HAL Id: tel-03140313
https://tel.archives-ouvertes.fr/tel-03140313
Submitted on 12 Feb 2021HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
PISA contrôlée par RAFT pour synthétiser des
nanofibres dans l’eau
Gaëlle Mellot
To cite this version:
Gaëlle Mellot. Combinaison de la chimie supramoléculaire et de la PISA contrôlée par RAFT pour synthétiser des nanofibres dans l’eau. Polymères. Sorbonne Université, 2019. Français. �NNT : 2019SORUS245�. �tel-03140313�
Thèse de doctorat de Sorbonne Université
Ecole doctorale 397 – Physique et Chimie des Matériaux
Institut Parisien de Chimie Moléculaire (IPCM – UMR 8232) / Equipe Chimie des Polymères
Combinaison de la chimie supramoléculaire
et de la PISA contrôlée par RAFT
pour synthétiser des nanofibres dans l’eau
Présentée par
Gaëlle MELLOT
Pour obtenir le grade de
Docteur de Sorbonne Université
Spécialité : Physique et Chimie des MatériauxDirigée par François STOFFELBACH et Jutta RIEGER
Soutenue publiquement le 16 septembre 2019 devant le jury composé de :
M. Guillaume DELAITTRE PR – FH Aachen, Université des Sciences Appliquées Rapporteur M. Mathias DESTARAC PR – Université Paul Sabatier Rapporteur
M. Olivier COLOMBANI MC – Le Mans Université Examinateur
Mme Laurence ROZES PR – Sorbonne Université Examinatrice
Mme Jutta RIEGER CR – Sorbonne Université Co-directrice de thèse M. François STOFFELBACH MC – Sorbonne Université Co-directeur de thèse
i
A mes parents,
A mes sœurs,
A Raphaël,
A Mehdi,
iii
« J'ai appris que la voie du progrès n'était ni rapide ni facile.»
v
R
EMERCIEMENTS
De nombreuses personnes ont été à mes côtés durant ces trois années de thèse et m’ont aidée à avancer tant sur le plan professionnel que personnel, il est donc important pour moi de leur exprimer toute ma gratitude.
Avant cela, je souhaite remercier les membres du jury qui m’ont fait l’honneur d’accepter de juger ce travail : Pr. Guillaume Delaittre et Pr. Mathias Destarac qui ont accepté de rapporter ce manuscrit mais également Pr. Laurence Rozes et Dr. Olivier Colombani qui ont accepté d’examiner ce travail.
Je remercie sincèrement Jutta Rieger et François Stoffelbach qui ont co-dirigé ce travail. Merci de m’avoir chaleureusement intégrée dans votre équipe et de m’avoir accordé votre confiance pour mener à bien ce projet. Je vous remercie tout particulièrement pour votre disponibilité au quotidien, vos nombreuses relectures et corrections et vos précieux conseils qui m’ont aiguillée durant ces trois ans. Jutta, je te remercie pour ta motivation communicative mais aussi pour tes nombreuses interrogations et idées qui nous ont ouvert la voie vers des pistes scientifiques qui n’auraient probablement pas été explorées sinon. Outre l’aspect scientifique, je te remercie pour ton écoute : tu as su déceler les moments difficiles et trouver les bons mots pour les dépasser. François, je vous suis reconnaissante pour tout le savoir tant expérimental que théorique que vous m’avez transmis. Merci également pour les heures de réflexions intenses qui ont permis de lever de nombreux verrous scientifiques. J’ai également acquis le sens du détail et de la rigueur en travaillant à vos côtés, on ne compte plus le nombre de fois où ces mots ont été prononcés : « c’est un détail, mais … ». Je suis à présent bien convaincue que c’est le détail qui fait toute la différence.
Je remercie tout particulièrement Laurent Bouteiller, non seulement pour son accueil au sein de l’Equipe Chimie des Polymères mais aussi pour son implication dans ce travail. Merci pour votre grande disponibilité malgré un emploi du temps déjà bien rempli. Je vous suis reconnaissante d’avoir pris le temps de répondre à toutes mes questions avec simplicité, beaucoup de patience et pédagogie.
Un grand merci également aux différents stagiaires qui ont contribué à l’avancée de ce projet : Noé, Hayley, Justine, Sabrine et Marcus. Une mention particulière est adressée à Justine pour sa grande motivation et ses nombreuses heures passées à la paillasse.
Je tiens également à remercier les personnes qui m’ont apporté un soutien technique considérable, notamment pour la caractérisation de mes polymères : Gaëlle pour ses nombreuses heures à bichonner la SEC DMF (un poil capricieuse !) et à refaire les calibrations au gré des sauts de pression de cette dernière. Je te remercie également pour ton soutien pendant la période de rédaction.
vi
Merci à Claire d’avoir pris le temps de répondre à mes questions et de m’avoir aidée à interpréter certains spectres RMN et tout simplement merci pour ta bonne humeur. Je remercie également Patricia Beaunier et Jean-Michel Guigner pour les nombreuses heures passées à imager mes polymères. Jean-Michel, je te remercie d’avoir à chaque fois trouvé un créneau pour analyser le fameux échantillon « urgent » (parce qu’il y en a toujours un !). Enfin, je remercie Jacques Jestin du Laboratoire Léon Brillouin (CEA) pour son aide précieuse lors des expériences de diffusion de neutrons sur le PA20.
De manière générale, je remercie toutes les personnes de l’Equipe Chimie des Polymères, permanentes ou de passage, qui ont contribué positivement au déroulement de ces trois années, et particulièrement, Nicolas I., Lydia, Cécile, Fabrice, David, Sandrine, Martine, Nicolas A. et Fanny. J’ai une pensée toute particulière pour André et Stéphanie qui m’ont accueillie très chaleureusement et avec lesquels j’ai partagé le bureau et le labo durant ma première année. Stéphanie, un grand merci pour ta gentillesse et ta bienveillance (et tes succulents gâteaux) durant toute cette période. André (Dédé les bonnes astuces !), je te remercie pour ton aide et toutes tes astuces, grâce à toi j’ai pu me bâtir une paillasse de compétition. Puis, de tes nombreuses expressions farfelues, on retiendra bien sûr que, peu importe le problème, « ça va toujours aller ».
Je remercie également Thomas, Léo et Julien qui ont notamment égayé nos pauses déjeuner avec leurs blagues toujours d’une grande finesse. Enfin, (last but not least !) des remerciements tout particuliers sont adressés à mes fameux « collègues » du 4e, qui m’ont épaulée au quotidien, chacun à sa façon et avec ses qualités respectives : Merci, Valentin, pour ta bonne humeur enfantine et communicative, Gwendoline, pour tes bonnes idées et ton regard « affuté », Laetitia, pour ta gentillesse et tes conseils avisés et Pauline, pour ton sourire, ton écoute, ta patience et tes nombreux discours de motivation. Vous m’avez tous les quatre montré que la thèse c’est certes de la recherche mais c’est aussi de beaux moments passés avec des personnes formidables. Je vous souhaite à tous les quatre, bon courage pour la suite de votre doctorat. Prenez garde aux calculs de concentration !
