Bacterial phosphoglycosyl transferases: initiators
of glycan biosynthesis at the membrane interface
The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters.
Citation Lukose, Vinita et al. "Bacterial phosphoglycosyl transferases: initiators of glycan biosynthesis at the membrane interface."
Glycobiology 27, 9 (September 2017): 820–833 © 2017 The Author(s)
As Published http://dx.doi.org/10.1093/glycob/cwx064
Publisher Oxford University Press (OUP)
Version Author's final manuscript
Citable link https://hdl.handle.net/1721.1/126236
Terms of Use Creative Commons Attribution-Noncommercial-Share Alike
Bacterial Phosphoglycosyl Transferases: Gatekeepers of Glycan
Biosynthesis at the Membrane Interface
Vinita Lukose,† Marthe T. C. Walvoort,†§ Barbara Imperiali*†
† Departments of Chemistry and Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
§ Current affiliation: Stratingh Institute for Chemistry, University of Groningen, the Netherlands
ABSTRACT
Phosphoglycosyl transferases (PGTs) initiate the biosynthesis of both essential and virulence-associated bacterial glycoconjugates including lipopolysaccharide, peptidoglycan, and glycoproteins. PGTs catalyze the transfer of a phosphosugar moiety from a nucleoside diphosphate sugar to a polyprenol phosphate, to form a membrane-bound polyprenol
diphosphosugar product. PGTs are integral membrane proteins and can have between 1 and 11 predicted transmembrane domains. Despite this variation, common motifs have been identified in PGT families through bioinformatics and mutagenesis studies. PGTs represent important antibacterial targets due to their significant role in initiating the biosynthesis of key bacterial glycoconjugates. Considerable effort has gone into mechanistic and inhibition studies for this class of enzymes, both of which depend on reliable, high throughput assays for easy
quantification of activity. This review summarizes recent advances made in the characterization of this challenging but important class of enzymes.
Introduction - Common Themes in Glycan Synthesis at the Membrane Interface in All Domains of Life
Cell-surface glycans play critical roles in bacterial physiology and in establishing interactions with host cells (Varki, A. 2017). Examples of glycoconjugates found associated with bacterial cell surfaces include capsular polysaccharide (CPS), lipopolysaccharide (LPS), wall teichoic acid (WTA), peptidoglycan and glycoproteins (Tytgat, H.L. and Lebeer, S. 2014). Although the identities of the sugars and the general structures of these glycans are highly diverse, the assembly of these complex molecules shares common themes.
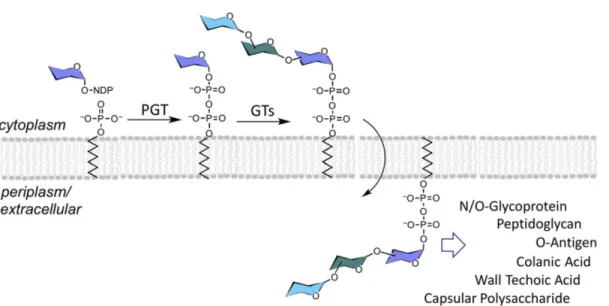
The biosynthesis of a variety of bacterial glycoconjugates, including glycolipids and glycoproteins, begins with the formation of a membrane-associated polyprenol diphosphosugar intermediate, which is catalyzed by a class of enzymes called phosphoglycosyl transferases (PGTs). These integral membrane proteins are also referred to as “priming” or “initiating” glycosyltransferases due to their roles in commencing glycoconjugate biosynthesis. The PGT reaction involves transfer of a C1′-phosphosugar from a nucleoside diphosphate sugar (NDP-sugar) donor to a polyprenol-phosphate (P) acceptor, forming a membrane-associated Pren-PP-sugar. In bacteria the products of PGT reactions are then further elaborated by glycosyltransferases (GTs), to produce complex Pren-PP-linked glycans as substrates for gycoconjugate assembly (Figure 1). In Gram-negative bacteria, the Pren-PP-linked glycan is ultimately flipped across the plasma membrane into the periplasm, where the glycan is transferred from the Pren-PP carrier to form glycoconjugates that may remain in the periplasm or become displayed at the cell surface. In Gram-positive bacteria, PGTs function on the cytoplasmic face of
the plasma membrane and the final glycoconjugate products are commonly found associated with the cell surface.
This review will focus on bacterial PGTs, however, it is important to note that PGTs are also found in eukaryotes. For example, a PGT catalyzes formation of Pren-PP-GlcNAc initiating the dolichol pathway towards N-linked protein glycosylation on the cytoplasmic face of the endoplasmic reticulum membrane (Burda, P. and Aebi, M. 1999). In eukaryotes the linear polyprenols are saturated at the a-isoprene unit and are referred to as “dolichols” (Hartley, M.D. and Imperiali, B. 2012). Interestingly, for selected archaea wherein glycoprotein biosynthesis pathways have been studied in biochemical detail, including Haloferax volcanii (Kaminski, L., Abu-Qarn, M., et al. 2010) and Methanococcus voltae (Larkin, A., Chang, M.M., et al. 2013) the biosynthesis of N-linked glycoproteins is not initiated with a PGT-catalyzed process. Instead, a GT catalyzes the transfer of a single carbohydrate from a NDP-linked sugar to a Pren-P to form an a-linked Pren-P-sugar.
Despite their importance in glycobiology, advances in structural and functional characterization of PGTs have proceeded at a relatively slow pace. This review describes the challenges associated with studying this class of integral membrane proteins, and highlights the progress made in the classification, isolation and characterization of bacterial PGTs since a comprehensive review in 2005 (Price, N.P. and Momany, F.A. 2005). A summary of the design and development of PGT inhibitors based on natural products is also presented.
An evolving classification of PGTs
Phosphoglycosyl transferases have been classified based on the structures of their nucleotide-activated sugar substrates, the predicted topologies of known PGTs, and based on whether the origin of the enzymes was prokaryotic or eukaryotic (Valvano, M.A. 2003). This
classification described two families of PGTs: polyisoprenol-phosphate hexose-1-phosphate transferases (PHPTs), which were observed to only occur in bacteria and polyisoprenol-phosphate N-acetylaminosugar-1-phosphate transferases (PNPTs), which were found in both prokaryotes and eukaryotes (Valvano, M.A. 2003). PHPT enzymes generate polyprenol-PP-hexose products, while PNPT enzymes produce polyprenol-PP-HexNAc products. This method of PGT classification was initially valuable for predicting the function of enzymes based on sequence similarity.
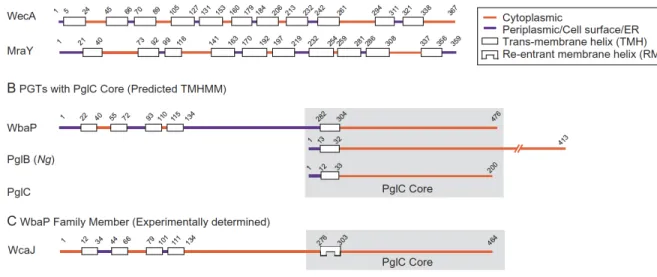
As PGTs with substrate preferences and predicted structures beyond the original definition continue to be discovered it is evident that the relationship between enzyme substrate and structure is more complex. Sequence homology and functional analyses reveal that the known PGTs are represented as two distinct superfamilies with divergent overall predicted topologies and mechanisms of action (see Fig. 2A and B). PGTs, including WecA and MraY, belong to a superfamily with a polytopic membrane architecture including 10-11 predicted transmembrane helices (TMHs) (Anderson, Eveland, Price 2000). Bacterial members of the WecA family transfer GlcNAc-1-phosphate from UDP-GlcNAc to a Pren-P acceptor, initiating the O-antigen and enterobacterial common antigen biosynthesis (Lehrer, J., Vigeant, K.A., et al. 2007). Eukaryotic members of the WecA family include the GlcNAc-1-phosphate transferases (GPTs) that initiate N-linked protein glycosylation at the beginning of the dolichol pathway by transferring GlcNAc-1-phosphate to dolichol phosphate (Burda, P. and Aebi, M. 1999, Lehrman, M.A. 1991). Members of the MraY family catalyze the transfer of 1-phospho-MurNAc-pentapeptide (L-Ala-γ-D -Glu-diaminopimelic acid/L-Lys-D-Ala-D-Ala, also called Park’s nucleotide) to undecaprenol phosphate, on the cytoplasmic face of the plasma membrane. The glycan component of the reaction product is then flipped across the membrane, and crosslinked with release of the Pren-PP to form peptidoglycan (Bouhss, A., Trunkfield, A.E., et al. 2008).
A second superfamily of PGTs with a distinct architecture based on primary structure is also prevalent (Hug, I. and Feldman, M.F. 2011, Lukose, V., Luo, L., et al. 2015, Saldias, M.S., Patel, K., et al. 2008). Key contributions defining this superfamily came from studies on a PGT designated as WbaP from Salmonella enterica, which initiates O-antigen biosynthesis in bacteria (Saldias, M.S., Patel, K., et al. 2008). WbaP is a polytopic membrane protein with 5 predicted TMHs, however, it has been shown that this PGT can be truncated, to remove four of the five N-terminal transmembrane helices, and still preserve both in vivo (Patel, K.B., Furlong, S.E., et al. 2010) and in vitro (Patel, K.B., Ciepichal, E., et al. 2012) activity. Therefore, the minimal structural and functional core comprises a single predicted membrane-associated domain (ca. 22 amino acids) and a small globular cytoplasmic domain (ca. 180 amino acids).
