HAL Id: hal-02360772
https://hal.archives-ouvertes.fr/hal-02360772
Submitted on 20 Nov 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Presenilin-Dependent Transcriptional Control of the
Aβ-Degrading Enzyme Neprilysin by Intracellular
Domains of βAPP and APLP
Raphaëlle Pardossi-Piquard, Agnès Petit-Paitel, Toshitaka Kawarai, Claire
Sunyach, Cristine Alves da Costa, Bruno Vincent, Sabine Ring, Luciano
D’adamio, Jie Shen, Ulrike Müller, et al.
To cite this version:
Raphaëlle Pardossi-Piquard, Agnès Petit-Paitel, Toshitaka Kawarai, Claire Sunyach, Cristine Alves
da Costa, et al..
Presenilin-Dependent Transcriptional Control of the Aβ-Degrading Enzyme
Neprilysin by Intracellular Domains of βAPP and APLP. Neuron, Elsevier, 2005, 46 (4), pp.541-554.
�10.1016/j.neuron.2005.04.008�. �hal-02360772�
of the A
-Degrading Enzyme Neprilysin
by Intracellular Domains of
APP and APLP
Raphaëlle Pardossi-Piquard,
1Agnès Petit,
1Introduction
Toshitaka Kawarai,
2Claire Sunyach,
1One of the two main histopathological hallmarks in
Alz-Cristine Alves da Costa,
1Bruno Vincent,
1heimer’s disease (AD) is the senile plaque, an
extracel-Sabine Ring,
3Luciano D’Adamio,
4,5Jie Shen,
6lular protein deposit composed in part by fibrillar
aggre-Ulrike Müller,
3Peter St. George Hyslop,
2gates of amyloid
β-peptides (Aβ) (
Haass and Selkoe,
and Frédéric Checler
1,*
1993
). A
β is a 40–42 amino acid peptide that is
gener-1
Institut de Pharmacologie Moléculaire et Cellulaire
ated from the
β-Amyloid Precursor Protein (βAPP) by
Centre National de la Recherche Scientifique
two sequential cleavages. The first of these cleavages
UMR6097 CNRS/UNSA
occurs in the extracellular domain and is mediated by
Valbonne 06560
a membrane-bound aspartyl protease termed
β-secre-France
tase (
Vassar and Citron, 2000
). The second set of
cleav-2
Centre for Research in Neurodegenerative Diseases
ages occurs at residues 40–42 (termed
γ-site) and at
Department of Medicine
residues 48–52 (termed
⑀-site) within the
transmem-University of Toronto and transmem-University Health Network
brane domain of the
βAPP stub generated by
β-secre-Toronto Western Hospital Research Institute
tase. The
γ-site cleavage generates Aβ, while the
con-6 Queen’s Park Crescent
current
⑀-site cleavage generates a cytosolic stub
Toronto, Ontario M5S 3H2
referred to as ICD (
Passer et al., 2000
) or AICD (
βAPP
Canada
IntraCellular Domain). The exact role of AICD remains
3
Institute for Pharmacy and Molecular Biotechnology
unclear.
University of Heidelberg
Both the
γ- and the ⑀-site cleavages are mediated by
69120 Heidelberg
presenilin (PS)-independent and dependent proteases
Germany
(
De Strooper et al., 1998; Armogida et al., 2001
). The
4
Albert Einstein College of Medicine
presenilin-dependent
γ-secretase and ⑀-site proteolytic
New York, New York
activities (which are often generically collectively termed
5
Dipartimento di Biochimica e Biotecnologie Mediche
γ-secretase) are dependent upon a multimeric complex
Universita’ Degli Studi di Napoli Federico II
of at least four different membrane proteins including
Napoli
Presenilin 1(PS1) or Presenilin 2 (PS2), nicastrin, Aph-1,
Italy
and Pen-2 (
Yu et al., 2000; Francis et al., 2002
). In these
6
Center for Neurologic Diseases
complexes, the presenilins have been proposed as a
Harvard Medical School
novel type of transmembrane aspartyl protease bearing
Boston, Massachusetts
the catalytic core of the
γ-secretase (
Wolfe et al., 1999
).
This novel type of intramembranous proteolysis
ap-parently governs the function of
βAPP and several Type
I transmembrane proteins including Notch, cadherins,
Summary
ErbB-4, CD44, or p75
NTR. Many of these proteins are
involved in a variety of vital cellular functions such as
Amyloid
-peptide (A), which plays a central role in
intracellular signaling in development and adulthood,
Alzheimer’s disease, is generated by
presenilin-cell adhesion, presenilin-cell growth and proliferation, and kinase
dependent
␥-secretase cleavage of -amyloid
precur-activities (for review see
Sisodia and St.
George-Hys-sor protein (
APP). We report that the presenilins
lop, 2002; Pollack and Lewis, 2005
). Thus,
γ-secretase
(PS1 and PS2) also regulate A
 degradation.
Preseni-cleavage of Notch releases an intracellular fragment
lin-deficient cells fail to degrade A
 and have drastic
called NICD (Notch IntraCellular Domain), which acts
reductions in the transcription, expression, and
activ-as a transcription factor mediating signal transduction
ity of neprilysin, a key A
-degrading enzyme.
Nepri-in the Notch-Delta pathway, a critical Nepri-intercellular
sig-lysin activity and expression are also lowered by
naling mechanism, during both embryonic
develop-␥-secretase inhibitors and by PS1/PS2 deficiency in
ment and adulthood (
Kopan et al., 1996; Shen et al.,
mouse brain. Neprilysin activity is restored by
tran-1997; De Strooper et al., 1998; Kopan and Goate, 2000
).
sient expression of PS1 or PS2 and by expression
Under normal conditions, A
β occurs as a soluble
of the amyloid intracellular domain (AICD), which is
fragment, the concentration of which is normally tightly
cogenerated with A
, during ␥-secretase cleavage of
controlled below the threshold for its self-aggregation
APP. Neprilysin gene promoters are transactivated
into
β sheet fibrils (
Burdick et al., 1992
). A
β is actively
by AICDs from APP-like proteins (APP, APLP1, and
degraded by several enzymes including neprilysin
APLP2), but not by A
 or by the ␥-secretase cleavage
(NEP), insulin-degrading enzyme (IDE), and
endothelin-products of Notch, N- or E- cadherins. The presenilin-
converting enzyme (ECE) (
Carson and Turner, 2002
).