Je suis également reconnaissante envers ma famille et mes amis qui m’entourent en dehors du monde professionnel : à commencer par mes parents et ma grand-mère qui m’ont toujours accompagnée avec beaucoup de bienveillance dans mon projet professionnel, mais également Victoria et Mehdi pour leur soutien sans faille, leur présence dans les moments de réussite comme dans les moments de doute ainsi que leur patience, en particulier pendant la rédaction. Je terminerai par remercier Chloé, une coloc et une amie en or qui a été présente pour moi à chaque moment ainsi qu’Andy dont l’humour plus que douteux et l’optimisme légendaire ont rythmé toutes mes journées.
vii
L
ISTE DES ABRÉVIATIONS
AA Acide Acrylique / Acrylic acid
ACPA Acide 4,4’-azobis(4-cyanopentanoïque) / 4,4’-Azobis(4-cyanopentanoic acid) AIBN 2,2′-Azobis(isobutyronitrile)
AM Acrylamide
APTS p-toluenesulfonic acid monohydrate
ATRP Polymérisation radicalaire contrôlée par transfert d’atome
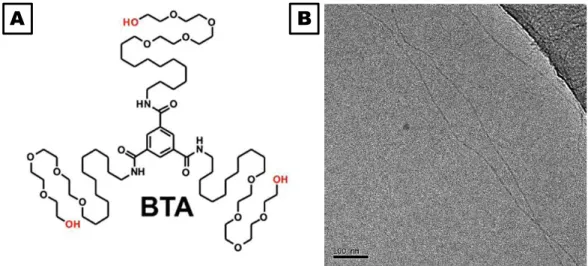
BTA Benzène-1,3,5-tricarboxamide
BzMA Méthacrylate de benzyle
CDSA Auto-assemblage induit par la cristallisation / Crystallization-driven self-assembly
CP Point trouble / Cloud point
Cryo-TEM Cryogenic transmission electron microscopy
CTA Cyclohexane-1,3,5-tricarboxamide (Chapitre 1) or Chain transfer agent CysMA Méthacrylate de cystéine
Ð Dispersité en masses molaires / Molar mass dispersity DAAm Diacetone acrylamide
DLS Diffusion dynamique de la lumière / Dynamic light scattering DMAc N,N-dimethylacrylamide
DMAEA Acrylate de 2-(N,N-diméthylamino)éthyle / (N,N-dimethylamino)ethyl acrylate
DMF N,N-Dimethylformamide
DMSO Dimethylsulfoxyde
DPn Degré de polymérisation moyen en nombre / Number-average degree of polymerization
DSC Differential scanning calorimetry
Dz z-average particle diameter
EG Ethylène glycol
Et2O Ether diéthylique / Diethyl ether
FTIR Fourier transformed infrared spectroscopy
GlyMA Méthacrylate de glycidyle GMA Méthacrylate de glycérol GSHMA Méthacrylate de gluthathion
HBA Acrylate d'hydroxybutyle
HBMA Méthacrylate de 4-hydroxybutyle H-Bonding Hydrogen-bonding
HEA 2-Hydroxyethylacrylate
HEMA Méthacrylate de 2-hydroxyéthyle HPMA Méthacrylate de 2-hydroxypropyle
ITC Calorimétrie de titration isotherme / Isothermal titration calorimetry LCST Lower critical solution temperature
MAA Acide méthacrylique
Macro-RAFT Agent de transfert macromoléculaire
MEA Acrylate de 2-méthoxyéthyle / 2-Methoxyethyl acrylate
viii
Mn Masse molaire moyenne en nombre / Number-average molar mass
Mw Masse molaire moyenne en poids / Weight-average molar mass
NAM N-4-acryloylmorpholine
Nano-DSC Nano-differential scanning calorimetry
NDI Naphtalène diimide
NMP Polymérisation radicalaire contrôlée par les nitroxydes
NMR Résonance magnétique nucléaire
oligoEG oligo(ethylène glycol)
p Paramètre d'empilement / Packing parameter
PAA Poly(acide acrylique) / Poly(acrylic acid)
PAM Polyacrylamide
PBzMA Poly(méthacrylate de benzyle) PCysMA Poly(méthacrylate de cystéine) PDAAm Poly(diacetone acrylamide)
PDMAc Poly(N,N-dimethylacrylamide)
PDMAEA Poly(acrylate de 2-(N,N-diméthylamino)éthyle) PDPMA Poly(méthacrylate de 2-(diisopropylamino)éthyle)
PEG Poly(éthylène glycol)
PEGA Acrylate de poly(éthylène glycol méthyléther) PEGMA Méthacrylate de poly(éthylène glycol méthyléther) PEHMA Poly(α-hydroxymethyl acrylate d’éthyle )
PGlyMA Poly(méthacrylate de glycidyle) PGMA Poly(méthacrylate de glycérol) PGSHMA Poly(méthacrylate de gluthathion) PHBMA Poly(méthacrylate de 4-hydroxybutyle ) PHEA Poly(2-hydroxyethylacrylate )
PHEMA Poly(méthacrylate de 2-hydroxyéthyle)
Photo-PISA Auto-assemblage induit par la polymérisation photo-amorcée PHPAm Poly(méthacrylamide de 2-hydroxypropyle)
PHPMA Poly(méthacrylate de 2-hydroxypropyle)
PISA Auto-assemblage induit par la polymérisation / Polymerization-induced self-assembly PMEA Poly(acrylate de 2-méthoxyéthyle) / Poly(2-methoxyethyl acrylate)
PMMA Poy(méthacrylate de méthyle) / Poly(methyl methacrylate) PNAM Poly(N-4-acryloylmorpholine )
PPEGA Poly(acrylate de poly(éthylène glycol méthyléther)) PPEGMA Poly(methacrylate de poly(éthylène glycol méthyléther)) PRC Polymérisation radicalaire contrôlée
PRDR Polymérisation radicalaire par désactivation réversible
PS Polystyrène
PtBA Poly(acrylate de tert-butyle)
PtBMA Poly(methacrylate de tert-butyle)
RAFT Polymérisation radicalaire contrôlée par transfert de chaînes réversible par addition-fragmentation / Reversible addition-fragmentation chain transfer
ix SANS Diffusion de neutrons aux petits angles / Small-angle neutron scattering SEC Chromatographie d'exclusion stérique / Size exclusion chromatography SPTP Phenyl-2,4,6-triméthylbenzoylphosphinate de sodium
SSDU Supramolecular structure-directing unit
Taux de solide / Solids content
tBA Acrylate de tert-butyle
tBMA Méthacrylate de tert-butyle
TEM Microscopie électronique en transmission / Transmission electron microscopy
Tg Température de transition vitreuse
THF Tétrahydrofurane
TIMT Temperature-induced morphological transition
TT Transition temperature
TTC Trithiocarbonate
UPy Uréido-pyrimidinone
V-50 2,2’-Azobis(2-amidinopropane) dihydrochloride
VA-044 2,2’-Azobis(N,N-dimethyleneisobutyramidine) dihydrochloride
η Viscosité dynamique
wt% Weight percent
xi
SOMMAIRE
REMERCIEMENTS
...
vLISTE DES ABRÉVIATIONS
...
viiINTRODUCTION GÉNÉRALE
...
1CHAPITRE 1
— Etude bibliographique...
7CHAPTER 2
— Beyond simple AB diblock copolymers: application of bifunctional and trifunctional RAFT agents to PISA in water...
87CHAPTER 3
— Using a symmetrical aromatic trisurea-functionalized RAFT agent: an efficient strategy to drive aqueous PISA towards the fiber morphology?...
121CHAPTER 4
— Bisurea-functionalized RAFT agent: a straightforward and versatile tool to prepare supra(macro)molecular cylindrical nanostructures in water ... 163CHAPTER 5
— Templated-PISA: driving Polymerization-Induced Self-Assembly towards the fiber morphology ... 219DISCUSSION
... 2553 Les scientifiques s’intéressent de plus en plus aux polymères afin de développer des nouveaux matériaux intelligents, toujours plus performants, qui permettent de relever les défis technologiques actuels tout en respectant les nouvelles réglementations environnementales. Dans ce contexte, les nanofibres polymères sont particulièrement intéressantes car leur facteur de forme élevé leur confère de nombreuses propriétés (propriétés de surface, mécaniques et/ou rhéologiques). En effet, les nanofibres peuvent être utilisées dans divers domaines notamment dans le biomédical et la santé (e.g. vecteurs de délivrance de médicaments), l’énergie (e.g. supercondensateurs pour le stockage d’énergie), la catalyse, et les matériaux (e.g. stabilisants d’émulsions de Pickering, additifs pour le renfort mécanique de films polymères).