Two other PGT families share a functional core with homology to WbaP and are therefore members of the second superfamily. The two families are represented by the Campylobacter jejuni PglC (Szymanski, Glover), which catalyzes the first membrane-committed step in a bacterial N-linked protein glycosylation pathway, and the Neisseria gonorrhoeae PglB (Hartley 2011), which is a bifunctional enzyme including an N-terminal PGT domain, with the same functional core as the full-length PglC, and a C-terminal amino-sugar acetyl transferase domain. The N. gonorrhoeae PglB functions at the initiation of a bacterial O-linked protein glycosylation pathway. In both N- and O-protein glycosylation pathways, the assembled glycan is ultimately transferred en bloc from the Pren-PP-sugar donor to a protein acceptor.
Recent bioinformatics analysis on 15,000 bacterial sequences from these three related PGT families underscores the relationships within this superfamily and highlights conserved residues (Lukose et al. 2015). As illustrated in Fig. 2B, the three superfamily members share a common domain that is represented in the simplest family member PglC, but may incorporate additional N-
or C-terminal domains. It was also evident from this study and previous analyses of the WbaP family (Patel, K.B., Ciepichal, E., et al. 2012) that all members of this PGT superfamily include a highly-conserved proline residue in the predicted transmembrane helix that is part of the minimal PgT functional core. Furthermore, this residue is critical for function (Lukose, V., Luo, L., et al. 2015). The membrane disposition of this conserved feature of the PGT sequences was recently examined in detail for the E. coli WcaJ, which is a homolog of WbaP that is involved in the colonic acid exopolysaccharide biosynthesis (Furlong, S.E., Ford, A., et al. 2015). By applying a combination of LacZ/PhoA reporter fusions and unique cysteine labeling using the substituted-cysteine accessibility method (SCAM)(Nasie, I., Steiner-Mordoch, S., et al. 2013) evidence was provided to show that the proline-containing helix common to this superfamily of PGTs is not a trans-membrane helix (TMH), but rather a re-entrant membrane helix (RMH) (Fig. 2C). This result underscores the importance of experimental determination of the membrane topology, since none of the five membrane-prediction algorithms employed in the study correctly predicted the reentrant helix (Furlong, S.E., Ford, A., et al. 2015). Given the common functional domain shared by the three families, the topological feature observed in WcaJ is likely to be shared amongst the corresponding domains in the related PglB(Ng) and PglC families. Therefore, similar analyses should be performed to clearly define the topology in native proteins in vivo to confirm the classification and topologies of PGTs.
Given the evidence to date, predicted membrane topologies and common structural cores together with biochemical validation are the most reliable determinants by which PGTs can be classified into superfamilies with unique topological and functional attributes. The structural diversity observed among bacterial PGTs represents an example of convergent evolution, where integral membrane proteins with vastly different structures catalyze the same basic reaction. From
a biochemical perspective, this structural variation raises important questions about the similarities and differences between the catalytic and substrate-binding domains present in inter-helix loops of the polytopic PGT superfamily and those present in cytoplasmic and membrane associated domains of the PglC-like PGT superfamily. The diversity in membrane topologies also raises questions about the roles of the membrane-associated domains in binding and catalysis, especially with respect to protein-protein interactions in the membrane and binding to the membrane-associated Pren-P substrate.
Common Motifs in PGT Families
Bioinformatics analysis and mutagenesis studies have provided information about motifs conserved within and among different PGT families. In particular, sequence alignments revealed conserved signature sequences in loops between the transmembrane helices in the WecA and MraY families. The significance of these motifs was investigated by selectively mutating conserved residues and analyzing the activity of the resulting proteins. For example, both families have a conserved DDXXD motif, in which the adjacent aspartate residues are believed to coordinate the essential metal ion cofactor (Lloyd, A.J., Brandish, P.E., et al. 2004). A recent crystal structure of MraY supports this hypothesis (Chung, B.C., Zhao, J., et al. 2013). Both families also have conserved motifs with multiple histidine residues (HIHH in the WecA family, and MAPIHHHFEL in the MraY family), which are thought to play a role in recognition of the carbohydrate substrate, although the exact interactions between these motifs and the substrates have not been established (Amer, A.O. and Valvano, M.A. 2001, Anderson, M.S., Eveland, S.S., et al. 2000). WecA and related family members also feature a conserved VFMGD motif, which is believed to contribute to the active site of the enzymes and also be involved in product release
(Furlong, S.E. and Valvano, M.A. 2012). Similar studies with the C-terminal domain of proteins in the WbaP family have also revealed residues important for catalytic activity (Patel, K.B., Furlong, S.E., et al. 2010). Recent bioinformatics studies and functional analysis (Lukose, V., Luo, L., et al. 2015) of members of the PglC, PglB(Ng) and WbaP families have also provided insight into residues required for the catalytic activity of these PGTs including a highly conserved Asp-Glu (DE) dyad. Importantly, the motifs identified in these studies are distinct from those found in the polytopic PGT family.
Sugar Substrate Specificity of PGTs
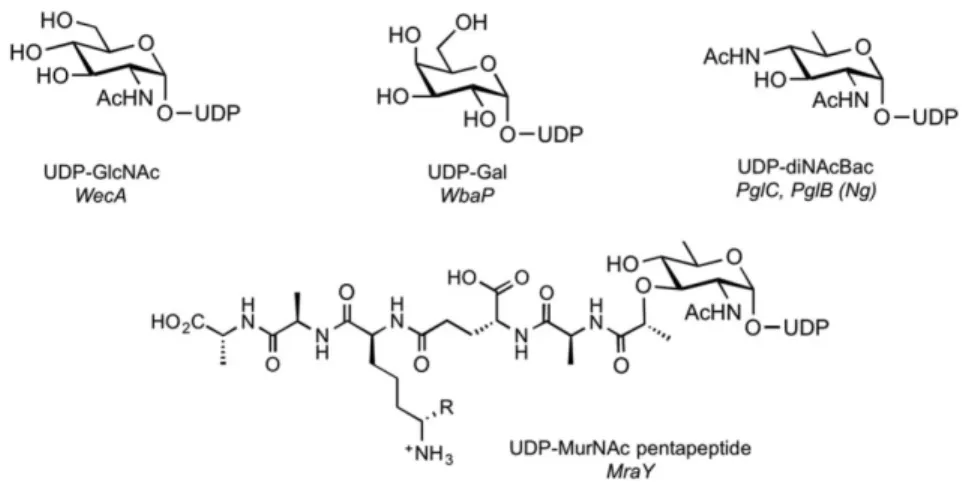
Protein sequence and topology can be useful to classify PGTs, but does not provide conclusive evidence of substrate specificity. Enzymes within the same family can have different sugar substrates, several PGTs have been reported that act on different UDP-sugars, some of which are highly modified substrates and synthesized only by bacteria (Figure 3) (Szymanski, C.M. and Wren, B.W. 2005). These UDP-sugar substrates may not be commercially available, which hampers also PGT characterization. For example, UDP-diNAcBac, the substrate for PglC, is biosynthesized in a three-step sequence (redox-dependent dehydration, amino transfer, and amino-acetylation) from UDP-GlcNAc. UDP-diNAcBac can be synthesized chemically (Weerapana, E., Glover, K.J., et al. 2005), however, chemoenzymatic synthetic strategies are more tractable and have allowed the characterization of enzymes that use this diNAcBac derivatives (Hartley, M.D., Morrison, M.J., et al. 2011, Morrison, M.J. and Imperiali, B. 2013, Olivier, N.B., Chen, M.M., et al. 2006). The MurNAc-pentapeptide substrate of MraY is a UDP-GlcNAc moiety decorated with a lactyl-pentapeptide chain at the C-3’ position, and also represents a complex donor substrate that
is not readily available commercially. In this case, a chemoenzymatic approach has provided good access to UDP-MurNAc-pentapepide (Kurosu, M., Mahapatra, S., et al. 2007).
Bioinformatics analysis can identify PGT enzymes based on sequence homology to characterized enzymes, however, the specificities of these enzymes cannot yet be predicted based on sequence alone, and the substrates of many of these PGTs have yet to be identified by biochemical techniques. If the sugar substrates of these newly identified PGTs are highly modified and not commercially available, they must be synthesized, which can add to the challenge of characterizing a PGT of interest and conclusively determining its substrate specificity.
Polyprenol specificity of PGTs
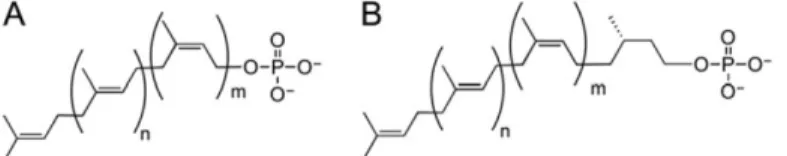
The products of PGT reactions are precursors for the biosynthesis of complex glycoconjugates containing the Pren-PP moiety, which serves both as a membrane anchor and an activating group in the ultimate transfer to acceptor substrates. The long-chain linear polyprenols that feature in glycan assembly pathways are classified based on the saturation state of the α-isoprene unit. Undecaprenol and related polyprenols of different lengths comprise a linear polymer of unsaturated isoprene units. In contrast, dolichols feature a saturated isoprene unit in the position of the polymer (Figure 4). Bacteria typically exploit polyprenols with unsaturated α-isoprene units, while archaea and eukarya use dolichols (Hartley, M.D. and Imperiali, B. 2012, Jones, M.B., Rosenberg, J.N., et al. 2009).