Al-dependent regulation of neprilysin, mediated by
though these A
β-degrading enzymes have been well
AICDs, provides a physiological means to modulate
characterized, very little is known about the regulatory
A
 levels with varying levels of ␥-secretase activity.
mechanisms that govern their expression and/or
activ-ity. Nevertheless, under normal physiological
circum-stances, the balance between the rates of production
*Correspondence: checler@ipmc.cnrs.frNeuron 542
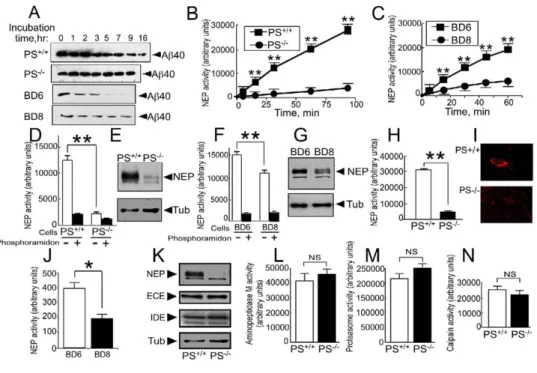
Figure 1. Neprilysin Expression and Activity Are Selectively Lowered in Presenilin-Deficient Cells
(A) Synthetic Aβ40 was incubated for various time periods with the indicated wild-type or PS-deficient cells; then Aβ-related immunoreactivity was analyzed after 16.5% Tris-tricine electrophoresis and Western blot with WO2.
(B–J) Neprilysin activity was measured in fibroblasts (B, D, and H) or blastocyst-derived (C, F, and J) homogenates (B–D and F) or intact cells (H and J). Neprilysin corresponds to total (white bars in [D] and [F]) or phosphoramidon-sensitive (B, C, H, and J) Suc-Ala-Ala-Phe-7AMC-hydrolyzing activity. Neprilysin-like immunoreactivity was monitored in whole homogenates (E and G) or by immunohistochemical labeling on intact fibroblasts (I). Bars represent the mean ± SEM of six (D), nineteen (F), three (H), or sixteen (J) independent determinations. *p < 0.001; **p < 0.0001.
(K–N) Neprilysin (NEP)-, endothelin-converting enzyme (ECE)-, and insulin-degrading enzyme (IDE)-like immunoreactivities were monitored in homogenates of PS+/+and PS−/−fibroblasts (K). Aminopeptidase M (L), proteasome (M), and calpain (N) activities were measured on the
indicated intact cells (L) or in fibroblast cell homogenates (M and N). Bars in (L)–(N) represent the mean ± SEM of ten (L) or three (M and N) independent determinations.
and clearance of A
β is likely to be delicately regulated,
neprilysin activity might be modulated, either directly
or indirectly, by the presenilins. This hypothesis was
di-breaking down only in circumstances that lead to the
rectly supported by the subsequent observation that,
onset of Alzheimer’s disease. We show here that
al-in comparison with wild-type cells, PS-deficient cells
though
γ-secretase cleavage produces Aβ, the other
displayed dramatically lower levels of total
neprilysin-product of
γ-secretase cleavage (AICD) specifically
like activity, phosphoramidon-sensitive activity, and
upregulates the transcription of NEP, which in turn,
ac-neprilysin protein expression. In
Figures 1
D and 1F,
celerates the degradation of A
β. This transcriptional
comparison of the white bars reveals the difference in
signaling pathway therefore provides a simple and
ele-total neprilysin-like activity: 12450 ± 934 versus 2148 ±
gant physiological mechanism for the regulation of A
β
326 for fibroblasts (
Figure 1
D; p < 0.0001) and 15450 ±
levels following physiological activation of
γ-secretase
754 versus 11160 ± 672 for blastocysts (
Figure 1
F; p <
cleavage of
βAPP.
0.0001). Similarly, phosphoramidon-sensitive activity is
lower in PS-deficient cells: 10480 ± 771 versus 1029 ±
Results
219 for fibroblasts (
Figure 1
D; p < 0.0001) and 13730 ±
749 versus 9149 ± 672 for blastocysts (
Figure 1
F; p <
Neprilysin Expression and Activity Are Reduced
0.0001). The same held for neprilysin protein
expres-in Cells Devoid of Presenilexpres-ins
sion: 29% ± 5.4% of control expression was observed
A
β40 immunoreactivity decreases in a time-dependent
in PS
−/−fibroblasts (
Figure 1
E, n = 7; p < 0.0001) and
manner upon exposure of exogenous A
β40 peptide to
51% ± 3% of control expression was observed in PS
−/−wild-type fibroblasts and blastocysts (PS
+/+and BD6,
blastocysts (
Figure 1
G, n = 11; p < 0.0001). Note that
Figure 1
A). This decrease could be blocked by phos-
kinetic analyses indicated that NEP activity was
sig-phoramidon (
Suda et al., 1973
), a specific inhibitor of
nificantly lower at all time points in PS-deficient
fibro-neprilysin (not shown). However, we observed that
blasts (
Figure 1
B; p < 0.0001) and blastocysts (
Figure
A
β40 was not efficiently degraded by PS-deficient fi-
1
C; p < 0.0001).
broblasts or by PS-deficient blastocysts (PS
−/−and
Because neprilysin is a typical type II
membrane-bound peptidase (
Roques et al., 1993
), we next
exam-BD8,
Figure 1
A). These results raise the possibility that
ined neprilysin activity on the surface of intact cells,
using a cell-impermeable fluorimetric substrate. In this
assay, the substrate is cleaved only by enzymes that
are present at the cell surface with their catalytic sites
facing the extracellular space. In agreement with the
studies on whole-cell lysates described above,
PS-deficient fibroblasts exhibited a significant 80%
reduc-tion of cell membrane neprilysin activity compared to
that in wild-type fibroblasts (31130 ± 582 versus 4609
± 359,
Figure 1
H; p<0.0001). Furthermore, neprilysin
im-munoreactivity was poorly detectable at the surface of
intact PS-deficient fibroblasts, although it was readily
detectable on the surface of wild-type fibroblasts (
Fig-ure 1
I). A similar reduction in cell membrane neprilysin
activity was also observed in PS-deficient blastocysts
(BD8), but not in wild-type blastocysts (BD6) (380.7 ±
34 versus 199 ± 25,
Figure 1
J; p < 0.001).
Presenilin Deficiency Selectively Affects Neprilysin
To assess whether PS deficiency specifically altered
neprilysin activity, or whether it also affected other
pu-tative A
β-degrading activities or proteases, we
mea-sured the expression of endothelin-converting enzyme
and insulin-degrading enzyme (
Figure 1
K) and the
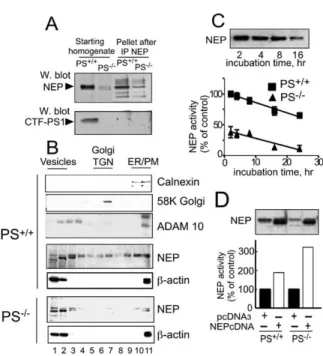
Figure 2. Presenilin Deficiency Does Not Affect Neprilysin at a
Post-activities of aminopeptidase M, another ectoenzyme
transcriptional Level
(
Figure 1
L) (
Checler, 1993
), proteasome (
Figure 1
M),
(A) NEP was immunoprecipitated from PS+/+and PS−/−fibroblast
and calpain (
Figure 1
N). In sharp contrast to the effects
homogenates. Immunological complexes were analyzed for their
of PS-deficiency on neprilysin, none of these other en-
endogenous NEP and like immunoreactivities. Note thatPS1-zymes were affected by the absence of PS1 and PS2.
like immunoreactivity corresponds to the C-terminal maturationproduct of PS1 (CTF-PS1;Checler, 1999).
