rl
SBo.
oo7
Rdpublique Argdrienne
Ddmocrati
q
ue et
popuraire
Ministdi..
d.,
I'
enseignement
supdrieur
f,'scurr6desMarhdm-Y*:n:,:lt"*"r?*:1ffi
.:""".de,aMariire
D6partement des Sciences
de la
Matiire
Mdmoire.du
projet
de
fin
d,dtude
ndme _
z
annee
master
spdcialitd
:
prlysleu'
DE
LA MATIERE coNDENsEE
Prdsentd
par
:
Guenouf
Soumia
STRUCTURE DE BANDES
DES
COMPOSES
BINAIRES
III-V
ET
IV-fV.
Sous
Ia
Direction
de
:DF.
S.
Djeroud
Dediectce
fe ffi{te
ce
memoire
di:
i{es
cddrsVarents.
fttesifreres
et
ti
ma
-reatr.
Toute
mafarniffr.
fous
mes
arrlk.
lour
qui
{o'nnent
une
contrifiutitn
fr
rdalTsation
ffi
Ce
mdmoire.
Remerciements
tre
tiens
a
remercier
trtr.A.Boufefel
professeur
d
l'universitd
de
Guelma
et
directeur
du
faborotoire
<<LPG
>>,Je
tiens d exprimer
mes
sincdres
remerciernents
d
mon
encadreur
modome
.S.Djeroud
moitre
de
conf,Lrence
d
l'universit6,
de
Guelmo
pour
ses
conseils
lelong
de ls rdolisotion de ce
travoil.
Je
tiens
d
exprimer
lgolernent
mq
reconnoissonce
d
Pr.B.Bennecer,
professeur d
f'universitd,
de
6uefmo.
Pr.N.Boukhorouba
,
professeur
d
f'universitd,
de
Guelmo. Dr.F.Kolorosse
mo?tre
de
conf6rence
d
f'univensit+d,
de
6uelrna.
Dr.
K.Zonct
mc?tre
de
conf6rence
a
l'universit
6,de
Guelmo.
Je
tiens
oussi
d
remercier
tous
les
membres
du
lsborotoire <LPG>
pour
leur
aide
tout
ou
long
dela
rdolisotion de ce
trovoil.
Ie
remercie
tous
mes collbgues
d
f'universitd,
de
Avzlmo
pour
feurs
R6sum6
R6sum6
Les propridtds structurales
et dlectoniques
des
dldmentsIII-V
et
IV-IV,
ces composdssont semi conducteurs cristallisant dans differentes skuctures : zine+lende, wurtzite
rocksatrt.
Les
calculsont
et6 effectu6spm la
mdthodede calcul
des ondesplares
augment6es(Fp-LAPW)
qui
se base sur Ia thdorie dela
fonctionnelle dela
densitd(DFT).
Nous avons utilis6 I'approximation dela
densite locale(LDA)
et l'approximation du gradient gsndralisd(GGA)
pour le
terme
du
potentiel
d'dchangeet de
corrdiation
(XC) pour
oalotrlerles
propri6tds structurales, bien que' pour les propri6t6s 6lectroniques (structure de bandes (lesgaps)),
on a
utilis6
LDA, GGA
. Les valeurs du paramdtre de rdseaud'dquilibre
sont en accord avec lesr6sultats expdrimentaux disponibles.
Gdndralement, les r6sultats obtenus sont
en
accord avec les mesures exp6rimentales et lescalculs th6oriques.
L'approximation
dela
densit6 localeLDA,
d I'inverse
dela GGA,
sousestime les paramdtres de maille mais surestime le module de compression et on peut conclure
Sommaire
Somm*ire
Inkoductiong*n€rale
...I
R€fErences ... 3chapitrre
r
: Th*orie
de rafondionnete
de ra densit*
DtrT.
r'1
inkoduction"""""'
...4
I'?
simplification
deI'*quation
de schrodingerd,un cristar
L3
L'approximationdeBorn-Oppenheimer
...,...;
L3.1Ddfinition
de laloi
variationnelle
... 6
L4 L'approximation d. un dleckon de
Halree_Fock...
...7
LS Thdorie de laFonctionnelle de
laDensiti
{DFf}
...9 L5.1 Prdambule : le moddle
de
Thomas_Fermiilg?4
... 9
I.5.?
LaDFT
selon Hohenberg Kohn etSham
...
ll
1.5.2.1 Thdordmes de Hohenberg,Kohn
...
tl
[5.1.1
Les 6quations de Kahnet
Sham
...
lI
L5.2.3
Lafonctionnelle
d'ichange_cordlation
... 13
L5.t.4
Rdsolution des dquations de Kohn-Sham
... 16
Rdferences
,... 18
chapitre
Ir
: Lesmt*hodes
decalcul
&
la
structure
de bandedlect*nique
tr.I
Intsoduction...
... 20
tr.2 M*thodes de calcul des shuctures de bandes €lectronique
...
...
Zl
tr.?.f
I"a mdthodecellulaire
...
Zl
tr
2.2 La mdthode des ondes planes orthogonalisds{OpW)
... ZZtr.2.3 Lamdthode
dupsendopotentiel
...22
tr-3
-?-Les {quations deKohn-Sham...
... g4 R€fdrences
...,...,.. 24
Sommaire
Chapitre
III
:L*
methodeslin6nire
des ondesphnes
augment6es et tinearisees[f,?-LAF}Iry
trI'l
La m€thodeAPw
... 25
trI.?
Principe de lamdthodeLAFW.
......27
trI.3
Les rdles des €nergies de lin€arisationEl
... 29trI.4
Ddveloppement en orbitaleslocales
... ZgtrL4.1Lam6thode
LAPIV+LO
...
...30trI.S Potentiel et densit€ de charge
...
...3l
trI.6
le code Wien?k...
...3l
trL6.1
L'initialisation
ducalcul
... 31trI-6-2
Le cycle du calculauto-compatible{ScF}
...g?,trI.6.3
Le calcnl depropridtds
^...3?,R€ferences
...rs
Chapitre
fV
: Resultets et. DiscussionIV.1
Propri€t€s structurales et ilectroniqueslapremidrr
s6rie...
...lg
IV.1.1
Ddtails decalculs
...40
IV.l
.2
Propri6t€sstructurales
...44IV.1 .3
Les propridtds{lecfoniques
...
...47
IV.1.3.1
Shuctue
debandes
...4?
IV.1.3.2
La densitd d'€tats{DOS)
... 49IV.1.3.3
Les densitds decharge
...j3
IV.?
Propri6t6s skucturales et 6lecFoniques la deuxiimes6rie...
... ssIV.2.1
Ddtails decalculs
...Ss
IV
.2.l
Propriftds
sto:ucturales
. .. . .. . .S5IV.? .3
Les propridtds dlectroniques...
... S?IV.2.3.1
Structure debandes
...57
IV.?.3.?
La densit€ d'€tafs{DOS)
... 58IV.?.3.3
Les densit6s decharge
.... 62IV
.3 Propridt€s sFucturales et ilectroniques la troisidme s€rie...