The availability of polyprenol phosphates with the desired chain length and cis/trans geometry can be a limiting factor in determining substrate specificities of PGTs. The isolation of pure polyprenol phosphates from the native organism is often challenging and low-yielding (Adair, W.L. and Keller, R.K. 1985). Furthermore, although some linear polyprenols are commercially
available, the high cost limits the scope of the biochemical and biophysical studies that are required to fully characterize enzyme activity. Interestingly, certain plants contain high levels of linear unsaturated polyprenols of varying length, and have proven to be a valuable source of isoprenoids for biochemical studies (Jankowski, W., Swiezewska, E., et al. 1994). Once isolated, the polyprenols can be regioselectively hydrogenated (Imperiali, B. and Zimmerman, J.W. 1988) to afford dolichols and/or phosphorylated using chemical or chemoenzymatic methods to obtain substrates that can be used to biochemically characterize PGTs from all domains of life (Hartley, M.D. and Imperiali, B. 2012).
The specificity of PGTs for their polyprenol phosphate substrates has been tested by varying the carbon chain length and saturation of the α-isoprene unit of these substrates. WecA from Thermatoga maritima is active with polyprenol phosphates with 10 and 35 carbons and shows reduced activity when tested with polyprenols longer than the native C55 substrate
(Al-Dabbagh, B., Mengin-Lecreulx, D., et al. 2008, Al-(Al-Dabbagh, B., Olatunji, S., et al. 2016). Interestingly, WecA appears to achieve 60% of the activity compared to undecaprenol-phosphate when tested with dolichol (C55) phosphate (Al-Dabbagh, B., Mengin-Lecreulx, D., et al. 2008).
MraY shows even broader substrate specificity, forming products with dolichol (C40) phosphate,
polyprenol (C35) phosphates, and even the shorter water-soluble prenyl substrates with as few as
10 carbons; however, the shorter prenol-diphosphate-linked substrates show much poorer turnover (Breukink et al. 2003). Analysis of the substrate specificity of the C. jejuni PglC reveals that the double-bond geometry and α-unsaturation at the alcohol terminus of the polyisoprenols are more important for substrate recognition than the number of isoprene units (Chen, M.M., Weerapana, E., et al. 2007). Studies with the PglC-like domain of WbaP demonstrate that this domain appears to be very selective for undecaprenol phosphate, and is unable to tolerate polyprenol phosphate
substrates with fewer than 50 carbons (10 isoprene units) (Patel, K.B., Ciepichal, E., et al. 2012). Both PglC and the C-terminal domain of WbaP show minimal activity with longer dolichol phosphates (Chen, M.M., Weerapana, E., et al. 2007, Patel, K.B., Ciepichal, E., et al. 2012).
Overexpression and Purification of PGTs
Phosphoglycosyl transferases represent potential antibiotic targets due to the significance of their glycoconjugate products, particularly in pathogenic bacteria (Tytgat, H.L. and Lebeer, S. 2014). However, PGTs are integral membrane proteins, and are particularly recalcitrant to efficient heterologous overexpression. Obtaining pure, stable protein remains a significant challenge in the characterization of these membrane-bound enzymes (Newby, Z.E., O'Connell, J.D., 3rd, et al. 2009). First, overexpression of membrane proteins in host organisms such as E. coli may result in the accumulation of large amounts of a non-native protein in cell membranes, which can be cytotoxic (Wagner, S., Baars, L., et al. 2007). Additionally, since integral membrane proteins contain many hydrophobic domains, they are extremely prone to aggregation over the course of protein overexpression and purification. The most destabilizing purification step is the extraction of proteins from native membranes and solubilization into detergent micelles, which always demands considerable optimization. Additionally, the best detergent for extraction may not necessarily be the detergent which affords the most active form of the protein; thus, optimization of detergents at multiple steps is required (Al-Dabbagh, B., Mengin-Lecreulx, D., et al. 2008). Cell-free protein expression shows promise as an alternative method for large-scale membrane protein production, as it avoids the challenges associated with cell toxicity and resolubilization into detergent micelles. This technique has been used to produce quantities of MraY (Henrich, E., Ma, Y., et al. 2016, Ma, Y., Munch, D., et al. 2011).
Due to the complications encountered in obtaining pure, stable protein, initial studies to characterize PGTs were performed with crude membrane preparations or detergent-solubilized membrane fractions. Although these studies provided insight into the PGTs enzymes, precise quantitative data often could not be obtained because the exact quantity and purity of the proteins was unknown. Challenges associated with obtaining stable protein have impeded crystallographic studies with PGTs, and, to date MraY is the only PGT that has been structurally characterized (Chung, B.C., Zhao, J., et al. 2013).
PGT activity assays
The PGT reaction involves the transfer of a phosphosugar from an aqueous-soluble nucleotide diphosphate sugar (NDP-sugar) substrate to a membrane-bound Pren-P acceptor. Assays have traditionally exploited this difference in solubility between the starting materials and products by analyzing the reaction using extraction-based and related separation techniques. Due to the potential of these enzymes as therapeutic targets, a great deal of effort has gone into developing high throughput assays to apply to inhibitor screening.
The transfer of NDP-sugars, which are radiolabeled in the carbohydrate moiety to unlabeled Pren-P acceptors can be quantified using liquid-liquid extraction or by thin layer and HPLC chromatography (Figure 5 B and C) (Behrens, N.H. and Tabora, E. 1978, Bouhss, A., Crouvoisier, M., et al. 2004, Patel, K.B., Ciepichal, E., et al. 2012, Patel, K.B. and Valvano, M.A. 2013, Stachyra, T., Dini, C., et al. 2004)The extraction approach has also been further developed into a high-throughput solid-liquid extraction-based assay using hydrophobic polymer beads. The beads are added to quenched reactions in a microwell plate format to selectively bind to the Pren-PP-linked substrate. After filtering and washing steps the bound product is quantified using
scintillation counting. This method can be applied in coupled assays and has been tested successfully with MraY, WecA, and a coupled MraY/MurG system (Hyland, S.A. and Anderson, M.S. 2003). However, the approach still relies on the availability of radiolabeled NDP-sugar substrates and thus would be limited with respect to commercially available substrates.
The majority of PGT assay development has focused on the reaction catalyzed by MraY, due to its importance as an antibiotic drug target. Initial assay strategies exploited fluorescently- labeled variants of Park’s nucleotide in which the e-amino group of the lysine residue was modified with a fluorophore. This approach has been most successful for MraY due to the modularity and relative promiscuity of the pentapeptide substrate structure, which allows for fluorophore modification of the substrate without affecting activity or substrate binding. When a dansyl group is used as solvatochromic fluorophore in the MraY substrate, a change in the emission wavelength is observed upon transfer of the fluorophore-conjugated glycopeptide moiety to a hydrophobic environment, providing a signal with which enzyme activity can be tracked (Weppner, W.A. and Neuhaus, F.C. 1977, Wohnig, S., Anatol, P.S.A., et al. 2016).Fluorescein isothiocyanate (FITC) has also been used successfully to label sugars for activity and inhibition assays, with the analysis performed after liquid-liquid extraction or chromatography (Mitachi, K., Siricilla, S., et al. 2016). Modifications to this assay include shorter polyprenols to increase water solubility. Prenols with as few as three isoprene units were found to be suitable substrate analogs for MraY and afford robust enzyme turnover (Mitachi, K., Siricilla, S., et al. 2015). A FRET-based assay using a BODIPY label as a fluorescence donor in the MurNAc-pentapeptide substrate and a fluorophore-labeled phospholipid as the acceptor has also been developed for high-throughput screening of MraY inhibitors (Shapiro, A.B., Jahic, H., et al. 2012).
Recently, the Imperiali group has adapted the UMP/CMP-Glo assay, which is a luminescence-based assay developed by PROMEGA that detects UMP (or CMP) release (Figure 5 A). Advantageously, this assay does not require substrate labeling, and can therefore be used to investigate PGT enzymes with diverse and unique carbohydrate specificities. The detection method is compatible with high throughput screening formats using 96- and 384-well plates. UMP/CMP-Glo has been used successfully to characterize the activities of PGTs with varying structures and UDP-sugar substrates, demonstrating the versatility of assay (Das, D., Walvoort, M.T., et al. 2016).