En raison de l’intérêt florissant pour ce type de structures, les méthodes de préparation des nanofibres à base de copolymères amphiphiles se sont considérablement développées (méthode du co-solvant, électrofilage, auto-assemblage induit par la température ou cristallisation dirigée, etc…). Bien que très utilisées, ces méthodes ne sont pas idéales puisqu’elles conduisent à des formulations ayant des concentrations en polymère souvent inférieures à 1 %mass., requièrent généralement l’utilisation d’un solvant organique et sont assez chronophages. Une méthode alternative plus directe et moins polluante, permettant de pallier tous ces inconvénients s’est récemment développée : la PISA (Polymerization-Induced Self-Assembly) en milieu aqueux. L’originalité de cette méthode repose sur le fait que la polymérisation et l’auto-assemblage ont lieu simultanément.
Par ailleurs, la PISA est associée à une technique de polymérisation vivante ou contrôlée permettant un bon contrôle de l’architecture et de la masse molaire des polymères synthétisés. Au vu du nombre accru de publications s’intéressant à ce sujet, il est à présent établi que la polymérisation radicalaire contrôlée par transfert de chaînes réversible par addition-fragmentation (RAFT) est la technique de choix pour mettre en œuvre un procédé PISA en milieu aqueux. En effet, elle permet de travailler avec une large gamme de monomères et dans des conditions très variées. Ainsi, la PISA contrôlée par RAFT s’est imposée comme étant une méthode robuste et efficace pour synthétiser des nano-objets ayant des morphologies bien définies telles que des sphères, des fibres ou des vésicules, dans l’eau, à des taux de solide élevés (10 – 60 %mass.) et cela sans tensioactif libre ni solvant organique. De nombreuses formulations, issues du procédé PISA contrôlée par RAFT dans l’eau, conduisant à la formation de nanofibres ont déjà été reportées dans la littérature démontrant l’efficacité de cette méthode.
4
Cependant, ce sont très souvent des mélanges de morphologies qui sont formés et pour un système donné, l’obtention de fibres « pures » nécessite de fastidieux ajustements. En effet, les diagrammes de phase obtenus à partir des copolymères amphiphiles, synthétisés par PISA contrôlée par RAFT dans l’eau, illustrent bien cette limitation puisque la fenêtre d’obtention des nanofibres « pures » occupe généralement un espace relativement étroit dans ces derniers.
Parallèlement, la chimie supramoléculaire qui est la chimie des interactions non covalentes, a suscité beaucoup d’intérêt et a notamment donné lieu à une nouvelle classe de polymères : les polymères supra(macro)moléculaires. Ces derniers sont le résultat de l’assemblage de (macro)molécules via des liaisons réversibles hautement directionnelles. Ces matériaux sont particulièrement intéressants puisqu’ils peuvent présenter des propriétés similaires aux polymères basés sur des liaisons covalentes ainsi qu’une sensibilité aux stimuli externes en raison des interactions non covalentes qui les composent. En particulier, les systèmes basés sur l’auto-assemblage via des liaisons hydrogène semblent être un bon moyen de synthétiser des assemblages supramoléculaires unidirectionnels (fibres supramoléculaires) fonctionnels dans l’eau. Un concept basé sur la combinaison de la chimie supramoléculaire et de la polymérisation contrôlée par RAFT, via la fonctionnalisation d’un agent RAFT par un motif associatif impliquant des liaisons hydrogène, a permis d’établir une méthode plus polyvalente de synthèse de nanofibres supramoléculaires dans l’eau. Cependant, une nouvelle fois, cette méthode requiert l’utilisation de solvants organiques et nécessite plusieurs étapes de synthèse et de purification.
Dans ce contexte, nous nous sommes intéressés à allier le concept de PISA contrôlée par RAFT et le concept de chimie supramoléculaire afin de tirer profit de leurs avantages respectifs et de concevoir une nouvelle stratégie de synthèse de nanofibres de différentes natures chimiques directement dans l’eau et à des taux de solide élevés.
Ce manuscrit de thèse s’articule en cinq chapitres. Dans un premier chapitre, une étude bibliographique permet d’aborder les principaux concepts utilisés dans ce projet afin de mieux comprendre l’intérêt de ce dernier. Le second chapitre, rédigé en anglais sous la forme d’un article, s’intéresse à l’influence de l’architecture du macro-RAFT sur la morphologie des nano-objets obtenus lors d’un procédé PISA dans l’eau.
5 Trois autres chapitres, rédigés selon le même format, permettent ensuite de décrire la stratégie mise en œuvre afin d’établir une nouvelle méthode originale de synthèse de nanofibres dans l’eau. Il s’agira principalement d’étudier comment l’introduction d’un motif associatif dans la structure du macro-RAFT impacte la morphologie des nano-objets obtenus et notamment si elle permet de favoriser la formation des fibres. Dans le chapitre 3, nous nous sommes tout d’abord intéressés à un motif associatif de type tris-urée aromatique. Ce dernier a été introduit au centre de la structure de copolymères amphiphiles symétriques synthétisés par PISA dans l’eau et nous avons évalué les paramètres qui permettent ou empêchent son impact sur la morphologie des nano-objets. Dans les deux derniers chapitres, c’est un motif associatif de type bis-urée aliphatique qui a été au cœur de l’étude et nous avons notamment montré comment il influence l’association des macro-RAFTs hydrophiles dans l’eau (chapitre 4) et la morphologie des nano-objets amphiphiles obtenus par PISA (chapitre 5). Enfin, une étude comparative des résultats obtenus avec le système basé sur le motif associatif tris-urée aromatique et ceux issus du système impliquant le motif associatif bis-urée aliphatique a été menée afin de mettre en lumière et de comprendre les différences entre ces deux systèmes.
— CHAPITRE 1 —
9
Sommaire
Introduction ... 10 I. Synthèse et auto-assemblage simultanés de copolymères amphiphiles sous forme de fibres dans l’eau ... 11
I. 1. Généralités sur l’auto-assemblage ... 11 I. 2. La polymérisation radicalaire contrôlée par RAFT ... 13 I. 3. La PISA contrôlée par RAFT en milieux aqueux ... 17 I. 4. Applications et mode d’obtention des nanofibres polymères dans l’eau ... 19 I. 4. 1. Exemples d’applications des nanofibres ... 19 I. 4. 2. Les différentes méthodes de préparation des nanofibres ... 23 I. 4. 3. Synthèse de nanofibres par le procédé PISA contrôlée par RAFT dans l’eau 24 II. Assemblages supramacromoléculaires sous forme de fibres dans l’eau ... 44 II. 1. Généralités concernant les polymères supramoléculaires ... 44 II. 2. Les différents motifs associatifs impliquant les liaisons hydrogène ... 45 II. 2. 1. Les motifs composés de fonctions amide ... 45 II. 2. 2. Le motif uréido-pyrimidinone (UPy) ... 54 II. 2. 3. Autres motifs faisant intervenir des liaisons hydrogène ... 57 II. 3. Utilisation du motif urée ... 59 II. 3. 1. Le cas des motifs urée aromatiques ... 59 II. 3. 2. Les motifs urée aliphatiques ... 63 II. 4. Combiner la chimie supramoléculaire et la polymérisation radicalaire contrôlée par RAFT en solution ... 71 Conclusion et stratégie ... 76 Références ... 78
10
Introduction
Etant donné que ce projet de thèse s’inscrit dans un contexte qui est à la convergence de deux concepts : la synthèse et l’auto-assemblage simultanés de copolymères à blocs amphiphiles par polymérisation radicalaire par désactivation réversible dans l’eau, d’une part, et la chimie supra(macro)moléculaire, d’autre part, ce chapitre bibliographique s’articulera en deux parties afin de présenter séparément ces concepts et de faire un état de l’art de chacun d’eux en se focalisant sur l’assemblage filamentaire. Dans la première partie, nous présenterons tout d’abord les grands principes de l’auto-assemblage des (macro)molécules amphiphiles. Puis, nous décrirons les différentes méthodes d’assemblages permettant de synthétiser des nanofibres dans l’eau, en détaillant en particulier le concept de PISA contrôlée par transfert de chaines réversible par addition-fragmentation (RAFT) en milieu aqueux. Dans une seconde partie, nous définirons la notion de polymère supra(macro)moléculaire, en portant une attention particulière aux systèmes basés sur les liaisons hydrogène. Nous présenterons ensuite les différents motifs associatifs à liaisons hydrogène donnant accès à des édifices supramoléculaires filamentaires en développant plus particulièrement le cas du motif urée. Enfin, en s’appuyant sur les différents exemples d’auto-assemblages filamentaires de (macro)molécules dans l’eau, décrits dans la littérature, nous clôturerons ce chapitre en proposant une stratégie de synthèse originale de fibres polymères dans l’eau.