... 64Sommaire
IV.3.Z
propridtis
sfucturales
64
IV
.g.g
Lespropriftds
dlectroniques...
tV-3 q
|
{rqra+,,-^ -r-r, ,
.... 65Iv.3.l.l
Shucture debandes
.'.'... 65...65
Iv.3.3.2
La densitd d'6tars{Dosi
... 6T
IV.3.3.3
Les densitds dechage
..". dB
IV.4
Propri6t6s stucturales et dlectroniquesla quatridnn
sirie...
... ?0
IV.4.1
D€tails decalculs
...70
IV.4
.A
propri6t6ssilucturales
...70
IV.4 .3
Les propridtds 6lectsoniques...
...
TI
IV.4.3.1
Shucture debandes
...71
Iv.4.3.2
La densitd d'6tats{Dos}
... Tz
IV.4.3.9
Les densitds decharge
....
Zl
Rdferences
... 7s
Liste
des
rv'34
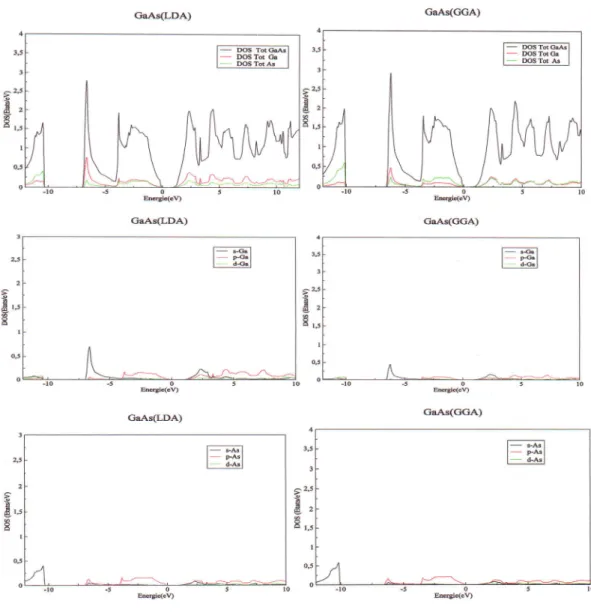
La densitdd'6tat(Dos)
totale et partielledu
Alp
avec les deux approximationsLDA
et
GGA
dans laphasezinc-blende
....67
rv'35
La
densit6
d'6tattotale et partielle
du
GaAs avec lesdeux
approximationsLDA
etGGA
dans laphasezinc-blende
...68
rv'36
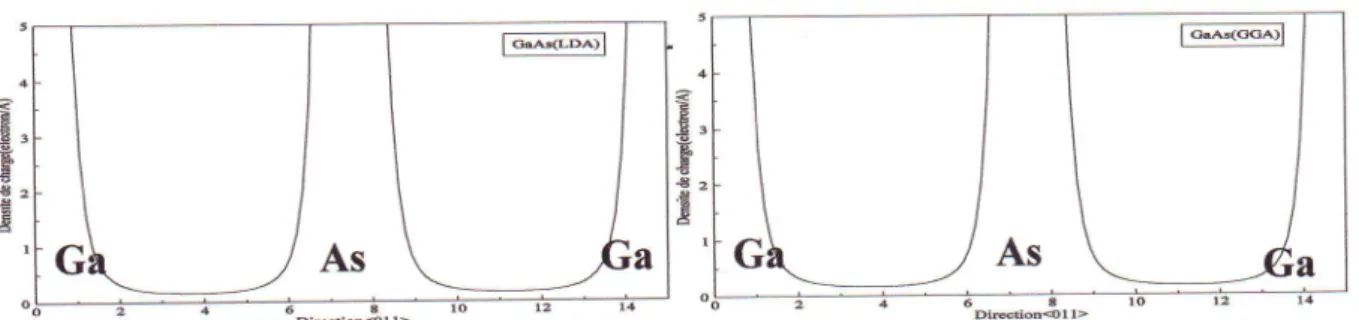
les
contours des densit6s de charge pour GaAs en phase zincblende, calcul6e par
LDA
et
GGA
...69
Iv'37les
contours des densit6s de charge pour GaAsen phase zinc blende, calcul6e par
LDA
et
GGA
...69
rv'38
Profils
de
la
densitdde
chargede GaAs
en phasezinc blende le
long de
liaisons, calcul6es avec
laLDA,
GGA
...69
rv'39
Profils
de
la
densitdde
chargede GaAs
en phasezinc blende le
long de
liaisons, calculdes avec laLDA,
GGA
...69
ry'40
La variation
del'dnergie
enfonction
duvolume du
Sic
dans les phases zinc-blende.calcul6e par
GGA
etLDA
...70
w'41La
structure de bandespour
Sic
obtenue parLDA
et
GGA
dans la phase zinc-blende ... 71lv'42
La
densitdd'ehtbtale
et partielle du SiC avec les deux approximationsLDA
et
GGA
dans la phase zinc-blende
...
...72
IV'43 les
contours des densit6s de chargepour
Sic
en phase zincblende, calcul6e par
LDA
et
GGA
...74
IJ'44
Profils de la densitd_de charge de GaAs en phase zincblende
le long de liaisons, calculdes avec la
LDA,
GGA
... ... .74
Liste
des
IV.IT
les
contours des densit6s de chaGGA
....:__"-'DrLEs
(re cnarge pour
AIN
en phase
wurtzite,
calculde parLDA
etIV
lfl
D-^fit^
r^
r- r
...54... )zlIV'18
Profils de la densitd de charge duAIN
en phase wurtzite le longde liaisons, calculdes avec la
LDA,
GGA.
.. .;. .
..
... ......54
ry'rg
les
contours des densitds de chargepour GaN en
phase
wurtzite,carcul6epar
LDA
etccA
...54
lv'20
Profils
de la densitd de charge de GaNen phase
wurtzite
le long de liaisons,calcur.es avec la
LDA'
GGA
... s4
rv'zt
Lavariation
de l'dnergie en fonctiondu volume dans phase zinc-blende
pour
Bp, BAs
et BSb calcule par
LDA
etGGA
... 56
rv'22
Les
structures des bandes desBN
(zinc
blende) etAlN,
GaN(wurtzite)
obtenue par
LDA
etGGA
... s7
rv'23
La
densitdd'6tats(DoS)
totale et partiellede
Bp
avec les deux approximationsLDA
et
GGA
dans la phasezinc-blende
... . ..59
Iy'24
La
densit6d'6tats(DoS)
totale et partielle deBAs
avec les deux approximationsLDA
et
GGA
dans la phasezinc-blende
.. ... ...60
rv'25
La
densitdd'dtats(Dos)
totale et partiellede BSb avec les deux approximations
LDA
et
GGA
dans laphasezinc-blende
...61
ry'26les
contours des densitds de charge pour BPen phase zinc blende, calcul6e par
LDA
et
GGA...
....,.,.62
N'27
Profils de la densitd de charge de BPen phase zinc blende le long de
liaisons, calculdes avec la
LDA'
GGA
...62
rv'28
les
contours de densitd de charge pourBAs
en phase zinc blende, calculde parLDA
etccA
...62
lv'29
Profils
de
la
densit6de
charge deBAs
en phasezinc blende le long
de
liaisons, calculdes avec laLDA'
GGA
...63
rv'30
Les
contours des densitds de charge pour BSb enphase zinc blende, calculde par
LDA
et
GGA....
. .. ... ...63
rv'31
Profils
de
la
densitdde
chargede BSb
en phasezinc blende le
long de
liaisons, calculdes avec laLDA,
GGA....
