Erreur ! Utilisez l'onglet Accueil pour appliquer Heading 7 au texte que vous souhaitez faire apparaître ici.Erreur ! Utilisez l'onglet Accueil pour appliquer Heading 7 au texte que vous souhaitez faire apparaître ici.
i
Département de
Génie de Procédés
Rapport de soutenance
En vue de l’obtention du diplôme
de Licence professionnalisant en :
Génie Chimique
Thème :
Réalisé par :
Mr BENATTOU Adel
Devant le jury composé de :
Pr BELALIA Fatiha
Président
Mme BENHOURIA Assia Examinatrice Mme DAIRI Nassima Encadreur
Année Universitaire 2017/2018
Fabrication et contrôle de qualité d’un médicament :
Dédicaces
A la mémoire de ma tendre chère mère
A mon cher père
A mes sœurs et frères
A mes amis et à tous ceux qui ont
contribué de prés ou de loin pour que
ce projet soit possible, je vous dis merci.
Remerciements
Je remercie en premier Dieu le tout puissant qui m’a donné le courage et la volonté pour réaliser ce travail.
En second lieu, je tiens à remercier mon enseignante et mon encadreur Mme N. DAIRI pour son aide et ses précieux conseils, pour sa disponibilité et pour son investissement constant.
Je remercie vivement les membres de Jury de ma soutenance qui ont accepté d’examiner mon rapport de stage.
Je tiens à remercier tout particulièrement Mr D. BAKHAT, mon superviseur professionnel, chef de production au sein du Site de production Gué de Constantine de SAIDAL, pour son accueil et la confiance qu’il m’a accordé dès mon arrivée à ce site.
Je remercie également toute l'équipe du laboratoire physico-chimique pour leur accueil, leur esprit d'équipe et en particulier Mr NOUASSE, qui m'a beaucoup aidé à comprendre le contrôle de qualité
de médicament.
J’adresse aussi mes remerciements, à tous les enseignants, qui m’ont donné les bases de la science et qui ont contribué à ma formation.
Je remercier enfin, ma famille et mes amis pour leur soutien sans limites et pour m’avoir encouragé tout au long de ce travail.
Sommaire
Sommaire
Liste d’abréviation Liste des tableaux Liste des figures
Introduction ……….………..1
Chapitre I : Synthèse Bibliographique I. Généralité sur le groupe SAIDAL ... 2
I.1. Présentation de groupe SAIDAL ... 2
I.2. Historique de groupe SAIDAL ... 2
I.3. Organisation de groupe SAIDAL ... 3
II. Généralité sur les médicamentes ... 5
II.1.Définition de médicament ... 5
II.2.Mise en forme du médicament ... 5
II.3.Dénomination d’un médicament ... 6
II.4.Voies d’administration des médicaments ... 6
II.5.Médicament antianémique ... 7
III. Contrôle de qualité (CQ) ... 7
III.1. Définition du CQ ... 7
III.2. Types de CQ ... 7
III.2.1. Contrôle physico-chimique ... 7
III.2.2. Contrôle Microbiologique………...8
III.2.3. Contrôle Toxicologique………….……… 9
Chapitre II : Fabrication et contrôle de qualité de ZANITRA®5mg I. Identification du médicament ... 10
I.1. Mode et voie d’administration ... 10
I.2. Effets thérapeutiques de ZANITRA®5mg ... 10
I.3. Principe actif (acide folique) ... 10
Sommaire
I.3.2. Structure chimique………...11
I.4. Présentation des excipients ... 11
I.4.1. Amidon de maïs………...11
I.4.2. Phosphate bicalcique………...12
I.4.3. Stéarate de magnésium………12
II. Fabrication des comprimés de ZANITRA®5mg ... 12
II.1. Formule de fabrication ... 12
II.2. Etapes de fabrication ... 13
III. Contrôle de qualité ... 16
III.1. Contrôle physico-chimique ... 16
III.1.1. Matières premières ... 17
A/ PA (Acide Folique) ... 17
B/ Excipient ... 19
III.1.2. Produit au cours de fabrication ... 22
III.1.3. Produit fini ... 24
III.2. Contrôle microbiologique du produit fini ... 26
III.3. Contrôle toxicologique du produit fini ... 27
Chapitre III : Résultats et Discussion I. Contrôle physico-chimique ... 29
I.1. Matières premières ... 29
I.1.1. Acide folique ... 29
I.1.1.1. Résultats………..……….29
I.1.1.2. Discussion ... 30
I.1.2. Amidon de maïs………..……….31
I.1.2.1. Résultats ... 31
I.1.2.2. Discussion………..………..32
I.2. Au cours de Fabrication……..………33
I.2.1. Résultats………..……….33
I.2.2. Discussion..………..33
Sommaire
I.3.1. Résultats…………..……….34
I.3.2. Discussion…………..………..34
II. Contrôle microbiologique du produit fini ... 35
II.1. Résultats………35
II.2. Discussion……….35
III. Contrôle toxicologique du produit fini ... 36
III.1.Résultat ... 36
III.2. Discussion...……….36
Conclusion…………..………..37 Références bibliographiques
Résumé
ZANITRA®5mg est un médicament antianémique sous forme de comprimé, fabriqué au sein de Groupe SAIDAL, filiale biotic (Gué de Constantine). Notre travail a porté sur le suivi de toutes les étapes de fabrication de ce comprimé ainsi que le contrôle de qualité de la matière première, produit au cours de fabrication et produit fini.
Les résultats obtenus ont révélé la conformité de ZANITRA®5mg aux spécifications de qualité des comprimés et la conformité de ce médicament aux critères de qualité, d’efficacité et de sécurité.
Mots clés : contrôle de qualité, comprimé, ZANITRA®5mg.
صخلم
ارتيناز 5 لاديصتكرش فرط نم تعىنصم صارقا مكش ىهع ودنا رقفن داضم ءاود ىه غم ) تنيطنسق رسج ( . يف تينولاا ةدامهن ةدىجنا تبقارم اذك و ءاودنا اذه عينصت ثاىطخ مك عابتإب انمق ممعنا اذه , و جاتنلاا محارم ءانثا جىتنمنا يئاهننا جىتنمنا , و ةءافكنا و ةدىجنا ريياعم عم ارتيناز ءاود صارقا ةدىج ثافصاىم تقباطم نيبت اهيهع مصحتمنا جئاتننا تملاسنا ) نملأا ( . ثحبلا تاملك : ةدىجنا تبقارم , صارقا , ارتيناز 5 غم .Liste des tableaux
Tableau 1 : Masse des matières premières utilisées pour un lot de 100g…………....………12
Tableau 2 : Résultats de l’acide folique………….………..………29
Tableau 3 : Résultats chromatographiques de la solution témoin………….…………..……30
Tableau 4 : Résultats chromatographiques de la solution essai………..….30
Tableau 5 : Résultats de l’amidon de maïs………..…31
Tableau 6 : Résultats du comprimés au cours de fabrication………..33
Tableau 7 : Résultats du produit fini………..…..34
Liste des figures
Figure 1 : Symbole du groupe SAIDAL………..………2
Figure 2 : Voies d’administration des médicaments……….………...6
Figure 3 : Boite du comprimé ZANITRA®5mg……….10
Figure 4 : Formule développée de l’AF……….…..11
Figure 5 : Cuiseur………...……13
Figure 6 : Mélangeur collette……….………..……13
Figure 7 : Calibreur………..……14
Figure 8 : Etuve à plateaux………..……14
Figure 9 : Comprimeuse……….………….…15
Figure 10 : Polarimètre………18
Figure 11 : Karel ficher……….……..18
Figure 12 : Appareil de HPLC……….19
Figure 13 : Pied à coulisse……….………..22
Figure 14 : Friabilimètre……….……….23
Figure 15 : Appareil de délitement………...……….……..23
Figure 16 : Duretémètre………...…………24
Figure 17 : Appareil de dissolution……….…….25
Figure 18 : Spectrophotomètre……….…………26
Figure 19 : Souri albinos de race SWISS………..…………..….28
Figure 20 : Chromatogramme de la solution témoin<b>……….30
Figure 21 : Chromatogramme de la solution essai……….………..30
Liste des abréviations
PCA : Pharmacie Centrale Algérienne
SNIC : Société Nationale des Industries Chimiques D.C.I : Dénomination Chimique Internationale PA : Principe actif
CQ : Contrôle de qualité MP : Matière première AF : Acide folique
SCR : Substance Chimique de Référence SA : Substance actif
UFC : Unité Formant Colonie BVT : Bactéries viables totales
VRBG : Gélose glucosée Biliée au cristal Violet et au Rouge neutre TBG : Tétra-thionate-Bile-vert-brillant
XLD : Xylose-lysine-Désoxycholate
BPLS : Gélose vert Brillant-rouge de Phénol-Lactose-Saccharose R :Pure.