(B) Endogenous cell distribution of NEP was analyzed by sucrose
Presenilin 1 and Presenilin 2 Affect
density gradient. Note that NEP immunoreactivity is drastically
Neprilysin Transcription
lower in PS−/−fibroblasts (while controlβ-actin is identical) and
The presenilins directly interact with several unrelated
that residual NEP behaves as in PS+/+, i.e., like the ectoenzymeproteins such as nicastrin, Aph-1, and Pen-2, and many
ADAM10.of these proteins are destabilized by the absence of the
(C) PS+/+and PS−/−fibroblasts were treated with cycloheximide toprevent NEP neosynthesis as described in theExperimental
Pro-presenilins (for review, see
De Strooper, 2003
).
How-cedures. At the indicated times, NEP activity was fluorimetrically
ever, six lines of evidence indicate that the reductions
recorded. Note the identical slopes (−1.65 ± 0.22 [PS+/+] and
in neprilysin in PS-deficient cells are not due to the loss
−1.35 ± 0.31 [PS−/−]), indicating a PS-independent similar decay of
of a direct, stabilizing interaction between the preseni-
NEP (each point is the mean ± SEM of three independentdetermin-lin and neprilysin proteins, but rather arise from a re-
ations). Upper panel shows NEP immunoreactivity decrease induction of neprilysin transcription. First, anti-NEP im-
PS+/+fibroblasts.(D) Forty-eight hours after transfection in PS+/+and PS−/−
fibro-munoprecipitates from wild-type fibroblasts do not
blasts, NEP expression (upper panel) and activity (lower panel)
contain PS1 (
Figure 2
A) or PS2 (not shown). Second,
were monitored as described in theExperimental Procedures. Bars
PS1 and PS2 immunoprecipitation does not deplete
su-represent the mean of two independent experiments carried out
pernatants of NEP activity (not shown). Third, residual
in duplicate.NEP expression in PS
−/−fibroblasts partitioned within
cell compartments with the same distribution as in
wild-type fibroblasts (
Figure 2
B), suggesting that PS
activity were not affected in cells devoid of either PS1
only or PS2 only. Thus, normal levels of neprilysin
deficiency did not alter trafficking of NEP in PS
−/−fibro-blasts. Fourth, neprilysin stability is not affected by PS
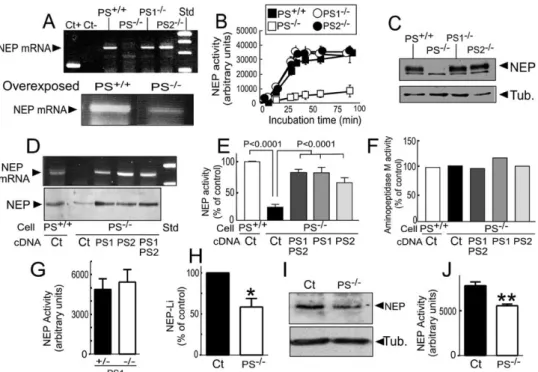
mRNA (
Figure 3
A), enzymatic activity (
Figure 3
B), and
protein expression (
Figure 3
C) were observed in PS1
−/−deficiency (
Figure 2
C). Fifth, both neprilysin expression
and cell surface neprilysin activity can be fully restored
fibroblasts (expressing only endogenous PS2) and in
PS2
−/−fibroblasts (expressing only endogenous PS1).
by transfection of neprilysin cDNA into PS-deficient
fi-broblasts (
Figure 2
D) and into PS-deficient blastocysts
Identical results were achieved when exogenous PS1
or exogenous PS2 were transfected into PS-deficient
(120% ± 4.6% above control mock-transfected cells;
p < 0.01; data not shown). Finally, quantitative RT-PCR
fibroblasts. Thus, neprilysin mRNA expression, protein
expression, and enzymatic activity were equivalently
analyses revealed an approximately 80% reduction in
neprilysin mRNA (
Figure 3
A) (note that overexposure of
and fully restored in PS-deficient fibroblasts by
tran-sient transfection of PS1 and PS2, PS1 only, or PS2
the gel [lower panel] shows residual mRNA expression).
Taken together, these data suggest that the loss of
only (
Figures 3
D and 3E). Control experiments indicate
that aminopeptidase M activity was not affected by
neprilysin expression in PS-deficient cells arises from
impairment in neprilysin transcription. Intriguingly, nep-
PS1 or PS2 complementation in PS-deficient
fibro-blasts (
Figure 3
F). These data were fully confirmed
rilysin transcription, protein expression, and enzymatic
Neuron 544
Figure 3. NEP mRNA Expression Is Reduced in PS-Deficient Cells and Restored by Either PS1 or PS2. NEP Is Reduced in Brain Tissue from Conditional Knockout Mice Lacking Both Presenilins
(A) Analysis of NEP mRNA expression by RT-PCR in PS+/+, PS1−/−, PS2−/−, and PS1−/−/PS2−/−(PS−/−) fibroblasts. Note that only the combined
depletion of PS1 and PS2 reduces NEP mRNA expression by 80% (lower panel, an overexposed gel analysis), while PS1 or PS2 invalidation does not affect NEP mRNA expression.
(B and C) PS1−/−and PS2−/−fibroblasts display unaffected NEP activity (B) and expression (C). Data in (B) represent the mean ± SEM of five
independent determinations.
(D and E) PS−/−fibroblasts were transiently transfected with PS1, PS2, or both (PS1/2) cDNAs, and then NEP mRNA and protein (D) or activity
(E) was monitored. Note that PS1 or PS2 cDNA alone fully restores NEP mRNA expression as well as NEP expression and activity. Bars in (E) represent the mean ± SEM of three independent determinations.
(F) In the same PS−/−transfected cells, aminopeptidase M activity remains unaffected. Bars represent the mean of two independent
determin-ations.
(G–J) PS1−/−(G) and double KO PS1−/−PS2−/−(H–J) mice brains were homogenized and examined for NEP activity (G and J) or expression (I).