...63
fv'32
La
variation de l'dnergie en fonction du volume duAlp
et GaAs dans les phases zinc-blende, calcul6e parGGA
etLDA
...
...64rv'33
La structure de bandes pourAIP
et GaAs obtenue parLDA
etGGA
dans la phasezinc-blende.
...66
Liste
des
figures
Liste
des
frgures
I.1
Cycle de calcule pour la r6solution des 6quations deKohn-Sham.
.. ...17II.1
Paftition de I'espace selon la m6thodeAPW
< PotentielMuffin-tin(MT)
>. .,...25III.1
La L'organigramme du codeWien2k.
... 34IV.l
Maille primitive
de la phasewurtzite..
. -. .. '...38W.2
Maille primitive
de la phasezinc-blende
...39IV.3
Maille primitive
de la phaseRocksalt
....39
IV.4
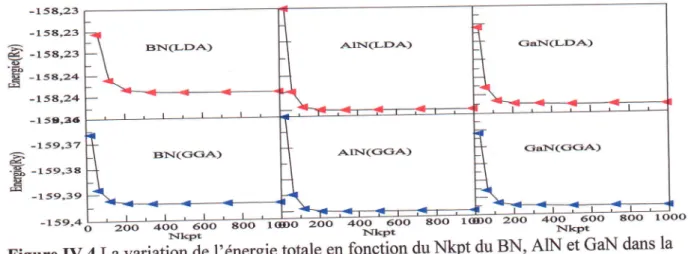
La variation de 1'6nergie totale en fonction duNkpt
duBN,
AIN
et GaN dans la phasezinc
blende
calculde parLDA
etGGA.
...."'411y.5
La
variation de l'6nergie totale en fonction duNkpt
duBN,
AIN
et GaN dans la phasewurtzitecalcul6eparlDAetGGA....
"'""""'41
IV.6
La variation de l'6nergie totale en fonction duNkpt
de GaN dans la phase zinc blende,Rocksalt calculde par
LDA
etGGA..
""
""'41
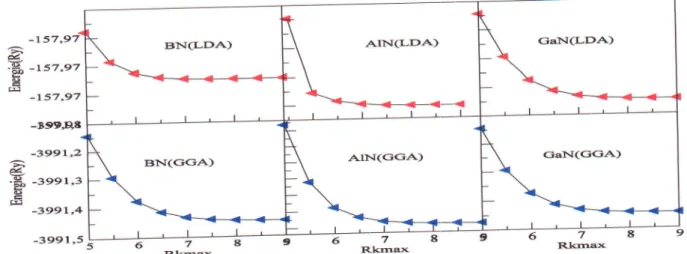
YY.1
Lavariation
del'{nergie
totale en fonction du Rkmax du BN,AIN
et GaN dans la phasezinc
blende
calcul6e parLDA
etGGA'
"""'42
IV.g
La variation de l'dnergie totale en fonction du Rkmax du BN,AIN
et GaN dans la phasewurtzite calculde par
LDA
etGGA
"""42
IV.9
La variation de 1'6nergie totale en fonction du Rkmax deBN,
AIN
et GaN dans la phaseRocksalt calcul6e par
GGA
etLDA
""42
IV.lg
La variation
de 1'6nergie enfonction
du volume duBN,
AIN
et Gal'{ dans les phases zinc-blende, wurtzite et Rocksalt calcul6e parGGA
etLDA
""""""'
47IV.l1
Les structures des bandes desBN
(zinc
blende) etAlN,
GaN (wurtzite)
obtenue parLDA
etGGA.
"""""""'
50IV.L2
La densit6 d'6tat (DOS) totale et partielle deBN
avec les deux approximationsLDA
etGGA
dans laphasezinc-blende
""'
51IV.13
La
densitdd'6tat(DOS)
totale et partielle deAIN
avec les deux approximationsLDA
et
GGA
dans la phase zincblende
" " "
"
'52IV.l4
La
densit6d,6tat(DOS)
totale et partielle du GaN avec les deux approximationsLDA
etGGA
dans la phasezinc-blende
"""""""""
53IV.15 les
contours
des densit6s de charge pourBN
en phase zinc blende, ca1cul6e parLDA
et
GGA
""'
53IV.16 Profils de
la
densitdde
chargede
BN
en
phasezinc
blende le
long de
liaisons'calcul6esavec1alDA,GcA..
''''"53
Liste de Tableau
Liste
deTableau
IV.l
Les valeurs deR*1*K^*
,K
points ,R6
deB
et Rmt deN
dans la structure zinc-blende,wurtzite et Rocksalt calcul6e par
LDA
et GGA..IV.2
Les valeurs deR*1*K-*
,K
points , R*1deAl
etR6
deN
dans la structure zinc-blende.wurtzite et Rocksalt calculde par
LDA
etGGA.
...43
IV.3
Les valeurs deR*1*K**
,K
points , R*1 de Ga etR6
deN
dans structure zinc-blende,wurtzite et Rocksalt calculde par
LDA
etGGA...
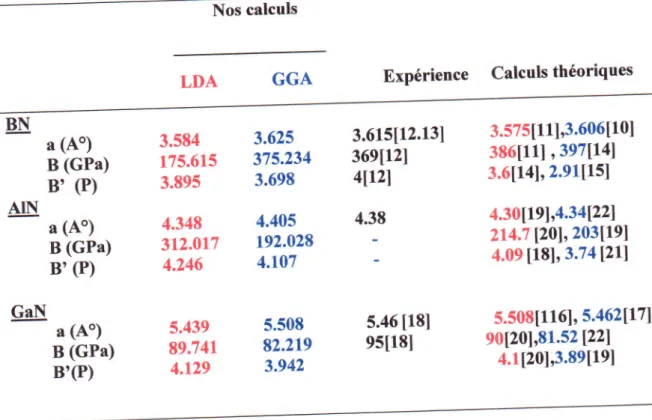
... ..43IV.4
Le paramdtre du rdseau a, module derigiditd
B et sa d6riv6eB'
de BN,AIN
et GaN dansla phase
zinc-blende
...44IV.5
Le paramdtre du r6seau a et c, paramdtre interne u, module derigiditd B
et sa ddriv6eB'
de BN,
AIN
et GaN dans la phasewurtzite
... ... ... ...45IV.6
Le paramdtre du r6seau a et c, paramdfre interne u, module derigiditd B
et sa ddrivdeB'
de BN,
AIN
et GaN dans laphaseRocksalt
...46
IV.7
Le
gap 6nerg6tique deAIN
et GaN
dansla
phasewurtzite et
BN
dansla
phase zincblende.