PE : Pharmacopée Européenne.
UICPA : Union Internationale de Chimie pure et Appliquée. Cp : Comprimé
Introduction
1
Introduction
L’industrie pharmaceutique est confrontée à la perte de nombreux brevets et à une concurrence de plus en plus précoce des génériques. En plus de cette pression financière, l’industrie pharmaceutique doit faire face à une demande croissante pour le contrôle de qualité des produits qu’elle fabrique. Ces tests de contrôle qualité en fin de processus de fabrication sont chers, c’est bien après sa production que la qualité d’un lot est contrôlée. Le lot contrôlé peut éventuellement s’avérer défectueux. Ainsi, faute de contrôle qualité en temps réel de fabrication, des lots entiers peuvent être détruits, ce qui représente un cout non négligeable.
L’assurance de la qualité appliquée à la fabrication des médicaments et garantissant notamment la bonne organisation des activités de production et des contrôles est essentielle pour que le consommateur reçoive des médicaments satisfaisant aux normes.
Ainsi, le rôle des laboratoires de contrôle de la qualité est de vérifier, par des essais appropriés, que les médicaments satisfont aux normes de qualité requises le but des bonnes pratique de fabrication est de régir la fabrication des produits pharmaceutiques.
L’assurance de la qualité des médicaments génériques et de leur conformité aux exigences réglementaires est un point très important pour leur efficacité thérapeutique.
L’objectif de notre travail est de faire le suivi de la fabrication et le contrôle de qualité de ZANITRA®5mg tout en appliquant les essais sur la qualité microbiologique, physico-chimique et toxicologique du produit fini. Ce contrôle a pour objectif de garantir la bonne qualité de ce médicament.
Le travail qui fait l’objet de ce rapport se divise en trois parties :
La première partie est consacrée à une étude bibliographique sur les médicaments et le contrôle de qualité des médicaments.
La deuxième partie porte la description des procèdes de fabrication des comprimés ZANITRA®5mg ainsi le détail des tests de contrôle de qualité effectués.
Chapitre I
Chapitre I Synthèse Bibliographique
2
I. Généralité sur le groupe SAIDAL
I.1. Présentation de groupe SAIDAL
SAIDAL est une société par actions, au capital de 2.500.000.000 dinars algériens, dont l’objectif primordial est d’accroitre, de créer et de distribuer des produits pharmaceutiques à usage humain et vétérinaire [2].
I.2.Historique de groupe SAIDAL
[3]SAIDAL a été crée en avril 1982 à la suite de la restructuration de la Pharmacie Centrale Algérienne (PCA) et a bénéficié, dans ce cadre, du transfert des usines d’El Harrach, de Dar El Beida et de Gué de Constantine. Il lui a été également transféré en 1988, le Complexe ‘’Antibiotiques’’ de Médéa dont la réalisation venait d’être achevée par la SNIC (Société Nationale des Industries Chimiques).
En 1989 et suite à la mise en œuvre des réformes économiques, SAIDAL devint une entreprise publique économique dotée de l’autonomie de gestion.
En 1993, des changements ont été apportés aux statuts de l’entreprise, lui permettant de participer à toute opération industrielle ou commerciale pouvant se rattacher à l’objet social par voie de création de sociétés nouvelles ou de filiales.
En 1997, la société SAIDAL a mis en œuvre un plan de restructuration qui s’est traduit par sa transformation en groupe industriel regroupant trois filiales (Pharmal, Antibiotical et Biotic).
Chapitre I Synthèse Bibliographique
3 En 2009, SAIDAL a augmenté sa part dans le capital de SOMEDIAL à hauteur de 59%. En 2010, elle a acquis 20 % du capital d’IBERAL et sa part dans le capital de TAPHCO est passée de 38,75% à 44,51%.
En 2011, SAIDAL a augmenté sa part dans le capital d’IBERAL à hauteur de 60%. En janvier 2014, SAIDAL a procédé par voie d’absorption, à la fusion de ses filiales détenues à 100% : Pharmal, Antibiotical et Biotic.
I.3. Organisation de groupe SAIDAL
[4]Les sites de production : le groupe SAIDAL compte 09 usines de production : a. Site de production de Dar El Beida
L’unité de Dar El Beida existe depuis 1958. L'activité de cette unité était limitée en la fabrication de quelques médicaments et produits cosmétiques, mais actuellement elle produit une gamme de médicaments très large dans plusieurs formes galéniques : comprimés, gélules, sirops (solutés buvables), forme pâteuses (pommades, gel, crème), suspension buvable, sels, et solution dermique.
Aussi l’usine est dotée d’un laboratoire de contrôle de la qualité chargé de l'analyse Physico-chimique et microbiologique et d’une surface de stockage de 6.600 m².
b. Site de production de Médéa
Le Complexe Antibiotiques, dont la production a démarré en 1988, produit les formes galéniques suivantes : injectables, gélules, pommades, sirops et comprimés. Le site est caractérisé par une capacité de production importante dans la fabrication de matières premières en vrac et des spécialités pharmaceutiques et des laboratoires d'analyse permettant le contrôle complet de la qualité.
c. Site de production de Constantine
L’usine de Constantine se compose de deux ateliers de sirops avec une capacité de production de 20 000 UV/jour.
d. Site de production d’El-Harrach
L’usine El-Harrach dispose de quatre ateliers, un atelier sirops, un atelier solutions, un atelier comprimés et dragées et un atelier pommades avec une capacité de production de 20 millions d’unités de vente.
Chapitre I Synthèse Bibliographique
4 e. Site de production de Cherchell
L’usine de Cherchell se compose d’un atelier de production avec une capacité de production de plus de 200.700 unités de ventes. Unique producteur algérien du concentré d’hémodialyse, il est doté d’un laboratoire contrôle de la qualité chargée du contrôle physico-technique, microbiologique et pharmaco-toxicologique.
f. Site de production de Batna
Spécialisé dans la production des suppositoires avec une capacité de production de 3 millions d’unités de vente par an.
g. Site de production d’Annaba
Cette usine est spécialisée dans la fabrication des formes sèches (comprimés et gélules), elle a été transférée auparavant à la filiale Pharmal suite à la dissolution de L‟ENCOPHRAM en date du 31 Décembre 1997.
h. Site de production de Constantine- unité d’Insuline
Spécialisé dans la production d’insuline humaine à trois types d’action : rapide (Rapid), lente(Basal) et intermédiaire (Comb 25).
i. Site de production du Gué de Constantine Il se compose de deux parties distinctes :
La première partie pour la fabrication des formes galéniques, suppositoires, ampoules buvables et comprimés.
Une autre partie dotée d’une technologie très récente est spécialisée dans la production des solutés massifs, poches et flacons.
Avec une capacité de production de plus de 18 millions d’unités de vente, Cette usine est dotée d’un laboratoire de contrôle de la qualité chargé de l’analyse physico-chimique, microbiologique et toxicologie et de la gestion technique et documentaire.
Chapitre I Synthèse Bibliographique
5
II.
Généralité sur les médicamentes
II.1. Définition du médicament
a) Juridique
On entend par médicament toute substance ou composition possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales. Un médicament exerce une action pharmacologique, immunologique ou métabolique [5].
b) Pharmacologique
Un médicament est composé d’une substance active et de (substances inertes) Convenables à la voie d’administration à laquelle il est acquis, appelées excipients. La substance active présente une concentration indiquée appelée dosage. Il dispose des propriétés pharmacologiques qu’il entreprend sur une cible organique ou fonctionnelle dans lequel les révélations curatives ou thérapeutiques s’ensuivront [6].
II.2. Mise en forme du médicament
Principe actif
Le principe actif, ou substance active, est la molécule entrant dans la composition d’un médicament qui lui confère ses effets curatifs. Un médicament peut comporter un ou plusieurs principes actifs [7].