Bars in (H) correspond to the densitometric analysis of NEP expression in three independent determinations; *p < 0.05. Bars in (G) and (J) correspond to the mean of three independent determinations; **p < 0.01.
in vivo because PS1-deficiency did not alter brain nep-
the
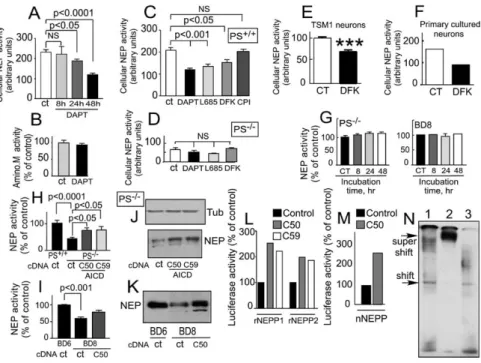
γ-secretase inhibitor DAPT (
Dovey et al., 2001
) led
to a 50% inhibition of neprilysin activity (p < 0.0001,
rilysin activity (
Figure 3
G), while neprilysin expression
Figure 4
A). The inhibitory effect was maximal at 48 hr
(
Figures 3
H and 3I) and activity (
Figure 3
J) were
simi-of treatment, a time point at which DAPT was inert on
larly and significantly reduced in brain tissue from
con-aminopeptidase activity (
Figure 4
B). Other
γ-secretase
ditional double knockout mice lacking both presenilins
inhibitors, namely L685,458 (
Shearman et al., 2000
) or
(41% ± 9% and 29% ± 2.3% inhibition of NEP
expres-DFK167 (referred to as MW167 in
Wolfe et al. [1998]
)
sion and activity, respectively, n = 3; p < 0.05 in PS
−/−also elicited significant reduction of neprilysin activity
versus control brain). These data therefore lead to the
(p < 0.001 and p < 0.05, respectively,
Figure 4
C), while
conclusion that PS1 and PS2 may have redundant roles
a control calpain inhibitor (CPI) was totally ineffective
in regulating neprilysin transcription, but depletion of
(
Figure 4
C). Importantly, DFK167 also lowers neprilysin
both PS1 and PS2 significantly reduces transcription of
activity in TSM1 neurons (30.4% ± 3.8% of inhibition
this enzyme.
versus control, n = 5; p < 0.0001,
Figure 4
E) and in
pri-mary cultured neurons (56.3% of inhibition, n = 2,
Fig-␥-Secretase Inhibitors Reduce Neprilysin Activity
ure 4
F). All inhibitors remain inactive on the residual
in Neuronal and in Wild-Type Cells but Not
neprilysin activity observed in PS-deficient fibroblasts
in PS-Deficient Fibroblasts
(
Figure 4
D) and do not affect in vitro NEP activity (not
The reduction in neprilysin transcription in PS-deficient
shown).
cells could arise from loss of presenilin-dependent
γ-secretase activity or from loss of some other putative
AICDs Upregulate Neprilysin Activity and
activity of the presenilin complexes. To resolve this ques-
Expression in PS-Deficient Fibroblasts and
tion, we examined whether neprilysin activity could be
Blastocysts and Neprilysin Promoter Transactivation
directly modulated in wild-type cells by
γ-secretase in-
The canonical
γ-secretase-mediated hydrolysis
liber-ates the C terminus of A
β40/42 and concomitantly
re-hibitors. Chronic treatment of wild-type fibroblasts with
Figure 4. Effect ofγ-Secretase Inhibitors and γ-Secretase-Derived βAPP Fragments on NEP Activity and Promoter Transactivation
(A–F) Wild-type (PS+/+) fibroblasts were chronically treated by successive additions of DAPT for a total time period of 8, 24, and 48 hr (see
Experimental Procedures) (A) or for 48 hr (B) with 2M of DAPT, and then NEP (A) or aminopeptidase M (B) activities were fluorimetrically recorded on intact cells. Error bars represent the mean ± SEM of three to six independent determinations. PS+/+(C), PS−/−fibroblasts (D),
TSM1 neurons (E), and primary cultured neurons (F) were treated with the indicated inhibitor (DAPT, 48 hr, 2M; L685,458, 8 hr, 1 M; DFK167, 8 hr, 100M; calpain inhibitor [CPI], 8 hr, 100 M), and then the NEP activity of intact cells was measured. Error bars represent the mean ± SEM of three to five independent determinations; ***p < 0.0001.
(G–K) PS-deficient (PS−/−) fibroblasts or PS-deficient (BD8) blastocyst-derived cells (G) were treated for various time periods with 10 ng/ml of
Aβ42, and then NEP activity was fluorimetrically assayed on intact cells. Error bars represent the mean ± SEM of three experiments. PS−/−
fibroblasts (H and J) or BD8 cells (I and K) were transiently transfected with empty vector or with the indicated AICD cDNA , and then NEP activity (H and I) or expression (J and K) was monitored. Bars in (H) and (I) represent the mean ± SEM of five independent experiments. (L and M) The indicated AICD was cotransfected in fibroblasts (L) or in TSM1 neurons (M) withβ-gal cDNA and either renal NEP promoters rNEPP1 and rNEPP2 (L) or neuronal NEP promoter nNEPP (M), and thenβ-galactosidase and luciferase activities were monitored.
(N) HEK293 cells were transfected with AICDC59, Fe65, and Tip60 cDNA , and then nuclear extracts and binding experiments with the 219 bp probe derived from rNEPP were carried out as described in theExperimental Procedures. Lane 1, labeled probe + nuclear extract; lane 2, labeled probe + nuclear extract + Anti-myc; lane 3, labeled probe + nuclear extract + unrelated antibody.
leases the 59 amino acid stub composed of the cyto-
neprilysin promoter elements upstream of a luciferase
reporter minigene. Both AICDs dramatically increased
plasmic C-terminal tail of
βAPP referred to as AICDC59
(see
Introduction
and
Figure 8
). An additional prese-
the transactivation of two renal neprilysin promoters,
rNEPP1 (−385 bp to +147 bp) and rNEPP2 (−263 bp
nilin-dependent proteolytic cleavage of
βAPP and
Notch occurs several amino acids downstream (re-
to +145 bp,
Figure 4
L) in fibroblasts. To confirm
AICD-induced transactivation of the neprilysin promoter in a
ferred to as
⑀ cleavage). This ⑀ cleavage event liberates
AICDC50 from APP and a Notch IntraCellular Domain
neural cell line, we also cloned the neuronal neprilysin
promoter and repeated the luciferase reporter assay. In
(NICD) from Notch (
Gu et al., 2001; Sastre et al., 2001;
Weidemann et al., 2002
). Because NICD is known to
agreement with the above observations, AICDC50 also
transactivated neprilysin promoter in TSM1 neurons
modulate the transcription of several genes, we
rea-soned that neprilysin transcription might be modulated
(
Figure 4
M). Supergel shift assay analysis
demon-strated that the AICD-potentiated transactivation of the
by one of the
γ/⑀-secretase-derived products.