11
I.
Synthèse et auto-assemblage simultanés de
copolymères amphiphiles sous forme de fibres dans
l’eau
I. 1. Généralités sur l’auto-assemblage
Depuis les travaux1, en 1913, de James William McBain portant sur l’agrégation des molécules de savon en solution aqueuse au cours desquels il instaura le concept de micelle, l’intérêt pour les molécules amphiphiles n’a pas cessé d’augmenter. De plus, l’accès à des copolymères à blocs amphiphiles synthétiques, qui a débuté que dans les années 1960 suite à la découverte de la polymérisation anionique « vivante » par Szwarc2 et son équipe, n’a fait qu’accroitre cet intérêt. Pour cause, il est à présent bien établi que l’assemblage de copolymères à blocs en solution est une méthode fiable et efficace d’obtention de nano-objets ayant des morphologies bien définies et variées menant à de nombreuses applications potentielles (e.g. utilisation des nano-objets en tant que vecteurs pour l’administration et le relargage de médicaments, agents antibactériens, lubrifiants pour des huiles de moteur, additifs pour le renfort de matériaux, etc…).3 Eisenberg4–7 a été l’un des pionniers dans le développement des méthodes d’auto-assemblage de copolymères amphiphiles dans l’eau. Il a notamment développé la méthode de « déplacement de solvant » aussi appelée méthode du « co-solvant ». Au cours de cette méthode, les copolymères à blocs amphiphiles sont généralement synthétisés dans un solvant organique qui est bon solvant de chacun des blocs. Puis, l’ajout progressif d’eau, non solvant de l’un des blocs, induit l’auto-assemblage des copolymères. Outre la morphologie sphérique initialement obtenue, d’autres morphologies5 telles que les fibres, les vésicules et les lamelles ont été mises en évidence. D’autres structures encore plus complexes7 telles que des vésicules multilamellaires, des micelles interconnectées ou des réseaux sont également accessibles. Israelachvili et al.8 ont développé une théorie, basée sur des modèles géométriques, qui permet de prédire la morphologie des assemblages (Figure 1). Cette théorie tient compte principalement des forces attractives et répulsives présentent au sein de l’assemblage et de celles-ci découle notamment le paramètre d’empilement p dont la valeur permet de prédire la morphologie.
12
Il se définit par la formule d’Israelachvili
: p =
Vao×lc avec V et lc, le volume et la longueur
maximale de la partie hydrophobe, et a0 l’aire interfaciale de la tête hydrophile dans le cas d’une
molécule amphiphile. Initialement développée dans le cadre de l’assemblage des tensioactifs, cette théorie a également démontré son efficacité dans le cadre de l’assemblage de copolymères amphiphiles et dans ce cas, l’aire interfaciale correspondà l’aire de la jonction entre les blocs. On note également que dans le cas des fibres, on a : 1/3 < p ≤ 1/2 (Figure 1).8,9
Figure 1. Représentation schématique de l’évolution de la morphologie en fonction du paramètre d’empilement p d’après la théorie d’Israelachvili (extraite de la référence 9).
13 La méthode de « déplacement de solvant », évoquée précédemment, est particulièrement mise en œuvre pour l’auto-assemblage de copolymères dont le bloc hydrophobe a une température de transition vitreuse élevée (Tg), cependant lorsque le bloc hydrophobe est un polymère de faible Tg l’auto-assemblage peut aussi se faire via la méthode dite de réhydratation de film.7,10 Comme le suggère son nom, cette méthode implique la préparation d’un film de copolymère par évaporation de solvant avant de le réhydrater afin d’induire l’auto-assemblage. En fait, les méthodes mises en œuvre pour assembler des copolymères amphiphiles dans l’eau sont nombreuses : par exemple via un changement de pH,11 une nano-précipitation,12 ou des techniques de microfluidique (préparation de nanostructures basée sur l’écoulement de fluides dans des systèmes de taille micro- et/ou nanométrique),13 etc… Toutes ces méthodes engendrent des procédés post-polymérisations et de ce fait exigent plusieurs étapes. De plus, bien que très utilisées, ces méthodes présentent de nombreux désavantages puisque qu’elles conduisent à des solutions ayant une faible concentration en particules (généralement <1 %mass.), risquent d’influencer fortement la morphologie obtenue, ou encore nécessitent l’utilisation de co-solvant organique, limitant ainsi leur développement à l’échelle industrielle.3,14 Afin de remédier à ces limitations, une méthode alternative plus directe s’est développée récemment : la PISA (Polymerization-Induced Self-Assembly), au cours de laquelle la polymérisation et l’auto-assemblage ont lieu simultanément. Généralement, ce procédé est basé sur l’utilisation de la polymérisation radicalaire contrôlée (PRC) et notamment le procédé de polymérisation contrôlée par RAFT (Reversible Addition-Fragmentation chain Transfer). Ainsi, avant de détailler le concept de la PISA, qui est au cœur de cette étude, nous commencerons par présenter le principe de polymérisation radicalaire contrôlée par RAFT.
I. 2. La polymérisation radicalaire contrôlée par RAFT
Ainsi comme introduit dans le paragraphe précédemment, la polymérisation radicalaire contrôlée par transfert de chaînes réversible par addition-fragmentation (RAFT) est une des techniques de polymérisation radicalaire contrôlée (PRC). Il est à noter que d’après les recommandations de l’IUPAC (International Union of Pure and Applied Chemistry), la PRC doit être à présent nommée : polymérisation radicalaire par désactivation réversible (PRDR),15 nous essaierons donc de privilégier l’utilisation de ce terme dans ce manuscrit.
14
Concrètement, la PRDR est un procédé qui repose sur l’introduction dans le milieu de polymérisation d’un agent de contrôle qui induit un équilibre entre les chaines dites actives, en croissance, et les chaines dormantes, principalement déplacé vers ces dernières.16 De cette façon, on peut accéder à une large gamme de polymères ayant une architecture contrôlée et bien définie,17 ce qui explique que la PRDR s’est largement développée ces dernières années.17–19 Classiquement, il existe deux méthodes de PRDR : celle basée sur une réaction de terminaison réversible et celle basée sur une réaction de transfert de chaine réversible. Les techniques les plus couramment utilisées sont la polymérisation radicalaire contrôlée par les nitroxydes (NMP),20 la polymérisation radicalaire contrôlée par transfert d’atome (ATRP)21 et la polymérisation radicalaire contrôlée par RAFT. La NMP et l’ATRP se basent sur la réaction de terminaison réversible alors que la polymérisation contrôlée par RAFT fait intervenir la seconde méthode.16
Depuis sa découverte en 1998, par une équipe du CSIRO (Commonwealth Scientific and Industrial Research Organization)22 et simultanément en industrie (Rhodia Chimie),23 le concept de polymérisation radicalaire contrôlée par RAFT a vu sa popularité augmenter. Il s’est imposé devant les autres techniques de PRDR en raison de sa polyvalence. En effet, il est facile à mettre en œuvre, adapté à une large gamme de monomères et peut être conduit en milieu aqueux, organique, homogène ou hétérogène.22 De plus, il ne nécessite que l’introduction d’un agent de transfert dans un système de polymérisation radicalaire classique (i.e. composé d’un amorceur et du monomère). Il est ainsi, d’un point de vue industriel, très attractif.19
Les trois principales étapes de la polymérisation radicalaire classique que sont l’amorçage, la propagation et la terminaison restent donc présentes au cours du procédé RAFT. L’agent de transfert, appelé agent RAFT (ou Xanthate, Z = O-alkyle) qui est introduit dans le milieu est un composé de type thiocarbonylthio (Figure 2), qui possèdent une double liaison C=S très réactive vis-à-vis de l’addition radicalaire.