Excipient
Tous les éléments entrant dans la composition d’un médicament, autres que le principe actif, sont généralement des excipients.
L’excipient est une substance d’origine chimique ou naturelle qui facilite l’utilisation du médicament mais ne présent pas d’effet curatif ou d’entrer dans la composition du vecteur contribuant ainsi à certaines propriétés du produit tel que la stabilité, le profil biopharmaceutique. L’aspect et l’acceptabilité pour le patient et facilité de fabrication [8].
Les excipients sont classés en plusieurs catégories à savoir
Diluants
Ils jouent un rôle de remplissage lorsque la quantité de PA est insuffisante pour faire un Cp de taille convenable.
Chapitre I Synthèse Bibliographique
6
Liants ou agglutinants : Leur rôle est de lier entre elles les particules qui ne peuvent l’être sous la seule action de la pression. Leur présence permet de réduire la force de compression.
Lubrifiants : ils jouent un triple rôle dans la fabrication des Cp
– Amélioration de la fluidité du grain donc du remplissage de la chambre de compression, ce qui est important pour la régularité de poids (pouvoir glissant) ;
– Diminution de l’adhérence du grain aux poinçons et à la matrice (pouvoir anti adhèrent) ; – Réduction des frictions entre les particules pendant la compression, ce qui assure une
meilleure transmission de la force de compression dans la masse du grain (pouvoir antifriction) ;
A ces trois rôles importants vient s’ajouter un intérêt supplémentaire des lubrifiants : ils donnent un bel aspect, brillant et non poussiéreux, aux Cp [9].
II.3. Dénomination d’un médicament
Tout médicament est caractérisé par la désignation chimique de son principe actif, Le nom chimique est l’interprétation exacte de la molécule chimique du médicament. Le nom de spécialité ou nom de marque de la molécule est donne par le laboratoire qui le commercialise. Le signe <®> qui joint les noms de spécialités désigne (Registred) en anglais, c’est-à dire Propriété commerciale [10].
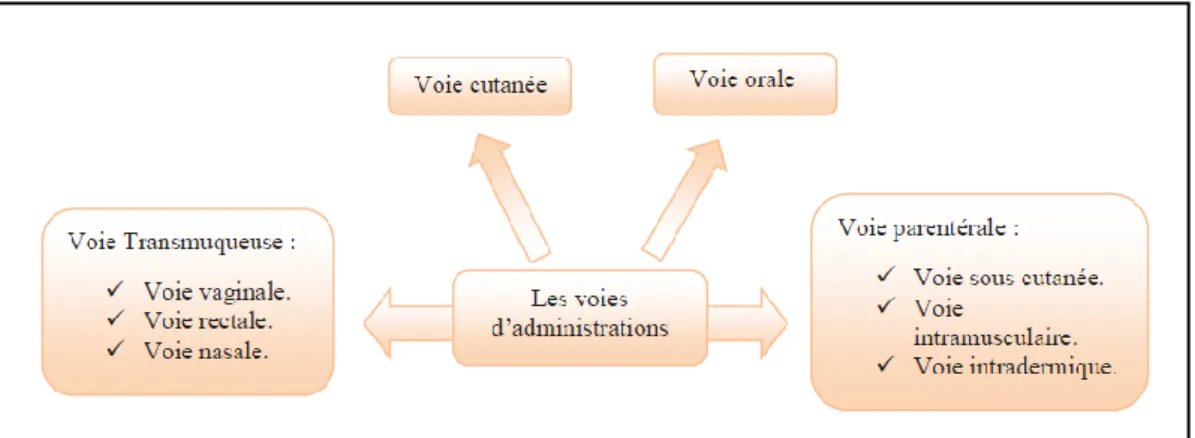
II.4. Voies d’administration des médicaments
Il existe plusieurs voies d’administration des médicaments [11]
Chapitre I Synthèse Bibliographique
7
II.5. Médicament antianémique
Les antianémiques sont des médicaments capables de stimuler la formation des globules rouges [12].
Ils perfectionnent la captation et le transport de l’oxygène par l’hémoglobine des globules rouges. Ils facilitent la synthèse de l’hémoglobine par un apport en fer, vitamine B12, acide folique, car l’absence de chacun de ces éléments joue un rôle crucial dans l’anémie [13].
III. Contrôle de qualité (CQ)
III.1. Définition du CQ
Le contrôle qualité des médicaments est un ensemble de mesures qui permet de savoir si les médicaments fabriqués ou vendus par une entreprise sont conformes.
• Aux exigences du marché ; • A la demande du client ; • Aux législations en vigueur ;
• Au cahier des charges de l'entreprise.
III.2. Types de CQ
III.2.1. Contrôle physico-chimique
Sert à vérifier la structure de la molécule et d’établir les propriétés physiques et chimiques. Il permet ainsi de vérifier et de s’assurer du bon usage de la substance annoncée
Dureté
Le test de dureté permet de s’assurer que les Cp présentent une résistance mécanique suffisante pour ne pas se briser lors de leurs manipulations ou d’étapes de production ultérieures.
Friabilité
Le test de friabilité permet de s’assurer que les Cp présentent une résistance mécanique suffisante, pour que leurs surfaces ne soit pas endommagées ou ne présentent pas
Chapitre I Synthèse Bibliographique
8 des signes d’abrasion ou de rupture, sous l’effet de toutes les manipulations (chocs mécaniques, frottements, attrition) qu’ils vont subir jusqu’au moment de leur utilisation.
Uniformité de masse
L’essai d’uniformité de masse des Cp permet de s’assurer qu’au cours de la fabrication, la répartition du mélange initial de poudre ou de granulés, en unités de prises (chaque Cp), a été suffisamment précise et uniforme pour garantir une même masse et donc une même teneur en PA pour l’ensemble des Cp d’un même lot.
Désagrégation
Le test de désagrégation des Cp non enrobés permet de s’assurer, que leur vitesse de désagrégation ne constitue pas le facteur limitant de la dissolution du PA qu’ils contiennent.
Dissolution
Le test de dissolution in vitro appliqué aux, permet de s’assurer, qu’une fois administrés, ces derniers libèreront le PA qu’ils contiennent, pour le mettre à la disposition de l’organisme, et ceci dans les limites de concentration et de vitesse déterminées, afin de garantir l’effet thérapeutique désiré.
Test d’identification du PA
L’identification d’un PA contenu dans un Cp, a pour but de s’assurer que ce dernier contient bel et bien le PA spécifié par le fabricant.
Les méthodes d’identification du PA les plus citées parles pharmacopées sont → Spectrophotométrie d’absorption dans l’infrarouge ;
→ Spectrophotométrie d’absorption dans l’ultraviolet et le visible ; → Chromatographie liquide à haute performance (HPLC) ;
→ Chromatographie sur couche mince ;
→ Réactions chimiques caractéristiques du PA (réactions colorées en tube par exemple). III.2.2. Contrôle Microbiologique
Les contrôles microbiologiques doivent permettre de garantir une bonne qualité hygiénique et marchande du produit fabriqué, et minimisent les pertes dues aux mauvaises conditions de fabrication.
Chapitre I Synthèse Bibliographique
9 III.2.3. Contrôle Toxicologique
Les molécules destinées à la thérapeutique humaine doivent subir avant tout essai clinique des tests de toxicité aigue et chronique sur les animaux.
Les études toxicologiques permettent d’éliminer de très nombreuses molécules dont les risques outrepassent les avantages [9].
Chapitre II
Fabrication et contrôle de
qualité de ZANITRA®5mg
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
10 Notre travail a été réalisé à l’unité BIOTIC de SAIDAL (Gué de Constantine-Alger) durant trois (03) mois.
L’objectif était de suivre la fabrication et le contrôle de qualité d’un médicament sous forme de comprimés.