Exoge-nous A
β42 did not modify neprilysin activity in PS-defi-
neprilysin promoter indeed appears to be mediated by
a direct physical interaction of AICD with the neprilysin
cient fibroblasts or in PS-deficient blastocysts (
Figure
4
G). However, transient transfections of AICDC50 or
promoter (
Figure 4
N).
AICDC59 cDNAs increased both neprilysin activity (
Fig-ures 4
H and 4I) and neprilysin expression (
Figures 4
J
AICD-Induced Complementation of Neprilysin
Activity Is Potentiated by Fe65 and Tip60
and 4K) in PS-deficient fibroblasts and blastocysts.
In order to link our observation of AICD-induced in-
in Fibroblasts and in HEK293 Cells
Several lines of evidence have indicated that the
adap-crease of neprilysin activity and expression to our
ob-servation of PS-dependent neprilysin mRNA upregula-
tor protein Fe65 modulates the stability of AICD (
Kim-berly et al., 2001; Kinoshita et al., 2002
), thereby
po-tion, we examined the effect of AICDC50 and AICDC59
Neuron 546
Figure 5. Effect of Fe65 and Tip60 on NEP in Fibroblasts and HEK293 Cells
(A–C) PS+/+([A], upper panel and [C]) and PS−/−(B and C) fibroblasts were transiently transfected with the indicated mix of cDNAs; then NEP,
ECE, IDE, Fe65, andβ−tubulin expressions were measured by Western blot. (Note that gel in [B] corresponds to a long exposure in order to visualize any putative effect of Fe65 and Tip60 on residual NEP). Densitometric analyses (C) indicate that transfection of Fe65 and Tip60 cDNAs alone increases NEP expression in PS+/+(p < 0.0005 when compared to vector alone) but not in PS−/−. Error bars in (C) represent the
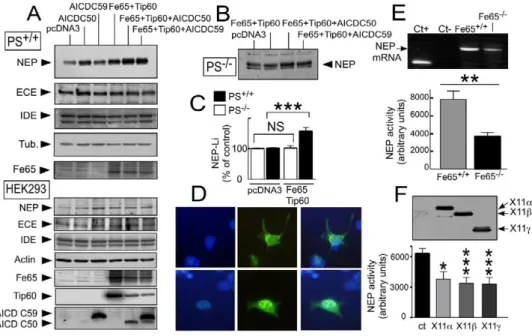
mean ± SEM of four independent determinations. (A, D, and F) HEK293 ([A], lower panel, [D], and [F]) were transiently transfected with the indicated mix of cDNAs (A) or indicated X11 cDNA (F); then NEP, ECE, IDE, Fe65, actin, AICDC50, AICDC59, and X11 expressions were measured by Western blot. In (D), AICDC59-like immunoreactivity was assessed by immunohistochemistry after transfection of AICDC59 alone (upper panels) or together with Fe65 and Tip60 (lower panels) in HEK293 cells. Note the increase of AICDC59 expression and the nuclear redistribution of AICDC59-like immunoreactivity (shown by merge with nuclear DAPI label [left panels]) triggered by Fe65 and Tip60 cDNA transfections. (E) NEP mRNA (upper panel) and activity (lower panel) are decreased by Fe65 deficiency in fibroblasts. Bars in (E) and (F) represent the mean ± SEM of four (E) or five (F) independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
interaction with the histone acetyltransferase Tip60
Tip60 (142.9% ± 19% and 174.9% ± 19% for AICDC50+
Fe65+Tip60 and AICDC59+Fe65+Tip60 versus control,
(
Cao and Südhof, 2001
). We therefore examined whether
Fe65 and Tip60 could (1) influence neprilysin by modu-
respectively, n = 3; p < 0.05). Fe65 and Tip60 (either
alone or in combination with AICDs) had no effect on
lating endogenous AICD in wild-type fibroblasts and in
HEK293 cells and (2) potentiate AICD-induced increase
the expressions of endothelin-converting enzyme and
insulin-degrading enzyme in PS
+/+fibroblasts and
in neprilysin activity.
Fe65 and Tip60 transfection enhanced neprilysin ex-
HEK293 cells (
Figure 5
A), a result that is in agreement
with the experiments described above showing that the
pression and activity in PS
+/+fibroblasts (
Figure 5
A
[up-per panel] and
Figure 5
C; p < 0.0005) and in HEK293
presenilin-dependent enhancement of A
β degradation
is specific to neprilysin (see
Figure 1
). In order to
exam-cells (
Figure 5
A [lower panel]), but not in PS-deficient
cells (
Figures 5
B and 5C), indicating that Fe65 and
ine whether Fe65 could be a limiting factor for
expres-sion of neprilysin, we examined the activity of neprilysin
Tip60 augment neprilysin expression through functional
interaction with an endogenous PS-dependent prod-
in p97Fe65-deficient mice fibroblasts (
Wang et al.,
2004
). Abolition of p97Fe65 diminished neprilysin
activ-uct. In wild-type PS
+/+fibroblasts, the cotransfection of
Fe65 and Tip60 with either AICDC50 or AICDC59 cDNA
ity (52% ± 5.9% of decrease in Fe65
−/−versus control
fibroblasts, n = 4; p < 0.01) (
Figure 5
E) and mRNA
ex-increased neprilysin expression when compared to
AICDs cDNA transfection alone (
Figure 5
A). This was
pression (27% ± 1.5% of decrease in Fe65
−/−versus
control fibroblasts, n = 3; p < 0.005) (
Figure 5
E).
accompanied by an augmentation of AICDC50 and
AICDC59 immunoreactivities (
Figure 5
A [lower panel])
Interestingly, the overexpression of X11
α, X11β, and
X11
γ that triggers opposite effects on Aβ recovery
and by a clear translocation of AICDC59 (
Figure 5
D)
and AICDC50 (not shown) into the nuclei of HEK293
when compared to Fe65 (
Borg et al., 1998; Sastre et al.,
1998; Lee et al., 2003
) decreased neprilysin activity in
cells and PS
+/+fibroblasts (not shown). Similar
potenti-ation of neprilysin expression by Fe65 and Tip60 trans-
HEK293 cells (40%, 46%, and 47% of inhibition of
nep-rilysin activity compared to control for X11
α, X11β, and
fection was also observed in AICD-transfected
PS-defi-cient fibroblasts (136% ± 6%, AICDC50+Fe65+Tip60
X11
γ, respectively, n = 5) (
Figure 5
F).
versus AICDC50 alone and 135% ± 6.2%, AICDC59+
Fe65+Tip60 versus AICDC59 alone; p < 0.0005). Inter-
APP and APLPs Complement Each Other
to Control Neprilysin In Vitro and In Vivo
estingly, AICDC50 and AICDC59 increase renal
nepri-lysin promoter transactivation in HEK293 cells (not
To test whether the endogenous PS-dependent
prod-uct controlling neprilysin activity was indeed AICD, we
shown), a phenotype further potentiated by Fe65 and
regulator of neprilysin transcription both in vitro and
in vivo. However, it should be noted that the extent of
inhibition of neprilysin activity appeared lower in
βAPP
−/−cells than in PS
−/−fibroblasts. Therefore, we
examined whether neprilysin activity and expression
could be controlled by other
βAPP-like proteins.