Mechanistic investigation of the PGT reaction
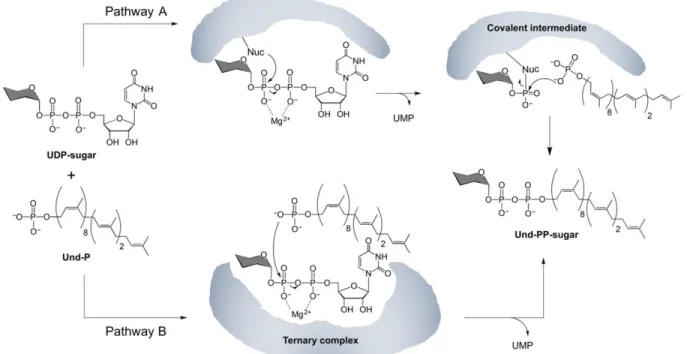
The PGT reaction is isoenergetic, involving the cleavage of a diphosphate bond in the UDP-sugar substrate, and formation of a diphosphate bond in the Pren-PP-linked product. The reaction formally involves nucleophilic attack of Pren-P on the a-linked phosphate of an NDP-sugar resulting in transfer of a NDP-sugar-1-phosphate to form a Pren-PP-linked carbohydrate. Two different mechanisms can be proposed for this bisubstrate reaction – a Bi Bi Ping Pong
mechanism with the formation of a covalent intermediate, or a Bi Bi random or ordered mechanism with intermediate formation of a ternary complex (Figure 6). Importantly, both mechanisms result in retention of stereochemistry at the anomeric center of the sugar since the reaction occurs at the adjacent phosphate center. Kinetic analysis of the eukaryotic GlcNAc-1-P transferase (GPT), a polytopic PGT in the WecA family, suggests a Bi Bi random mechanism, with formation of a ternary complex for this enzyme(Keller, R.K., Boon, D.Y., et al. 1979). In contrast, early studies with a crude solubilized membrane fraction of MraY initially suggested that transfer of the MurNAc-pentapeptide occurred via formation of an acyl phosphate
intermediate involving an aspartic acid residue in MraY (Lloyd, A.J., Brandish, P.E., et al. 2004). However, these studies were performed with crude membrane protein fractions, as purification strategies for these polytopic membrane proteins had not been established at the time. More recently, both WecA and MraY have been purified to homogeneity and now, studies with purified protein samples strongly support that both enzymes proceed through the formation of a ternary complex mechanism, without formation of a covalent intermediate (Al-Dabbagh, B., Olatunji, S., et al. 2016), suggesting that this may be a common feature of the polytopic PGT superfamily. In contrast, a recent investigation of the reaction mechanism of PglC from C. concisus supports a ping-pong mechanism involving a covalent intermediate (Das, D., Kuzmic, P., et al. 2017). In this case evidence is provided to support a mechanism, which proceeds through a two-step mechanism involving first nucleophilic attack by an active site aspartic acid residue on the a-linked phosphate of the NDP-sugar substrate to form a covalent phosphosugar intermediate. The key aspartic acid is part of the Asp-Glu dyad that is highly conserved in the 15,000-sequence analysis of the superfamily of PGTs that share the PglC-like structural core (Lukose, V., Luo, L., et al. 2015). In the second step of the mechanism the covalent enzyme intermediate is subject to attack by Pren-P to complete the PGT reaction.
PGT inhibitors based on nucleoside antibiotics
Inspired by the centrality and essentiality of PGTs in the assembly of bacterial glycoconjugates, these enzymes are important targets for inhibitor design with the goal of developing novel antimicrobial and antivirulence agents. Currently, there is only limited structural information on PGTs, due to their challenging topologies (vide supra) and poor expression in heterologous systems. Therefore, efforts towards the development of PGT inhibitors have centered
on using the structures of nucleoside antibiotics, which have emerged from natural product screening campaigns, for inspiration (Elshahawi, S.I., Shaaban, K.A., et al. 2015). Many of these structurally complex compounds are secondary metabolites produced by Streptomyces species, which display a wide variety of biological activities, including antibacterial, antifungal, antiviral and antitumor activity (Niu, G. and Tan, H. 2015). For the purpose of this review, we will focus on a selection of well-known natural products with an emphasis on their structural differences and enzyme target selectivities. The potential of nucleoside antibiotics to serve as inspiration for novel antimicrobials, and synthetic efforts towards derivatives thereof have recently been reviewed (Ichikawa, S., Yamaguchi, M., et al. 2015, Serpi, M., Ferrari, V., et al. 2016, Wiegmann, D., Koppermann, S., et al. 2016).
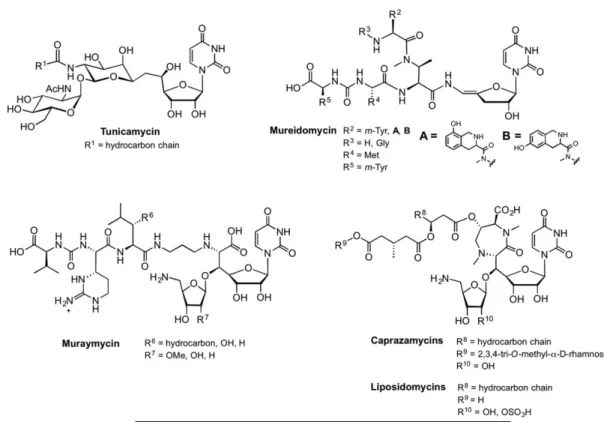
Tunicamycin was the one of the first nucleoside antibiotics to be discovered (Figure 7) (Takatsuki, A., Arima, K., et al. 1971), and since its discovery in the 1970s it has been used in numerous biological studies due to its potent inhibition of protein glycosylation in eukaryotic cells. The structure of tunicamycin reveals the 11-carbon dialdose sugar, tunicamine, which includes a uridine moiety, connected via a carbon-carbon linkage to a central pseudo-GalNAc moiety. Tunicamine is in turn connected to GlcNAc through a 1,1’-linkage, and N-acylated with a long-chain fatty acid. Tunicamycin has high affinity for the GlcNAc-1-phosphate transferase (GPT), also known as Alg7, which is the eukaryotic PGT that initiates the dolichol pathway (Heifetz, A., Keenan, R.W., et al. 1979). Specifically, tunicamycin arrests N-linked glycoprotein biosynthesis in the endoplasmic reticulum by inhibiting formation of dolichol diphospho-GlcNAc (Dol-PP-GlcNAc), and therefore is highly toxic to humans (Scocca, J.R. and Krag, S.S. 1997, Zeng, Y. and Elbein, A.D. 1995). Tunicamycin also potently inhibits WecA (Al-Dabbagh, B., Mengin-Lecreulx, D., et al. 2008), which is the bacterial PGT that initiates the biosynthesis of enterobacterial
common antigen and O-antigen lipopolysaccharide (LPS) (Meier-Dieter, U., Barr, K., et al. 1992). In contrast to the potent inhibition of Alg7 and WecA, tunicamycin displays only moderate inhibition of MraY, which transfers 1-phospho-MurNAc-pentapeptide (Inukai, M., Isono, F., et al. 1993). The tunicamycins are structurally related to the streptovirudins and corynetoxins, which differ in the fatty acid composition (R1 in Figure 7).
The mureidomycins were characterized in 1989, and contain a unique enamide moiety attached to a 3’-deoxyribose unit (Figure 7) (Isono, F., Inukai, M., et al. 1989). The mureidomycins share structural features with the pacidamycins, sansanmycins, and napsamycins, which display alternative side chains at the R2 to R5 positions, and none has a fatty acid moiety (Ichikawa, S.,
Yamaguchi, M., et al. 2015). Mureidomycin potently inhibits MraY, and a putative mode of binding has been described based on competition studies with both carbohydrate and lipid substrates (vide infra). Interestingly, mureidomycin has low affinity for the eukaryotic GPT, suggesting target specificity for bacterial PGTs that could be exploited in the quest for new antibiotics. However, the enamide linkage has limited general applications of the mureido scaffold in medicinal chemistry approaches.
Of all nucleoside antibiotics, the muraymycins have gained the most attention from the scientific community because of their antibiotic potential through inhibition of MraY. First isolated in 2002, muraymycins are characterized by a glycyl-uridine motif, an aminoribose, a urea moiety and a fatty acid (Figure 7) (McDonald, L.A., Barbieri, L.R., et al. 2002). The active site of MraY is located in the cytoplasm, and the fatty acids of the muraymycins are thought to facilitate uptake to confer antimicrobial activity.
Finally, the caprazamycins and liposidomycins share a unique diazepanone ring, adjacent to the aminoribose unit and a fatty acid (Figure 7) (Igarashi, M., Nakagawa, N., et al. 2003,
Igarashi, M., Takahashi, Y., et al. 2005, Isono, K., Uramoto, M., et al. 1985). These natural products are known to inhibit MraY activity, and the caprazamycins have also shown inhibition of WecA and TagO, which is a GlcNAc-1-phosphate transferase involved in teichoic acid biosynthesis (Ishizaki, Y., Hayashi, C., et al. 2013).
Together, the nucleoside antibiotics form an interesting class of complex structures to inspire the next generation of PGT inhibitors. However, care has to be taken in interpreting the results from PGT inhibition studies. The reported antimicrobial potencies of the nucleoside antibiotics against different target PGTs are difficult to compare, as the assays on membrane-bound PGTs are not standardized. Data on whole cells, solubilized membrane fractions, and purified proteins cannot be compared, as nonspecific interactions as well as variable cellular uptake may greatly influence potency. Moreover, while IC50 values are commonly cited, these can
only be compared as relative values within a set of identical assays as they are condition-dependent parameters that are influenced by substrate concentrations and enzyme preparations. In addition to challenges with expression and purification, as presented previously PGT activity is difficult to assess, since the assays are largely labor-intensive, and do not allow straightforward inhibitor screening. Ultimately, perhaps the most reliable comparison is the minimum inhibitory concentrations (MIC values) against the same bacterial strains, but that assumes prior validation of the mode of action and comparable bioavailability.