I. Identification du médicament
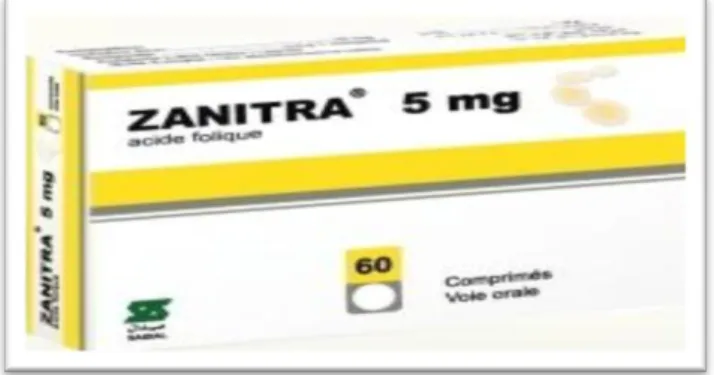
Le produit choisi pour notre stage est un comprimé : une boite de 60 comprimés portant l’inscription ZANITRA®5mgcommele montre la photo ci-dessous.
I.1. Mode et voie d’administration
Voie orale, les comprimés doivent être avalés tels quels avec une boisson (par exemple eau, lait, jus de fruits).
I.2. Effets thérapeutiques de ZANITRA®5mg
ZANITRA® est un antianémique. Il est indiqué en cas de - Anémies macrocytaires par carence en acide folique ;
- Troubles chroniques de l’absorption intestinale quelle que soit leur origine ; - Carences d’apport : malnutrition, alcoolisme chronique ;
- Grossesse, en cas de carence prouvée.
I.3. Principe actif (acide folique)
I.3.1. Définition
ZANITRA® est le nom commercial de la vitamine B9 fabriqué par SAIDAL où l’acide folique est le principe actif dosé à 5 mg. L’acide folique ou la vitamine B9, composé organique synthétisé par les plantes et les micro-organismes et non par l'homme.
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
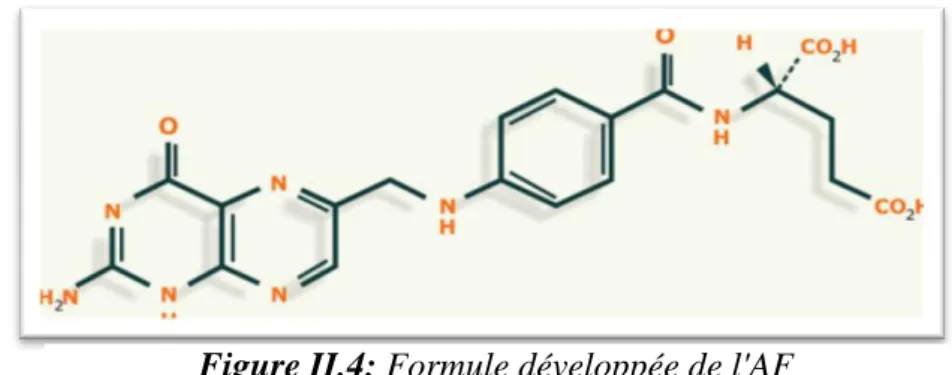
11 I.3.2. Structure chimique
𝐅𝐨𝐫𝐦𝐮𝐥𝐞𝐛𝐫𝐮𝐭𝐞 :
𝐶
19𝐻
19𝑂
6𝑁
7 Nom UICPA :(2S)-2-[[4-[[(2-amino-4-oxo-1,4-dihydroptéridin-6 yl) méthyl] amino]benzoyl] amino] pentane dioïque.
I.4. Présentation des excipients
I.4.1. Amidon de mais
Caractère
Aspect: poudre d’un blanc mat à faiblement jaunâtre, très fine, qui crisse sous la pression des doigts.
Solubilité : pratiquement insoluble dans l’eau froide et dans l’éthanol à 96 %.
Le rôle a. Un diluant :
En rouge : principe actif. En blanc : excipients.
Le principe actif occupe généralement un volume trop petit pour pouvoir réaliser un comprimé ; alors les diluants permettent d’augmenter ce volume afin de pouvoir réaliser un comprimé de taille raisonnable.
b. Un liant:
Ils permettent de lier les particules entres elles (agrégation) comme de la colle sous forme d’Empois d’amidon. Les liants peuvent être choisis de manière à jouer en même temps le rôle de diluants comme l’amidon de maïs dans notre cas.
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
12 I.4.2. Phosphate bicalcique
Caractère
Aspect: poudre cristalline, blanche ou sensiblement blanche.
Solubilité: pratiquement insoluble dans l’eau et dans l’éthanol à 96 %. La substance à examiner se dissout dans l’acide chlorhydrique dilué et dans l’acide nitrique dilué (Pharmacopée Européenne, 2014).
Le rôle
Le Phosphate bicalcique joue le rôle d’un diluant. I.4.3. Stéarate de magnésium
Caractère
Aspect: poudre blanche, très fine, légère, onctueuse au toucher.
Solubilité: pratiquement insoluble dans l’eau et dans l’éthanol anhydre (Pharmacopée Européenne, 2014).
Le rôle
Le Stéarate de magnésium joue le rôle d’un lubrifiant antifriction
Anti collage: évite que la poudre ne colle sur les poinçons de la machine à comprimer. Anti grippage : évite l’érosion des poinçons par la poudre, en général ils sont intégrés
dans le mélange avant la compression, ils donnent un bel aspect brillant et non poussiéreux au comprimé.
II.
Fabrication des comprimés de ZANITRA®5mg
II.1. Formule de fabrication
Pesée des matières premières : pour la fabrication d’un lot de 100 kg correspondant à 1 250 000 Cp.
Tableau 1 : Masse des matières premières utilisées pour un lot de 100 kg.
Matières premières Masse (Kg)
Acide folique 5
Phosphate bicalcique 68.4
Amidon de mais 25.6
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
13
II.2. Etapes de fabrication
1) Préparation de la solution de mouillage
Dissoudre une masse d’amidon de maïs dans de l’eau distillée, mettre la solution obtenue dans un cuiseur qui contient de l’eau bouillante sous agitation pendant 5 minutes afin d’avoir une solution homogène. Verser la solution obtenue dans une cuve mise dans un bain marie pendant une heure et demi au minimum.
2) Préparation du mélange à sec
Cette étape consiste à mélanger le PA avec les excipients. Introduire dans un Mélangeur Collette des quantités qui sont déjà pesées du PA avec les diluants (Amidon de maïs, Phosphate bicalcique), maintenir le mélange sous agitation pendant 10min.
3) Mouillage
Cette opération consiste à ajouter l’eau distillée et la solution de mouillage préparée précédemment au mélange à sec dans le Mélangeur Collette sous agitation pendant 5min.
Figure II.5: Cuiseur
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
14 La solution de mouillage préparée lors de la première étape a permis de lier les grains du mélange préparé à sec lors de la deuxième étape. C’est pour cette raison cette méthode est appelée la méthode de granulation par voir humide.
4) Granulation
C’est une opération qui consiste à transformer les grains obtenus de grande taille en une taille inférieure.
La granulation a été effectuée en utilisant un Calibreur de façon à obtenir des grains de taille inférieure à 2.5 mm.
5) Séchage
Les grains obtenus sont séchés dans l’étuve à 50°C pendant 6 heures après leur répartition sur des plateaux en inox. Le but de ce séchage est de diminuer le taux d’humidité des grains.
Figure II.7: Calibreur
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
15 6) Calibrage
A l’aide d’un Calibreur, les grains du mélange séché sont calibrés sur un tamis de 1.6 mm de diamètre.
7) Lubrification
Introduire dans le Mélangeur Collette, le mélange de particules de taille inférieure à 1.6 mm et les lubrifiants : le Stéarate de magnésium. Laisser le tout sous agitation pendant 5min.
8)Compression
C’est une technique utilisée en pharmacie pour former les Cp. Elle consiste à éliminer l’air libre qui trouver entre les grains du médicament.
La formation des Cp est faite par une machine rotative qui s’appelle Comprimeuse KILIN 150, son fonctionnement est le suivant :
Après le remplissage du silo de Comprimeuse par le mélange final, les grains vont remplir l’espace libre de la matrice, la Comprimeuse se fait par les poinçons supérieurs. L’éjection des Cps est réalisée par le poinçon inferieur.
Un contrôle de l’aspect et du poids ainsi que l’épaisseur du comprimé est réalisé tous les 30 min. Les contrôles de friabilité et le temps de délitement trois fois, au début, au milieu et en fin de la compression.
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
16 9) Conditionnement
A/ Conditionnement primaire
Le conditionnement primaire ce fait à l’aide d’une machine blisttirage, cette étape consiste à mettre les comprimés dans les blistères.