Inter-estingly, APLP2-deficiency in fibroblasts triggers a
de-crease in both neprilysin activity and expression that
is similar to the reductions observed in
βAPP
−/−cells
(
Figures 6
A–6D). Neprilysin activity is fully restored by
APLP2 cDNA transfection in APLP2
−/−fibroblasts
(125.1 ± 6.9 of control, n = 3; p < 0.05;
Figure 7
A).
How-ever, the absence of both
βAPP and APLP2 in
fibro-blasts resulted in an even more dramatic reduction in
neprilysin activity and expression (80% ± 2.6%
reduc-tion of activity in homogenate, n = 8; p < 0.0001;
Figure
6
A; 87% ± 2.% reduction of activity on intact cells, n =
6; p < 0.0001;
Figure 6
B; and 92% ± 0.7% reduction of
expression in APP
−/−APLP2
−/−versus control
fibro-blasts, n = 3; p < 0.0001;
Figures 6
C and 6D), while
ECE-like and IDE-like immunoreactivities remained
un-affected (not shown). Importantly, double
βAPP/APLP2
deficiency also led to decreased NEP mRNA
expres-sion (30% reduction in two independent experiments,
Figure 6
B [inset]). Of most interest is our observation
that cotransfection of Tip60 and Fe65 cDNAs
drasti-cally increase neprilysin activity in wild-type fibroblasts,
but not in APP
−/−APLP2
−/−doubly deficient fibroblasts
(
Figure 7
B). However, AICDC50 still potentiates
trans-activation of rNEPP1 and rNEPP2 promoters in APP
−/−APLP2
−/−fibroblasts (
Figure 7
C).
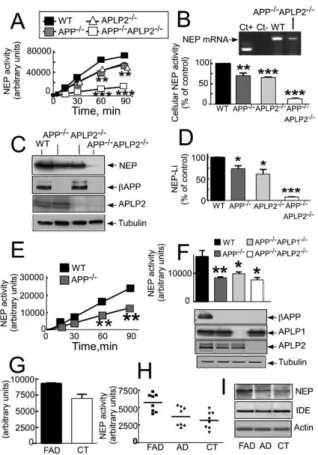
Figure 6. NEP Expression and Activity Are Affected by APP, APLP1,
This apparently synergistic effect led us to
hypothe-and APLP2 Deficiencies In Vitro hypothe-and In Vivo hypothe-and by FAD Mutationssize that APP and APLP2, which have homologous C
in Brain Tissuestermini, could partially complement each other for the
NEP activity in homogenates (A and E–H) or intact cells (B) andcontrol of neprilysin expression. However, it should be
expression (C, D, and I) were monitored as described in theExperi-noted that the extent of inhibition of neprilysin activity
mental Proceduresin the indicated single or multiple KO fibroblasts
in brain (52% ± 9.8% in APP
−/−APLP2
−/−versus control,
(A–D) or in mice (E and F) or Alzheimer’s (G–I) brain tissues. (G) NEP
n = 4; p < 0.05;
Figure 6
F) was lower than that observed
activity in L392V-PS1 and control brain. Activity (H) and expressionof NEP and IDE (I) in L235P-PS1 and F386S-PS1 cases (FAD), two
in the corresponding APP
−/−APLP2
−/−double knockout
sporadic cases (AD), and control brains (CT). Bars in (B), (F), and
fibroblasts. This could be due to another protein that
(G) represent the mean ± SEM of three to seven independent deter-would complement APP and APLP2 function in brain
minations. Insert in (B) corresponds to RT-PCR NEP mRNA analysisbut not in fibroblasts. In this context, it is noteworthy
in wild-type (WT) and APP−/−APLP2−/−fibroblasts. Bars in (D)corre-that, unlike in the brain, fibroblasts totally lack the
βAPP
spond to the densitometric analysis of NEP expression andrepre-family member APLP1 (not shown). Therefore we
exam-sent the mean ± SEM of three determinations. *p < 0.05; **p <ined whether the
γ-secretase-derived fragments of
0.005; ***p < 0.0001.APLP1 (ALID1) and APLP2 (ALID2) could complement
neprilysin activity in APP
−/−APLP2
−/−fibroblasts.
In-examined neprilysin activity and expression in
βAPP-
deed, both ALID1 and ALID2 significantly increase
nep-deficient fibroblasts.
βAPP
−/−fibroblasts exhibit a
simi-rilysin activity in APP
−/−APLP2
−/−fibroblasts (142.1 ± 8,
larly significant reduction of neprilysin activity (30% ±
n = 3; p < 0.005 for ALID1 and 138.8, n = 3; p < 0.005 for
7.6% [n = 7; p < 0.005;
Figure 6
A] and 31% ± 6.7% [n =
ALID2, versus control;
Figure 7
D). The fact that APP
−/−,
3; p < 0.001;
Figure 6
B] reduction in homogenates and
APP
−/−APLP1
−/−, and APP
−/−APLP2
−/−brains all display
intact cells, respectively, versus control activity) that
similar reductions in neprilysin activity (
Figure 6
F)
indi-fully matched the reduction in NEP expression (26.1% ±
cate that all the members of the APP family control
ce-6.9% reduction, n = 3; p < 0.05;
Figures 6
C and 6D).
rebral neprilysin transcription in vivo. It should be
Neprilysin activity is fully restored by
βAPP cDNA trans-
noted, however, that the mice brains devoid of APP,
fection in APP
−/−fibroblasts (125.6 ± 6 of control, n =
APLP1, and APLP2 do not exhibit enhanced neprilysin
4; p < 0.01;
Figure 7
A). It is of interest that loss of
βAPP
decrease when compared to double KO brains (not
expression in APP
−/−mouse brain triggers a significant
shown). This suggests that, besides
AICD/ALID-regu-reduction in neprilysin activity (47% ± 3.5% in APP
−/−lated neprilysin expression, there exists also a
constitu-versus control, n = 4; p < 0.005;
Figures 6
E and 6F). This
tive APP/APLP-independent cerebral NEP activity.