...49
IV.8
Le paramdtre du r6seau a, paramdtre interne u, module derigiditd
B
et sa d6riv6eB'
deBP, BAs et BSb dans laphase zinc
blende
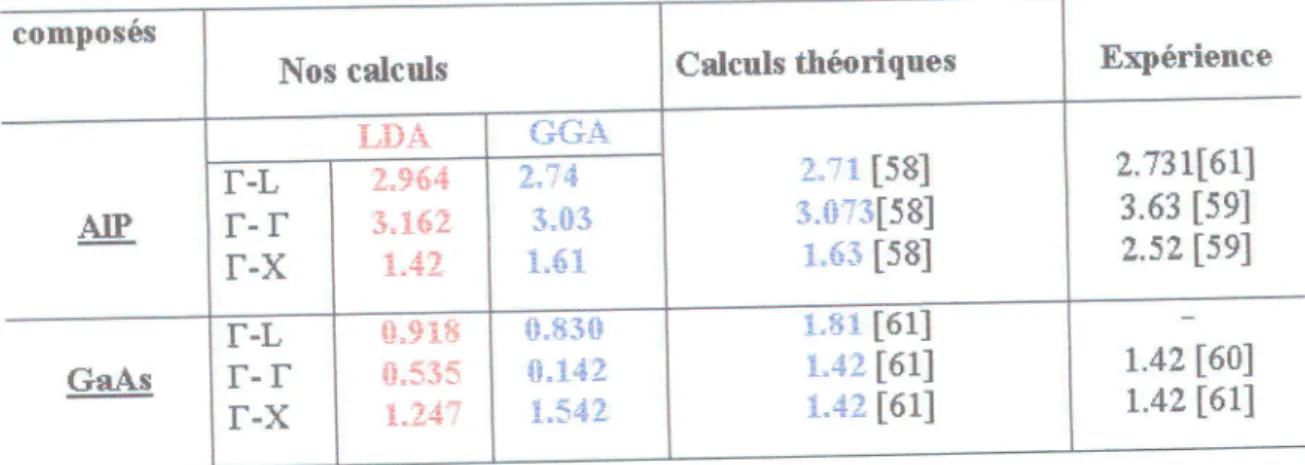
...56IV.9 Le gap 6nerg6tique
d'AlP
et GaAs dans laphase zinc-blende...
...58
Iv.10
Le paramdtre du r6seau a, module derigidit6
B
et sa d6rivdeB'
du SiC dans la phase zinc-blendeIV.ll
Le gap 6nerg6tique du SiC dans la phase zinc-blende, calculde parGGA
etLDA
...67
1V.12
Le paramdtre du r6seau a, module derigidit6
B
et sa ddriv6eB'
du SiC dans la phasezinc-blende
...71IV.13 Le
gap 6nerg6tique du SiC dans la phase zinc-blende, calculde parGGA
etLDA...
87Introduction gin€rale
Introduction
g6n6rale
La
physique dela
matidre condens6e etla
science des mat6riauxjouent
unrole
de plus enplus
important
dansles
applications technologiques,et
ce
r6le
ne fera
que progresser dansbeaucoup de domaines.
Avant
d'employer les mat6riaux (solides) dansl'industrie,
il
faut s'assurer de la qualit6 de leurs propri6t6s structurales, 6lectroniques, m6caniques, optiques...etc.Les propri6t6s physiques
d'un
solide sont 6froitement li6es au comportement des 6lectrons qui le probldme de la structure dlectronique des solides. La thdorie de la strucfure 6lectroniqueest
utile
i
la
fois pour
comprendreet
interprdter les rdsultats exp6rimentaux, etpour
servircomme moyen de pr6diction.
Pour une compr6hension fondamentale de
la
structure dlectronique et par consdquent despropri6t6s des matdriaux, les thdoriciens ont d6veloppd des mdthodes bas6es sur des moddles dits : semi-empirique, de tels moddles comportent souvent de nombreux paramdhes ajustables
aux
donn6es
exp6rimentales.D'autres
mdthodes
de
calcul
plus
rigoureuses
et
plus sophistiqu6esdites ab-initio
[1],
bas6essur
la
th6orie
quantique
fondamentale, utilisentseulement
les
constantesatomiques comme
paramdtresd'entr6s
pour
la
r6solution
deI'dquation de
Schr0dinger. Ces mdthodes sont devenuesaujourd'hui
un outil
de base pourl'6tude
des propri6t6s structurales, 6lechoniques, mdoaniques,optiques,...
des mol6cules et des mat6riaux. Elles sont aussiun
outil
de choixpour l'6tude
de certains effetsdifficiles
ouimpossibles
de
ddterminer
par voie
exp6rimentaleet
pour
la
pr6diction
de
nouveauxmatdriaux,
et
elles
ont
parfois
pu
remplacer des exp6riencestrds
couteusesou
m6me irr6alisables en laboratoire.la
puissancedes calculs
ab-initio
a
pour origine
le
formalisme
de
la
thdorie de
la fonctionnelle de densitd(DFT) [2],
et ses deux approximations de 1'6nergie d'6change et decorrdlation
:
l'approximation
dela
densit6 locale(LDA)
l2l,
et l'approximation du
gradient gdndralisd(GGA)
f2l,le
formalisme de base de la DFT est bas6 sur le thdordme de Hohenberg etKohn
(1964)[3], qui
repose sur la considdration que 1'6nergie totaled'un
systdme est unefonctionnelle de la densit6 dlectronique.
Il
y
aplusieurs
m6thodes thdoriquesqui
peuvent €tre utilisdespour
calculerla
structure des bandes (propridtds 6lectroniques), la mdthode cellulaire, la mdthode des ondes planes, lar-t
f
ntroduction
96n6rafe
mdthode des ondes planes augmentdes
(APW),
la
mdthode des ondes planes orthogonalisds
(OPW), la mdthode du pseudopotentiel t41....
Parmi
les
mdthodesab-initio,
la
mdthodeFp-LApw [4],
est l,une
desprus
pr.cises,actuellement' pour le calcul
de la skucture dlectronique des solides dans le cadre de la (DFT).
Dans notre
travail'
nous avons 6fudi6lespropri6tds dlectroniques des eompos6s
III-v
etIV
et les calculs ont 6td rdalisds en utilisant Ie codewien
2k [5],
qui est une impl6mentation de lamdthode
FP-LAPW
dansle
cadrede la DFT.Alors
le travail
que nous prdsentons dans cemdmoire est composd de
deux
parties avec une
inkoduction
et conclusion gdndrale.Le premier est prdsente la th6orie de bandes.
Le deuxidme chapitre est destin6 au fondement
de
la DFT'
LDA'
et GGA, on
discutera
les
dif,fbrentes nr6thodes.Le
troisidme
chapitre pr6sente la m6thode
Fp-LApW.
La deuxidme partie r6sume nos r6sultatg
leurs interpr6tations ainsi qu,une comparaisoR avec certains travaux th6oriques et expdrimentaux disponibles dans la litt6rature.
|---.'l
i2j
Introduction
g6n6rale
R6f6renees
[1]
G.C.Flecher,the
electron band
theory
of
solids, North-Holland pubtishing
company Amsterdam London.t2l
R.G. Pan
W.Yang, Density-Funetional Theoryof
Atoms
and moleeules(oxford
ssienoepublication, 1989.
[3]
P.Hohenberg and W.kohn,phys.rev.Bl36
(1964)564.[4]
Davidj.
Singh, plane waves, pseudopotentials and theLAPW
Method,kluwer
Academicpublishers, Boston/ Dordrecht/London.
[5]
P. Blaha
,
K.
Schwarz,G.K.
H
.Madsen,D
. Kvasnicka
andJ.
Luitz,
Wient
2k,
an augmented plane-wave+local orbitals program fior calculating crystal properties(
KartheinzS chwarz, Techn. Universitat Wienna, Austria),200 I .