PGT inhibitors: Mechanism of action
Nucleoside antibiotics are potent inhibitors of PGTs, and a great deal of effort has gone into exploring the mechanisms of inhibition by these diverse compounds. In particular, tunicamycin is proposed to be a bisubstrate analogue inhibitor of GlcNAc-1-phosphate transferases, because it includes features from both the sugar-nucleotide and polyprenol substrates
(Keller, R.K., Boon, D.Y., et al. 1979). However, detailed studies have revealed that the binding mechanism is less straightforward. In one study, Brandish et al investigated the mode of inhibition of MraY by tunicamycin, mureidomycin and liposidomycin, using substrate competition studies (Brandish, P.E., Kimura, K.I., et al. 1996). In these analyses, it was revealed that, although these three classes of compounds shared some common structural features, the mechanism of inhibition of these compounds varied greatly. For example, with MraY, tunicamycin was found to be competitive for the UDP-MurNAc pentapeptide substrate, but not for the Pren-P substrate, even though it has a fatty acyl component that could potentially bind in a polyprenol binding site. Interestingly, mureidomycin was found to be competitive for both substrates, and liposidomycin was only competitive for the Pren-P substrate (Brandish, P.E., Kimura, K.I., et al. 1996). Using molecular modeling of the lowest energy conformations, the different inhibitors have been superimposed with UDP-MurNAc, starting with the common uridine motif (Dini, C., Collette, P., et al. 2000). The studies suggested that the GlcNAc moiety of tunicamycin might superimpose with the MurNAc moiety. Also, the diphosphate functionality in UDP-MurNAc may be mimicked by the pseudo-GalNAc moiety in tunicamycin, and similarly by the aminoribose unit in liposidomycin. In the case of tunicamycin, NMR studies corroborated the suggestion that the pseudo-GalNAc moiety serves as a diphosphate mimic, because it is ideally positioned to coordinate to the divalent metal ion in the PGT active site (Xu, L., Appell, M., et al. 2004). A similar binding mechanism was proposed for mureidomycin, in which a protonated amine binds the DDxxD motif, instead of the divalent metal ion (Howard, N.I. and Bugg, T.D. 2003). Major advances in the field include the recently published structures of MraY co-crystallized with muraymycin D2 (Chung, B.C., Mashalidis, E.H., et al. 2016) and tunicamycin (Hakulinen, J.K., Hering, J., et al. 2017). In comparison to the MraY apo structure published previously (Chung,
B.C., Zhao, J., et al. 2013), the inhibitor-bound structures revealed that MraY undergoes a large, but similar conformational change upon binding of either inhibitor. In contrast to tunicamycin, muraymycin seems to have little interaction with the catalytically important amino acid residues. Docking studies based on the co-crystal structures suggested that the uracil and ribose moieties of UDP-GlcNAc overlap well with these functionalities in tunicamycin (Hakulinen, J.K., Hering, J., et al. 2017), as proposed previously (Dini, C., Collette, P., et al. 2000).
With regard to GPT, which has a homolog in Saccharomyces cerevisiae designated as Alg7, there are contradicting reports on the mechanism of inhibition by tunicamycin. In one report, tunicamycin was found to be competitive with the UDP-GlcNAc substrate (Keller, R.K., Boon, D.Y., et al. 1979), while another report suggested that tunicamycin is noncompetitive with respect to the concentrations of either substrate, suggesting an alternative mode of inhibition (Heifetz, A., Keenan, R.W., et al. 1979). Sequence alignment of human GPT with crystallized MraY revealed distinct amino acid differences in the regions involved in substrate/inhibitor binding (Hakulinen, J.K., Hering, J., et al. 2017). This reinforces the possibility that a different mechanism of inhibition of GPT may be relevant, and creates opportunities to selectively target MraY over GPT. The contradicting biochemical results illustrate the difficulties associated with comparing inhibition data; the two GPT studies use different sources of solubilized GPT-containing cell fractions and different activity assays, which may have led to different conclusions. Interestingly, the different binding modes of tunicamycin and muraymycin to MraY illustrate the challenge in understanding the mode of PGT inhibition by the nucleoside antibiotics.
Efforts to utilize nucleoside antibiotics as lead structures and inspiration for inhibitor development have mainly focused on MraY as the target for two reasons (Winn, M., Goss, R.J., et al. 2010); MraY has an essential role in bacterial survival, and a fluorescently-labeled versions of Park’s nucleotide are available, allowing quantification of enzyme activity using fluorescence-based approaches, which are not readily applicable to PGTs that act on simple NDP-sugar substrates.
From the previous section it is clear that it is experimentally quite challenging to decipher the exact mode of inhibition of the nucleoside antibiotics. Chemical synthesis has played a large role in understanding the binding modes of these compounds by generating derivatives to test in inhibition experiments. Because of the complex chemical structures of all classes of natural product-based nucleoside antibiotics, only few reports describe efforts towards the total synthesis of nucleoside antibiotics, including muraymycin (Mitachi, K., Aleiwi, B.A., et al. 2016, Tanino, T., Ichikawa, S., et al. 2010), tunicamycin (Li, J. and Yu, B. 2015, Myers, A.G., Gin, D.Y., et al. 1993a, Myers, A.G., Gin, D.Y., et al. 1993b, Myers, A.G., Gin, D.Y., et al. 1991, Suami, T., Sasai, H., et al. 1983, Suami, T., Sasai, H., et al. 1984), and pacidamycin (Okamoto, K., Sakagami, M., et al. 2012b). Rather, chemists have started with the minimal scaffolds needed to obtain binding, and have elaborated on these using various methods. The majority of efforts have focused on derivatives of muraymycin, and some synthetic strategies will be highlighted here.
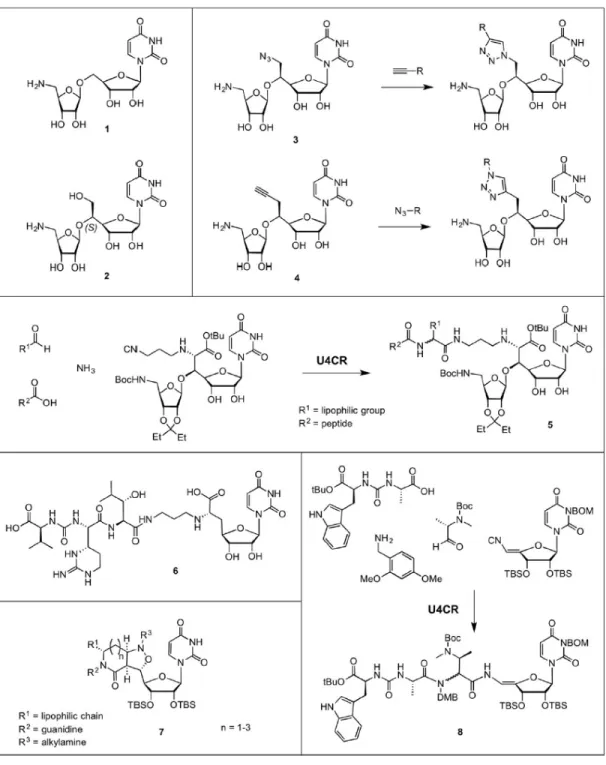
Starting from muraymycin, the minimal scaffold needed for inhibition was found to be the aminoribosyl-O-uridine scaffold 1 (Dini, C., Collette, P., et al. 2000), where an additional hydroxymethyl group with the (S)-configuration increased inhibitory potency (2, Figure 8). The group of Gravier-Pelletier reported a diversity-oriented synthesis method to easily obtain various analogues of scaffold 2, in which the hydroxymethyl moiety was replaced by an azide or alkyne
(3 and 4, Figure 8) (Fer, M.J., Bouhss, A., et al. 2015, Fer, M.J., Olatunji, S., et al. 2013). A subsequent copper(I)-catalyzed cycloaddition reaction with diverse alkynes or azides (CuAAC), respectively, allowed for screening of various lipophilic moieties. Inhibitory activities (IC50) were
found to be in the low micromolar range, and seemed to be competitive for the nucleotide substrate. Matsuda and co-workers developed an activity-driven approach to increase the antibacterial activity of muraymycin D2, which was found to be highly potent against purified MraY in vitro (IC50 ~ 0.01 µM), but not in whole cell assays (Tanino, T., Ichikawa, S., et al. 2010, Tanino, T.,
Yamaguchi, M., et al. 2014). Using an Ugi four-component reaction (U4CR), muraymycin D2 derivatives (5, Figure 8) were assembled that displayed various lipophilic side chains (R1) and
peptidic components (R2). While the inhibitory activity with purified MraY decreased by 10-fold,
the antibacterial activity, especially for Gram-negative bacteria, was significantly increased in different bacterial strains by the introduction of a lipophilic moiety (Takeoka, Y., Tanino, T., et al. 2014). Ducho and co-workers revealed that the amino-ribose moiety is not necessary for antibacterial activity, and developed an elegant synthesis towards a simplified muraymycin-based scaffold (6 in Figure 8) that is a promising lead for further development (Spork, A.P., Buschleb, M., et al. 2014). Ichikawa and co-workers have developed a lactam-fused isoxazolidine scaffold as a simplified structural analogue of muraymycin and caprazamycin (7, Figure 8) (Tanino, T., Yamaguchi, M., et al. 2014). When connected to a uridine moiety and a lipophilic side chain, comparable MIC values to the parent compounds were obtained.