Articles de conditionnement
- Une bobine d’aluminium imprimé avec le nom du médicament, sa dose et le PA. - Une bobine de PVC transparent.
Etapes de conditionnement
a/Les chambres des comprimés ont été réalisés par thermoformage du film de PVC à une température 150°C ;
b/ Remplissage des chambres par les Cp à l’aide de distributeur ;
c/ Collage de la couche d’aluminium avec le film de PVC qui contient les comprimés sous l’effet de la température ;
d/Découpage en blistères à 30 comprimés ;
e/Thermoformage de N° de lot, la date de fabrication et la date de péremption. B/ Conditionnement secondaire
Consiste à mettre les blistères et la notice dans les étuis avec la vignette, mettre le N° de lot, la date de fabrication et la date de péremption.
III. Contrôle de qualité
Assurer la qualité des médicaments fabriqués est fondamentale dans tout système de santé. En effet, un médicament de mauvaise qualité pourrait mettre en péril la vie des citoyens d’un pays donné. Par ailleurs, la garantie de la qualité médicaments s’appuie sur le contrôle qualité qui est effectué pendant tout le processus de fabrication et jusqu’à la vente publique. Ce contrôle s’effectue sur la base des normes de qualité qui fournissent des descriptions détaillées des caractéristiques du médicament et des techniques analytiques à mettre en œuvre.
III.1. Contrôle physico-chimique
Le protocole suivi et les techniques utilisées pour le contrôle physico-chimique sont ceux proposés par la Pharmacopée Européenne (2014).
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
17 III.1.1. Matières premières
A/ PA (Acide Folique)
L’objectif de ces tests est de confirmer l’identité d’une substance par une comparaison avec les normes.
a) Caractère
Aspect: Poudre cristalline jaunâtre ou orangée.
Solubilité :La solubilité de l’Acide Folique a été testée dans de l’eau distillée, dans l’alcool 96%, dans HCl dilué (1/10) et dans NaOH (0,1M).
Le résultat montre que l’AF est insoluble dans l’eau et dans la majorité des solvants organiques, et il est soluble dans les acides dilués et dans les solutions alcalines.
b) Identification
Le test d’identification consiste à déterminer le pouvoir rotatoire de l’AF par un polarimètre.
- Dissoudre 0,5g de l’acide folique (AF) dans 50 ml d’une solution d’hydroxyde de sodium NaOH (0,1M) ;
- Effectue un essai à blanc avec la solution d’hydroxyde de sodium (0.1 M) ; - Examiner la solution préalablement préparée.
La lecture est faite après la stabilité de l’angle de rotation. Le pouvoir rotatoire est ensuite déterminé par la formule suivante
𝑎 = 𝑎 × 𝑉 × 100
𝑃𝑒 × (100 − 𝑇𝑒)… … … . . 𝟎𝟏
[a]: la déviation de la lumière à laquelle l’acide folique commence à se dégrader. a: Angle de rotation en degrés.
V: Le volume de solvant (50ml).
Pe: La masse de la prise d’essai de l’acide folique (proche le plus possible de 0.5g).
100 : les 100% de l’acide folique.
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
18 c) Essai limite
Teneur en eau
La détermination de la teneur en eau (Te) est faite par un appareil appelé≪ Karl Fischer ≫, dont lequel on introduit 0.15 g d’acide folique.
Dosage par chromatographie liquide à haute performance (HPLC) Préparation de la phase mobile
La phase mobile est constituée de 120ml du méthanol (96%),de 880ml d’un mélange d’une solution de phosphate mono potassique (11,16g/l) et de phosphate di-potassique (5,5g/l).
Préparation de la solution à examiner
- Dissoudre 0,1g de l’AF dans 5ml d’une solution de carbonate de sodium R à 28,6g/l dont 100 ml de la phase mobile ont été rajoutés ;
- Diluer la solution de l’AF préparée par la phase mobile (2ml de cette solution avec 10ml de la phase mobile) ;
Figure II.10 : Polarimètre
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
19 Préparation de la solution témoin
Solution témoin <a>
Préparer la solution témoin < a > de la même manière que la solution à examiner, en utilisant l’AF SCR.
Solution témoin <b>
Dissoudre 20mg de l’AF SCR dans 5ml d’une solution de carbonate de sodium R (28,6g/l), ajuster jusqu’à 100ml avec la même solution. Prendre 1ml de cette solution et 1ml de solution témoin <a> et compléter jusqu’à 100ml par la phase mobile.
Conditions chromatographiques
Dimensions de la colonne : i=0,25m, ∅=4mm. Débit : 0,6ml/min.
Détection : spectrophotomètre à 280nm.
Injection : 5𝜇l de solution à examiner et de solution <b>.
B/ Excipient
L’excipient qui a été contrôlé est l’Amidon de Maïs. a) Caractères
Aspect : Poudre d’un blanc mat à faiblement jaunâtre, très fine, qui crisse sous la pression des doigts.
Solubilité : 50 mg d’amidon de maïs ont été mises dans 10ml d’eau froide, une même masse est mise dans l’éthanol à 96%.
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
20
Le résultat montre que l’Amidon de Maïs est insoluble dans l’eau froide et dans l’éthanol à 96%.
b) Identification Chauffe à ébullition
- Chauffer à ébullition une suspension de 1g d’amidon de maïs dans 50ml d’eau pendant1min.
Après refroidissement, il se forme un empois trouble et liquide. Coloration par l’iode
- Ajouter 0,05 ml d’une solution d’iode à 1 ml de l’empois d’Amidon de Maïs formé.
Le mélange a pris une couleur bleue foncée.
c) Essais limite
Détermination du pH
- Dissoudre une masse de 5 g d’amidon de maïs dans 25 ml d’eau exemptée de dioxyde de carbone. Après la dissolution complète, le pH de la solution préparée a été déterminé.
Fer
Dissoudre 1,5 g d’amidon de maïs dans l’acide chlorhydrique dilué (1/10) ;
Après dissolution, ajuster le volume de la solution à 15mlaveclemême solvant ;
Après agitation et filtration de la solution précédente, ajouter 2ml d’une solution d’acide citrique à 200 g/L, après homogénéisation, compléter la solution à 20ml avec de l’eau distillée ;
Le témoin est préparé dans la même condition en remplaçant la prise d’essai d’amidon de maïs par 10ml d’une solution à 1ppm de fer.
La lecture se fait par examen visuel après 5 minutes.
Substances oxydantes
- Introduire dans une fiole 4g de l’Amidon de Maïs et ajouter 50ml d’eau R, agiter pendant 5 min à l’aide d’un agitateur magnétique et filtrer ;
- Faire passer la solution à travers un filtre dans une autre fiole de 50ml ;
- Transvaser 30mldusurnageantlimpidedansunefiolede125ml, ajouter 1ml d’acide acétique glacial R et 0,5 à 1g d’iodure de potassium (KI) R, boucher, agiter manuellement et laisser reposer à l’obscurité pendant 25 à 30min ;
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
21 - Ajouter 1ml de la solution d’amidon R (le filtrat obtenu) et titrer par le Na2S2O3 (0.002M)
jusqu'à disparition de la coloration brune de l’iode ;
- Effectuer un essai à blanc, le virage de l’indicateur ne nécessite pas plus de 1,4ml de Na2S2O3 (0.002M) pour la solution d’essai.
1ml de Na2S2O3correspond à 34µg de substances oxydantes.
Perte à la dessiccation
- Peser un flacon vide préalablement desséché à l’étuve (𝑀𝑣).
- Mettre 1g d’amidon de maïs dans le flacon taré et placer l’ensemble dans une étuve à 130°C pendant 90min.
- Après refroidissement de 15 min dans un dessiccateur, peser le flacon à nouveau (𝑀𝑓) . Le pourcentage de la perte à la dessiccation est calculé par la formule suivante
𝑃 % =(𝑀𝑣+ 𝑃𝑒) − 𝑀𝑓
𝑃𝑒 × 100 … … … . 𝟎𝟐
P(%) : pourcentage de la parte à la dessiccation. 𝑴𝒗 : Masse à vide du flacon (g).
𝑷𝒆 : Prise d’essai (g).
𝑴𝒇 : Masse finale du flacon (g).
