HAL Id: tel-01624788
https://tel.archives-ouvertes.fr/tel-01624788
Submitted on 26 Oct 2017HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Mécanismes de l’immunogénicité des cellules cancéreuses
hyperploïdes
Norma Bloy
To cite this version:
Norma Bloy. Mécanismes de l’immunogénicité des cellules cancéreuses hyperploïdes. Cancer. Univer-sité Paris Saclay (COmUE), 2017. Français. �NNT : 2017SACLS239�. �tel-01624788�
REMERCIEMENTS
Je souhaiterais tout d’abord remercier Isabelle Cremer d’avoir accepté de présider ce jury de thèse, Filippo Rosselli et Antoine Tesnière d’avoir accepté d’être rapporteurs de ma thèse, et Ilio Vitale et Michaela Semeraro pour avoir accepté d’examiner ce travail.
Je remercie ensuite le Pr Guido Kroemer de m’avoir accueillie dans son laboratoire pendant mes 4 années de thèse, de m’avoir guidée, conseillée et soutenue tout au long de la réalisation de ce projet. Réaliser mon doctorat dans son laboratoire a été une formidable opportunité. Je le remercie également pour la tradition des fêtes de saisons, qui ont toujours été des évènements conviviaux et mémorables.
Le Dr Laura Senovilla pour sa supervision de ce projet de thèse, pour m’avoir beaucoup appris de manière générale. Son aide a été précieuse à chaque étape de ma thèse, particulièrement dans l’élaboration des articles, du manuscrit et dans la préparation de la soutenance.
Aitzi pour toute son aide et son amitié durant ma thèse, je le remercie en particulier d’avoir pris en charge une partie très importante de mon travail à un moment où j’en avais besoin.
Lorenzo pour son soutien continu et généreux durant toute ma thèse, pour tous ses conseils scientifiques extrêmement pertinents, sur mon projet, les articles scientifiques, le manuscrit de thèse et la présentation. Mais aussi et surtout pour son amitié à toute épreuve, et cela dans des moments où j’en ai eu particulièrement besoin.
John P pour son infinie gentillesse, sa disponibilité permanente et constante, concernant particulièrement la planification et la réalisation de toutes les expériences d’immunologie, pour lesquelles son aide m’a été indispensable.
Je remercie tout particulièrement Céline Laumont pour sa disponibilité, sa gentillesse et sa collaboration avec nous depuis le Canada, à chaque fois que nous en avons eu besoin.
Kees Melief pour ses conseils, sa bienveillance et sa collaboration avec nous pour la mise en place de nos protocoles de vaccination.
Fede pour toute son aide et sa générosité, ses réponses à toutes mes questions, et ses conseils à tout moment.
Allan pour sa patience et sa gentillesse dans notre collaboration, et pour sa personnalité flamboyante capable de donner le sourire en permanence et avec qui il est impossible de s’ennuyer.
Je remercie chaleureusement Chiara d’avoir volé à mon secours à de nombreuses reprises, en trouvant à chaque fois des solutions inespérées à mes problèmes.
Je salue également l’aide précieuse d’Erika dans certains de ces moments de crises, et ses conseils précieux pour mon manuscrit.
Kariman pour sa collaboration dans la mise au point de l’algorithme d’analyse morphométrique, et pour son aide précieuse dans d’autres domaines par la suite.
Didier, Yohann puis Isabelle pour leur aide en cytométrie, plus particulièrement Didier pour les séances de psychothérapies gratuites, et Isabelle pour sa disponibilité et sa gentillesse jamais démenties.
Gautier pour toutes ses explications en bio-informatique (non, ce n’est pas mon futur métier !), ses débats contradictoires, et sa générosité.
Lelly pour son aide spontanée, ses incroyables talents de gestion, et également Juliette et Franci, lorsque j’en ai eu besoin.
Naoufal pour ses conseils scientifiques très pertinents, et nos discussions enrichissantes.
Julia, la pro des coupes pour sa disponibilité, son aide et sa gentillesse.
Taka pour tous ses conseils, et sa consultation pour les expériences d’immunologie, toute son aide généreuse, ainsi que tous les bons moments passés en dehors du laboratoire.
Jono, pour sa gentillesse et sa disponibilité dans notre collaboration.
Adrien, le pro de DC-lamp.
Je remercie l’ensemble des personnes du laboratoire sans qui l’atmosphère n’aurait pas été si enrichissante et stimulante. En particulier Lelly, Franci Sarah, Juliette H, Juliette P et Shaoyi pour leur amitié et leur originalité, Lynda, Sylvie, Medhi, Sarah, Juliette H et Juliette P pour nos débats politiques passionnants, Florine avec qui j’ai débuté ma thèse, pour tous les bons moments que l’on a partagés, Gautier, Yohann, Didier pour nos apéros et Lucillia, Ligia, Sica, Chema, Maria C, Lorella, Maria P, Adriana, Amena, Friedo, Kris, Oliver, Kévin, Guo, Heng, Thierry, David, Sylvère, Valentina I, Valentina A, Marion, Noélie, Laura M, Sabrina, Peng, Liwei, Barbara et tous les autres qui ont contribué à rendre ces années si spéciales.
Je remercie Arash Nanbakhsh pour sa supervision, son aide indispensable et généreuse durant mon stage de Master 2 qui m’a permis de continuer mon parcours. Et pour les liens d’amitié que nous avons gardés depuis, avec tout ce qu’il m’a apporté.
Roy Golsteyn de m’avoir accueillie dans son laboratoire au Canada en Master 1, en m’offrant ainsi une expérience qui m’a profondément enrichie pour la suite de mon parcours.
Nicolas Guilbaud pour tous ses conseils extrêmement précieux et les opportunités qu’il m’a offertes depuis mon entrée à l’université.
Je remercie aussi tout particulièrement Brigitte Cyrille pour son aide, sa compréhension, sa disponibilité et sa gentillesse dans toutes les étapes de ma thèse (et surtout les étapes finales !).
Je remercie enfin ma famille pour leur soutien indispensable tout au long de mes études et de ma vie personnelle, leur accompagnement et leur aide à chacune des étapes de ma vie, qui m’ont permis d’arriver jusqu’ici. Et Julien pour sa présence réconfortante à mes côtés, son aide et son soutien.
INDEX
I. ABREVIATIONS 5
II. INTRODUCTION 8
1 Ploïdie 8
1.1 Physiologie de cellules tétraploïdes et polyploïdes 8
1.2 La génération de cellules tétraploïdes 10
1.3 Physiopathologie de cellules tétraploïdes et polyploïdes 13
2 Caractéristiques du cancer 15
2.1 Instabilité génomique et tétraploïdie : la répression des cellules tétraploïdes
et leur implication dans la cancérisation 15
2.2 Le système immunitaire 21
2.2.1 Le système immunitaire inné et adaptatif 21
2.2.2 Réponse immunitaire anti tumorale 23
2.2.2.1 Reconnaissance : antigènes tumoraux et signaux de danger 24
2.2.2.2 Mécanismes effecteurs des réponses immunitaires innées : NK, NKT, T γδ et adaptative : CD4, CD8, Interferons (IFNs) 26
2.2.3 Antigénicité et adjuvanticité 27
2.2.4 L’immunoédition: l’immunosurveillance des tumeurs 30
2.2.5 Mort cellulaire immunogène (ICD) 33
2.2.6 Stress du RE et exposition de la CALR à la surface des cellules mourantes 39
2.2.7 Perception de l’exposition de la CALR par les effecteurs immunitaires 44
3 Hyperploïdie et système immunitaire 45
3.1 Hyperploïdie, exposition de CALR et stress du RE 45
3.2 Immunosurveillance des cellules tétraploïdes 47
3.3 Données cliniques 48
4 Immunothérapie : les vaccins peptidiques 49
4.1 Mécanismes de l’immunité anti tumorale induite par les vaccins peptidiques 52
4.2 Les agonistes des Toll-like receptor (TLR) comme adjuvants 54
4.3 Peptides longs ou peptides courts 55
5 Le stress du RE affecte la transformation des peptides associés au CMH
de classe I 56
III. HYPOTHESES ET OBJECTIFS 57
IV. MATERIEL ET METHODE 58
1 Immunosurveillance contre les cellules cancéreuses hyperploïdes - impact
sur l'exposition de la calréticuline et l'immunopeptidome 69
1.1 Adjuvanticité 69
1.2 Antigénicité 74
2 Analyse morphométrique de l'immunosélection contre les cellules cancéreuses
hyperploïdes 78
2.1 Comparaison des cellules tumorales diploïdes et hyperploïdes par des
méthodes immunocytochimiques 78
2.2 L’hyperploïdie et l’hyperphosphorylation de eIF2α des tumeurs non
immunosélectionnées 83
2.3 Comparaison immunohistochimique de cancers non sélectionnés et
immunosélectionnés 86
VI. DISCUSSION 88
VII. REFERENCES 93
VIII. RESUME 128
ABREVIATIONS
ADN Acide désoxyribonucléique
ANXA1 Annexine A1
APC Adenomatous polyposis coli
ATF4 Activating transcription factor 4 ATF6 Activating transcription factor 6 ATP Adenosine triphosphate
AURKA Aurora kinase A
BAK Bcl-2 family members BCL2-antagonist/killer 1 BAP31 B-cell receptor-associated protein 31
BAX Bcl-2 family members BCL2-associated X protein
BBC3 BCL2 Binding Component 3
BCR-ABL Breakpoint Cluster Region- Abelson
BiP Binding immunoglobulin protein
BRCA1 Breast cancer 1
CALR Calreticulin
CD Cluster de différenciation
CDAMs Cell death-associated molecules CDKN1A Cyclin-dependent kinase inhibitor 1 CHEK1/ CHK1 Checkpoint kinase 1
CLIP Peptide Ii associé CMH de classe II CMH Complexe majeur d'histocompatibilité
CTL Lymphocyte T cytotoxique
DAMPs Damage associated molecular pattern »,
DC Cellules dendritique
eiF2α Eukaryotic Initiation Factor 2α ERAD ER-associated degradation
ERp57 ER protein of 57 kDa
FACS Fluorescence-activated cell sorter GRP78 78 kDa glucose-regulated protein HLA Human leucocyte antigen
HMGB1 High-mobility group box 1
HPV Human papillomavirus
ICD Immunogenic cell death
IFN Interféron
IL Interleukine
LATS2 Large tumor suppressor kinase 2 LPS Bacterial lipopolysaccharides IRE-1 Inositol-requiring enzyme 1
MAMP Microbe associated molecular patterns
MCA Methylcholanthrène
MDSCs Myeloid-derived suppressor cells MAPK14 Mitogen-activated protein kinase 14 MIIC MHC class II compartment
MICA/B MHC class I chain-related proteins A and B MULT1 Mouse UL16 binding protein-like transcript 1
NK Natural killer
NO Nitric oxide (monoxyde d’azote)
NOD Non-obese diabetic
Non-ICD Non immunogenic cell death PRR Pattern recognition receptor
P2RX7 Purinergic receptor P2X, ligand-gated ion channel, 7
PDT Photodynamic therapy
PERK RNA-activated (PKR)-like ER kinase PI3K Phosphoinositide-3-kinase
Prkdc Protein kinase, DNA activated, catalytic polypeptide
PS Phosphatidylserine
RAET-1 Retinoic acid early transcript 1 RAG Recombination activating gene
RB1 Retinoblastoma 1
RLR RIG-I-like receptors
ROS Reactive oxygen species
SERCA Sarco/endoplasmic reticulum Ca2+ ATPase SLP Synthetic long peptide
TAP Transporter associated with antigen processing TDLN Tumor-draining lymph node
TLR Toll-like receptor
UL16 Binding proteins
UPR Unfolded protein response
vSNAREs v N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) attachment receptor (SNARE)
WT Wild type
INTRODUCTION
1 Ploïdie
Les organismes et les cellules polyploïdes contiennent plus de deux paires de chromosomes homologues. La plupart des espèces nucléées (eucaryotes) sont diploïdes (2n), ce qui signifie qu'elles possèdent une paire de chromosomes, chaque chromosome hérité de chaque parent. Cependant, la polyploïdie se retrouve dans certains organismes et elle est particulièrement fréquente chez les plantes. En outre, la polyploïdie se produit dans certains tissus d'animaux diploïdes, tels que les tissus musculaires humains (Parmacek and Epstein 2009). Cela s'appelle l’endopolyploidie.