15 Dans la structure de cet agent RAFT (Figure 2), le groupe Z joue le rôle à la fois d’activateur et de stabilisant et le groupe R, est un groupe partant. Ils peuvent être très différents d’un agent RAFT à l’autre comme le montre les exemples donnés dans le Tableau 1. Il convient de les choisir judicieusement pour que l’agent RAFT soit efficace dans le contrôle de la polymérisation radicalaire d’un monomère donné.
Tableau 1. Noms, structures et exemples des différents types d’agents RAFT.24
Nom Structure (R’, R’’ = groupement aliphatique ou aromatique) Exemples Dithioester Trithiocarbonate Dithiocarbonate (= Xanthate) Dithiocarbamate
Le mécanisme complet de la polymérisation contrôlée par RAFT est donné en Figure 3. Dans le cas de l’utilisation d’un amorceur thermique conventionnel (e.g. l’azobisisobutyronitrile (AIBN)), suite à un apport d’énergie suffisant, l’amorceur, noté A, se dissocie et des radicaux sont créés, il s’agit de la première étape d’amorçage. Au cours de la seconde étape d’amorçage, les radicaux primaires formés vont réagir avec une molécule de monomère pour former l’adduit IM●
16
Les radicaux formés I●, IM● ou les chaines en croissance, Pm●, peuvent s’additionner sur l’agent RAFT (1), induisant la formation d’un radical intermédiaire (2). L’équilibre mis en place s’appelle le pré-équilibre. La fragmentation de cette espèce intermédiaire peut potentiellement se faire de deux façons mais si l’agent RAFT est correctement choisi, la fragmentation de cette espèce intermédiaire a lieu majoritairement en faveur de la libération de l’espèce réactive R●
, capable de réagir avec une molécule de monomère (formant P1● puis après propagation Pn●), ainsi qu’avec une chaine Pm-S-C(S)-Z (3) momentanément inactive. Il se met alors en place un équilibre, dit équilibre principal entre les chaines actives en croissance et les chaines dormantes.25,26 Grâce à l’établissement de cet équilibre, les chaines croissent quasiment simultanément et de façon contrôlée. Tout comme lors d’une polymérisation radicalaire classique, la croissance des chaines peut se terminer par des réactions de terminaison irréversibles (dismutation, combinaison ou transfert). Il est important de noter que ces réactions de terminaison irréversibles en cours de polymérisation ne sont pas supprimées24 mais elles sont largement minoritaires.
17 En termes de performance, l’efficacité d’un agent RAFT se transcrit via les valeurs de deux constantes de transfert, Ctr1 et Ctr2, associées respectivement au pré-équilibre et à l’équilibre principal. Elles sont définies de la façon suivante Ctr1 = ktr1
kp et Ctr2 =
ktr2
kp, avec ktr1 et ktr2 les constantes de vitesse de transfert du pré-équilibre et de l’équilibre principal, respectivement, et kp la constante de vitesse de propagation (Figure 3).24,26,28 Ctr1 influence l’évolution du degré de polymérisation moyen en nombre (DPn) en fonction de la conversion. Idéalement, elle doit être très supérieure à 1, de cette façon l’agent RAFT est rapidement consommé. Ctr2 impacte la vitesse de l’échange entre les chaines dormantes et les chaines actives en croissance. C’est donc d’elle que dépend la dispersité en
masses molaires (Mw
Mn) (= Ð). En somme, si ces deux constantes sont suffisamment élevées, le
contrôle de la polymérisation est bon. Tous ces paramètres sont en fait directement liés au choix de l’agent RAFT. En choisissant l’agent RAFT, il faut garder à l’esprit que le groupe Z qui joue le rôle à la fois d’activateur et de stabilisant, a une influence sur la vitesse d’addition des radicaux propagateurs sur la fonction thiocarbonylthio et sur celle de fragmentation des radicaux intermédiaires. Le groupe R, quant à lui, doit être un bon groupe partant et doit être capable d’amorcer une nouvelle chaine.24,26,27 Le DP
n, quant à lui, est fonction du rapport entre les concentrations initiales en monomère ([M]0) et en agent RAFT ([Agent RAFT]0) et évolue idéalement, linéairement avec la conversion. Si l’on néglige la fraction des chaines issues de l’amorceur, il peut être calculé en fonction de la conversion (ρ), de la façon suivante : DPn =
[M]0
[Agent RAFT]0× ρ.
27,28 Un autre rapport important est celui entre la concentration initiale en
amorceur et celle en agent RAFT car il conditionne la proportion de chaines amorcées par l’amorceur plutôt que par le groupement R●, en d’autres termes les chaines « mortes », il faut donc veiller à le minimiser.28
I. 3. La PISA contrôlée par RAFT en milieux aqueux
Nous allons à présent détailler le principe de la PISA, découvert il y a une quinzaine d’années.29,30 Bien qu’elle soit encore très récente, cette technologie est déjà très utilisée pour la préparation de nano-objets tels que des fibres ou des vésicules. Elle peut être mise en œuvre dans différents milieux,31,32 nous nous limiterons ici à décrire le cas de la PISA contrôlée par RAFT en milieux aqueux. Typiquement, la PISA en milieu aqueux repose sur l’extension d’un homopolymère fonctionnel hydrosoluble par un second bloc qui devient progressivement insoluble dans le milieu de polymérisation au cours de sa croissance.
18
Ainsi du fait de l’affinité de chacun des blocs avec le solvant, ici l’eau, l’auto-assemblage du copolymère amphiphile est induit par la polymérisation conduisant à la formation de nanoparticules dont la morphologie dépend notamment de la taille et de la nature chimique de chacun des blocs (Figure 4).33,34 La PISA sous-entend que la polymérisation a lieu principalement en milieu hétérogène où le premier bloc polymère joue le rôle de stabilisant stérique ou électro-stérique. Ce procédé ne nécessite donc pas l’emploi de tensioactif libre ce qui est un des avantages de cette méthode.
On distingue deux types de polymérisation lorsque l’on utilise le procédé PISA: lorsque le monomère choisi pour obtenir le bloc hydrophobe est soluble dans l’eau, la PISA a lieu en dispersion,17,35 à l’inverse, lorsque le monomère est très peu soluble dans l’eau (et que l’on travaille à une concentration en monomère supérieure à sa solubilité dans le solvant), le procédé PISA se fait en émulsion.17,36 La PISA s’impose donc comme étant un bon moyen de synthétiser des copolymères à blocs par polymérisation radicalaire contrôlée, en milieu hétérogène dans l’eau et en l’absence de tensioactif libre.37–40
Figure 4. Représentation schématique de la synthèse de nano-objets par PISA dans l’eau.
La PISA est associée à une technique de polymérisation vivante ou contrôlée, permettant ainsi d'accéder à des chaines polymères ayant une architecture prédéfinie et contrôlée. Parmi les différentes voies de contrôle en polymérisation radicalaire, la polymérisation RAFT s’est imposée comme étant la technique de choix pour mettre en œuvre un procédé PISA car elle permet notamment de travailler avec une large gamme de solvants et de monomères et dans différentes conditions (voir partie I. 2).41
PAn PAn-b-PBm Copolymères amphiphiles Polymère hydrosoluble Polymérisation en dispersion ou en émulsion B Auto-assemblage in situ H2O H2O H2O H2O H2O H2O H2O Nano-objets
19 Ainsi, il est établi aujourd’hui que la PISA contrôlée par RAFT en milieu aqueux est une méthode fiable et efficace pour la synthèse de nano-objets à base de copolymères à blocs amphiphiles ayant des morphologies bien définies telles que des sphères, des fibres et des vésicules, et ceci à des taux de solide élevés (10 – 60 %mass.).34,40–43 Dans la partie qui suit nous détaillerons plus particulièrement la façon d’obtenir des fibres par PISA contrôlée par RAFT en milieu aqueux.