ISBN
3 -95 0 I 03 I - I -2.-''r
l3l
Chapite
I
Thdorie de laFonctionnelle de laDensitdDFf
Chapitre
I
Th6arie
de Iafonctionnelle
dela
densit6Df'T
Llintrodnrtion:
La
description quantique non-relative d'un systdme moldculaire oucristallin
est bas6e surl'dquation de schrddinger.une
infoduction d
ceformalisme
ddbute ndcessairementpar
Ia
prdsentation de
l'dquation
de Schrddinger exacte {<< dquationi
plusieurs corps >>)qui
serasimplifide
ultfrierrement par
diverses approxinmtionsde
maniire
i
ce qn'elle
puisse 6tre rdsolue. Le kaitement de ce << probldmeiplusieu's
corps >> en mfcanique quantique consiste drrechercher les solutions de l'dquation de Schrddinger. Malheureusemen!
les
dlechons et lesnoyaux
qui
composentles
matdriaux
constituerdun
systdmei
plusieurr
cofps
fortement interagissant et ceci rendlardsolution
del'fquation
de Schrddinger extremementdifiicile.
I.2
simplilication
del'6quation
deSchriidinger
d'un cristal
: lesapproximations
:Les solides sont constituds par une association de particules dldmentaires
:
les ions et les dlectrons. Leproblime
thdorique fondamental#
la
physique des solides est de conryrendreI'organisation
intime de
cesprticules
et I'origine de
leurs propri€tds.Mais
dansce
cas,Ia
mdcanique classique s'avdre insuffisante
et
il
faire
appeli
la
mfcanique quantiquedont
la
base est lardsolution de l'dqnation de Schrddinger :
Hv
=
Ettt
Tel
queE
est l'dnergie del'6tat
fandamental du cristalt1l.
Le
problime
gfn6rale peut Stre posd souslaforme
d'une dqudion du mouvement de toutes les particules prisentesdms le
cristal
L'Hamiltonien
exact ducristal
(non relativiste) r€sulte de la pndsence des forces dlecfostatiquesd'intermtion
: soit repulsion ouattaction
suivantla
chmge des particules {ions. 6lectrons).
H:I"
+
fr,J+
V*-"
*
Vs-N*
VN-NDans laquelle les termes T",TN,Ve-e,Vr-N
et
lljv-jv correspondent respectivement :Q
:L'€nergie
cin{tique des dlecffons.-hZ
T":
*ErA,
I*
:L'fnergie
cin6tique des noyaux.-EZ
T*:#E*
a*
I/*
-"
L' dnergie potentielle d'interaction r6p ul sive (6lectron- € lectron).{r.2}
(r.1)
ddcrit
pr
la fonction
9rdu
cristal{r3)
Theorie de Ia
fonctionnelle
dele
denrit6DtrT
V"-"
:jX,,ro,
U,;
=
jX,,ro,G+=;
(r.5)
I4v-,iv : L'€nergie potentielle d'interaction euke les
noyarx.
lf_. :J1'
,r _ 1r,
ezzpz2vff-ff
-
rL,E,r#E utet
=
;Ia,i*r;ffi
(r.6)e: la charge de €lectron.
m
: Iamasse del'6lecfon.
Ti,Ti
i
ddfinissent les positions des drectsons{i)
effi},
respectivementFs,
ftr
: ddfinissent les positions des noyaux(k)
et (r), respectivementzn,zt:
soit
les nombres atomiques des noyaux{k) et
{l},
respectivemenl L'dquation{I'1},
dquation de schnidinger{1926} avec
H
{Hamiltonien totale} s,est r6v6lde6ke
eractementdifficile
f,
rdsoudre, m6me dansles
cas
les plus
sirnples.
Efectivement
lorsque
le
nombre d'atomes
augmente,les difficultds
du
caleul
angmentaienfde
fafo'
exponentielle.Ainsi,
lmsqueI'on
considdre rm certain nombred'dlecfons
N,
ces fonctionsd'ondes d€pendent de leurs
N
coordonndes tridimensionnelles.par
suite,la
fonction globaleddpend de 3N variables.
Les diverses mdthodes de
calcul
dela
shrctr"re de bandesilechoniques
des matgriauxi
I'dtat
solide mises aupoint
au cours des dernieres ddcennies reposent surufi
certain nombre d'approximations reparties sur Fois niveaux[ZJ.
1.
I'approximation de Born-Oppenheimer.2.
I'approximation de Hartree-Fock ou le formalisme delaDFT.
3.
L'approximation inhdrente irIarisolution
des iquations.I.3
L'approximation
deBorn-Oppenheimer
:L'approximalion
de
Born-Oppenheimer,
dite
adiabatique
est
la
premidre
desapproximations
utilisde pour
la rfsolution
del"dquation
de Schnidingerpour
les
systdmescomplexes contenant
plus d'un oir
deux €lectrons, ellesdpre
le
mouvement desilecfons
efdes noyaux
en se
basantsur
la
diftrence
de
rnasseenFe
les
noyaux atomique
et
lesdlectronsltf.Fr
}}
m".
En gdn€rale la simplification de I'dquation de
sclrddinger
i
deux niveaux :(i) i
condition
quela
gdomdFiede
la
strucfuresoit
conservde,
le
terurc de repulsiou internuclfaire I/,"-r, peut€fe
considdrf comme constant L'avantage de ceci reside dans lefait
Ch*pitre
I
Thmrie
del*
fondiannelle
dela
densitdDf,'T
que
I'qjout
d'une constanteiun
opdrateurn'apas d'effet
sur ses fonctions propres. En cas dechangement de
gdomikie,
ce terme doit r66valuer.(ii)
la vitesse des noyaux estnulle,
il
en est de m6me pour leur impulsionet
leur dnergie cindtique.Elle
conduit donci
la sdparation duI'Hamilbnien
totale en une partieilecfonique
et une partie nucldaire relide ar$( noyaux, ces derniers sanffxes.
Les 6tats propres
du systime
sont alors carartdrisdspar
des fonctions d'ondeproduit
d'uRefonction d'onde 6leckonique par une fonction d'onde nucldaire.
t/r{ft,r}
:
!&rviff}
tltrtr}
L' dquation de Schrddinger
s'6crit:
t4
+
rr-r*v*_"
*
F*_r**
Irrv-ivlfjY{n}
*"tB,r}:Etply{fr} $"(R,r}
Ce qui amine aux deux €quations indfpendantes :
tL
+
l"jv*
tr"-nl
*"tR,r):E*
*r{R,r}
trrv
+
Iriv_ivlfiv {fr}
:
Ef,v{fr}
(r.8)
f.e)
fl.10)
(r.1 1i
Pour
les
6tatsd'fnergie flecfoniques du cristal
on
n'utilise
que l'dquation
{L10},
les noyau:( 6tant supposds fixes a leur positiond'fquilibre.
I.3.1
l)6finition
de laloi vari*tionnelle
:Dans
le
cadre deI'approximation
de Born-Oppenheimer,le
syst&men'est ddcrit
que parI'opErateur Hamiltonien €lechonique. L'dquation de Sclrrtidinger est donc r66crite de lafagon zuivante:
H,
!h*:
E, $"
ir.12)
Dans cette
eryression,
E*
est
I'dnergie
correspondant
i
I'dtat
quantique
**
{ryr2,rs...