La polyploïdie se réfère à des mutltiples de l’ensemble complet des paires de chromosomes. Les organismes dans lesquels un chromosome particulier ou un segment chromosomique sont sous ou surprésentés sont eux considérés comme aneuploïdes (des mots grecs signifiant «non», «bon» et «pli»). Par conséquent, la distinction entre aneuploïdie et polyploïdie est le fait que l'aneuploïdie se réfère à un changement numérique dans une partie de l'ensemble chromosomique, alors que la polyploïdie se réfère à un changement numérique dans l'ensemble des chromosomes (Griffiths 1999). Les cellules aneuploïdes peuvent être considerées comme des cellules derivées des cellules tétraploïdes. Les cellules tétraploïdes présentent le double de chromosomes par rapport aux cellules diploïdes, c’est à dire deux paires (4n) (au lieu d’une pour les cellules diploïdes) de chromosomes homologues. La tétraploïdie n’est pas pas physiologique chez l’Homme, sauf dans les mégacaryocytes et le foie (Parmacek and Epstein 2009). En effet, les cellules tétraploïdes meurent normalement par apoptose grâce au point de contrôle de la tétraploïdie, impliquant TP53.
Enfin, notre laboratoire utilise le terme hyperploïde pour désigner des cellules possédant un ensemble de chromosomes supérieur à celui des cellules tétraploïdes (>4n), sans qu’il ne représente nécessairement un set complet de tous les chromosomes.
1.1 Physiologie de cellules tétraploïdes et polyploïdes
Par contraste avec certaines espèces végétales et animales (comme les insectes), les mammifères ne tolèrent pas la polyploïdisation des cellules germinales (Van de Peer, Mizrachi et al. 2017). Cependant des modifications de la ploïdie des cellules somatiques (endopolyploïdisation) participe au programme développemental de plusieurs tissus et organes
(Lee, Davidson et al. 2009) (Figure1). Par conséquent, bien que la majorité des cellules de mammifères soient diploïdes, des cellules multinucléées et endoploïdes peuvent être retrouvées à une fréquence relativement haute (0,5-20%), particulièrement dans les tissus hautement prolifératifs comme le placenta (Zybina and Zybina 2005). L’endopolyploïdisation qui constitue parfois un mécanisme permettant d’augmenter la taille cellulaire, a été documentée dans de multiples types cellulaires incluant les megakaryocytes et les neurones. Dans des conditions physiologiques, l’endopolyploïdisation peut constituer une réponse adaptive au stress (Lee, Davidson et al. 2009). Dans certains scénarios physiologiques (comme le repeuplement du foie par des cellules dérivées de la moelle osseuse), des intermédiaires polyploïdes générés par des fusions cellulaires hétérotypiques peuvent retrouver un génome diploïde et proliférer (Wang, Willenbring et al. 2003). De même sous certaines conditions spécifiques le pathogène humain Candida albicans peut accomplir son cycle de vie qui inclus des intermédiaires tétraploïdes mais se termine avec des cellules diploïdes ou presque-diploïdes capables de se reproduire (Bennett and Johnson 2003). Ainsi, la dépolyploïdisation des cellules tétraploïdes peut aussi constituer un processus physiologique. Cependant, la polyploïdisation illicite des cellules prolifératives a été associée à des maladies humaines comme le cancer (Storchova and Kuffer 2008). Dans ce contexte, les cellules polyploïdes, spécialement celles qui voient le jour après des dysfonctionnements mitotiques, sont considérées comme contribuant à la tumorigenèse ou la progression cancéreuse, en favorisant l’aneuploïdie et l’instabilité chromosomique.
- d’une fusion cellulaire (Figure 2) générant des cellules multinucléées arrêtées en phase G1 du cycle cellulaire (Ogle, Cascalho et al. 2005)
- une endoréplication (Figure 3), durant laquelle les cellules effectuent deux séries consécutives de réplication d’ADN non séparées par une mitose (Lee, Davidson et al. 2009)
- ou des échecs de certaines étapes de la mitose, donnant lieu à une une endomitose (Figure 4), durant laquelle les cellules avec leurs chromosomes dupliqués n’arrivent pas à effectuer leur division nucléaire (karyokinèse) ni leur division cytoplasmique (cytokinèse) et avortent la mitose (Lee, Davidson et al. 2009).
L’endo-polyploïdisation par fusion cellulaire a été documentée dans certains types cellulaires comme les myocytes (Rochlin, Yu et al. 2010). L’endoréplication (dont l’exemple le plus frappant est donné par les cellules géantes trophoblastiques qui se développent dans le tropectoderme murin (Zybina and Zybina 2005)) exerce des fonctions critiques pour le développement normal, l’efficacité de l’assimilation et du stockage, et de la différenciation (Lee, Davidson et al. 2009). L’endomitose survient dans les mégacaryocytes et dans certains hépatocytes (Lee, Davidson et al. 2009).
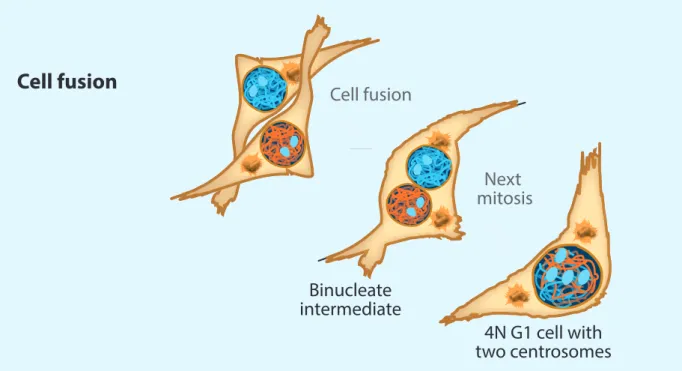
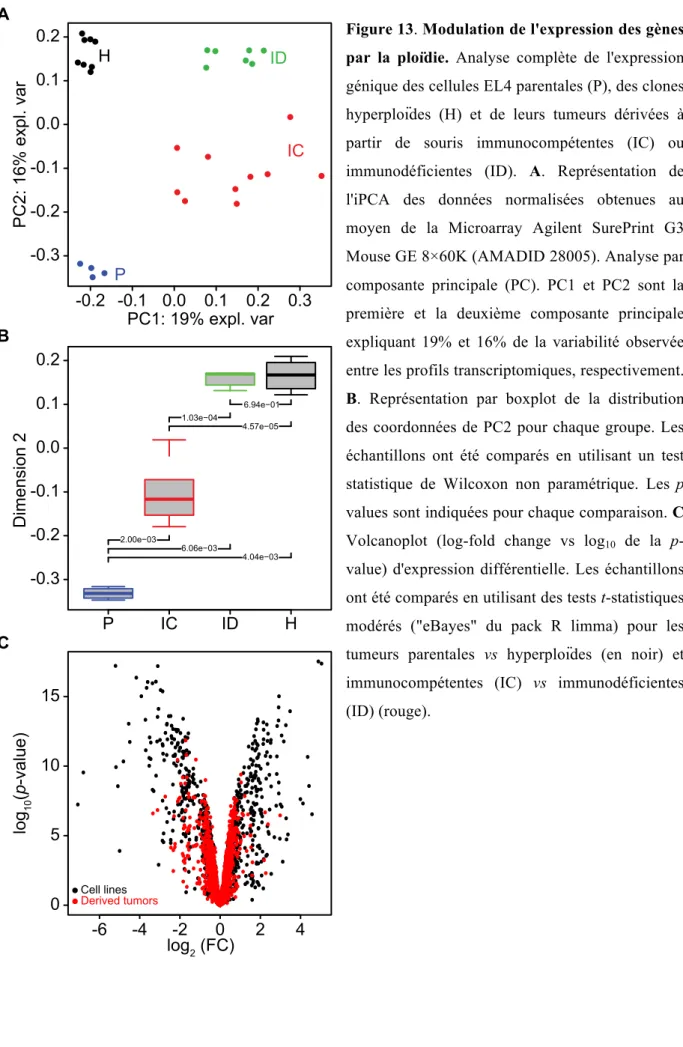
Figure 2 La fusion entre cellules comme générateur de tétraploïdie. A Des cellules tétraploïdes binucléées (2x 2n, 2C) peuvent apparaître en présence de stress mécanique, ou de protéines virales fusogènes. (Adapté de (Davoli and de Lange 2011)).