suggests that derivatives of
βAPP might also control
In order to establish whether the control of neprilysin
cerebral neprilysin in vivo and supports the notion that
activity was restricted to the APP-related ICDs, we next
a presenilin-dependent,
γ-secretase-mediated cleav-
examined the putative effect of other PS-dependent
γ-secretase-mediated products. Thus, NICD is the
Neuron 548
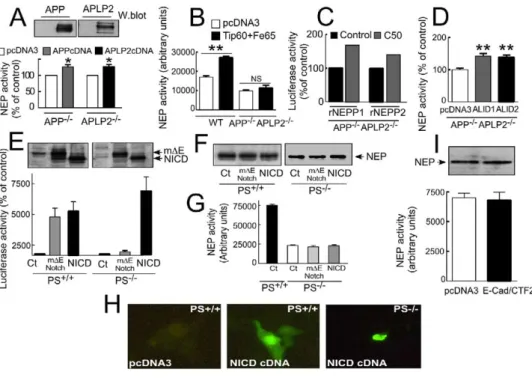
Figure 7. Neprilysin Activity Is Modulated by ALID1 and ALID2 but Not by NICD or E-Cad/CTF2
APP−/−(A), APLP2−/−(A), or APP−/−APLP2−/−(B–D) fibroblasts were transfected with empty pcDNA3 vector or indicated cDNAs; then NEP
activity (A, B, and D) or transactivation of rNEPP1 and rNEPP2 promoters (C) were monitored as described in theExperimental Procedures. Error bars in (A), (B), and (D) represent the mean ± SEM of three to four independent experiments. *p < 0.05; **p < 0.005. (E–G) The indicated fibroblasts were transiently cotransfected with 4XCBF-luciferase andβ-gal cDNAs in combination with either empty vector (Ct) or cDNAs encoding myc-tagged m⌬ENotch or myc-tagged NICD, and then m⌬ENotch and NICD (E) or NEP (F) expressions were assayed by Western blot in the indicated cell lines, and luciferase (E) or NEP (G) activities were measured as described in theExperimental Procedures. Bars in (E) and (G) represent the mean ± SEM of three to four independent experiments. NICD expression was also estimated by immunohistochem-istry in PS+/+and PS-deficient fibroblasts (H). Note a clear dense label of NICD in the nucleus. (I) E-Cad/CTF2 does not modulate NEP
expression and activity in HEK293 cells. Bars in (I) represent the mean ± SEM of seven independent experiments.
tracellular fragment liberated from Notch upon
γ-secre-
activity in PS
−/−cells. Taken together, these data
indi-cate that the complementation of neprilysin function by
tase cleavage. To test whether NICD might also affect
neprilysin activity, we created fibroblast- and blasto-
γ-secretase-derived products is specifically elicited by
ICDs of the
βAPP family members.
cyst-derived cell lines transiently expressing the
4XCBF-luciferase reporter gene. In agreement with previously
published works (
Herreman et al., 2000; Zhang et al.,
Neprilysin Expression and Activity Are Increased
in Alzheimer’s Brains of Genetic Origin
2000
), the transient cotransfection of m
⌬ENotch (which
generates NICD upon
γ-secretase cleavage) led to a ro-
We have examined the neprilysin activity of control
hu-man brains and compared them to brain samples of
bust transcriptional activation in wild-type fibroblasts
(
Figure 7
E) and blastocyst-derived cells (not shown),
sporadic or genetic AD origin. Neprilysin activity (
Fig-ures 6
G and 6H) and expression (
Figure 6
I) were not
but not in the equivalent PS-deficient cells (
Figure 7
E).
However, transfection of NICD itself into wild-type and
statistically different between control (CT) and sporadic
AD brains, while Familial Alzheimer’s disease (FAD)
PS-deficient cells induced NICD protein expression
(
Figure 7
E), favored nuclear localization of NICD (
Figure
brains harboring PS1 mutations displayed higher
activ-ity (
Figures 6
G and 6H) and expression (
Figure 6
I).
Inter-7
H), and increased luciferase activity (
Figure 7
E). These
control experiments therefore established that NICD
estingly, insulin-degrading enzyme (IDE) was not
al-tered in FAD brains (
Figure 6
I).
was indeed functionally expressed in our experimental
system. However, regardless of the cell system used,
m
⌬ENotch and NICD were unable to restore neprilysin
Discussion
expression (
Figure 7
F) or neprilysin activity (
Figure 7
G)
in PS-deficient cells. Furthermore, the
γ-secretase-
Several lines of evidence strongly suggest that
nepri-lysin (
Hauss-Wegrzyniak and Wenk, 2002; Hama et al.,
derived C-terminal products of E-Cadherin (E-Cad/
CTF2,
Figure 7
I) (
Marambaud et al., 2002
) and N-Cad-
2001; Marr et al., 2003; Leissring et al., 2003; Iwata et
al., 2001
), endothelin-converting enzyme (ECE;
Eckman
herin (N-Cad/CTF2, not shown) (
Marambaud et al.,
2003
) were also unable to affect neprilysin expression
et al., 2003
), and insulin-degrading enzyme (IDE;
Farris
et al., 2003
) could participate in the catabolism of A
β
and activity. Thus, unlike AICDs and ALIDs, NICD and
expression, and enzymatic activity are dramatically re-
drastically reduces both A
β production and Notch
sig-naling (
De Strooper et al., 1998
). We hypothesize that in
duced in non-neuronal and neuronal cells when they
are devoid of PS1 and PS2 and that this phenotype can
PS1
−/−fibroblasts and brain tissue, APLP1 and APLP2
could undergo
γ-secretase-like cleavage by residual
be mimicked by
γ-secretase inhibitors. We have shown
that the presenilins control NEP activity at a transcrip-
endogenous PS2, thus complementing AICD function,
and thereby control neprilysin expression and activity.
tional level and that PS deficiency does not affect the
expression of IDE, ECE, or other widely distributed in-
Because APLPs lack the A
β sequence (although Aβ-like
peptides seem to be generated from APLP2), their
PS2-tracellular (calpain and proteasome) or ectopeptidase
(aminopeptidase M) activities. We have also shown that
dependent cleavage would not compensate for A
β
re-duction that is triggered by PS1 deficiency. In this
neprilysin expression and activity are significantly
re-duced in brain tissue from mice lacking PS1 and PS2
context, the resulting phenotype would be a reduction
in A
β production but with unaltered or weakly modified
(
Saura et al., 2004
), indicating that the presenilins also
control cerebral neprilysin transcription, expression,
neprilysin levels. Alternatively, we hypothesize that
there is likely to be both a constitutive
(AICD-indepen-and activity in vivo. This highly specific (AICD-indepen-and
physiologi-cal relationship between the presenilins and neprilysin
dent) basal expression of NEP and an inducible
(AICD-dependent) expression of NEP, as suggested by
resid-strongly argues for a major evolutionary and functional
role of neprilysin in the physiological catabolism of A
β,
ual NEP activity in triple KO (APP
−/−APLP1
−/−APLP2
−/−)
mice brains (see
Results
). In PS1
−/−fibroblasts and in
but does not preclude a role for other enzymes in A
β
catabolism.