Taux de cendres sulfuriques (CS) (Déterminé sur 1g d’amidon de maïs)
- Chauffer un creuset de silice à 600°C pendant 30min dans un four ; - Laisser refroidir dans un dessiccateur sur du gel de silice puis peser ;
- Dans le même creuset, introduire la prise d’essai d’1g d’amidon de maïs, puis peser ; - Humecter la substance à examiner avec un 1ml d’acide sulfurique, chauffer doucement, à
une température aussi faible que possible, jusqu’à carbonisation complète de l’échantillon ; - Après refroidissement, humecter le résidu avec 1 ml d’acide sulfurique, chauffer
doucement jusqu’à ce qu’il n’y ait plus de dégagement de fumées blanches ;
- Calciner à 600°C jusqu’à incinération complète du résidu jusqu’à l’absence flammes ; - Laisser refroidir le creuset dans un dessiccateur, peser à nouveau et calculer le pourcentage
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
22 Le taux des cendres sulfuriques est calculé par la formule suivante
𝐶𝑆 % =(𝑀𝑒 − 𝑀𝑣) 𝑃𝑒
× 100 … … … 𝟎𝟑
CS(%) : Pourcentage des cendres sulfuriques.
𝑴𝒇 : Masse à vide (g). 𝑷𝒆: Prise d’essai (g). 𝑴𝒇: Masse finale (g).
III.1.2. Produit au cours de fabrication A/ Au cours de séchage
Test d’humidité
Mettre une masse de 5g du mélange dans un dessiccateur à une température de 65°C pendant 5min.
Le séchage est arrêté lorsque le taux d’humidité est compris entre 2 et 3%, dans le cas contraire, le séchage sera prolongé.
B/ Au cours de la compression Dimensions
L’épaisseur et le diamètre ont été déterminés à l’aide du pied à coulisse.
Epaisseur : 2,1 ± 0,2mm.
Diamètre : 6mm.
Masse moyenne
Peser 10 comprimés ensemble et chaque comprimé séparément. La masse moyenne est calculée par la formule suivante
𝑀𝑚 = 𝑀𝑖 10 10 𝑖=1
. . . . … … … . … … … . … … … . . 𝟎𝟒
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
23 Mm : Masse moyenne (g).
Mi : Masse de comprimé (g).
Friabilité
Mettre une masse (mi) de 6.5 g de comprimés dans le testeur de friabilité pendant 4 minutes en faisant des rotations en cylindre 25 trs/min, tout en vérifiant que les comprimés font des chutes régulières. La masse finale des comprimés après le test est (mf).
Le pourcentage de friabilité est déterminé par la formule suivante 𝐹 % = 𝑚𝑖− 𝑚𝑓 𝑚𝑖 × 100. … … . … … … . . 𝟎𝟓 F(%) : Pourcentage de Friabilité. 𝒎𝒊 : Masse initiale (g). 𝒎𝒇 : Masse finale (g). Délitement
Prélever 20 comprimés, introduire 1 comprimés par tube, chaque tube est émergé dans un bêcher qui contient de l’eau distillée chauffée à 37°C. Relever le temps de désagrégation.
Figure II.14 : Friabilimètre
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
24 Dureté
Le test consiste à mesurer l’intensité de force qui lui est diamétralement appliquée pour provoquer sa rupture par écrasement. La dureté mètre comporte 2 mâchoires, l’un se déplace vers l’autre. On place un Cp entre les deux mâchoires et on lance le test.
III.1.3. Produit fini a) Caractère
Aspect : Comprimes jaunes, ronds, aux bords chanfreines, lisses et brillants. b) Essais limite
Masse moyenne
La masse est calculée par la formule (04) en suivant le protocole. Test de dissolution
Le test de dissolution appliqué au comprimé permet de s’assurer, qu’une fois administrés, ces derniers libèreront là SA qu’ils contiennent pour le mettre à la disposition de l’organisme, et ceci dans les limites de concentration et de vitesse déterminées afin de garantir l’effet thérapeutique désiré.
Solution témoin
Peser 55.56mg d’acide folique dans une fiole de 100ml, diluer jusqu’au trait de jauge par le milieu tampon phosphate (0,2M), de pH 7,5.
Solution à examiner
- Remplir les 6 vases de l’appareil de dissolution par 900 ml d’une solution de KH2PO4dans chacun ;
- La température de ces vases doit atteindre 37°C ;
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
25 - Placer 1 comprimé dans chaque vase des 6 cloches, les comprimés doivent être au fond des
cloches ;
- Au bout de 45 min de dissolution, prélever 05ml, filtrer les 06 solutions avec un filtre de 0,45µm, laisser refroidir le filtrat ;
- Déterminer les absorbances de la solution témoin et de la solution à examiner à une longueur d’onde égale à 280 nm contre un blanc (milieu tampon phosphate pH 7,5) ;
La teneur en acide folique dissout est donnée par la relation
𝑄 % = 𝐴𝑒 × 𝑃𝑡 × 900
𝐴𝑡 × 100 × 100 × 𝐷× 𝑡𝑖𝑡𝑡𝑟𝑒 … … … . . … … … . 𝟎𝟔
𝑨𝒆 : Absorbance de la solution examiné. 𝑨𝒕 : Absorbance de la solution témoin. 𝑷𝒕 : Prise d’essai de la solution témoin (g).
Titre : Titre standard (c’est l’acide folique pure de la prise d’essai de la solution témoin, il est déterminé par HPLC).
D : Dose théorique de chaque comprimé (5mg).
Q : Quantité spécifiée de substance active passée en solution.
Dosage de l’acide folique
Solution à examiner
- Dissoudre 200mg de la poudre de comprimé dans une fiole de 100ml dans une solution aqueuse de NaOH (0,1N), compléter le volume jusqu’au trait de jauge ;
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
26 - Filtrer et effectuer une dilution au 1/10ml à l’aide de NaOH (0,1N).
Par le spectrophotomètre, lire l’absorbance de la solution à λ= 256 nm.
Solution témoin
Sur une prise d’essai de 100mg de principe actif (acide folique), procéder de la même façon que la solution examinée en faisant une dilution au 1/100.
La teneur en acide folique est donnée par la relation :
𝑄 % =𝐴𝑒 × 𝑃𝑡 × 100 × 1 × 𝑚𝑚𝑜𝑦 𝐴𝑡 × 𝑃𝑒 × 10 × 100
× 𝑇𝑒 … … … 𝟎𝟕
Q(%) : Teneur en acide folique (°mg/cp). 𝑨𝒆 : Absorbance de la solution examinée. 𝑨𝒕 : Absorbance de la solution témoin. 𝑷𝒕: Prise d’essai du témoin (92.96 mg). 𝑷𝒆: Prise d’essai à examiner (200.5 mg). mmoy: Poids moyen (101.86 mg).
𝑻𝒆 : Pourcentage de l’acide folique anhydre (après lyophilisation) dans la prise d’essai de la solution témoin (99.69%).
III.2. Contrôle microbiologique du produit fini
Afin de faciliter le contrôle microbiologique, la Pharmacopée Européenne (2014)a préconisé des protocoles de travail pour les différents types de médicaments.
Dans notre travail, on se limite à décrire le protocole suivi pour rechercher l’Escherichia coli.
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
27
Recherche de d’Escherichia Coli
Escherichia coli est une bactérie à gram négatif de la famille des entérobactéries, de
forme bâtonnet, vivant normalement dans l’intestin, mais pouvant parfois provoquer diverses infections (génitales féminines, intestinales ou urinaires). Cette bactérie est aussi utilisée dans l’expérimentation en biologie moléculaire.
a- Pré-enrichissement
- Peser 100 comprimés qui correspondent à une masse de 10g ; - Diluer les 10 g dans 90 ml de bouillon lactose, bien homogénéiser ; - Incuber à 37°C pendant 4 heures pour revivification ;
- Après incubation, prélever 10ml de la solution et les mettre dans90mld’un milieu liquide à base de peptone de caséine de Soja, incubéà37°Cpendant24 heures.
b- Enrichissement
- Prendre 1ml de la solution obtenue après agitation, l’ensemencer dans 9ml du milieu liquide de Mac Conkey, incubation à44°C pendant 24 heures.
c- Isolement
- A partir des tubes positifs (présentant un trouble), prendre quelques gouttes de la solution et faire un ensemencement par étalement sur une boite de pétri contenant la gélose Mac Conkey, incuber à37°C pendant 72heures.
d- Lecture
Absence des colonies
III.3. Contrôle toxicologique du produit fini
Principe
Le test consiste à administrer par voie intra-gastrique (orale) à des souris albinos de race SWISS une dose du produit relativement élevée par rapport à la dose thérapeutique conseillée afin de déceler la présence d’une ou plusieurs anomalies de nature variée du produit : l’apparition des symptômes d’une maladie donné ou le déroulement d’une fatalité.