Cell fusion
4N G1 cell with two centrosomes Binucleate intermediate Next mitosis Cell fusionFigure 3 L’endoréplication
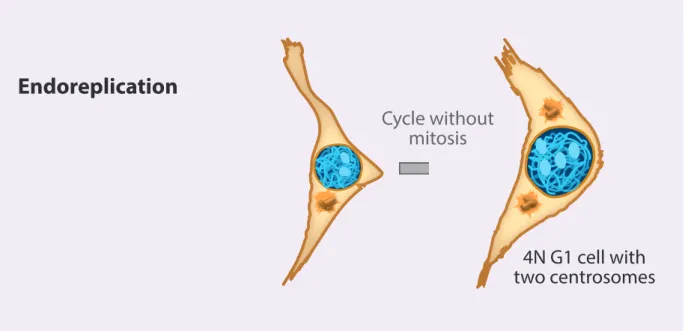
L’endoréplication survient lorsqu’une cellule diploïde ayant normalement dupliqué son ADN et ses centrosomes, n’entre pas en mitose. Cela entraine la formation d’une cellule tétraploïde mononuclée, possédant deux centrosomes (adapté de (Davoli and de Lange 2011)).
Endoreplication
4N G1 cell with two centrosomes
Cycle without mitosis
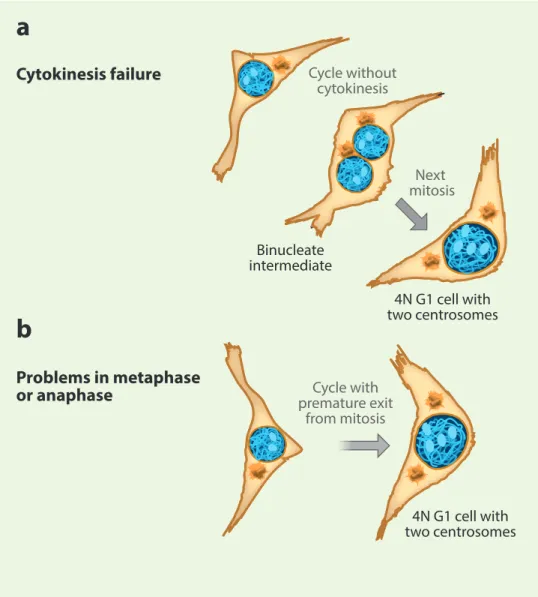
Figure 4: Les échecs mitotiques
Le non achèvement de la division cellulaire suite à l’échec de la cytokinèse (a) ou à la sortie précoce de la mitose (b) entraine la génération de cellules tétraploïdes (adapté de (Davoli and de Lange 2011)).
1.3 Physiopathologie de cellules tétraploïdes et polyploïdes
Dans les tissus blessés ou malades, des cellules polyploïdes somatiques peuvent également être générées accidentellement. En effet, les macrophages peuvent fusionnent entre eux durant l’inflammation (Helming, Tomasello et al. 2008), les cellules hématopoïétiques peuvent fusionner avec plusieurs types cellulaires comme les hépatocytes et les neurones en réponse à un stress des organes ou du corps entier (Johansson, Youssef et al. 2008), (Nygren, Liuba et al. 2008). Des virus oncogéniques peuvent éliciter une fusion entre cellules somatiques (Duelli and Lazebnik 2007) et les leukocytes peuvent fusionner avec des cellules cancéreuses sous l’influence de l’inflammation, générant ainsi des hybrides au potentiel métastatique (Pawelek
Problems in metaphase or anaphase 4N G1 cell with two centrosomes Cytokinesis failure
a
Binucleate intermediate 4N G1 cell with two centrosomes Next mitosis Cycle without cytokinesis Cycle with premature exit from mitosisb
and Chakraborty 2008).
De plus la tétraploïdisation peut, se produire par erreur, notamment dans les cellules tumorales dans certaines conditions comme :
- un échec de la mitose (ou adaptation mitotique) (Figure 4), un phénomène par lequel les cellules ne peuvent pas terminer la mitose à cause de l’activation continue du point de contrôle d’assemblage du fuseau « spindle assembly checkpoint » (SAC), et basculent ainsi dans la suite du cycle cellulaire dans un état tétraploïde (Brito and Rieder 2006)
- un échec de la cytocinèse, dû à une régression du sillon mitotique (à cause de ponts de chromatine obstruant le sillon de clivage ou à cause de défauts des protéines de la cytokinèse) et aboutissant à la génération de cellules binucléées (Steigemann, Wurzenberger et al. 2009). La non ségrégation des chromosomes (une erreur de l’anaphase aboutissant à la ségrégation de deux paires des chromosomes dans la même cellule fille) favorise l’apparition de la tétraploïdie plutôt que l’aneuploïdie (Shi and King 2005), indiquant que qu’une ségrégation correcte des chromosomes est requise pour effectuer la cytocinèse. Récemment, un lien a été établi entre la tétraploïdie et les lésions d’ADN pré-cancéreuses induites par l’activation d’un oncogène (Ichijima, Yoshioka et al. 2010) ainsi qu’une nouvelle voie d’endoréplication, déclenchée par une signalisation prolongée de dommages à l’ADN. Il y a donc des voies multiples et indépendantes de polyploidization.
Les points de contrôle préviennent les progressions incorrectes dans le cycle cellulaire pour s'assurer que les cellules conservent l'intégrité de leur génome. Au cours de la mitose, le point de contrôle de la métaphase suppervise l’alignement correct de tous les chromosomes sur le fuseau mitototique avant de la sortir de la mitose. Pourtant, des erreurs mitotiques se produisent dans des cellules de mammifères avec une fréquence relativement élevée. La métaphase représente le dernier point de contrôle de la mitose. Une fois que la cellule entre en anaphase, il n'y a pas de points de contrôle pour détecter des défauts dans la ségrégation des chromosomes. Il existe néanmoins un point de contrôle chez les cellules de mammifères qui agit après les erreurs mitotiques pour bloquer la prolifération des cellules qui sont entrées en phase G1 avec un statut tétraploïde. Cet arrêt dépend à la fois de TP53 et de la protéine du rétinoblastome (RB1), qui jouent ainsi un rôle important en tant que suppresseurs de tumeurs (Margolis, Lohez et al. 2003), (Andreassen, Lohez et al. 2001), (Borel, Lohez et al. 2002), (Andreassen, Lohez et al. 2003), (Margolis 2005). En effet la tétaploïdie non physiologique est
considérée comme un état intermédiaire entre la diploïdie et l’aneuploïdie, caractéristique des cancers (Andreassen, Martineau et al. 1996).
2 Caractéristiques du cancer
Jusqu'à récemment, les études sur la nature du cancer se focalisaient strictement sur les cellules cancéreuses et sur le cancer en tant que maladie génétique. Ceci est illustré dans l'article très cité de Hanahan et Weinberg (Hanahan and Weinberg 2000) publié en 2000 qui, après avoir examiné un grand nombre de recherches sur le cancer, a proposé six caractéristiques qui pourraient être utilisées pour définir une cellule comme cancéreuse (Finn 2012). Les caractéristiques comprenaient la capacité de maintenir une signalisation proliférative, de résister à la mort cellulaire, d'induire l'angiogenèse, de permettre l'immortalité réplicative, d'activer les capacités d’invasions et de métastases, et d'éviter les suppresseurs de croissance. Une décennie plus tard, Hanahan et Weinberg ont présenté de nouvelles caractéristiques qui incluent notamment l’instabilité chromosomique (Hanahan and Weinberg 2011) qui génère la diversité génétique permettant aux cellules cancéreuses d’accélérer l’acquisition de ces mêmes caractéristiques, essentielles à leur développement et leur dissémination. Les anomalies géniques peuvent porter sur la structure des chromosomes, regroupant les duplications, les délétions ou les translocations de fragments de chromosomes, ou peuvent aussi concerner le nombre de chromosomes, donnant lieu à des caryotypes sub diploïdes (<2n) ou bien hyperploïdes (>4n). Les progrès de ces 10 dernières annéesont également permis de mettre en évidence l’échappement à la destruction par le système immunitaire comme caractéristique essentielle de la progression tumorale (Hanahan and Weinberg 2011). Un nombre croissant de données tend à démontrer que l’échappement à la surveillance immunitaire (Hanahan and Weinberg 2011), (Schreiber, Old et al. 2011) et les interventions thérapeutiques à base de composés cytotoxiques ou d’agents de thérapies ciblées n’ont de succès que lorsqu’elles sont capables de réétablir un contrôle immunitaire sur la croissance tumorale (Delahaye, Rusakiewicz et al. 2011), (Zitvogel, Kepp et al. 2011).