The Ugi four-component reaction was also successfully applied to the pacidamycin scaffold (Okamoto, K., Sakagami, M., et al. 2012a), which is a structural analogue of mureidomycin. Using cleverly designed building blocks, pacidamycin D (8, Figure 8) and an arsenal of derivatives were generated, which provided detailed information on the
structure-activity relationship of MraY inhibition, and Pseudomonas aeruginosa MIC values. In an alternative “mutasynthesis” (Weissman, K.J. 2007) approach Goss and co-workers have reported a biosynthetic approach to pacidamycin derivatives by hijacking the natural product synthesis machinery in Streptomyces coeruleorubidus (Gruschow, S., Rackham, E.J., et al. 2009, Ragab, A.E., Gruschow, S., et al. 2010). When S. coeruleorubidus cultures were supplemented with tryptophan or tyrosine analogues, the amino acids were incorporated into newly synthesized pacidamycins. Therefore, this strategy provides an alternative platform for tailoring new pacidamycin analogues.
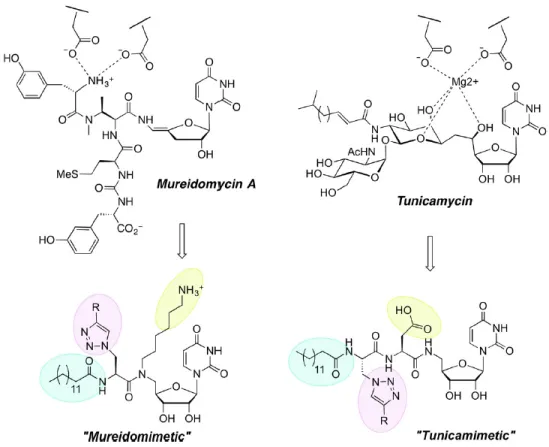
Recent research from the Imperiali group revealed that the combination of specific chemical moieties together yielded inhibitors with micromolar IC50 values (Walvoort, M.T.,
Lukose, V., et al. 2016) (Walvoort, Lukose, Imperiali 2016). The inhibitors were inspired by the structures and postulated binding modes of mureidomycin A (Gentle, C., Harrison, S., et al. 1999) (Gentle et al. 1999) and tunicamycin (Figure 9) (Price, N.P. and Momany, F.A. 2005), and were designed in such a way that they displayed 1) a uridine moiety, 2) a primary amine or carboxylate, to substitute or coordinate the metal in the active site, 3) a side chain (R) to occupy the carbohydrate-binding site, and 4) an alkyl chain to occupy the prenyl moiety in the acceptor substrate. The resulting scaffolds are highly modular and can be easily be differentiated to investigate structure-activity relationships and target selectivity.
Taken together, all of these examples illustrate the power of organic chemistry in understanding the features of the scaffolds that are important for the generation of PGT inhibitors.
PGTs are an important class of enzymes, which catalyze the first membrane-committed step in glycoconjugate biosynthetic processes in both prokaryotes and eukaryotes. Despite their significance and potential as therapeutic targets, the PGT class of enzymes has been underexplored, largely due to difficulties in expressing, purifying and assaying these integral membrane proteins. Nonetheless, the advancement of technologies such as Nanodiscs (Bayburt, T.H. and Sligar, S.G. 2010) and lipid super-structures (Barauskas, J., Johnsson, M., et al. 2005)(Barauskas, Johnsson, Tiberg 2005) as alternative platforms for stabilizing membrane proteins in a more native-like environment provide promising opportunities for the characterization of PGTs.
Bioinformatics analysis and mutational studies have provided insight into conserved residues found in two PGT superfamilies (Amer, A.O. and Valvano, M.A. 2001, Lukose, V., Luo, L., et al. 2015). Furthermore, advances in genome sequencing have dramatically increased the pool of data available for such types of analyses, and have demonstrated that the PGTs discussed in this review are representative of thousands of homologs in other bacterial glycoconjugate biosynthesis pathways. Additionally, the successful structure determination of MraY, both with and without bound inhibitors bound, has tremendously increased our understanding of the functionally-significant residues in the polytopic PGT superfamily. Therefore, inhibitor development may now be guided by structural information, which will accelerate the process of finding novel classes of antimicrobials. Additionally, the progress made in identifying members of a second major PGT superfamily has paved the way for our understanding of PGTs that have yet to be discovered. There is no doubt that future structural and mechanistic studies will also form an important foundation for inhibitor development targeted at the broad emerging superfamily that includes the common PglC-like core.
Finally, PGTs offer promising opportunities in the field of glycoengineering. Bacterial genes encoding enzymes responsible for the synthesis and processing of Pren-PP-linked glycans are generally clustered in operons in bacterial genomes. Using bioinformatics analysis, these gene clusters can be identified (Kumar, M. and Balaji, P.V. 2011) and expressed heterologously in tractable bacterial expression systems for large-scale biosynthesis of complex glycoconjugates. Together, a detailed structural and functional characterization of PGTs and related glycosyltransferases in pathways from both symbiotic and pathogenic bacteria could provide both novel antimicrobial targets and diverse sets of glycoconjugates for a variety of biotechnological applications.
Acknowledgements
The authors thank the NIH (NIH GM-039334) for support of their research on PGTs and bacterial protein glycosylation.
Figures and figure captions
Figure 1. Phosphoglycosyl transferases (PGTs) initiate the biosynthesis of glycoconjugates at the
surface of cellular membranes by forming polyprenol-PP-linked sugars that are elaborated by glycosyltransferases (GTs). During biosynthesis the polyprenol-PP-linked glycans may be actively flipped across the membrane to serve as substrates for transfer to acceptor molecules, such as lipids or proteins. The complex glycoconjugates from these pathways participate in a variety of important biological processes.
Figure 2. Predicted and experimental topologies of PGT superfamilies. Residue numbers are
indicated above the representation of each enzyme. (A) Predicted topologies of the polytopic PGT superfamily using the TMHMM prediction tool. (B) Predicted topologies of the PGTs that share a common PglC-like domain (highlighted in grey) using the TMHMM prediction tool. (C) Experimentally determined topology of WcaJ (a WbpA homolog).
Figure 3. Structures of UDP-sugar substrates of PGTs. The PGTs that use each substrate depicted
Figure 4. Structures of polyprenol phosphate substrates of PGTs. (A) Fully unsaturated polyprenol
phosphate and (B) Dolichol phosphate. The length of the polyprenol and the number of isoprene units varies. The ‘m’ denotes the number of cis isoprene units and ‘n’ is the number of trans isoprene units. The value of ‘m’ ranges from 12 - 37 in eukaryotes, and the value of ‘n’ ranges between 2-3. For example, bacterial undecaprenol double bonds are m = 8 and n = 2 and plant-derived undecaprenols are m = 7 and n = 3.
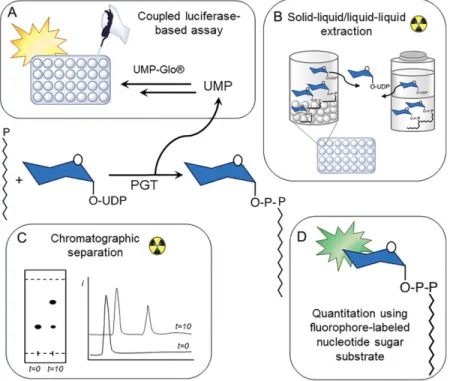
Figure 5. Summary of assays used to study PGT activity. A general PGT reaction is shown in the
center. (A) The UMP by-product of the reaction is used in a coupled luciferase-based assay. (B) Substrates are separated from products using solid/liquid or liquid/liquid extractions. (C) Substrates are separated from products using chromatography. (D) The sugar substrate is modified with a fluorophore that produces a signal upon transfer to the polyprenol-phosphate carrier.
Figure 6. Potential phosphoglycosyl transferase reaction mechanisms. (A) Ping Pong mechanism
with formation of a covalent enzyme-phospho-sugar intermediate. (B) Bi Bi random (or sequential) mechanism with the intermediate formation of a ternary complex.
Figure 9. Bottom-up approach to inhibitor scaffold development, based on the postulated binding
References
Adair WL, Keller RK. 1985. Isolation and assay of dolichol and dolichyl phosphate. Methods Enzymol, 111:201-215.
Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A. 2008. Purification and characterization of the bacterial UDP-GlcNAc:undecaprenyl-phosphate GlcNAc-1-phosphate transferase WecA. J Bacteriol, 190:7141-7146.
Al-Dabbagh B, Olatunji S, Crouvoisier M, El Ghachi M, Blanot D, Mengin-Lecreulx D, Bouhss A. 2016. Catalytic mechanism of MraY and WecA, two paralogues of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily. Biochimie, 127:249-257.
Amer AO, Valvano MA. 2001. Conserved amino acid residues found in a predicted cytosolic domain of the lipopolysaccharide biosynthetic protein WecA are implicated in the recognition of UDP-N-acetylglucosamine. Microbiology, 147:3015-3025.
Anderson MS, Eveland SS, Price NP. 2000. Conserved cytoplasmic motifs that distinguish sub-groups of the polyprenol phosphate:N-acetylhexosamine-1-phosphate transferase family. FEMS Microbiol Lett, 191:169-175.
Barauskas J, Johnsson M, Tiberg F. 2005. Self-assembled lipid superstructures: Beyond vesicles and liposomes. Nano Lett, 5:1615-1619.
Bayburt TH, Sligar SG. 2010. Membrane protein assembly into Nanodiscs. FEBS Lett, 584:1721-1727.