Chapitre II Fabrication et contrôle de qualité de ZANITRA®5mg
28
Mode opératoire
Le test est pratiqué sur 5 souris (par lot) de race SWISS, sélectionnées au hasard et pesées le jour de l’essai, leurs poids doit être compris entre 17 et 24g, elles doivent être du même sexe (dans l’expérience à laquelle on a assisté, c’étaient des femelles). Les souris sont privées de nourriture, sauf de l’eau pendant 12 heures.
- Dans un mortier, broyer 6 comprimés de ZANITRA ® 5 mg, ajouter 10 ml de NaCl à 0,9% puis homogénéiser la solution ;
- Administrer 0,5 ml de cette solution à chaque souris à l’aide d’une sonde de gavage gastrique.
Chapitre III
Chapitre III Résultats et Discussion
29
I.
Contrôle physico-chimique
I.1. Matières premières
I.1.1.Acide folique I.1.1.1. Résultats
Lesrésultatsducontrôlephysico-chimiquedel’acidefoliquesontreprésentésdansle tableauci-dessous
Tableau 2 : Résultatsde l’acide folique.
Test Lecture Norme Résultat
Car ac tè re Aspect Poudre cristalline jaunâtreouorangée Poudre cristalline
jaunâtre ou orangée Conforme Solubilité dans : - Eau -Solvants organiques - Acides dilués - Solutionsalcalines -Insoluble -Insoluble -Soluble -Soluble Insoluble dansl’eau et les solvants organiques mais
soluble dans les acides diluéset les solutionsalcalines Conforme Id en tif icat io n Pouvoirrotatoire ∝= 0,17 0,17 × 25 × 100 0,2503 × 100 − 7,95 = 18,44°𝑚𝑙/𝑔 [18-22] Conforme E ssai s li m ite s
Teneur en eau 7,95% [ 5 -8,5] Conforme
Chapitre III Résultats et Discussion
30 Résultats de l’analyse HPLC
Tableau 3: Résultats chromotagraphiques
du témoin. Tableau 4 :Résultatschromotagraphiques de la solution essai. I.1.1.2. Discussion L’étudedel’aspectdel’acidefoliqueadonné uneidéeinitialesurlanatureetlaqualitédela matièreanalysée. Concernantlasolubilité, l’acidefolique(vitamineB9)estclasséparmilesvitamines hydrosolubles,il seprésentesousformed’unepoudrejauneorangée.Ilestsolubledans les acides dilués, peusolubledansl’eau,insolubledansl’éthanol,l’acétone,l’étheretlechloroforme.
Nom Temps Surface Surface (%) 1 1,764 44614 0,28 2 1,956 49451 0,31 3 2,415 17182 0,11 4 3,645 32246 0,20 5 A.F 5,226 15726216 98,83 6 8,310 42238 0,27
Nom Temps Surface Surface (%) 1 1,768 71101 0,45 2 1,961 44846 0,28 3 2,416 29395 0,18 4 3,626 9211 0,06 5 A.F 5,205 15754283 98,90 6 8,067 5225 0,03 7 8,268 15094 0,09
Figure III. 20 : Chromatogramme de la
Solution témoin « b »
Chapitre III Résultats et Discussion 31 D’aprèsnotrerésultatdupouvoirrotatoire,nousconcluonsqu’ilexisteuncarboneasymétriquequ ireprésentedoncuncentredechiralitéauniveaudelastructuredel’acidefolique. Lerésultatdelateneureneaunousrenseignesurlabonnedéshydratationetconservationdu principeactif,cequiréduitlesrisquesdécontaminationmicrobienneparladiminutiondel’activité del’eaucarlesmicro-organismesexigentpourleurcroissanceuncertainseuild’humiditésinonilsne sedéveloppentpas. Ledosageduprincipeactifestaussifondamentalcarilpermetdes’assurerdelaprésencedu médicamentenconcentrationsimilaireauxnormesduproduit.
Parconséquent,àpartirdurésultatévoquésurletableau,ledosageparHPLC nous a indiqué la conformitédedosedelasubstanceactiveàlanormedécritedanslaPharmacopéeEuropéenne (2014),ainsilapuretédecederniervul’absencedepicsparasites.
I.1.2. Amidon de mais I.1.2.1. Résultats
Lesrésultatsobtenuslorsdel’analysedel’amidondemaïssontreprésentésdansletableau
Tableau 5 : Résultats de l’amidon de mais.
Test Lecture Norme Résultat
Car
ac
tè
re
Aspect
Poudre d’un blanc mat à faiblement jaunâtre, très fine,
qui crisse sous la pression des doigts.
Poudre d’un blanc mat à faiblement jaunâtre, très fine, qui crisse sous la pression des doigts.
Conforme Solubilité dans : - Eau froide - Ethanole à 96% -Insoluble -Insoluble - Insoluble - Insoluble Conforme Id en tif icat ion
Chauffage à ebullition à Formationd’unempois troubleetliquide de couleurblanchâtre
Formation d’un empois trouble et liquidede
couleur blanchâtre
Conforme
Coloration parl’iode Coloration rouge-orange à bleu foncé qui disparait
par chauffage
Colorationrouge-orangeàbleu foncé qui disparait par chauffage
Conforme E ssai s li m ite s pH 5,35 [4 - 7] Conforme
Chapitre III Résultats et Discussion
32 Fer Coloration rose éventuelle
de la solution à examiner n’est pas plus intense que
celle du témoin.
Coloration roseéventuelle de la solution à examiner n’est
pas plus intense que celle du témoin. Conforme Perte à la desiccation 67,5446 + 1,0043 − 68,43471,0043 = 11,37% ≤ 15% Conforme Cendressulfuriques 20,1498 − 20,1476 1,0058 × 100 =0,22% ≤ 0,6% Conforme Les substances oxydantes 𝑉𝑒𝑞(Na2S2O3)=0,15ml 1ml de Na2S2O3=34µg de sub.oxy 0,15ml Na2S2O3=5,1µg de sub.oxy Le teneur en sub.oxy = Msub.oxy/Veq(Na2S2O3)= 5,1/0,15×103 =0,034µg/ml =0,034ppm ≤ 20ppm Conforme I.1.2.2. Discussion
L’étude ducaractère a les mêmes intérêts que ceux cités pour le principe actif.
Concernantlasolubilité,l’amidondemaïsestinsoluble
dansl’eaufroide.Ilsesolubiliseavecl’augmentationdelatempérature.
LerésultatdupH(quiestégaleà5,35),indiquel’absenced’impuretésalcalinesouacides au niveau del’excipient.
Lerésultatdufertémoignedel’absenced’impuretésdenatureferriqueainsiquelefaible taux de la perte à la dessiccation (11,37%) indique la bonne déshydratation et la bonne conservationdel’amidondemaïs.
Quantaufaibletauxdescendressulfuriques,ilnouspermetdedéduirequel’amidonde maïsnecontientpasd’impuretésminérales.
L’ensembledesrésultatsobtenus,vapermettredecertifierquecetexcipientestd’une qualitésatisfaisanteetrépondauxnormesdelaPharmacopéeEuropéenne(2014).
Chapitre III Résultats et Discussion
33
I.2.1. Résultats
Lesrésultatsobtenuslorsdel’analysesontregroupés danscetableau
Tableau 6 : Résultats du comprimés au cours de fabrication.
Test Lecture Normes Résultat
S
éc
h
age Humidité 2 ,3% [2 -3] Conforme
C ompr essi on Epaisseur 2,2mm [1,9 - 2,3] Conforme Masse moyenne (MM) La masse de 10 comprimés =1020,7mg MM=1020,7/10= 102,07mg [92,5 -107,5] Conforme Friabilité Mi=6,6 mg, Mf=6,58 mg 𝐹 % =6.6 − 6,58 6,6 × 100 𝐹(% = 0,3% ≤1% Conforme
Délitement 2min ≤15min Conforme
Dureté 4,99kP [2 - 6] Conforme
I.2.2. Discussion
Lerésultatdela masse moyenneconformeaux
normes,cequiindiquel’homogénéitédescomprimés.