2.1 Instabilité génomique et tétraploïdie : la répression des cellules tétraploïdes et leur implication dans la cancérisation
Les cellules cancéreuses accumulent des multiples altérations génétiques et épigénétiques, une condition d’instabilité génomique qui favorise l’acquisition de phénotypes agressifs. Un des
mécanismes de l’instabilité génomique implique une phase transitoire de polyploïdisation, et dans la plupart des cas de tétraploïdisation.
Environ un cinquième des cancers humains sont caractérisés par une aneuploïdie (c’est à dire un nombre anormal de chromosomes) avec un nombre de chromosomes supérieur à la normale (Gordon, Resio et al. 2012). L’aneuploïdie, qui est une caractéristique presque constante des cancers, peut être le résultat d’un événement de doublement du génome, suivi de par la perte progressive de chromosomes, due à une mauvaise ségrégation (Vitale, Galluzzi et al. 2011). De multiples de données vont dans ce sens. D’abord une étude de caractérisation extensive du génome de multiples tumeurs solides a révélé qu’approximativement 40% de ces cancers, incluant les tumeurs possédant une contenu en ADN presque-diploïde, ont connu au moins un événement de doublement du génome durant leur évolution (Zack, Schumacher et al. 2013), (Lundberg, Jin et al. 2013), (Dewhurst, McGranahan et al. 2014). Ensuite, les cellules tétraploïdes, sont retrouvées dans des stades précoces de cancers ainsi que dans des lésions pré malignes comme l’œsophage de Barret (Gordon, Resio et al. 2012).
Dans la plupart des types cellulaire, des variations significatives du statut diploïde ne sont pas tolérées, et les cellules hyperploïdes meurent aussitôt qu’elles sont générées (Fujiwara, Bandi et al. 2005). Cependant, l’état de tétraploïdie a pu être observé dans des stades précoces de cancers bronchiques, de l’œsophage, gastriques, mammaires, colorectaux, ovarien, du col de l’utérus et de la prostate, corrélé avec l’inactivation du gène suppresseur de tumeur, TP53, et de retinoblastoma 1 (RB1) (Davoli and de Lange 2011). La tétraploïdie peut agir comme un générateur stochastique d’instabilité génomique car les cellules tétraploïdes sont prônes à gagner ou perdre progressivement des chromosomes durant des cycles successifs de mitoses bipolaires puis multipolaires (Figure 5) (Vitale, Galluzzi et al. 2011); (Vitale, Galluzzi et al. 2011).
Une telle cascade de "polyploïdisation-dépolyploïdisation" est normalement évitée par des mécanismes de contrôles cellulaires autonomes oncosuppressifs (Vitale, Galluzzi et al. 2011); (Vitale, Galluzzi et al. 2011); (Duelli and Lazebnik 2007); (Ganem and Pellman 2007). Par conséquent l’inactivation de TP53 (Margolis 2005) ainsi que la perte d’autres protéines oncosuppressives comme la « cyclin-dependent kinase inhibitor 1 » (CDKN1A, plus connue sous le nom de p21) (Waldman, Lengauer et al. 1996), la « Bcl-2–associated X protein » (BAX) (Castedo et al. 2006, EMBO J), l’ « adenomatous polyposis coli » (APC) (Tighe, Johnson et al. 2004), BRCA1 (Schlegel, Starita et al. 2003), et la « large tumor suppressor kinase 2 »(LATS2) (Aylon, Michael et al. 2006) rend possible la tétraploïdisation. En accord avec cette notion, la surexpression de facteurs oncogéniques comme MYC (Yin, Grove et al. 1999), l’Aurora kinase A (AURKA) (Wang, Zhou et al. 2006) et la protéine E6 du virus du papillome humain (HPV-E6 de l’anglais « human papilloma virus E6 ») (Incassati, Patel et al. 2006) permet également la tétraploïdisation. Une des interprétations possibles de ces résultats est l’existence d’un « point de contrôle de la tétraploïdie » qui préviendrait la génération et la propagation des cellules tétraploïdes (Andreassen, Lohez et al. 2001). En effet, des défauts dans les points de contrôle du cycle cellulaire sont impliqués dans l’oncogenèse à différents niveaux. Par exemple, notre laboratoire a montré que l’inhibition de la protéine « checkpoint kinase 1 » (CHEK1, mieux connue sous le nom de CHK1) est particulièrement toxique pour les cellules cancéreuses tétraploïdes, qui sont relativement résistantes aux agents génotoxiques. Les mécanismes rendant compte dans l’effet préférentiel des inhibiteurs de CHK1 sur les cellules tétraploïdes impliquent le blocage du point de contrôle de l’assemblage du fuseau mitotique, provoquant une entrée prématurée catastrophique en mitose, associée à l’activation de la « mitogen-activated protein kinase 14 » (MAPK14, plus connue sous le nom de p38MAPK) et de TP53.
En retour, cela provoque la transactivation des gènes pro-apoptotiques ciblant TP53, dont le « BCL2 Binding Component 3 » (BBC3, plus connu sous le nom de PUMA), entrainant l’activation de la voie mitochondriale de l’apoptose (Vitale, Galluzzi et al. 2007), (Vitale, Senovilla et al. 2008) (Figure 6).
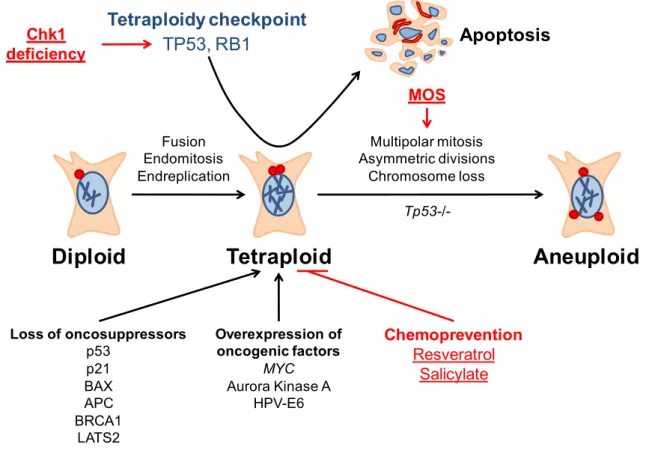
Figure 6. Cascade de polyploïdisation/dépolyploïdisation dans l’oncogenèse. Les cellules diploïdes peuvent devenir tétraploïdes suite à une endoréplication, des erreurs mitotiques et des fusions entre cellules, un phénomène qui est favorisé par certains oncogènes comme AURKA et la perte de certains gènes suppresseurs de tumeur. Nous avons montré que l’inhibition de CHK1, la sous expression de MOS, ainsi que l’administration de resvératrol ou de salicylate sont impliqués dans la destruction préférentielle des cellules tétraploïdes. De plus, nous avons trouvé que l’absence de TP53 est non seulement permissive pour la survie mais aussi pour les divisons asymétriques multipolaires des cellules tétraploïdes via la surexpression de MOS.
Un oncogène en particulier, MOS, est strictement nécessaire pour la première division méiotique des oocytes (Sagata, Daar et al. 1989), et exerce une fonction critique de point de contrôle durant la métaphase II (Sagata, Watanabe et al. 1989). Jusque récemment, très peu de choses étaient connues sur le rôle de MOS dans les cellules somatiques, et sur les mécanismes par lesquels MOS promouvait l’oncogenèse. Notre laboratoire a mis en lumière un mécanisme dépendant de MOS et de TP53 à travers lequel des divisions asymétriques provoquent une instabilité chromosomique chez les cellules tétraploïdes. La surexpression de MOS, ce qui se produit de manière spontanée dans les cellules, inhibe le regroupement des centrosomes, favorisant ainsi les divisions multipolaires et par conséquent la réduction du génome tétraploïde à un état sub-tétraploïde. Les mitoses multipolaires sont en effet plus fréquentes
Diploid
Tetraploid
Aneuploid
Apoptosis Tetraploidy checkpoint Multipolar mitosis Asymmetric divisions Chromosome loss Fusion Endomitosis Endreplication TP53, RB1 Loss of oncosuppressors p53 p21 BAX APC BRCA1 LATS2 Overexpression of oncogenic factors MYC Aurora Kinase A HPV-E6 Chk1 deficiency MOS Chemoprevention Resveratrol Salicylate
Tp53-/-lorsque TP53 est sous exprimée et MOS est surexprimé. En accord avec ces observations, le knockdown de MOS permet d’éviter les évènements de mitoses multipolaires parmi les cellules TP53-déficientes, exerçant ainsi des effets de stabilisation du génome (Vitale, Senovilla et al. 2010).
Des données robustes, mettant en évidence une implication directe de la tétraploïdie dans l’oncogenèse, ont été obtenues dans des expériences impliquant la tétraploïdisation de cellules épithéliales primaires in vitro. Ces cellules étaient par la suite capables de former des tumeurs
in vivo (Padilla-Nash, Hathcock et al. 2012). Notre laboratoire a étudié la réponse suite à la
tétraploïdisation des cellules épithéliales de glande mammaire primaires (MMECs de l’anglais « mouse mammary epithelial cells ») ou d’organoïdes des cryptes de l’intestin, obtenues de souris wild type (WT) ou Trp53-/-, ainsi que leur capacité à induire la tumorigenèse. Il a été observé que les MMECs TrTP53-/- et les cultures de colonocytes contenaient un pourcentage de cellules polyploïdes (>4n) – presque tétraploïdes – plus élevé que leurs homologues WT. Cette différence est exacerbée par l’administration d’agents tétraploïdisants, indiquant que l’absence de TP53 est permissive pour la polyploïdisation des cellules primaires (Senovilla, Vitale et al. 2009), (Boileve, Senovilla et al. 2013). De plus, les cultures de MMECs Trp53 -/-présentent un taux plus faible de cellules mourantes ou mortes que les cellules WT, suggérant un lien possible entre la résistance à l’apoptose et une plus grande susceptibilité à la polyploïdisation (Senovilla, Vitale et al. 2009). Notre laboratoire ainsi que d’autres équipes on précédemment observé une résistance aux dommages à l’ADN chez les cellules tétraploïdes, comparé à leurs homologues parentales diploïdes (Castedo, Coquelle et al. 2006), (Shen, Moran et al. 2008). Conformément à ces observations, notre laboratoire a trouvé que des MMECs Trp53-/- récemment isolées, étaient significativement moins tuées par les anthracyclines que leurs précurseurs diploïdes, corroborant la notion que la tétraploïdie augmente la résistance à la chimiothérapie (Senovilla, Vitale et al. 2009).