I. 4. Applications et mode d’obtention des nanofibres polymères
dans l’eau
I. 4. 1. Exemples d’applications des nanofibres
Parmi les différentes morphologies accessibles lors de l’auto-assemblage de copolymères dans l’eau, la morphologie filamentaire suscite un grand intérêt. Différents termes sont employés pour caractériser ce type de structures : fibre, filament, vermicelle, nanotube, micelle cylindrique ou filamentaire (filomicelle), etc… Ils permettent notamment de distinguer la forme des nano-objets : longs, courts, sous forme de tube ou de vers, etc… Nous utiliserons ici le terme général de fibres (ou nanofibres) pour évoquer les nano-objets de morphologie filamentaire, les autres termes seront employés en fonction des études présentées. Les fibres ont un facteur de formea élevé ainsi qu’une large surface accessible, potentiellement fonctionnalisable et de plus, leur nature chimique est facilement modulable.44,45 Elles peuvent ainsi présenter différentes propriétés, comme nous le verrons dans la suite de ce paragraphe, et donc jouer un rôle dans de nombreuses applications potentielles. Pour des raisons de clarté et de concision, nous nous sommes principalement intéressés dans le cadre de cette étude aux fibres à base de copolymères amphiphiles et à leurs applications, mais il est important de noter que l’intérêt pour ce type de structures s’étend aux fibres au sens large du terme (fibres de carbone,46 fibres à base de protéines ou de cellulose,47 etc…). Il ne s’agit pas ici d’être exhaustif quant aux différentes utilisations envisageables des fibres dans notre quotidien cependant, il est intéressant de mettre en évidence la grande variété de domaines dans lesquels elles peuvent intervenir. Par exemple, dans le domaine biomédical et de la santé, l’utilisation des nanofibres est très courante et en particulier dans le cadre de l’ingénierie tissulaire et de la médecine régénérative.45
20
Trois paramètres jouent un rôle essentiel pour la création de tissus : les cellules, les marqueurs biologiques et les supports.45 Les nanofibres constituent de très bons candidats pour faire office de support puisqu’elles offrent une architecture spécifique et une myriade de compositions. Une autre application principale des nanofibres, toujours dans le domaine biomédical, est leur utilisation en tant que vecteurs dans des systèmes d’administration et de relargage contrôlé de médicaments.45,47 Une étude menée dans l’équipe de Discher en 2007 a notamment montré qu’outre la nature chimique des nano-objets, la forme et la taille de ces derniers jouent un rôle important dans leur efficacité.48 Ainsi, des longues filomicelles persistent plus longtemps dans la circulation sanguine et sont plus efficaces pour la délivrance de médicaments anticancéreux, tel que le paclitaxel, en comparaison à des filomicelles courtes ou à des micelles sphériques de même nature chimique. Ce résultat repose principalement sur la structure filamentaire flexible des filomicelles : sous l’effet du flux de la circulation sanguine les particules filamentaires s’alignent, n’adhèrent que très peu aux cellules phagocytes et les contournent alors que les micelles sphériques s’accrochent aux cellules et sont assimilées.
Plus récemment, l’équipe de Perrier s’est intéressée à la synthèse d’hybrides polymère-peptide cyclique biocompatibles qui s’auto-assemblent sous forme de nanotubes.49,50 De cette façon, ils ont été capables d’élaborer un vecteur d’administration de médicaments anticancéreux efficace : dans un premier temps, ils ont tiré profit de la forte persistance des nanotubes dans la circulation sanguine pour atteindre les organes cibles puis ils ont exploité la capacité que présente le système à se désassembler pour éviter l’accumulation du vecteur dans les organes et ainsi faciliter son élimination. Au travers de cet exemple, on note que les nanofibres peuvent être facilement fonctionnalisées pour acquérir diverses propriétés. Dans cette optique, on peut également citer les travaux de l’équipe de Armes qui s’est intéressée à la synthèse de nanofibres thermosensibles pour la préparation d’hydrogels stérilisables pouvant, par exemple, être utilisés en tant que support injectable pour la croissance de cellules souches.51
Outres les nombreuses problématiques auxquelles peuvent répondre les fibres dans le domaine biomédical, elles interviennent également dans le domaine de la science des matériaux. Dans plusieurs études menées ces dernières années par l’équipe de Armes, il a notamment été montré que les longues nanofibres possèdent des propriétés interfaciales intéressantes pour la stabilisation d’émulsions de Pickering.52–55 En effet, ils ont mis en évidence que les nanofibres sont capables de s’adsorber fortement à la surface des gouttelettes, et ainsi conduire à une émulsion stable.
21 Au-delà de l’adsorption, qui est plus forte que dans le cas des nano-objets sphériques, la stabilité de l’émulsion est également liée à la taille des gouttelettes formées : les gouttelettes stabilisées par des nanofibres sont généralement plus petites que celles stabilisées par des sphères. Maruyama et al.56 se sont également intéressés à ce type de systèmes afin de créer des microréacteurs. Dans cette étude, l’émulsion a été stabilisée par des nanofibres supramoléculaires formées directement à la surface des gouttelettes suite à la réaction à l’interface huile/eau de précurseurs (Figure 5A). La fusion des gouttelettes a été induite via l’application d’un stimulus thermique au système donnant ainsi accès à des microréacteurs, pour des réactions de chimie « click » par exemple (Figure 5B).
Figure 5. Représentations schématiques de (A) la formation des nanofibres à l’interface huile/eau et (B) de la fusion des gouttelettes stabilisées par les nanofibres, induite par un stimulus, donnant lieu à des micro-réacteurs au sein desquels ont lieu des réactions chimiques (adaptée de la référence 56)
Récemment, Zhu et al.57 ont montré que les nanofibres sont également très efficaces pour stabiliser des émulsions de Pickering riches en phase dispersée (jusqu’à 84% du volume). Les émulsions riches en phase disperséeb, aussi appelées émulsions gels nécessitent très souvent des quantités importantes de tensioactif pour être stables. Dans cette étude, ils ont montré que 0,3 à 2 %mass. de nanofibres suffisent pour stabiliser efficacement une émulsion de ce type. Dans une autre étude de l’équipe de Armes, il a été montré que les fibres pouvaient aussi jouer le rôle de floculant.58 Dans cette étude, ils ont préparé des fibres chargées (via un bloc cationique) et réticulées, et ils ont évalué leur efficacité en tant qu’agent de floculation d’une suspension de silice. S’il est vrai que, dans ce cas, le caractère cationique des fibres est très important pour permettre l’interaction avec les particules de silice chargées négativement, l’étude montre surtout ici que la réticulation est nécessaire. En effet, elle permet de garder les fibres intactes après l’adsorption sur les particules de silice, ce qui est un point clé puisque l’efficacité des fibres en tant qu’agent floculant repose sur le fait que la longueur des fibres est comparable au diamètre des particules de silice.
b Ces émulsions sont très connues sous le terme HIPE = high internal phase emulsion
22
Outre leur mission de stabilisation ou floculation, les nanofibres peuvent également impacter la rhéologie du milieu dans lequel elles sont dispersées.59 Cette fonction de modificateur de rhéologie a notamment suscité un grand intérêt à l’échelle industrielle.60,61 En effet, des particules sphériques (à 19,6 %mass. dans la dispersion) ne modifient que très peu la viscosité dynamique de la dispersion (η(à 10-1s-1) = 8 mPa.s comparée à η(à 10-1 s-1) = 1 mPa.s pour l’eau distillée), alors que la viscosité d’une dispersion de particules filamenteuses (également à 19,6 %mass.) est élevée (η(à 10-1 s-1) = 870 000 mPa.s). De plus, une telle dispersion se comporte comme un rhéofluidifiant (η(à 10 s-1) = 23 000 mPa.s), ce qui est intéressant notamment pour la formulation de produits cosmétiques ou de peintures.60 Par la suite, il a également été montré que l’utilisation d’un viscosifiant à base de particules filamenteuses permet d’améliorer la résistance au vieillissement thermique de la formulation.61 Plus récemment, dans l’équipe de Rieger, il a été montré que les nanofibres peuvent être utilisées pour renforcer des films polymères acryliques.62 A cette fin, des fibres à base de polystyrène (bloc hydrophobe) qui possède une Tg élevée (~ 100 °C), ont été synthétisées et incorporées en tant qu’additif dans des latex industriels afin d’obtenir des films polymères renforcés. Les études de traction mécanique ont montré que les films contenant les fibres (à 5 ou 10 %mass.) sont plus rigides que leurs homologues sans additif ou contenant des sphères (à 5 ou 10 %mass.), tout en conservant une certaine élasticité. Une nouvelle fois, c’est de leur facteur de forme élevé que les fibres tirent leur avantage. En effet, de par cette forme particulière, elles sont capables de former un réseau percolant rigide au sein de la matrice polymère ce qui confère au matériau de meilleures propriétés mécaniques et ceci même pour des teneurs assez faibles en additif (≤ 10 %mass.).