...,Tn}dfcrirmrt
le systdme desN
dlectrons plonges dans le potentiel des noyaux.On peut ddduire aisdment de
(I.1?)
quel'fnergie
dusystime
s'e4prime sousla forme
dela
fonctionnelle suivante.
E[+]:ffi
Otr
lanotion
deDirac
a 6td adopt6e [3,4].Theorie de
la fonctionnelle
dela densitdIltrT
D'ryrts
le principe selon lequel unsystime
tend fl minimiser son dnergie totale, l,dnergie E
relative
i
un 6tat rp quelconque est supdrieureon
6galei. r,dnergie Escorrespondao1 el,6tat
fondamentat rpn du systdme. Ceci nous conduit
i
l,in6galit6 sui'ante.
E[*l
>
so(r.l4i
Autrement
dit' l'fnergie
calculfe
flparfir
d'unefonction d,onde
rf
quelconque du systdmeest supdrie're
i
l'dnergiewaie
de l'dtat fondamentar du systdme[z].
E
[Ss]:
Es:
min([+]]
on
enhevoit
ici
mdthodepour
ddterminer Iiiitd
fondamentald'un
systdme donn6. En appliquant
le principe
variationnelqui
consistei
minimiser
l,dnergie totale par rapport d.Ia
fonction d'onde
(I-rs),
il
est possible de d€terminer les Es et r/rs exacts.On s'int€resse donc
dr6sou&e
l,dquation :as[U:
n du
Au
lien
de
l'equation
de
$chrddinger sous
la
forme fi-12).L,ryplication
de
Ia
loi
variationnelle permet de calculer
Es et dr6pour n'importe quel systime composd d,€lecFons et
de noyaur.
cependant la
fonction
tfis sousla
forme exacfe est beaucoup complexei
manipuler (c,est
une fonction de 3N variables avec
N-+
mlet
on peut danslapratique
lasimplifier
n.flcei
une apprcximation.Four r€soudre de
tels
systimes flN
particules,il
existe principalement deux grands tSpesde mdthodes : m€thode Hartree-Fock(HF)
[5],
et les mdthodes basdes surIaDFT
[Z].
f.4
L'epprnximation
i
un dlectron
delfaftree_X,ock:
cetfe
approximation propos6epar
Hartree-Fock[?,s],
consisted
supposerque
chaquedlecfon
se ddplace inddpendamment dansun
champ moyen cr66par
les aphes dlectrons Et
noyaux.
L'Hamiltonien
peut
6be
6cris
comme 6lechon.H:
Xi
ffi
une sofilme des
Hamiltonien dicrivant un
seulfi.15)
Avec
nzHt:
h+
Ir{r)
Ofl
{r{r}
possddela p6riodicit6
dus aux ionsavec tous les
aufes
flecbons.(r.16)
{r.17)
(r18)
Ch*pitre
I
Theorie
dele fonctionnelle
dela
densitd Dtr'TLa fonction d'onde du systSme €lectronique
i
lafcrme
d'unpro&rit
de fonction d'onde dechacun des
dlecbons, et
l'dnergie de ce
systAme 6lectsonique est 6gale drla
somme desfnergies de tous les flecFons.
$* (rr,rr,rr...
...,ro):
tF,(rr)
*,
VrJS.
(rrJ
....
tl"
(A)
E:8"
=
EL+
Ez*
E3 .... . EnAvec
Hi !&i:S,
tli
H*
*":E*
rh"Le champ de Hartree-Fock permet de ramener l'€quation
multiple
i
un systimed"iquation
d'un seul 6leckon.
(r.1e) (r.20)
(r.21)
{r.22)
{r.23}
lS
a,*v--iv(ri)+
tr/"-"{ri)l
*tU}-
4
{k}{1{ri
%-,,v{ri} L'€nergie potentielle de
I'Slecfon{i)
dars le ehamp de tous les noyaux.Vr-*(rJ
Est le champeffectifde Harfee.
Mais tant
que l'dlectron est
un
fermion donc
la
fonction d'onde totale
doit
0fe
antisymdfique
par
rapport
ir
I'dchangede
deuxparticules
quelconquequi
est ndglig€ par Hartree. Pourcorriger
ce d€faut,Fock
[6]
aproposf d'appliquer
Ieprincipe
d'exclusion dePauli,
doncla fonction
d'ondedlecfonique s'6erit
sousla
formed'un
ddterminant de Slatert6l.
{r.24}
Ot
+
est la constant de normalisation-vivlLe
syst*me d'€quations{I-23)
serfsout
denani*re
auto-cohdrente dansla
mesureoir
lepotentiel ddpend des fonctions d'onde. La m€thode de Hartree-Fock basfe sur
I'hlpothdse
des.
I
rrDr{rr}
$tkr}...
lD,(A}
g*
{ryr2,r3...
... ,rrr}:
# :l
*z
tr}
*z
{rz}...
. .Sz
{A}
Yrr
i
Chapitre
f
Thdorie dela
fonrtionnelle
dela
densit6 Df,,Tles moldcules, mais pour les solides, elle est moins precise et
diffcile.
Cependantil
existe unemfthcde
moderne et certainement plus puissmte qui est la Th6orie de la Fonctionnelle dela
Densit€
(DFf).
L5 Th6orie
de la tr'onctionnelle de la Densitdfl)X,T)
:Comme son non
I'indique, c'est
une th€orie quiutilise la
densitd dlecfronique en tant quefonction'
fondamentale aulieu
de lafonction
d'onde comme c'estle
cas dansla mithode
deHartree-Fock.
L'intdret
de laDFT
est desimplilier
larisolution
del'dquation
de Schnidinger en plagantla
densitd dleckonique
p (r)
au cenke ducalcul.
Cefte quantitd estddfinie
dela
fagon suivante TZJ.p
(rli=+{
Jtout r,"spr." . . -Jtoot resp*e**
(r t, rz ...ro}*
{rt
rz
...rn}dr2drn
Ori
N{toott'"rp"." p
{r}
&
fl.25)
(r.26)
L'ing€niositd
de cette approche est ainsi de passff du calcul d'rme fonctioni
3N variables*{rurz...r"}
au calcul d'une fonction beaucoupphs
simplei
3 variables p {r).Avant
d'e4poserles
principes g6ndraux dela
DFT,
nous allons succinctementddcrire
une m€thodeplus
ancienne, basdeelle
aussi surle
calcul dela
densitd, mais dontle
formalismes'articule
autourd'une rypmche
statistiquedu
probldme.Il
s'agit du modile
de
Thomas-Fermi, modile qui
initi4
les
ryproches
DFT et
est
destindi
calculer
ta
disfibution
des€lectrons dans un atqme.
I.5.1Prenmbule
: le moddlede
Thomas-Fermi
{lgTfi
zEnfait, I'idde d'utiliser ladensitf
€leckoniqueapour
origine lesdfbuts
de lamdcaniqueavec les travaux de Thomas
[9]
et Fermi[0]
qui ont d'e4primer l'6nergie tatale d'un systimeen fonction de sa
densitd
€lecboniqueen
repndsentantson
dnergiecinetique selon
unefonctionnelle de cetfe grandeur.