De plus notre lanoratoire a montré que les cellules cancéreuses tétraploïdes sont résistantes, par rapport aux cellules cancéreuses diploïdes, à une série d'agents induisant des dommages à l'ADN, à savoir le cisplatine, l'oxaliplatine, la camptothécine et l'irradiation par rayons gamma et UVC. Cette augmentation de la résistance est corrélée à une réduction des paramètres apoptotiques (comme la dissipation du potentiel transmembranaire mitochondrial et la dégradation de l'ADN nucléaire) dans les cellules tétraploïdes par rapport aux cellules diploïdes soumises à des dommages à l'ADN. En outre, les cellules tétraploïdes présentaient un
niveau d'activation élevé de TP53. L'inhibition de TPp53 a supprimé la différence de susceptibilité entre les cellules cancéreuses diploïdes et tétraploïdes à l'apoptose induite par les dommages à l'ADN. Ces données indiquent une résistance intrinsèque des cellules tétraploïdes contre la radiothérapie et la chimiothérapie ciblée par l'ADN qui peuvent être liées au statut de TP53 (Castedo, Coquelle et al. 2006), (Castedo, Coquelle et al. 2006).
Connaissant le rôle central de cascade de polyploïdisation/dépolyploïdisation dans l’oncogenèse et la résistance des cellules tétraploïdes à l’apoptose, il semble fondamental de développer des stratégies pour éradiquer sélectivement les cellules tétraploïdes. En utilisant un screening de phénotypes, notre laboratoire a identifié le resvératrol et d’autres activateurs de l’ « AMP-activated protein kinase » (AMPK) tel que le salicylate, comme des composés tuant les cellules tétraploïdes plus efficacement que leurs homologues parentales en stimulant la voie intrinsèque de l’apoptose. Le resvératrol et le salicylate limitent tous les deux la formation de cellules tétraploïdes ou de polyploïdie d’ordre supérieur, à partir de cultures cellulaires de carcinome du colon humain TP53-/- ou de cellules épithéliales primaires de souris lorsque ces cellules sont soumises à la présence d’agents anti mitotiques. De plus, l’administration orale du resvératrol ou du salicylate diminue l’accumulation des cellules épithéliales intestinales tétraploïdes dans les modèles murins de carcinomes du colon ApcMin/+. Nous avons émis l’hypothèse selon laquelle, l’action chimiopréventive du resvératrol et du salicylate entrainent une élimination des précurseurs cancéreux tétraploïdes (Lissa, Senovilla et al. 2014).
2.2 Le système immunitaire
2.2.1 Le système immunitaire inné et adaptatif
Le système immunitaire d'un organisme est un système biologique constitué d'un ensemble d'éléments de reconnaissance et de défense qui discriminent le soi du non-soi, afin de protéger l’organisme.
Le système immunitaire inné, comprend les cellules et les mécanismes qui défendent l'hôte contre une infection par d'autres organismes (Grasso 2002). Les cellules du système inné reconnaissent et répondent aux agents pathogènes d'une manière non spécifique, et contrairement au système immunitaire adaptatif, elles ne fournissent pas une immunité durable
à l'hôte (Alberts B 2002), (Janeway 2001). Les principales fonctions du système immunitaire inné des vertébrés comprennent: le recrutement de cellules immunitaires sur des sites d'infection, par la production de facteurs chimiques, y compris des médiateurs chimiques spécialisés, appelés cytokines ; l’identification et élimination des substances étrangères présentes dans les organes, les tissus, le sang et la lymphe, par des globules blancs spécialisés ; l’activation du système immunitaire adaptatif par un processus connu sous le nom de présentation antigénique ; et agir comme une barrière physique et chimique aux agents infectieux.
Les cellules effectrices du système immunitaire inné sont les mastocytes, les phagocytes, les macrophages, les neutrophiles, les cellules dendritiques (DCs, de l’anglais « dendritic cells », les basophiles et éosinophiles, les cellules tueuses naturelles (NK, de l’anglais natural killer), et les cellules T γδ.
Les DCs sont très importantes dans le processus de présentation des antigènes et servent de lien entre le système immunitaire inné et adaptatif (Alberts B 2002).
Le système immunitaire adaptatif, également connu sous le nom de système immunitaire spécifique, est composé de cellules hautement spécialisés qui éliminent les agents pathogènes ou empêchent leur croissance. L'immunité adaptative crée une mémoire immunologique après une réponse initiale à un pathogène spécifique et conduit à une réponse améliorée aux rencontres ultérieures avec ce pathogène. Ce processus d'immunité acquise est à la base de la vaccination. Comme le système immunitaire inné, le système adaptatif comprend à la fois des composants d'immunité humorale et des composants d'immunité à médiation cellulaire. Les cellules qui effectuent la réponse immunitaire adaptative sont des globules blancs appelés lymphocytes. Les deux principales fonctions - production d'anticorps et réponse immunitaire à médiation cellulaire - sont également portées par deux types de lymphocytes différents (cellules B et cellules T). Dans les réponses aux anticorps, les cellules B sont activées pour sécréter des anticorps, qui sont des protéines également appelées immunoglobulines. Les anticorps se déplacent à travers le flux sanguin et se lient à l'antigène étranger pour l'inactiver, ce qui ne permet pas à l'antigène de se lier à l'hôte (Alberts B 2002). Dans l'immunité acquise, les récepteurs spécifiques des agents pathogènes sont « acquis » au cours de la durée de vie de l'organisme, alors que chez les immunitaires innés, les récepteurs spécifiques du pathogène appelés « pattern recognition receptors » (PRR) tels que les « toll like receptor » (TLR), sont déjà codés dans la lignée germinale. Le système adaptatif est hautement adaptable en raison de l'hypermutation somatique (un processus de mutations somatiques
accéléré) et de la recombinaison V (D) J (une recombinaison génétique irréversible des segments de gènes du récepteur antigénique). Ce mécanisme permet à un petit nombre de gènes de générer un grand nombre de récepteurs antigéniques différents, qui sont ensuite exprimés de manière unique sur chaque lymphocyte. Étant donné que le réarrangement du gène entraîne un changement irréversible de l'ADN de chaque cellule, tous les descendants de cette cellule héritent des gènes qui codent le même récepteur spécifique, y compris les cellules B mémoires et les cellules T mémoires qui sont les effectrices clés de la longévité de l’immunité adaptative.
Les effecteurs immunitaires de l’immunité adaptative sont les lymphocytes T : lymphocytes T CD8+ cytotoxiques, lymphocytes T « Helper » CD4+, les lymphocytes T γδ ; et les lymphocytes B producteurs d'anticorps (Alberts B 2002).
Les antigènes sont des macromolécules naturelle ou synthétique qui sont reconnues par des anticorps ou des cellules du système immunitaire et sont capables de déclencher une réponse immunitaire. Les antigènes sont généralement des protéines, des polysaccharides et leurs dérivés lipidiques. L'antigène peut provenir de l'organisme («auto-antigène») ou de l'environnement externe («non-soi»). Le système immunitaire ne réagit généralement pas aux auto-antigènes dans des conditions homéostatiques normales en raison de la sélection négative des cellules T dans le thymus. Il est censé identifier et attaquer uniquement au «non-soi» des pathogènes du monde extérieur ou des substances nocives présentes dans le corps dans des conditions de stress (Gallucci, Lolkema et al. 1999).
Les cellules présentatrices d'antigène comme les DCs, présentent des antigènes sous forme de peptides sur des molécules du complexe majeur d'histocompatibilité (CMH). Les cellules T du système immunitaire adaptatif reconnaissent les antigènes. Selon l'antigène et le type de la molécule d'histocompatibilité, différents types de lymphocytes T sont activés. Pour la reconnaissance du récepteur des cellules T (TCR pour « T cells receptor »), le peptide doit être transformé en petits fragments à l'intérieur de la cellule et présenté par le CMH (Parham 2009).
2.2.2 Réponse immunitaire anti tumorale
Depuis qu'Ehrlich, en 1909, a proposé l'idée que les cellules transformées naissantes se développent continuellement dans notre corps et que le système immunitaire analyse et éradique ces cellules transformées avant qu'elles ne se manifestent cliniquement, la surveillance immunitaire a été un sujet controversé dans l'immunologie tumorale (Ehrlich
1909), (Kim, Emi et al. 2007). Dans la moitié du 20ème siècle, des preuves expérimentales que les tumeurs pouvaient être réprimées par le système immunitaire ont été apportées par des modèles de transplantation de tumeurs. Les résultats de ces modèles ont fortement suggéré l'existence d'antigènes associés à une tumeur et ont servi de base à la théorie de la surveillance immunitaire, qui a été postulée par Burnet et Thomas (Burnet 1970), (Burnet 1957). Par la suite Polly Matzinger a émis en 1994, la « théorie du danger », selon laquelle le système immunitaire est capable de différencier non seulement le soi du non soi, mais également les signaux de dangers émis par des cellules potentiellement dangereuses (Matzinger 2002). Après cela, le rôle fonctionnel des cellules présentatrices d'antigène dans l'activation des lymphocytes T a été démontré, et le modèle de surveillance immunitaire contre le cancer a été développé. Cependant, l'idée de la surveillance immunitaire contre le cancer a résisté à une acceptation répandue jusqu'à la fin des années 1990, lorsque des modèles animaux expérimentaux utilisant des souris knock-out ont validé l'existence d'une surveillance immunitaire contre le cancer dans les tumeurs spontanées et chimiquement induites (Dunn, Bruce et al. 2002), (Dunn, Bruce et al. 2005). Et depuis, de nombreuses preuves précliniques et cliniques ont montré que les cellules malignes peuvent être reconnues, et éliminées dès les premiers stades du développement tumoral (Burnet 1970), (Fridman, Pages et al. 2012), (Schreiber, Old et al. 2011).