Behrens NH, Tabora E. 1978. Dolichol intermediates in the glycosylation of proteins. Methods Enzymol, 50:402-435.
Bouhss A, Crouvoisier M, Blanot D, Mengin-Lecreulx D. 2004. Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan
biosynthesis. J Biol Chem, 279:29974-29980.
Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. 2008. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol Rev, 32:208-233.
Brandish PE, Kimura KI, Inukai M, Southgate R, Lonsdale JT, Bugg TD. 1996. Modes of action of tunicamycin, liposidomycin B, and mureidomycin A: inhibition of
phospho-N-acetylmuramyl-pentapeptide translocase from Escherichia coli. Antimicrob Agents Chemother, 40:1640-1644.
Burda P, Aebi M. 1999. The dolichol pathway of N-linked glycosylation. Biochimica et biophysica acta, 1426:239-257.
Chen MM, Weerapana E, Ciepichal E, Stupak J, Reid CW, Swiezewska E, Imperiali B. 2007. Polyisoprenol specificity in the Campylobacter jejuni N-linked glycosylation pathway. Biochemistry, 46:14342-14348.
Chung BC, Mashalidis EH, Tanino T, Kim M, Matsuda A, Hong J, Ichikawa S, Lee SY. 2016. Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature, 533:557-560.
Chung BC, Zhao J, Gillespie RA, Kwon DY, Guan Z, Hong J, Zhou P, Lee SY. 2013. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science, 341:1012-1016.
Das D, Kuzmic P, Imperiali B. 2017. Analysis of a dual domain phosphoglycosyl transferase reveals a ping-pong mechanism with a covalent enzyme intermediate. Proc Natl Acad Sci U S A, in press.
Das D, Walvoort MT, Lukose V, Imperiali B. 2016. A rapid and efficient luminescence-based method for assaying phosphoglycosyltransferase enzymes. Scientific reports, 6:33412.
Dini C, Collette P, Drochon N, Guillot JC, Lemoine G, Mauvais P, Aszodi J. 2000. Synthesis of the nucleoside moiety of liposidomycins: elucidation of the pharmacophore of this family of MraY inhibitors. Bioorg Med Chem Lett, 10:1839-1843.
Elshahawi SI, Shaaban KA, Kharel MK, Thorson JS. 2015. A comprehensive review of glycosylated bacterial natural products. Chem Soc Rev, 44:7591-7697.
Fer MJ, Bouhss A, Patrao M, Le Corre L, Pietrancosta N, Amoroso A, Joris B, Mengin-Lecreulx D, Calvet-Vitale S, Gravier-Pelletier C. 2015. 5'-Methylene-triazole-substituted-aminoribosyl uridines as MraY inhibitors: synthesis, biological evaluation and molecular modeling. Org Biomol Chem, 13:7193-7222.
Fer MJ, Olatunji S, Bouhss A, Calvet-Vitale S, Gravier-Pelletier C. 2013. Toward analogues of MraY natural inhibitors: synthesis of 5'-triazole-substituted-aminoribosyl uridines through a Cu-catalyzed azide-alkyne cycloaddition. J Org Chem, 78:10088-10105.
Furlong SE, Ford A, Albarnez-Rodriguez L, Valvano MA. 2015. Topological analysis of the Escherichia coli WcaJ protein reveals a new conserved configuration for the polyisoprenyl-phosphate hexose-1-polyisoprenyl-phosphate transferase family. Scientific reports, 5:9178.
Furlong SE, Valvano MA. 2012. Characterization of the highly conserved VFMGD motif in a bacterial polyisoprenyl-phosphate N-acetylaminosugar-1-phosphate transferase. Protein Sci, 21:1366-1375.
Gentle C, Harrison S, Inukai, M., Bugg TD. 1999. Structure-function studies on nucleoside antibiotic mureidomycin A: Synthesis of 5 '-functionalised uridine models. JCS Perkin Trans 1:1287-1294.
Gruschow S, Rackham EJ, Elkins B, Newill PL, Hill LM, Goss RJ. 2009. New pacidamycin antibiotics through precursor-directed biosynthesis. Chembiochem, 10:355-360.
Hakulinen JK, Hering J, Branden G, Chen H, Snijder A, Ek M, Johansson P. 2017. MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat Chem Biol, 13:265-267. Hartley MD, Imperiali B. 2012. At the membrane frontier: a prospectus on the remarkable evolutionary conservation of polyprenols and polyprenyl-phosphates. Arch Biochem Biophys, 517:83-97.
Hartley MD, Morrison MJ, Aas FE, Borud B, Koomey M, Imperiali B. 2011. Biochemical characterization of the O-linked glycosylation pathway in Neisseria gonorrhoeae responsible for biosynthesis of protein glycans containing N,N'-diacetylbacillosamine. Biochemistry, 50:4936-4948.
Heifetz A, Keenan RW, Elbein AD. 1979. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry, 18:2186-2192. Henrich E, Ma Y, Engels I, Munch D, Otten C, Schneider T, Henrichfreise B, Sahl HG, Dotsch V, Bernhard F. 2016. Lipid requirements for the enzymatic activity of MraY translocases and in vitro reconstitution of the lipid II synthesis pathway. J Biol Chem, 291:2535-2546.
Howard NI, Bugg TD. 2003. Synthesis and activity of 5'-uridinyl dipeptide analogues mimicking the amino terminal peptide chain of nucleoside antibiotic mureidomycin A. Bioorg Med Chem, 11:3083-3099.
Hug I, Feldman MF. 2011. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology, 21:138-151.
Hyland SA, Anderson MS. 2003. A high-throughput solid-phase extraction assay capable of measuring diverse polyprenyl phosphate: sugar-1-phosphate transferases as exemplified by the WecA, MraY, and MurG proteins. Anal Biochem, 317:156-165.
Ichikawa S, Yamaguchi M, Matsuda A. 2015. Antibacterial Nucleoside Natural Products Inhibiting Phospho-MurNAc-Pentapeptide Translocase; Chemistry and Structure-Activity Relationship. Curr Med Chem, 22:3951-3979.
Igarashi M, Nakagawa N, Doi N, Hattori S, Naganawa H, Hamada M. 2003. Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J Antibiot (Tokyo), 56:580-583.
Igarashi M, Takahashi Y, Shitara T, Nakamura H, Naganawa H, Miyake T, Akamatsu Y. 2005. Caprazamycins, novel lipo-nucleoside antibiotics, from Streptomyces sp. II. Structure
elucidation of caprazamycins. J Antibiot (Tokyo), 58:327-337.
Imperiali B, Zimmerman JW. 1988. Synthesis of dolichols via asymmetric hydrogenation of plant polyprenols. Tetrahedron Lett, 29:5343-5344.
Inukai M, Isono F, Takatsuki A. 1993. Selective inhibition of the bacterial translocase reaction in peptidoglycan synthesis by mureidomycins. Antimicrob Agents Chemother, 37:980-983.
Ishizaki Y, Hayashi C, Inoue K, Igarashi M, Takahashi Y, Pujari V, Crick DC, Brennan PJ, Nomoto A. 2013. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J Biol Chem, 288:30309-30319.
Isono F, Inukai M, Takahashi S, Haneishi T, Kinoshita T, Kuwano H. 1989. Mureidomycins A-D, novel peptidylnucleoside antibiotics with spheroplast forming activity. II. Structural
Isono K, Uramoto M, Kusakabe H, Kimura K, Isaki K, Nelson CC, McCloskey JA. 1985. Liposidomycins: novel nucleoside antibiotics which inhibit bacterial peptidoglycan synthesis. J Antibiot (Tokyo), 38:1617-1621.
Jankowski W, Swiezewska E, Sasak W, Chojnacki T. 1994. Occurrence of polyprenols and dolichols in plants. J Plant Physiol, 143:448-452.
Jones MB, Rosenberg JN, Betenbaugh MJ, Krag SS. 2009. Structure and synthesis of polyisoprenoids used in N-glycosylation across the three domains of life. Biochimica et biophysica acta, 1790:485-494.
Kaminski L, Abu-Qarn M, Guan Z, Naparstek S, Ventura VV, Raetz CR, Hitchen PG, Dell A, Eichler J. 2010. AglJ adds the first sugar of the N-linked pentasaccharide decorating the Haloferax volcanii S-layer glycoprotein. J Bacteriol, 192:5572-5579.
Keller RK, Boon DY, Crum FC. 1979. N-Acetylglucosamine-1-phosphate transferase from hen oviduct: Solubilization, characterization, and inhibition by tunicamycin. Biochemistry, 18:3946-3952.
Kumar M, Balaji PV. 2011. Comparative genomics analysis of completely sequenced microbial genomes reveals the ubiquity of N-linked glycosylation in prokaryotes. Mol Biosyst, 7:1629-1645.
Kurosu M, Mahapatra S, Narayanasamy P, Crick DC. 2007. Chemoenzymatic synthesis of Park's nucleotide: Toward the development of high-throughput screening for MraY inhibitors.
Tetrahedron Lett, 48:799-803.
Larkin A, Chang MM, Whitworth GE, Imperiali B. 2013. Biochemical evidence for an alternate pathway in N-linked glycoprotein biosynthesis. Nat Chem Biol, 9:367-373.