Les
hlpothdses
sur les
quelles repose
ce
moddle
sont
une r6partition
uniforme
desdleckons dans I'espace
et
le
fait
que
chaque
dlecfon
subisse
un
potentiel effectif
€leckostatique
dil
au noyau d'une part, eti
ta distribution des aufues dleckonsd,aufe
part
De plus, la ddtermination de l'dnergie cindtique est basfe sur lemodile du
gazde fermions libres.Theorie
de Iafonctionnelle
deta densitdDf,'T
En
considdrant que I'espace estdiviss
en cellulescubiques 6l6mentaires,
il
est possible de ddterminer l'€nergie cindtique totare (E4rrldesdlecfom
dans une celre
[z].
'
_3.hz
t3$t2
Ecin:
*A(;f"
,s/t
73
$.27)
Le
ddveloppement permettantd'aboutir
d
cette erpressionest
dom€
an
n6cessaire desommer sur toutes les cellules.
on
en arrive donc anpoint
central de ce paragraphepuisqu,il
est possible de cette fa+on, de ddterminer cin€tique du systime;
c,est une fonctionnelle dela
densit6 qui prend
laforme
suivante (donnde enunitis
domiques] :5
Ir.p:
C Jtootr"rp"."pr{f}
{I.28)
Avec:
c:
fr{r''r}tn
Compte tenu des
hlpothises
mentionndesau d€bnt
de
paragraphe,il
est
maintenant possible&
ddterminer l'dnergie totale du systime qui elleune fonctionnelle de la densitd :
rrrh
{r)l
:
c
J,oo,r"a.."pi(r}
z
ffF
*r4
f ##
dr dr,
(r.2e)Le premier terme
du
membre dedroite
estle
terme d'dnergie cindtique,le
second refldte I'interantion entre lesflecfons. Ainsi,
en minimisantr1,p[p{r}],il
est enfinle
troisidme estI'interaction
€lectrostatiquede
I'dtat
fondamentaldu
systdme.Le
nombrede
parficules dusystdme 6tant constant,
minimisation
de$-27)
sefait
enincorpormt
cetteconfainte par
lebiais
d'un multiplicateur de Lagrange b.on
resors alorsl,iquation
suivante :6
{Err
[p
{r}]
-
blp
{r}dr
-
NI}
:
0Les
rdsultants donndspar
cettethdorie sont
enrialitd
assezmediocres
li.,ll!,
et
cecimalgrf
de nombreuses tentatives d'amdliorafion. On peut citer parmi celles-ci, celle deDirac
qui
consistef,
introduire un terme
d'dchange dansla
fonctionnelle
E'pptp{rl
[12J.
Cefte approche approximative renconFe, en effet, de s€rieux probldmes[11]
puisgge d,gne part, lesfonctions d'ondes obtenues
n'ont
par le bon conportementqualitatif
au noyau et que d'autrepart' elle
ne permet pas defaiter
les ions ndgarifs. Les moldcules[13,14,
l5].
pogr plus dedftails,
consultezle
chryihe
6
du
liwe
de
Par
et
Yang [ZJ,
qui
prdsentece
moddle
etdiffirentes
variantes ou anx rerrues de Gombas[16]
et March[l{
portant surI'application
decette th€orie aux atomes et
molfcules.
Ce moddle est cependant int6ressant dansle
sens oriil
constitue
le
premier par
vers unethdorie
oir le
calcul
compliqudde
la
fonction d'onde
estremplac€ par celui d'rme simple, la densit6 €lecfonique.
I!g"i*
de Iafonrtionnelle
dele
dcnsitdDf,T
on
gdndrale'la
prdcision obtenue dansle
moddle de Thomas etFermi
itait
inferieure
dcelle
de Harhee'Focki
carse de I'absence duterme d'dchange-corretation.
Dirac
a
arn6li,*6
cettethdorie
en qioutant au unefnergie
d'dchangefonctionnelle
de
la
densitd 6lectronique.Mais le terme correlation flecbonique 6tait toujours absent dans cette
nouvelle approche.
I.5,2
La
IIFT
selonIfohenberg,
Kohn
et
Sham :1.5,2.1
Th6orimes
deHohenb*rg,
Kohn
:Le formalisme de
IaDFT
est basd sur les deur thiordmes de Hohenberg,Kohn [z,rgJ.
Fremisrement,
Hohenberg,Kohn
ont
montrdqu'il
existe une coffespondancebinnivoque
entse
le
potentiel extdrieur et densitd €leckoniquep
ir)
permettant de reprisenterle
premiercomme une fonctionnelle
de
l'6td
fondamenia"lest igalement une
fonctionnelle
uniqueuniverselle de la densitd dlechonique, soit :
E:E
[p
(ri]
(r.31i
Ce
thdorime
est drla
base dela DFT
et e4pliqueI'appellation qui
lui
a
6t6 donnee.ceci
diffare de lam€thode
Harfee-Fock,
dans laquelleI'inergie
totale dn systdme est fonctionnelle
de
lafonction
d'onde.une
cons€quenceimmddide
de ce thdorime est quela
densit6dlecbonique d6termine de
fagon unique I'opdrateur Hamiltonien du systdme et
f,travers
ce Hamiltonien. Les difl?rentes propri€t€s du mat€riau peuvent calcul6es.Deuxi&mement, Hohenberg
et
Kohn
ont
mortr€ qu'un
potentiel
v**set
un
nombre d'dlecFonsN
donn6s, l'4nergie totale du systdme atteint savaleur minimale lorsquela densit6 p
(r)
conespond d la densitd exacte deI'itat
fondarnental s'dcrit comme suit :Po (r).
g{po):minE(p}
La fonctionnelle de l'dnergie totale de I'dtat fondement
s'6crit
comme suit:E
Ip
(ril
=FHr[p
(r]l+J
Vext F{r}
d3rof
reprisetrteV**,
le potentiel externe agissant sur les particuleset
Flyplp{rl
reprdsentela
fo ncti onnel I e rmiversel I e de Hohenberg, Kohn.
Avec
Frirlp
(r)l
: {*lr+
rhp}
[.34]
(r.33)$.33)
llIleorie
d!
la fonctionnelle
dela
deasitf
DtrT
ta
connaissance de cette fonctionnellepermet de ddterminer l,6nergie totale et
densit.
de
charge de I'dtat fondamental externe donn6, en utilisant le
principe variationnel. Nous
conduit
en thdoriei
toutes les propridtds plrysico-chimiques de l,dtatfondamental
d'un
systdmedonn6'
cependant,il
reste un probldmede
taille
d
rdgler:
il
n,existepar
defermions en interaction
est
inconnue. Par ailleurs,trous avons que
modile
deT-F
constitue une approche trop brutalepour
obtenir des rdsulhts satisfaisants.il
ftut
donc sediriger
dans une auhe direction et c'est d ce stade queKohn
et sham ont proposd une approche tout dfait
ingdnieuse pour coRtourner ce probldme.