2.2.2.1 Reconnaissance : antigènes tumoraux et signaux de danger
Les cellules tumorales bien qu’étant à l’origine, des cellules du « soi », sont caractérisées par une grande instabilité génétique qui favorise l’apparition de nouvelles protéines (néoantigènes, c’est à dire des peptides entièrement absents du génome humain normal (Schumacher and Schreiber 2015)) ou l’expression ectopique ou en quantité anormale de molécules les différenciant du « soi ».
Un des objectifs clés de l'immunothérapie est d’identifier des antigènes tumoraux, exprimés de façon stable par la tumeur, et exprimés de manière moindre dans les tissus normaux, afin de diriger le système immunitaire contre ces cellules tumorales par un processus de vaccination. Plusieurs molécules clés, ont été identifiées comme étant des antigènes tumoraux, notamment une famille d’antigènes spécifiques, les antigènes "cancer-testicules" (CTA), des protéines exprimées par les cellules tumorales de différents types tissulaires mais absentes des cellules saines, à l'exception des cellules germinales et des trophoblastes (Scanlan, Simpson et al. 2004). Cela a révélé le rôle important de la ré-activation du programme génétique des gamètes, normalement silencieux dans les cellules somatiques, dans la tumorigénèse (Simpson,
Caballero et al. 2005). La liste des CTA a été ensuite considérablement élargie par le développement de nouvelles techniques, notamment celle développée par le groupe Pr Pfreundschuh (Sahin, Tureci et al. 1995). Elle a permis d’identifier plus de 20 CTA différents grâce à l’immunocriblage du sérum de patients autologues et à des bibliothèques d’expression d’ADNc phagique recombinant (Scanlan, Gure et al. 2002). Les principaux antigènes tumoraux identifiés comme étrangers par le système immunitaire ont été classés en cinq catégories et résumés dans le tableau suivant.
Tableau 1. Classification des antigènes tumoraux définis.
Abréviations: Her2/neu, human epidermal growth factor receptor 2 ; Wilm's Tumor 1 (WT1) and Ephrin receptor (Eph3) proteins ; prostate specific membrane antigen (PSMA) ; Tyrosinase Related Protein-1 (TRP)-1 ; MAGE Melanoma-associated antigen, gp glycoprotein (Adapté (Srinivasan and Wolchok 2004)).
Le système immunitaire inné est également capable de discriminer les cellules saines des cellules transformées grâce l'interaction du récepteur d’activation « natural killer group 2 member D » (NKG2D), exprimé sur les cellules NK, les cellules T γδ et les cellules T CD8+ αβ, avec des ligands spécifiques de la tumeur, exprimés à la suite d’un stress génotoxique, une infection virale ou par transformation tumorale (Bauer, Groh et al. 1999).
Les ligands de NKG2D ont un niveau d‘expression normalement faible dans les cellules saine et élevé dans les cellules tumorales ou infectées, chez l’Homme comme chez la souris. Ces ligands sont répartis en deux grandes familles de protéines, structurellement proches des molécules du CMH de classe I mais n’interagissant pas avec la β2 microglobuline: les familles « MHC class I chain-related proteins A et B » (MICA et MICB) et « retinoic acid early transcript 1 » (RAET-1). Les familles MIC et « UL16 binding proteins » (ULBPs) uniquement présente chez l’Homme, et la famille RAET-1 qui est en revanche retrouvée dans les deux espèces (Gasser and Raulet 2006). MIC A et MIC B sont des protéines fréquemment exprimées à la surface des carcinomes primaires du poumon, du rein, des ovaires, du colon (Groh, Wu et al. 2002) et du foie (Jinushi, Takehara et al. 2003) mais aussi des mélanomes (Vetter, Groh et s e c n e r e f e R s e l p m a x E s n e g i t n A
Unique antigens (usually caused by mutations) p53, ras, β-catenin, CDK4, CDC27, αactinin-4 32,34,36,37,38,39 0 0 1 p g , 2 P R T , 5 7 p g / 1 P R T , e s a n i s o r y T s n e g i t n a n o i t a i t n e r e f fi
D , Melan-A/MART1, gangliosides, PSMA 42, 43, 44, 48, 50, 51
2 6 , 1 6 , 0 6 , 9 5 , 5 5 0 2 D C , R F G E , 3 A h p E , 1 T W , 2 R E H s n e g i t n a d e s s e r p x e r e v O , E G A M s n e g i t n a s i t s e t -r e c n a C BAGE,GAGE,NY-ESO-1 63,64 0 2 , 9 1 n i v i v r u S , e s a r e m o l e T s n e g i t n a l a s r e v i n U
al. 2002). Chez la souris, NKG2D se lie aux protéines RAE-1, à l’antigène mineur d’histocomptatibilité H60 (Diefenbach, Jensen et al. 2001), (Cerwenka, Baron et al. 2001) et au « mouse UL16 binding protein-like transcript 1 » (MULT1) (Carayannopoulos, Naidenko et al. 2002). L’expression de ces trois ligands de NKG2D a été observée dans un nombre important de tumeurs murines et est suffisante pour induire le rejet de plusieurs tumeurs transplantables (Diefenbach, Jamieson et al. 2000).
2.2.2.2 Mécanismes effecteurs des réponses immunitaires innées : NK, NKT, T γδ et adaptative : CD4, CD8, Interferons (IFNs)
Le processus d'élimination des cellules tumorales est à la fois le résultat de l’activation de l’immunité innée et de l’immunité adaptative. La réponse immunitaire innée est la première à se déclencher : plusieurs cellules effectrices telles que les cellules NK, NKT et les lymphocytes T γδ sont activées par des cytokines inflammatoires (tumor necrosis factor (TNF)-α, transforming growth factor (TGF)-β, interleukin (IL)-1β, IL-6, IL-10, IL-12) libérées par les cellules tumorales, ainsi que les macrophages et les cellules stromales qui les entourent (Yamamoto, Kimura et al. 2003). Les cellules immunitaires recrutées produisent à leur tour des cytokines pro-inflammatoires comme l’IL-12 et l’interféron (IFN)-γ. La lyse des cellules tumorales par les cellules NK libère ensuite des antigènes tumoraux permettant l’activation d’une réponse immunitaire adaptative (Smyth, Thia et al. 2000), (Mori, Jewett et al. 1997), (Takeda, Hayakawa et al. 2001). L’ingestion des antigènes tumoraux par cellules dendritiques (DCs) promeut leur maturation et leur migration aux nœuds des ganglions lymphatiques drainants (TDLNs de l’anglais « tumor-draining lymph node ») où elles effectuent leur présentation antigénique aux cellules T naïves, et induisent l’expansion clonale des lymphocytes T cytotoxiques (CTLs de l’anglais « cytotoxic T lymphocytes »). Les lymphocytes T spécifiques des antigènes tumoraux sont recrutés au niveau de la tumeur et sont capables de tuer directement les cellules tumorales (Zitvogel, Terme et al. 2006). Le processus d’élimination s’effectue selon les 4 phases suivantes:
1- Reconnaissance des cellules tumorales par le système immunitaire inné : lorsqu’une tumeur solide atteint plus de 2 ou 3mm, elle nécessite un afflux sanguin plus important ainsi qu’une réorganisation du stroma afin de poursuivre sa progression tumorale. Cet environnement produit des signaux pro inflammatoires entrainant le recrutement des cellules NK, NKT, T γδ, des macrophages et des DCs au site tumoral (Matzinger 1994), (Smyth,
Godfrey et al. 2001). Les cellules transformées exprimant des antigènes tumoraux peuvent ainsi être reconnues par ces cellules productrices d’INFγ (Street, Hayakawa et al. 2004).
2- Maturation et migration des DCs et présentation croisée aux cellules T : l’IFN-γ exerce un contrôle anti-prolifératif (Gollob, Sciambi et al. 2005), angio-statique (Qin, Schwartzkopff et al. 2003) et proapoptotique (Wall, Burke et al. 2003) sur les tumeurs. Certaines chimiokines provenant de la tumeur ou du microenvironnement comme CXCL10 (1P10), CXCL9 (MIG) et CXCL11 (I-TAC) inhibent la néovascularisation de la tumeur, tout en ayant une activité anti tumorale (Angiolillo, Sgadari et al. 1995). Les cellules tumorales nécrotiques sont ingérées par les DCs immatures qui maturent en présence de molécules pro inflammatoires et migrent vers les TDLNs.
3- Production de cellules T spécifiques des antigènes tumoraux: les cellules NK infiltrant la tumeur et les macrophages producteurs d’IL-12 et d’IFN-γ, détruisent les cellules tumorales grâce l’activation de mécanismes cytotoxiques comme la perforine, TRAIL et la production d'espèces réactives de l'oxygène (ROS de l’anglais « reactive oxygen species »), respectivement (Ikeda, Old et al. 2002), (Shi, Evans et al. 2003). Dans les TDLNs, les DCs présentent les antigènes tumoraux aux cellules T CD4+ naïves qui se différencient en cellules T CD4+ helpers permettant le développement des lymphocytes T CD8+ spécifiques des antigènes tumoraux et leur expansion clonale.
