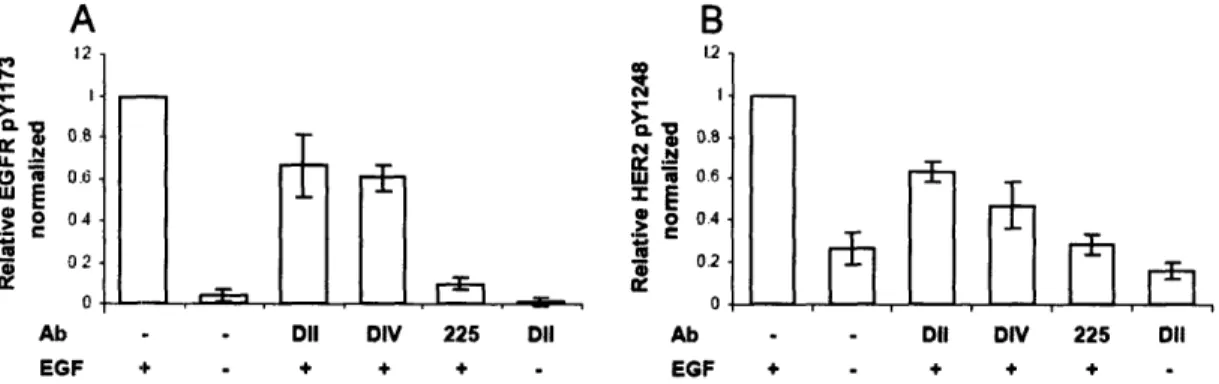
Documents relatifs
The effect of synthesized amino acid LDH fillers on the chain extending and molecular weight evolution was measured using melt rheology.. The rheological data were plotted
This study shows for the first time that WR consumption results in major biological modifica- tions –increased plasma and liver n-3 EPA and DHA levels and improved gut microbiota
For any integer ∆, there exists a robot with the property that for any graph G of degree bounded by ∆, it is possible to color the nodes of G with two colors (or alternatively,
In our context each expert is a Kalman filter fed by a subset of sensors, and a gating network serves as a mediator between individual filters, basing its decision on sensor inputs
Report (National Research Council of Canada. Radio and Electrical Engineering Division : ERB), 1962-08. READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING
During volume controlled ventilation with small tidal volume and constant inspiratory flow in hypercapnic patients with ALI or ARDS, this study shows that a 20% post-inspiratory pause
To establish the effects of blocks of linked genetic polymorphisms on gene transcription, we repeated fat transcriptome profiling in a series of reciprocal BN.GK and GK.BN
Lors de la lecture d’un objet X, le client doit déterminer si la version de X stockée dans le centre de données est sûre à lire (c’est-à-dire qu’elle reflète toutes les mises