Enfin, une autre application notable des nanofibres est leur utilisation en tant que nanocatalyseur. Guler et al.63 se sont intéressés à la synthèse de nanofibres peptidiques fonctionnalisées par du palladium. Cette structure hybride a été utilisée en tant que catalyseur d’une réaction de couplage de Suzuki-Miyaura. L’étude a mis en évidence que ce système a une bonne activité catalytique, ceci dans des conditions douces, et que le nanocatalyseur peut être facilement régénéré.
En résumé, ces quelques exemples illustrent bien le potentiel intéressant des nanofibres dans différents domaines d’application, allant de la nanomédecine aux matériaux en passant par des applications de chimie plus spécifiques, ce qui justifie sans aucun doute l’intérêt accru des scientifiques pour ce type de structure.
23 I. 4. 2. Les différentes méthodes de préparation des nanofibres
Comme nous venons de le voir, les fibres polymères sont utilisées dans de nombreux domaines et cela depuis leur découverte, que nous estimons à 1932 avec les travaux de Carothers et Hill portant sur le Nylon (6-6).64 Bien que notre propos se limite aux fibres polymères synthétiques, il est important de noter que les fibres naturelles sont présentes dans la nature depuis des millénaires et sont également très utilisées dans le domaine des matériaux ou du textile par exemple.65 De ce fait, plusieurs méthodes de préparation des fibres polymères ont été développées.44 Certaines méthodes de synthèse de nano-objets, notamment basées sur l’auto-assemblage de copolymères, ont déjà été citées précédemment telles que la méthode du co-solvant ou la méthode de réhydratation de film. Ces dernières ne sont cependant pas exclusives à la préparation des nanofibres et conduisent également à d’autres morphologies ; à l’instar de la méthode de réhydratation de film qui est particulièrement utilisée pour former des vésicules géantes.7 D’autres méthodes plus spécifiques à la préparation des fibres ont alors été mises au point : l’électrofilage (« electrospinning »),66,67 la réplication de particules dans des modèles non mouillants (« Particle Replication In Non-wetting Templates », PRINT),68 l’étirage de films,69 l’auto-assemblage induit par la cristallisation dirigée (« Crystallization-Driven Self-Assembly », CDSA),70–73 etc… L’auto-assemblage induit par la cristallisation (CDSA) est une technique récente qui présente un avantage particulier : elle permet de contrôler la longueur des fibres. Cette technique se base sur l’auto-assemblage de copolymères semi-cristallins : au cours du procédé l’un des blocs est soluble dans le solvant choisi et assure la stabilité de l’assemblage alors que l’autre bloc cristallise ; c’est la cristallisation de ce bloc qui dirige l’auto-assemblage. Généralement, la méthode mise en œuvre est la suivante : des micelles cylindriques préformées, de taille aléatoire, sont fragmentées en micelles cylindriques très courtes qui jouent ensuite le rôle de semence. L’ajout de polymère non assemblé dans le milieu conduit ensuite à la formation de longues fibres de taille contrôlée via la croissance de la semence. Ce procédé est très efficace mais il faut noter qu’il reste limité par le choix des polymères puisque l’un des prérequis est l’utilisation de copolymères semi-cristallins.
Enfin, une autre technique qui permet également la synthèse de nanofibres est la PISA. Pour les raisons évoquées dans les parties précédentes, la PISA est de plus en plus utilisée pour la synthèse et l’auto-assemblage de copolymères amphiphiles sous forme de nanofibres dans l’eau à des hauts taux de solide. Dans la partie qui suit, nous nous attacherons donc à présenter les différents systèmes décrits jusqu’à présent dans la littérature permettant la synthèse de nanofibres par PISA dans l’eau et nous évoquerons aussi leurs limitations.
24
I. 4. 3. Synthèse de nanofibres par le procédé PISA contrôlée par RAFT dans l’eau
S’il est vrai qu’il existe une myriade de monomères disponibles pour la synthèse du bloc hydrophile stabilisant – appelé dans le cas de la polymérisation radicalaire contrôlée par RAFT, le « macro-RAFT » – le nombre de monomères pour la synthèse du second bloc hydrophobe reste encore limité. Les exemples de systèmes permettant de produire des fibres par PISA contrôlée par RAFT dans l’eau que l’on trouve actuellement dans la littérature concernent principalement la polymérisation en dispersion du méthacrylate de 2-hydroxypropyle (HPMA) et du N-(1,1-diméthyl-3-oxobutyl) acrylamide (DAAm), couramment appelé diacétone acrylamide, et la polymérisation en émulsion du styrène, du méthacrylate de méthyle (MMA) et du méthacrylate de benzyle (BzMA) dont les structures se trouvent en Figure 6. De nombreuses études basées sur ces monomères ont été décrites dans la littérature et les systèmes sont maintenant bien maitrisés comme on le verra dans toute cette partie. Plus récemment, d’autres systèmes basés sur des monomères encore très peu exploités pour la synthèse de fibres par PISA dans l’eau ont vu le jour (e.g. fibres à base de poly(acrylate de 2-méthoxyéthyle) (PMEA), de poly(méthacrylate de glycidyle) (PGlyMA), de poly(acrylate de tert-butyle) (PtBA) ou de polymères ioniques), nous les évoquerons succinctement en clôture de cette partie.
Figure 6. Structures des principaux monomères permettant la formation de fibres par PISA contrôlée par RAFT dans l’eau.
i) Synthèse de nanofibres par polymérisation radicalaire contrôlée par RAFT en dispersion
Le système le plus étudié pour la synthèse de nanofibres se base sur la polymérisation en dispersion aqueuse de l’HPMA en présence d’un macro-RAFT à base de méthacrylate de glycérol (GMA). L’équipe de Armes a particulièrement étudié ce système et a montré que de nombreux paramètres influencent la morphologie des nano-objets formés, notamment la taille de chacun des blocs et la concentration en copolymère.74 Pour obtenir une morphologie donnée, il est nécessaire de moduler les différents paramètres et ceci de façon concomitante.
25 En effet, dans le cas des copolymères diblocs PGMA-b-PHPMA, il a été montré que lorsque le bloc hydrophile est court (DPn = 47) la concentration du copolymère ne semble avoir que peu d’influence sur la morphologie (Figure 7A) et dans ce cas c’est uniquement la variation de la taille du bloc hydrophobe qui détermine la morphologie. Alors que lorsque le bloc hydrophile est plus long, il y a une forte dépendance de la morphologie à la concentration en copolymère (Figure 7B). Ainsi, pour un copolymère de composition donnée, la morphologie évolue avec l’augmentation de la concentration. A une concentration en copolymère donnée, par exemple 25 %mass., la morphologie évolue des sphères vers les fibres puis vers les vésicules avec l’augmentation de la taille du bloc hydrophobe.