4- Migration des cellules T CD4+, CD8+ spécifiques des antigènes tumoraux au site tumoral, élimination des cellules par les CTLs, amplifiée par l’IFN-γ et sélection des cellules présentant une faible immunogénicité (Shankaran, Ikeda et al. 2001), (Kim, Emi et al. 2007).
2.2.3 Antigénicité et adjuvanticité
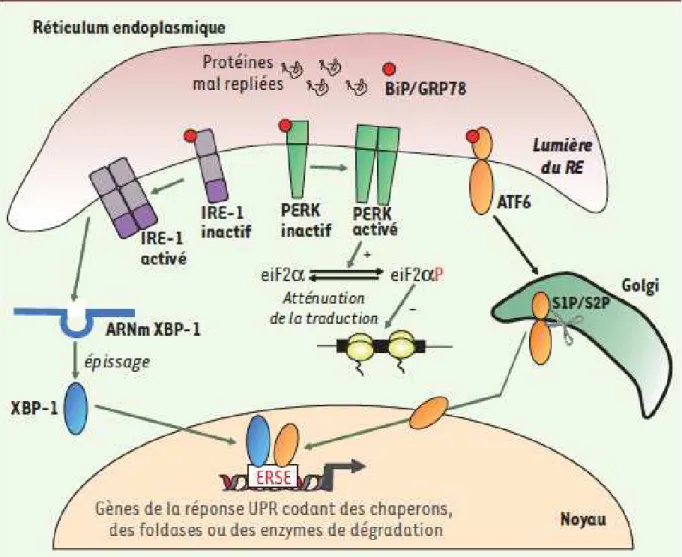
Les cellules cancéreuses peuvent activer le système immunitaire par deux mécanismes : l’antigénicité et l’adjuvanticité. L’antigénicité représente la capacité des cellules à activer le système immunitaire (cellules T et B) en présentant à leur surface des peptides antigéniques sur les molécules de CMH de classe I. L’adjuvanticité représente la capacité à stimuler la réponse immunitaire par l’expression à la surface des cellules de molécules de co-stimulation, comme les motifs moléculaires associés aux dégâts cellulaires (DAMPs de l’anglais « damage associated molecular pattern ») ou la calréticuline (CALR), qui seront décrits plus loin.
Selon l’hypothèse du « danger » (Matzinger 2002) les réponses immunitaires sont déclenchées de manière optimale si le stimulus comporte deux caractéristiques distinctes. D’abord, il est
important que les cellules cancéreuses (ou les cellules infectées par un pathogène) soient antigéniquement différentes de leurs homologues sains.
Le profil antigénique des cellules cancéreuses reflète :
(1) la transformation maligne, qui est associée à des changements génétiques et épigénétiques qui sont reflétés dans la synthèse de protéines mutantes ou de fusion mais influence aussi l’expression génique au niveau global (Wick, Webb et al. 2014), (Overwijk, Wang et al. 2013), (Kreiter, Castle et al. 2012), (Castle, Kreiter et al. 2012).
(2) une instabilité génique qui entraine l’accumulation de modifications génétiques (plus ou moins importantes) et augmente ainsi la quantité de polypeptides immunogénique (Vitale, Galluzzi et al. 2011), (Vitale, Galluzzi et al. 2011), (Negrini, Gorgoulis et al. 2010), (Sieber, Heinimann et al. 2003), (Gorgoulis, Vassiliou et al. 2005).
(3) les facteurs intracellulaires et micro environnementaux qui entrainent des réponses adaptatives liées à des changements dans la traduction des protéines et leur dégradation par le protéasome, et donc agissant directement sur l’immunopeptidome, c’est à dire l’ensemble des peptides présentés à la surface des cellules par les molécules de CMH I (Kumari, Walchli et al. 2014), (Caron, Vincent et al. 2011), (Admon and Bassani-Sternberg 2011).
De ce fait, les cellules malignes expriment pratiquement toujours des antigènes tumoraux (TAAs pour « tumor associated antigens »), c’est à dire des antigènes qui différent qualitativement ou quantitativement de ceux exprimés par les cellules normales de même type
(Mufson 2006), (Boon, Cerottini et al. 1994), (Coulie, Van den Eynde et al. 2014). On peut noter que les TAAs peuvent être viraux dans le cas des tumeurs induites par des virus (comme les protéines E6 et E7 des papillomavirus humains type 16 et 18, qui sont associés aux cancers du col de l’utérus et de l’oropharynx) ; uniques lorsqu’ils reflètent une altération génique propre à une seule cellule et ses clones (par exemple le récepteur des cellules B exprimé par certaines formes de lymphomes) ; tumeur-spécifiques lorsqu’ils ne sont pas exprimés par les cellules normales mais sont trouvés dans différentes tumeurs du même type (par exemple la protéine de fusion Breakpoint Cluster Region- Abelson (BCR-ABL), qui est exprimée par une grande majorité de leucémie lymphoïdes chroniques) ; ou bien partagés lorsqu’ils sont exprimés par les cellules malignes ainsi que par certains types de cellules non transformées
(Mufson 2006), (Boon, Cerottini et al. 1994), (Coulie, Van den Eynde et al. 2014), (Nizard, Sandoval et al. 2013). Les TAAs partagés incluent des antigènes fœtaux, exprimés uniquement durant le développement embryonnaire, ainsi que les antigènes des cancers des testicules, qui sont normalement exprimés uniquement par les cellules germinales mâles dans les testicules
(Nizard, Sandoval et al. 2013), (Kirkin, Dzhandzhugazyan et al. 2002), (Simpson, Caballero et al. 2005), (Fratta, Coral et al. 2011), (Clark and Vonderheide 2006).
Il existe une corrélation entre l’efficacité des inhibiteurs de points de contrôles immunitaires et le nombre de mutations, signifiant que les cancers présentant un haut niveau de mutations (comme les mélanomes, les cancers non à petites cellules, ou les carcinomes colorectaux à microsatellites instables) sont particulièrement sensibles à l’immunothérapie (Woo, Corrales et al. 2015), (Snyder, Makarov et al. 2014). Cependant la présence d’antigènes tumoraux ou associés à des pathogènes n’est pas entièrement suffisante pour susciter une réponse immunitaire contre les cellules tumorales ou infectées. Ces cellules doivent également émettre des signaux de danger sous forme de DAMPs qui agissent comme des adjuvants (McGranahan, Furness et al. 2016). Dans le contexte infectieux, les DAMPs peuvent être considérés comme des motifs moléculaires associés aux microbes (MAMPs de l’anglais « microbe associated molecular patterns ») qui sont des molécules étrangères au soi (Bianchi 2007), (Mackey and McFall 2006). Néanmoins, dans le contexte du cancer, les DAMPs sont plutôt des molécules endogènes qui apparaissent à la surface des cellules ou qui sont libérée dans l’espace péricellulaire en tant que molécules associées à la mort cellulaire (CDAMs de l’anglais « cell death-associated molecules ») (Zitvogel, Ayyoub et al. 2016). Les CDAMs les plus importantes incluent les facteurs de chemio-attraction des cellules dendritiques comme l’adénosine triphosphate (ATP) qui est un ligan des récepteurs purinergiques et la protéine annexine A1 (ANXA1) qui un ligand du récepteur-1 au peptide formyl (Vacchelli, Ma et al. 2015), (Zitvogel, Kepp et al. 2010), (Elliott, Chekeni et al. 2009), le signal « mange-moi » émis par la calréticuline (CALR) qui est exposée à la surface des cellules stressées et/ou mourantes et facilite le transfert des TAAs à l’intérieur des cellules dendritiques (Michaud, Martins et al. 2011), (Gardai, McPhillips et al. 2005), et le signal de maturation des cellules dendritiques de la « high mobility group B1 protein » (HMGB1), un ligans de Toll-like receptor-4 (Obeid, Tesniere et al. 2007), comme les interférons de type 1 (IFN) qui élicitent une cascade d’évènements permettant le recrutement local des cellules T (Apetoh, Ghiringhelli et al. 2007). De plus, plusieurs types de chimiothérapie efficaces sont capables de déclencher l’émission de CDAMs par les cellules cancéreuses, induisant ainsi leur mort cellulaire immungène (ICD de l’angalis « immunogénic cell death ») (Sistigu, Yamazaki et al. 2014), (Krysko, Garg et al. 2012), (Kroemer, Galluzzi et al. 2013), (Kepp, Senovilla et al. 2014). En effet le succès des chimiothérapies aux antracyclines et à l’oxaliplatine contre les carcinomes mammaires ou colorectaux, respectivement, apparait être largement médié par une réponse immunitaire anticancéreuse, à la fois dans les modèles animaux et dans les patients atteints de cancer
(Vacchelli, Ma et al. 2015), (Galluzzi, Buque et al. 2017), (Casares, Pequignot et al. 2005), (Ghiringhelli, Apetoh et al. 2009), (Ghiringhelli, Apetoh et al. 2009), (Sistigu, Yamazaki et al. 2014), (Tesniere, Schlemmer et al. 2010).