Concernantlafriabilité,lerésultatestde0,3%,cefaibletauxdonneunefortesécurité contre les chocs mécaniques au moment du conditionnement, du transport et de la distribution dumédicament.
Letempsdedélitementesttrèscourt,onpeutdoncdirequelescompriméssedélitentfacilement,ce quifavoriseladispersionetl’absorptionduprincipe actif.
Chapitre III Résultats et Discussion
34 I.3.1.Résultats
Le tableau 7 regroupe les résultats physico-chimiquesdu produit fini
Tableau 7 :Résultats du produit fini.
Test Lecture Norme Résultat
Car ac tè re Aspect
Comprimés jaunes, ronds aux bords chanfreinés lisses et
brillants.
Comprimés jaunes, ronds aux bords chanfreinés lisses et brillants. Conforme E ssai s li m ite s Masse moyenne Pesée de 10 comprimés M= 1002,8mg Mm= 1002,8/10= 100,28mg [92,5 - 107,5] Conforme Dissolution 𝑃𝑡 = 56,1 𝑚𝑔 𝐴𝑡 = 0,348 Titre (%)=100,3 D=5mg Q(%)= 𝐴𝑒×𝑃𝑡×900 𝐴𝑡×100×100×𝐷× 𝑡𝑖𝑡𝑟𝑒 essai Abs Q(%) 1 0,358 104,19 2 0,331 96,33 3 0,351 102,15 4 0,328 95,46 5 0,345 100,40 6 0,348 101,28 ≥ 75% Conforme Dosage 𝐴𝑒 = 0,590 , 𝐴𝑡 = 0,545 𝑃𝑒 = 200,5 𝑚𝑔 , 𝑃𝑡 = 91,81𝑚𝑔 𝑀𝑚 = 100,3 𝑚𝑔 𝑇𝑒 = 99,69 % Q(%)=𝐴𝑒×𝑃𝑡×100×1×𝑀𝑚 𝐴𝑡×𝑃𝑒×10×100 × 𝑇𝑒 = 4,95% [4,5 - 5,5] Conforme I.3.2. Discussion
L’étude du caractèrea les mêmes intérêts que ceux du principe actif.
Lesrésultatsdela masse moyenne sontconformesaux
normes,cequiindiquel’homogénéitédescomprimés.
Le pourcentage du principe actif dissout montre l’efficacité relative de ZANITRA®.
Lerésultatdudosagedel’acidefoliqueprouved’unepart,labonnemaitriseduprocessus de fabrication.
L’ensembledesanalysesphysico-Chapitre III Résultats et Discussion
35 chimiquesvapermettredejugerleproduitfiniZANITRA®5mg comme étant d’une bonne qualité physico-chimique.
II.
Contrôlemicrobiologiqueduproduitfini II.1. RésultatII.2. Discussion
Le test microbiologique montre une absencetotaledel’Escherichia coli.
Lefaitquelemédicamentsoitsousformedecomprimésanhydres,empêchetoute proliférationmicrobienne. CecinouspermetdoncdedéduirequelescomprimésZANITRA®5mgsontd’unebonne qualitémicrobiologique.Etcel’estdûàplusieursfacteurs,àsavoir L’efficacitédeladésinfectiondumatérieletdeslocaux ; L’absencedecontaminationlorsdelafabricationetduprélèvement d’échantillons ; L’absence d’impuretésdanslesmatièrespremières ; Lerespectdesbonnespratiquesd’hygiènes.
Figure III.22 : Absence d’Escherichia
Chapitre III Résultats et Discussion
36
III.
Contrôle toxicologiqueduproduitfiniIII.1. Résultat
Le résultat du contrôle toxicologiqueest représenté dans le tableau
Tableau 8: Résultats du produit fini.
Test Lecture Norme Résultat
Toxicité Absence d’anomalie et de mortalité Absence d’anomalie et de mortalité Conforme III.2. Discussion L’absencedemortalitéainsiquel’étatnormaldessourisindiquequ’iln’existepas d’impuretésdanslamatièrepremièrenid’additionaccidentelleoucriminelled’unautre principeactifoud’unélémenttoxique.Celatémoignequelesconditionsdefabrications ontétéstrictementrespectées,permettantainsid’obtenirunproduitfinisatisfaisantet conformeauxnormespréconiséesparlaPharmacopéeEuropéenne(2014).
Conclusion
37
Conclusion
Le suivi de la fabrication nous a permis de mettre le point sur toutes les étapes de fabrication de ZANITRA®5mg et ainsi d’acquérir une bonne connaissance sur les bonnes pratiques de fabrication et d’enrichir nos connaissances dans le domaine pharmaceutique.
Ce travail nous a permis également de mettre le point sur toutes les méthodes de contrôle appliquée sur ZANITRA®5mg au niveau du laboratoire de contrôle de qualité de SAIDAL, filiale BIOTIC (Guéde Constantine) selon les méthodes de la pharmacopée européenne 2014.
Les analyses réalisées sur le principe actif et excipients nous ont permis de vérifier l’identité, la qualité et la pureté des matièrespremières avant d’entamer la formulation.
Sur le plan microbiologique, nous avons révélé une absence totale des germes recherchés tels qu’Escherichia coli.
Du point de vue pharmaco technique, tous les résultats étaient conformes aux spécifications de la PE en ce qui concerne les critères de qualité des comprimés.
Sur le plan toxicologique, le test d’innocuité sur le produit fini n’a révélé aucune mortalité sur la population étudié.
L’ensemble des analyse et divers contrôle effectués révélé la conformité de ZANITRA®5mg, un médicament générique sous forme de comprimé qui répond à tous les critères de qualité, d’efficacité et sécurité.
Références bibliographiques
Références bibliographiques
[1] CH.Bakahoum, TINA Samia ; "le suivi de fabrication et de contrôle de qualité du médicament générique ZANITRA®5mg "; Master; Université A.M.Oulhadj bouira ; (2016) . [2]M .BERROUAG, H.GANDI ;"Contrôle de qualité physico-chimique, microbiologique et toxicologique de l’Acide folique " ; Master ; Université M.Bougara Boumerdes ;(2017). [3]https://www.saidalgroup.dz/fr/notre-groupe/historique.
[4]A.Ounissi ; "Etude de l'évolution des ventes prévisionnelles des médicaments del'entreprise SAIDAL" ; Master ; Université Abou Bekr Belkaid-Tlemcen ; (2014).
[5]J.M.Aiache, E.Beyssac, J.M.Cardot, V.Hoffart et R.Renoux ;"Initiation à la connaissance du médicament "; 5ème Edition Elservier Masson SAS ; p413 ; (2008).
[6]D.Stora ;"Pharmacologie BP, Classes pharmacologiques ";4ème Edition WoltersF.Kluwer ; p415; (2010).
[7]C.Meunier, "Opinion vis-à-vis des médicaments génériques, Faculté Mixte de Médecineet de Pharmacie de Rouen" ;(2003).
[8]Lehir ;"Pharmacie galénique, bonne pratique de fabrication des médicaments"; 7ème édition ; Masson ; Paris ; p 269 ;(2000).
[9]K.Joel Franck ; "Contrôle de qualité des comprimés non enrobes, cas d’un générique et d’un principe de doxycycline ";Thèse de doctorat eu pharmacie ;universitéMohammed V ; Maroc ; (2008).
[10]L.Aveline ;O.Cartier ;P.Cuer ;P.Daucé ;C.March ;E.Désévédavy ;P.Dovillez ;N.Duchetet autres ;"Gériatrie. Estem (éditions scientifiques, techniques et médicales)" ; p359 ;(2000). [11]A.AH.Noura ; A.Razika ; B.Malia ; "contrôle de qualité d’un médicament non obligatoirement stérile :cas de comprimé <HISTAGAN> ; Mémoire de fin d’étude ; Université M’hamedBougara de Boumerdes ; (2016).
[12]D.Stora ; "Pharmacologie BP, Classes pharmacologiques "; 4ème Edition F.Wolters Kluwer ; p415 ; (2010).
[13]D.Stora ; "Pharmacieetsurveillanceinfirmière.5èmeEdition" ;F.Wolters Kluwer ; p372 ;(2008).