Figure 7. Diagrammes de phase pour une série de copolymères PGMAx-b-PHPMAy synthétisés par
polymérisation contrôlée par RAFT en dispersion aqueuse, à 70 °C à différentes concentrations en copolymère et les clichés obtenus par microscopie électronique en transmission (TEM) correspondants pour x = 47 (A) et x = 78 (B) et pour différentes valeurs de y (DP du bloc de PHPMA). S = sphères, W = fibres, BW = fibres branchées, V = vésicules (adaptée à partir de la référence 74).
En compilant leurs nombreux travaux sur ce système,51,74–77 ils ont notamment dressé un diagramme de phase pour une série de copolymères diblocs PGMAx-b-PHPMAy synthétisés à 70 °C, à un taux de solide de 20 %mass. (Figure 8).
A
26
Là encore, à une taille de bloc hydrophile (PGMA) donnée, par exemple DPn ~ 45, la morphologie évolue des sphères vers les fibres puis vers les vésicules avec l’augmentation de la taille du bloc hydrophobe (PHPMA) comme le prévoit le paramètre d’empilement. Ce diagramme de phase permet notamment de mettre en évidence les compositions qui conduisent à la formation de fibres.78
Figure 8. Diagramme de phase d’une série de copolymères diblocs PGMAx-b-PHPMAy synthétisés à
70 °C, à un taux de solide de 20 %mass., à l’exception des copolymères ayant un bloc hydrophile avec un
DPn ≤ 47, synthétisés à 10 %mass.. PGMA DP = x ; PHPMA DP = y (adaptée à partir de la référence 78).
Au cours de ces études, ils ont également mis l’accent sur le comportement thermosensible de ces fibres.51,75,79 En effet, en abaissant la température de 25 °C à 5 °C, une transition de morphologie des fibres vers les sphères est observée ce qui est dû à la plastification du bloc PHPMA, partiellement hydraté, qui induit une diminution du paramètre d’empilement. Récemment, ils ont montré que ce caractère thermosensible peut être supprimé en incorporant des unités de méthacrylate de glycidyle (GlyMA) plus hydrophobes que celles de l’HPMA, au sein du bloc hydrophobe.79 Dans cette étude, ils ont synthétisé une série de copolymères PGMA56-b-P(HPMAy-co-GlyMAz), avec y+z = 144 et z = 0, 7, 14, 22 ou 29 et dans un premier temps, ils ont montré que la morphologie filamentaire est bien obtenue dans tous les cas. Par la suite, ils ont mis en évidence via des études rhéologiques que le caractère thermosensible du système est progressivement supprimé en augmentant la teneur en GlyMA dans le bloc hydrophobe. Sachant que le GlyMA est plus rapidement consommé que le HPMA durant la copolymérisation, augmenter le teneur en GlyMA conduit à enrichir la quantité de GlyMA à la jonction entre le bloc hydrophile et le bloc hydrophobe et puisque le GlyMA est plus hydrophobe que l’HPMA la température nécessaire à la plastification du bloc hydrophobe s’en trouve abaissée.
27 Durant leurs travaux, ils ont également montré qu’avec ce système la morphologie filamentaire peut être induite par un stimulus. Par exemple, en fonctionnalisant le bout de chaine des copolymères PGMA56-b-PHPMAy par une fonction acide carboxylique les nano-objets sont sensibles au pH : une transition de morphologie sphère-fibre est observée via l’acidification du milieu et ceci de façon réversible.80 Un autre moyen d’induire une transition de morphologie est l’utilisation d’un acide boronique. En effet, l’ajout d’un acide boronique dans le milieu permet d’induire une transition de morphologie vésicule-fibre par complexation des fonctions hydroxyle du bloc stabilisant (PGMA) avec l’acide boronique.81,82 La transition de morphologie est le résultat de la création de liens covalents de type ester boronique, qui induit une réduction du paramètre d’empilement.
Plus récemment, Zhang et al.83 se sont également intéressés à ce système en changeant le type d’amorçage de la polymérisation. En effet, ils ont étudié la polymérisation amorcée par irradiation lumineuse, i.e. photo-amorcéec, en dispersion dans l’eau du HPMA en présence d’un macro-RAFT à base de GMA. Dans un premier temps, ils ont examiné l’influence du type d’amorçage sur la cinétique de la polymérisation en comparant la cinétique d’une polymérisation conduite à 70 °C amorcée thermiquement via la décomposition de l’acide 4,4’-azobis(4-cyanopentanoïque) (ACPA) et celle d’une polymérisation photo-amorcée, par le phenyl-2,4,6-triméthylbenzoylphosphinate de sodium (SPTP), conduite à 25 °C, sous une irradiation à 405 nm. Ils ont ainsi montré que la polymérisation est bien plus rapide dans le cas d’un photo-amorçage – la conversion complète du monomère est atteinte en 15 min, contre 2 h dans le cas de la polymérisation amorcée thermiquement – tout en conservant un bon contrôle de la polymérisation. Ceci résulte en fait du choix de l’amorceur puisque le SPTP se décompose très rapidement à 405 nm, son temps de demi-vie est estimé à 17 min dans ces conditions, alors que le temps de demi-vie de l’ACPA est de 10 h à 70 °C. Outre cette étude, ils ont ensuite investigué l’influence de la température sur la cinétique de la polymérisation photo-amorcée et sur la morphologie des nano-objets formés. Ils ont ainsi montré que la cinétique de la polymérisation photo-amorcée en présence de SPTP est la même à 25 °C, 50 °C et 70 °C et que la polymérisation peut également être conduite à 0 °C tout en conservant une cinétique relativement rapide (une conversion > 98 % est atteinte en 40 min).
28
Afin d’évaluer l’influence de la température sur la morphologie, ils ont établi et comparé les diagrammes de phase des copolymères PGMA68-b-PHPMAy synthétisés à 25 °C et 70°C, à différentes concentrations en monomère (Figure 9.). Ces diagrammes montrent clairement que la morphologie des nano-objets est dépendante de la température. A 25 °C, la morphologie sphérique est prédominante au dépend des morphologies d’ordre supérieur : la fenêtre d’obtention des vésicules « pures »d n’occupe qu’un petit espace sur le diagramme et celle des fibres pures n’existe pas. A 70 °C, la phase des vésicules pures est considérablement plus grande et on observe également que des fibres pures sont obtenues dans le cas du copolymère PGMA68-b-PHPMA200 à 19 et 20 %mass. en monomère. Ainsi, l’augmentation de la température semble favoriser l’obtention de morphologies d’ordre supérieur ce qui s’explique par le fait qu’en augmentant la température les chaines de PHPMA sont plus mobiles et la fusion sphère-sphère, nécessaire à la formation des fibres et des vésicules, est facilitée.83 Dans la même étude, ils montrent que les fibres peuvent tout de même être obtenues à 25 °C, en diminuant la longueur du bloc stabilisant notamment.
Figure 9. Diagrammes de phase pour une série de copolymères PGMA68-b-PHPMAy synthétisés par
polymérisation radicalaire contrôlée par RAFT, photo-amorcée sous une irradiation à 405 nm en présence de SPTP, en dispersion aqueuse à différentes concentrations initiales en monomère (HPMA), à 25 °C (A) et 70 °C (B). S = sphères, W = fibres, Mixed = phase mixte, V = vésicules ; DP of PHPMA = y (adaptée à partir de la référence 83).
De la même façon, dans une autre étude, ils ont étudié la polymérisation photo-amorcée de l’HPMA en présence de SPTP et d’un macro-RAFT poly(ethylene glycol) (PEG), à 25 °C dans l’eau.84 L’établissement du diagramme de phase d’une série de copolymères PEG
113 -b-PHPMAy a montré que différentes morphologies sont accessibles avec ce système (sphères, lamelles, vésicules uni- et multi-lamellaires, etc …) et notamment des fibres dans le cas du copolymère PEG113-b-PHPMA100 synthétisé à un taux de solide de 25 %mass..
d La notion de morphologie/phase « pure » indique que c’est la seule morphologie qui est obtenue, en opposition
aux phases mixtes qui sont composées d’un mélange de morphologies.