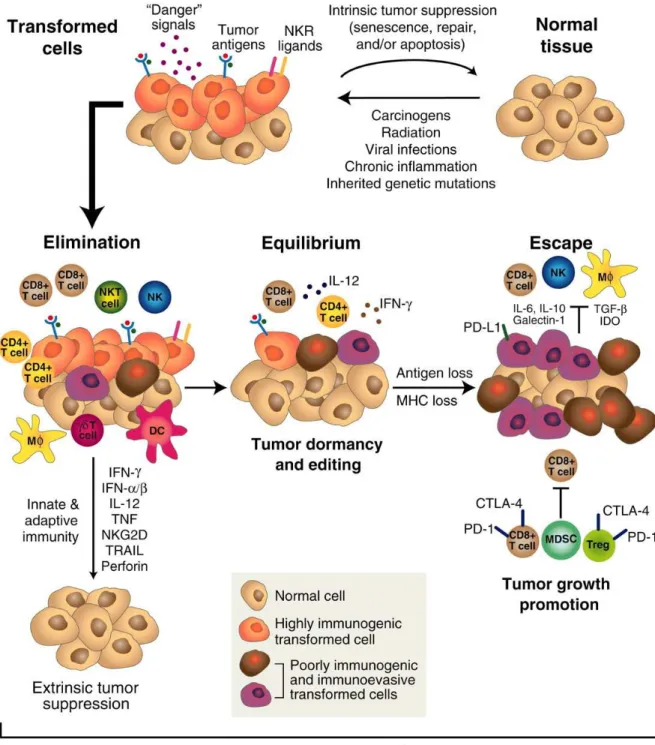
2.2.4 L’immunoédition: l’immunosurveillance des tumeurs
En 2002 le groupe de Robert Schreiber a élucidé le rôle paradoxal du système immunitaire, entre protection tumorale et protection de l’hôte. La pression continue exercée par le système immunitaire sur les tumeurs fait qu’elles doivent devenir « silencieuses » immunologiquement pour survivre (Dunn, Bruce et al. 2002). De ce fait, comme le système immunitaire est capable de promouvoir la croissance de tumeurs capables d’éviter leur détection, le concept original de l’immunosurveillance a été revisité avec la notion de « cancer immunoédition ». La théorie de l’immunoédition implique 3 phases : l’élimination, l’équilibre et l’évasion (Figure 7) (Dunn, Bruce et al. 2002), (Dunn, Old et al. 2004), (Koebel, Vermi et al. 2007). L’hypothèse de l’immunoédition postule que le système immunitaire est initialement capable d’éradiquer des cellules potentiellement tumorigènes jusqu’à ce qu’elles acquièrent progressivement des mutations leur permettant de contrôler le système immunitaire ou de lui échapper (Schreiber, Old et al. 2011).
Figure 6. Les trois E de l’immunoédition du cancer. Durant phases les précoces du développement tumoral, des molécules de danger (telles que l’IFN) et de surface sont capables d’activer le système immunitaire, et lui permettent ainsi de reconnaitre et d’éradiquer la tumeur avant qu’elle ne soit détectable cliniquement. Cette phase est appelée l’élimination ou immunosurveillance. Les rares variants cellulaires qui n’auront pas été éradiqués durant la phase d’élimination peuvent entrer dans la phase d’équilibre, une phase de dormance de la tumeur dans laquelle les cellules cancéreuses et le système immunitaire établissent un équilibre dynamique qui empêche la croissance tumorale. L’accumulation de mutations génétiques et l’acquisition de la capacité à échapper à une éradication immune sous la pression constante de la sélection immunitaire, rend les cellules cancéreuses invisibles
pour l’immunité adaptative et insensibles aux mécanismes effecteurs. De plus, cette dernière phase d’échappement est caractérisée pour un nombre important de mécanismes immunosuppresseurs mis en place pour atténuer la réponse immune anti tumorale (Schreiber, Old et al. 2011).
Les cancers ne peuvent se développer que dans un contexte de défaillance du système immunitaire. Les tumeurs sont ainsi plus fréquentes et progressent plus rapidement dans des souris immunodéficientes que dans des souris immunocompétentes (Vesely, Kershaw et al. 2011). De plus, les tumeurs ayant poussé dans des souris immunodéficientes sont généralement incapables de proliférer lorsqu’elles sont transplantées dans des souris immunocompétentes (Vesely, Kershaw et al. 2011), (Koebel, Vermi et al. 2007), (Shankaran, Ikeda et al. 2001). Dans les cas où les cellules cancéreuses provenant d’une souris immunodéficientes génèrent des tumeurs dans des souris immunocompétentes, les caractéristiques des cellules malignes diffèrent de la tumeur originale. Ce processus est appelé « l’immunoédition » (O'Sullivan, Saddawi-Konefka et al. 2012). Les cellules cancéreuses immunoéditées perdent l’expression de leurs antigènes tumoraux (réduisant ainsi leur antigénicité) (Matsushita, Vesely et al. 2012) ou bien de molécules de danger (DAMPs, pour danger-associated molecular patterns) qui stimulent les réactions immunitaires (et donc diminuent leur “adjuvanticité”) (Yamazaki, Hannani et al. 2014), (Obeid, Tesniere et al. 2007). De plus les cellules cancéreuses immunoéditées ont souvent acquis la capacité de supprimer activement la réponse immunitaire (Su and Anderson 2014). Quel que soit le mécanisme moléculaire précis, les cellules précancéreuses doivent déjouer l’immunosurveillance pour pourvoir générer de véritables néoplasies.
Cependant le concept d’immunosurveillance a été vraiment adopté par les médecins en oncologie quand les données se sont accumulées pour montrer que :
(1) les cancers qui sont fortement infiltrés par les cellules T CD8+ cytotoxiques (ou d’autres effecteurs immunitaires positifs comme les cellules T CD4+, les cellules NK et les cellules dendritiques activées) et qui contiennent relativement peu des cellules T régulatrices FOXP3+, ou d’autres cellules immunosuppressives telles que les cellules myéloïdes suppressives (MDSCs de l’anglais « myeloid-derived suppressor cells ») ont un meilleur pronostic (Fridman, Pages et al. 2012), (Becht, Giraldo et al. 2016)
(2) les agents chimiothérapeutiques ou de thérapie ciblées tels que les inhibiteurs de tyrosine kinase comme l’imatinib sont particulièrement efficaces s’ils arrivent à déclencher une réponse immunitaire anticancéreuse locale (Vacchelli, Ma et al. 2015), (Rusakiewicz, Semeraro et al. 2013), (Zitvogel, Rusakiewicz et al. 2016)
(3) et les inhibiteurs de points de contrôles immunitaires à savoir les anticorps ciblant CTLA-4, PD-1 ou PD-L1 ont un effet sans précédant contre différents types de cancers et réussissent même à guérir certains patients atteints de mélanomes métastatiques (Sharma and Allison 2015), (Topalian, Wolchok et al. 2015).
En effet, les variants tumoraux ayant survécus peuvent entrer dans la « phase d’équilibre », dans laquelle leur croissance est prévenue par des mécanismes immunologiques. Durant cette phase seule l’immunité adaptative (les cellules CD4+ et CD8+) et des molécules effectrices (comme l’IFNg et l’IL-12) sont requis pour maintenir les cellules tumorales dormantes. Cependant, sous pression sélective constante du système immunitaire, les variants tumoraux ne sont plus reconnus par le système immunitaire adaptatif (perte d’antigènes, ou acquisition de défauts dans le proccessing ou la présentation des antigènes) ; deviennent insensibles aux mécanismes immunitaires effecteurs ; et établissement un microenvironnement immunosuppresseur (immunosubversion) (Schreiber, Old et al. 2011), (Dunn, Bruce et al. 2002).
Le résultat est la génération, via un processus de sélection darwinien, des variants tumoraux très peu immunogènes, indétectables par le système immunitaire et par conséquent capable de se développer progressivement, (Dunn, Bruce et al. 2002), (Schreiber, Old et al. 2011), (Vesely, Kershaw et al. 2011).
De plus, un nombre croissant de données montrent que le succès de certaines thérapies anti-cancéreuses, incluant les molécules cytotoxiques traditionnelles, la radiothérapie ainsi que les agents de thérapie ciblée, repose (au moins en partie) sur l’activation de réponses immunitaires anti cancéreuses (Galluzzi, Senovilla et al. 2012).
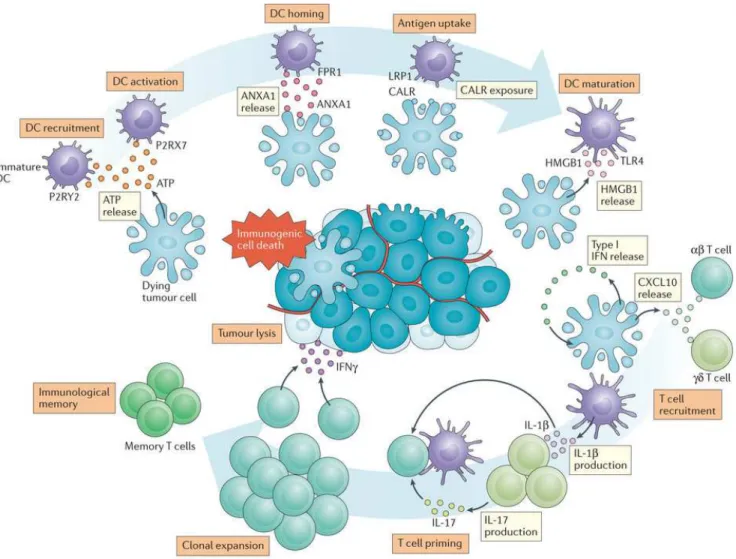
2.2.5 Mort cellulaire immunogène (ICD)
L’apoptose immunogène in vivo a été mise en évidence grâce à plusieurs études démontrant qu’une mort dépendante des caspases produit une vaccination anti-tumorale, basée sur l’activation des DCs qui ingèrent, processent et présentent les antigènes des cellules tumorales mourantes, ainsi que plusieurs populations de lymphocytes T (Obeid, Tesniere et al. 2007). En particulier, il a été montré dans les souris que l’induction de l’ICD dépend des récepteurs TLR4 (un TLR, appartenant à la famille des PRRs) et P2RX7 (un récepteur purinergique de l'ATP) ainsi que chez l’homme, des défauts dans ces gènes compromettant la réponse clinique