L5.2.2
Les
6quations del{ohn
et
Sham :Kohn
et
$ham
[19]
ont
inhoduit
un
ddveloppement suppldmentaire
qgi
consistei
remplacer
le
systdmerdel
interactif en un systcmefictif
noninteractif
.cette approche
r*alise
une coffespondance exacte de fermions non interactifs placds dans un
potentiel
efectif
et
le systime r6el dplusieurs dlecbons en interaction sournis au potentielrdel. De ce
fait,
la densit.{lecfonique
et I'dnergie du systdme r6el sont conservdes dans ce syst6mefictif
Four ce
systdmefictif,
les thdorimes
de Hohenberget
Kohn. s,appliquent 6galemen[ La
fonctionnelle de
la
densitdF[p]
Porn le systdmeinteractifpeut
€fe
e4primde parl,e:
ression suivante :F
[p(rl
:
Io[p(rl
+ ga[p(r]J
+E*"[p(r[
+H**s[p{rl
ofi
:
rs[p(rr
est l'dnergie
cin6tique
du
gaz
d,dlecFons
non
interagissant,Eri
tp(rr
d€signe
le
terme
de
Harbee,
E*"[p(r]J
est une fonctionnelle
additionnelle
qui
ddcritI'interaction
inter-dlechonique appelde 6nergie
d'echange-corrdlationet
t/"*1[p(r)J inclut
I'interaction
coulombienne des dlechons auec les noliauxet celle
des noyaux
enke
eux. Le terme de Harhee etcelui
del'fnergie
cindtiquejouent
unrdle
iruportant dansIa
descriptiondes itats des dlechons. la diffErence enhe
I'inergie
d'interaction r6elle et celle de Hartree sontprises en compte dans l'dnergie d'dchange et corr€lation l/**s[p{r)J.
L'€quation de schrtidinger d risoudre dans le cadre de I'approche
de Kohn
et Sham est delaforme:
tSa,*
r"rr(rlJld,(r)
>:
Elfi(r)
>,
i:r,....N
Ori le potential
effectifest
delaforme:
V"rr:V**t[#fu,
rV^"
(r.35)
(r.36)
Chapitre
I
Ihrryrie
dela fonctionnelle
dele densitfIIF,T
Le potential d'dchange et corrslation
est donn6
pr
rafonctionnere
derive:V*"(r\
= d(arclP(r)l)dp(r) (r.38)
Et la densit6 est donnde par une somme sur r,ensembre des orbitares occupdes
:
p
(r):x[1h]{r}12
(r.3e) L'6quation
(I'35)
correspondmt aux fquationsde Kohu et sham et doivent etre resolues de
fafon
auto-cohdrente,c'est- d- dire
endibutant
d
parfir
d'une certaine densitdinitiale,
un
potentiel est
obtenn
pour
lequel l'dquation
fi-35)
est
rdsolue
et
une nouvelle
densitddlectronique est
alors
ddterminde.A
partir
de
cettenouvelle
densit6,ufi
nouveau potentiel effectif peut Ghe calculd.ce
processus estrdpdti
de faqon auto-cohrirente[l]
jusqu,i
ce
quela
convergence soitatteinte.
L5.2.3
La fonctionnele
d'6change_corrdation
:
L'dlaboration
des fquations deKohn
et shamapermis
demdfe
en rividence lefait
quela
seule
fonctionnelle
d'dchange-corr6lationE*"[p{rI.
Ainsi,
pour
rdsoudreles
dquations deKohn et sham, diverses fonctionnelles d'6cha4ge-correlation ont 6t6 envisag6e
[z].
Les effets qui rdsultent des interactions
enfe
les dlectrons sont dehois
cafdgories : l,dnergie,la
correldion
dynamique et lacorrilation
non dynamique.r
I'effet
d'6change r6sultede
l'antisymdfie
de
la
fonction
d,ondetotale
vis-i-vis
del'echange des coordonndes dlectronique.
rl
correspand auprincipe
depagli
qui stipuleque deux dleckons
de
m6mespin
ont
rmeprobabilitd nulle
de sebouver au
memeendroit'
cet
effet est inddpendant dela
charge del'dlectron
et estpris
en compte dansla
thiorie
de
Hartree-Fock
d
cause
de I'antisymdfie
du
dfterminant
de
slater
reprfsentant la fonction d'onde.
r
I'efet
de
correlalion
ddsignela
corrdlation
entseles
mouvements dlectroniquesrdsultant
de
la
rdpulsion
inter-dlectsonique coulombienne"nfr.
Il
correspondessentiellement d'
l'efet
decordlation
pur
des €lecbons decaur.
il
est ind6pendant du spin. cet effet est n6grig6 par ra thdorie de Harbee-Fockr
le
troisidme
effet provient
du
fait
que les fonctions
d"onde
dlecfoniques
sontformulEes en termes de
particules
ind6pendants.Il
s'agit
deIa
correction de<<
self-interaction >> qui doit conduire
dun
comptage correct du nombrede paires d,€leckon.
Chapftre
f
I!yt*,,1.=8r.f":
l3en.
efi
densitoDF,r
La
fonctionnelled'ichange-corrilation
doit t+nir compte, en prus
de ce
qui
a dt6 $nonc€,de
Ia diftrence
d'dnergie cin6tique enhe
re
ryetdmefictif
noninteractif et re systime
noninteractifet
le systime r6el.Ainsi' le
calcul
deI'inergie
et dupotentiel d'ichange-
corrdration repose sur un cerfain nombre d' approximations.
a.
L'approximation
dela densitdloc*Ie
(LIIA)
:rl
est supposd quela
densit6 dlectronique peutGhe traitde rocalement sous ra
forme d,un gaz
d'dlecffons uniforme' Le mot <<locale r> indique
q,e
la fonctionnelleF[p]
ddpend seulement der
il travers la densitd p.ce
qui revienti
effectuer les deux h5pothdses suivantes :
cette approximatiou consiste donc
i
considdrer qre la contribution de
E*"[p(r[
d l,6nergie totale dusystime
peut6be
additionnde de fa4on
cumulie
i
partir
de chaqueportion
du gaz non uniforme comme
s'il
6tait localement uniforme.sj8*tpiril=Ip{r)
E}fd[pir]ld3r
ir.40)
Dans
laquelle
E'ffa[p{r}]
representel'dnergie
d'dchangeet
de
corrdlationpar *recfon
dans un gae d'*lechrons donf ra dishibution
est srrypos*e uniforme
[?].
A partir
deE'*8Ah{r}l
le potentiel d'dchange-corrfrationr,|.f*trl
peut 6o.e obtenu d,unefagon variationnelle selon l,dquation
:
tr/*TA
[rl:
d (P(r) E*84 tp(r)])o(p(r)
{r41}
Por:r
les
systdmes
magnitiques,
le
spin
elechronique
inkoduit un
degr6
de
libertd
zuppldmentaire
et
la LDA doit
€he alors
dtendue
i
l'ryproximdion
de
la
densitd
locale
despin
[2]'
otr l'6nergie d'dchange etcorelation
estdeux densit6s de spin haut et bas :
EiEo[pt,ptJ
:
lp(r]
Elpr{r),
ps{r}drr
0n
g6n€ralela
LDA
supposeque
la
dnergie est divisde en deux termes.
Ex"[p(rJ]
=
Ex[
p(rf+r"[
p{r!
(L4zj
foncfionrrelle
![Figure IV.1 Maille primitive de la phase wurtzite [2]'](https://thumb-eu.123doks.com/thumbv2/123doknet/14920199.662559/54.892.319.611.627.837/figure-iv-maille-primitive-de-phase-wurtzite.webp)
![Figure IV.3 Maille primitive de la phase Rocksalt [2]'](https://thumb-eu.123doks.com/thumbv2/123doknet/14920199.662559/55.892.326.586.199.408/figure-iv-maille-primitive-phase-rocksalt.webp)