Lehrer J, Vigeant KA, Tatar LD, Valvano MA. 2007. Functional characterization and membrane topology of Escherichia coli WecA, a sugar-phosphate transferase initiating the biosynthesis of enterobacterial common antigen and O-antigen lipopolysaccharide. J Bacteriol, 189:2618-2628. Lehrman MA. 1991. Biosynthesis of N-acetylglucosamine-P-P-dolichol, the committed step of asparagine-linked oligosaccharide assembly. Glycobiology, 1:553-562.
Li J, Yu B. 2015. A modular approach to the total synthesis of tunicamycins. Angew Chem Int Ed Engl, 54:6618-6621.
Lloyd AJ, Brandish PE, Gilbey AM, Bugg TD. 2004. Phospho-N-acetyl-muramyl-pentapeptide translocase from Escherichia coli: catalytic role of conserved aspartic acid residues. J Bacteriol, 186:1747-1757.
Lukose V, Luo L, Kozakov D, Vajda S, Allen KN, Imperiali B. 2015. Conservation and
covariance in small bacterial phosphoglycosyltransferases Identify the functional catalytic core. Biochemistry, 54:7326-7334.
Ma Y, Munch D, Schneider T, Sahl HG, Bouhss A, Ghoshdastider U, Wang J, Dotsch V, Wang X, Bernhard F. 2011. Preparative scale cell-free production and quality optimization of MraY homologues in different expression modes. J Biol Chem, 286:38844-38853.
McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. 2002. Structures of the muraymycins, novel peptidoglycan biosynthesis inhibitors. J Am Chem Soc, 124:10260-10261.
Meier-Dieter U, Barr K, Starman R, Hatch L, Rick PD. 1992. Nucleotide sequence of the
Escherichia coli rfe gene involved in the synthesis of enterobacterial common antigen. Molecular cloning of the rfe-rff gene cluster. J Biol Chem, 267:746-753.
Mitachi K, Aleiwi BA, Schneider CM, Siricilla S, Kurosu M. 2016. Stereocontrolled Total Synthesis of Muraymycin D1 Having a Dual Mode of Action against Mycobacterium tuberculosis. J Am Chem Soc, 138:12975-12980.
Mitachi K, Siricilla S, Klaic L, Clemons WM, Jr., Kurosu M. 2015. Chemoenzymatic syntheses of water-soluble lipid I fluorescent probes. Tetrahedron Lett, 56:3441-3446.
Mitachi K, Siricilla S, Yang D, Kong Y, Skorupinska-Tudek K, Swiezewska E, Franzblau SG, Kurosu M. 2016. Fluorescence-based assay for polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA) and identification of novel antimycobacterial WecA inhibitors. Anal Biochem, 512:78-90.
Morrison MJ, Imperiali B. 2013. Biosynthesis of UDP-N,N'-diacetylbacillosamine in
Acinetobacter baumannii: Biochemical characterization and correlation to existing pathways. Arch Biochem Biophys, 536:72-80.
Myers AG, Gin DY, Rogers DH. 1993a. A convergent synthetic route to the tunicamycin antibiotics - synthesis of (+)-tunicamycin-V. J Am Chem Soc, 115:2036-2038.
Myers AG, Gin DY, Rogers DH. 1993b. Synthetic studies of the tunicamycin antibiotics - preparation of (+)-tunicaminyluracil, (+)-tunicamycin-V, and 5'-epi-tunicamycin-V. J Am Chem Soc, 116:4697-4718.
Myers AG, Gin DY, Widdowson KL. 1991. Silicon-mediated reductive coupling of aldehydes and allylic alcohols - a stereoselective synthesis of tunicaminyluracil. J Am Chem Soc, 113:9661-9663.
Nasie I, Steiner-Mordoch S, Schuldiner S. 2013. Topology determination of untagged membrane proteins. Methods Mol Biol, 1033:121-130.
Newby ZE, O'Connell JD, 3rd, Gruswitz F, Hays FA, Harries WE, Harwood IM, Ho JD, Lee JK, Savage DF, Miercke LJ, et al. 2009. A general protocol for the crystallization of membrane proteins for X-ray structural investigation. Nat Protoc, 4:619-637.
Niu G, Tan H. 2015. Nucleoside antibiotics: biosynthesis, regulation, and biotechnology. Trends Microbiol, 23:110-119.
Okamoto K, Sakagami M, Feng F, Takahashi F, Uotani K, Togame H, Takemoto H, Ichikawa S, Matsuda A. 2012a. Synthesis of pacidamycin analogues via an Ugi-multicomponent reaction. Bioorg Med Chem Lett, 22:4810-4815.
Okamoto K, Sakagami M, Feng F, Togame H, Takemoto H, Ichikawa S, Matsuda A. 2012b. Total synthesis and biological evaluation of pacidamycin D and its 3'-hydroxy analogue. J Org Chem, 77:1367-1377.
Olivier NB, Chen MM, Behr JR, Imperiali B. 2006. In vitro biosynthesis of UDP-N,N'-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein glycosylation system. Biochemistry, 45:13659-13669.
Patel KB, Ciepichal E, Swiezewska E, Valvano MA. 2012. The C-terminal domain of the Salmonella enterica WbaP (UDP-galactose:Und-P galactose-1-phosphate transferase) is
sufficient for catalytic activity and specificity for undecaprenyl monophosphate. Glycobiology, 22:116-122.
Patel KB, Furlong SE, Valvano MA. 2010. Functional analysis of the C-terminal domain of the WbaP protein that mediates initiation of O antigen synthesis in Salmonella enterica.
Patel KB, Valvano MA. 2013. In vitro UDP-sugar:undecaprenyl-phosphate sugar-1-phosphate transferase assay and product detection by thin layer chromatography. Methods Mol Biol, 1022:173-183.
Price NP, Momany FA. 2005. Modeling bacterial UDP-HexNAc: polyprenol-P HexNAc-1-P transferases. Glycobiology, 15:29R-42R.
Ragab AE, Gruschow S, Rackham EJ, Goss RJ. 2010. New pacidamycins biosynthetically: probing N- and C-terminal substrate specificity. Org Biomol Chem, 8:3128-3129.
Saldias MS, Patel K, Marolda CL, Bittner M, Contreras I, Valvano MA. 2008. Distinct
functional domains of the Salmonella enterica WbaP transferase that is involved in the initiation reaction for synthesis of the O antigen subunit. Microbiology, 154:440-453.
Scocca JR, Krag SS. 1997. Aspartic acid 252 and asparagine 185 are essential for activity of lipid N-acetylglucosaminylphosphate transferase. Glycobiology, 7:1181-1191.
Serpi M, Ferrari V, Pertusati F. 2016. Nucleoside derived antibiotics to fight microbial drug resistance: New utilities for an established class of drugs? J Med Chem, 59:10343-10382. Shapiro AB, Jahic H, Gao N, Hajec L, Rivin O. 2012. A high-throughput, homogeneous,
fluorescence resonance energy transfer-based assay for phospho-N-acetylmuramoyl-pentapeptide translocase (MraY). J Biomol Screen, 17:662-672.
Spork AP, Buschleb M, Ries O, Wiegmann D, Boettcher S, Mihalyi A, Bugg TD, Ducho C. 2014. Lead structures for new antibacterials: stereocontrolled synthesis of a bioactive muraymycin analogue. Chemistry, 20:15292-15297.
Stachyra T, Dini C, Ferrari P, Bouhss A, van Heijenoort J, Mengin-Lecreulx D, Blanot D, Biton J, Le Beller D. 2004. Fluorescence detection-based functional assay for high-throughput
Suami T, Sasai H, Matsuno K. 1983. Synthetic approach towards antibiotic tunicamycins. 5. Synthesis of methyl hexaacetyl-tunicaminyl uracil. Chem Lett:819-822.
Suami T, Sasai H, Matsuno K, Suzuki N, Fukuda Y, Sakanaka O. 1984. Synthetic approach toward antibiotic tunicamycins. 6. Total synthesis of tunicamycins. Tetrahedron Lett, 25:4533-4536.
Szymanski CM, Wren BW. 2005. Protein glycosylation in bacterial mucosal pathogens. Nat Rev Microbiol, 3:225-237.
Takatsuki A, Arima K, Tamura G. 1971. Tunicamycin, a new antibiotic. 1. Isolation and characterization of tunicamycin. J Antibiot (Tokyo), 24:215.
Takeoka Y, Tanino T, Sekiguchi M, Yonezawa S, Sakagami M, Takahashi F, Togame H, Tanaka Y, Takemoto H, Ichikawa S, et al. 2014. Expansion of Antibacterial Spectrum of Muraymycins toward Pseudomonas aeruginosa. ACS Med Chem Lett, 5:556-560.
Tanino T, Ichikawa S, Al-Dabbagh B, Bouhss A, Oyama H, Matsuda A. 2010. Synthesis and Biological Evaluation of Muraymycin Analogues Active against Anti-Drug-Resistant Bacteria. ACS Med Chem Lett, 1:258-262.
Tanino T, Yamaguchi M, Matsuda A, Ichikawa S. 2014. Function-oriented synthesis of liponucleoside antibiotics. Eur J Org Chem, 9:1836-1840.
Tytgat HL, Lebeer S. 2014. The sweet tooth of bacteria: common themes in bacterial glycoconjugates. Microbiol Mol Biol Rev, 78:372-417.
Valvano MA. 2003. Export of O-specific lipopolysaccharide. Front Biosci, 8:s452-471. Varki A. 2017. Biological roles of glycans. Glycobiology, 27:3-49.