PS1
−/−brain tissue, this constitutive level of expression,
together with the cumulative effects of residual AICD
The molecular mechanisms by which the presenilins
regulate neprilysin transcription would appear, from the
and ALIDs generated from APP, APLP1, and APLP2 by
the PS2
γ-secretase activity, could be sufficient to
data presented here, to involve the C-terminal
intracel-lular domains of APP and APLP proteins (AICDs and
maintain neprilysin expression and activity in PS1
−/−cells. In contrast, for Notch, there is both absence of
ALIDs). Thus, we have clearly established that
defi-ciency of
βAPP, APLP1, or APLP2 drastically reduces
constitutive basal expression of Notch-targeted genes
and a lack of redundancy in alternate signaling
mole-neprilysin expression and activity in both fibroblasts
and in brain tissues. We have specifically shown that
cules. Another possibility could be that NEP
tran-scription is preserved in PS1
−/−cells because APLPs
A
β42 and the γ-secretase-derived products of Notch,
E-cadherins, and N-cadherins do not support this activ-
γ-secretase cleavage is not affected to the same extent
as APP by PS1 deficiency. However, to our knowledge,
ity. Furthermore, the Fe65 adaptor protein (which binds
to the GYENPTY motif in the cytoplasmic tail of
βAPP/
there is currently no data to support the notion that
PS1 and PS2 have differential/preferential effects on
AICD [
King and Turner, 2004
]) and Tip60 clearly
potenti-ate the AICD-induced upregulation of neprilysin in wild-
γ-secretase cleavage of APP, APLP1, and APLP2.
The experiments described here do more than simply
type fibroblasts but not in PS
−/−and APP
−/−/APLP2
−/−cells.
confirm the previously and widely held suspicion that
AICD, like NICD, might act as a signaling molecule
in-It has been previously reported that Fe65 and Tip60
stabilize AICD and favor its translocation to the nucleus
volved in transcriptional activation. The experiments
here depict an elegant and unusual mechanism by
(
Kimberly et al., 2001; Kinoshita et al., 2002
). Our
exper-iments have shown that AICD translocates to the nu-
which A
β levels are controlled. Thus, βAPP is
proteolyti-cally processed by presenilin-dependent
γ-secretase,
cleus in the presence of Fe65 and Tip60 and
transacti-vates neuronal and renal neprilysin promoters in TSM1
which generates two distinct types of catabolites, A
β
and AICD (
Figure 8
). One of these products, AICD, then
neurons, HEK293 cells, and APP
−/−/APLP2
−/−fibro-blasts, respectively. Furthermore, Supergel shift analy-
controls the lifetime of the other product, A
β, by
selec-tively activating the transcription of an enzyme
(nepri-sis indicates that AICDC59 interacts physically with the
neprilysin promoter in the presence of Fe65 and Tip60.
lysin) capable of degrading that other product. Thus,
γ-secretase activity directly controls Aβ production and
Interestingly, X11, which clearly reduces
γ-secretase
cleavage of
βAPP, also drastically reduces neprilysin
then indirectly modulates its degradation. To our
knowl-edge, a similar “self-contained” mechanism for
regulat-activity. Therefore, Fe65 and X11, which trigger
oppo-site effects on A
β production, also elicit an opposite
ing the degradation of enzyme products has not been
described previously; (NB: this does not preclude
phenotype in neprilysin regulation.
Altogether, the similar decrease in neprilysin activity
AICDs from also activating other genes).
If A
β production and degradation are tightly linked,
and expression triggered by the absence of PS or APP/
APLPs, the analogous absence of control of neprilysin
this raises the question of why A
β accumulates in AD.
The net accumulation of A
β in AD pathology likely
re-by Fe65 and Tip60 in these invalidated fibroblasts, and
the opposite phenotype observed with Fe65 and X11
flects the cumulative effect of multiple events acting on
production, fibrillogenesis, and degradation. In many
all lead to the firm suggestion that the endogenous
γ-secretase-dependent fragments controlling neprilysin
forms of AD, especially the late-onset sporadic forms,
it has not been shown that there is increased
β- and
are indeed AICD/ALIDs.
The redundant role of PS1 and PS2 in regulating nep-
γ-secretase activity. In fact, some have suggested that
these forms may reflect defective degradation of A
β
rilysin transcription contrasts with their overlapping but
not redundant roles in modulating A
β peptide and
(
Iwata et al., 2001; Leissring et al., 2003
). Therefore,
AICD levels are likely to be unchanged in these
late-Notch signaling. Thus, PS2 deficiency alone does not
affect
γ-secretase-mediated production of Aβ or Notch
onset forms of AD, and as a result, the AICD-mediated
ability to upregulate neprilysin activity would not be
ef-in vivo (
Herreman et al., 1999
). However, PS1 deficiency
Neuron 550
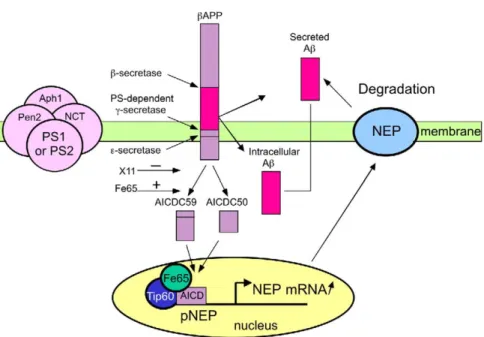
Figure 8. Model for PS-Dependent Transcriptional Activation of NEP by theβAPP-Intracellular Domain
βAPP undergoes proteolysis by various secretases. Aβ peptide is released by the sequential cleavages triggered by β- and γ-secretases in
the intracellular compartment from which it can be secreted. Once secreted, Aβ is degraded by the ectopeptidase named neprilysin (NEP), a type II integral protein with catalytic domain facing the extracellular space. Presenilin-dependentγ-secretase cleavage is triggered by a multiproteic complex composed of presenilin 1 (PS1) or presenilin 2 (PS2), nicastrin (NCT), Aph1, and Pen2.γ-secretase contributes to an additional cleavage, referred to as⑀ cleavage, that takes place slightly downstream from the γ-secretase site. The action at γ and ⑀ sites gives rise to AICDC59 and AICDC50, respectively. AICDs interact with Fe65 and Tip60 to form an active complex activating NEP mRNA transcription. Increased NEP activity, in turn, leads to increased Aβ degradation.
ficiently brought into play to protect the brain. In con-
AICD. Upregulation of neprilysin by transgenic
overex-pression, at least to modest levels, appears to be
suffi-trast, in those cases of AD arising from mutations in
APP and PS1, which activate
γ-secretase and AICD
cient to reduce brain A
β levels and to pose few toxic
side effects (
Leissring et al., 2003
). This strategy would
production, the principal effect is to produce longer A
β
isoforms such as A
β42. However, although Aβ40 is effi-
also circumvent the other side effects of
γ-secretase
inhibitors, including the potentially self-defeating effect
ciently degraded by NEP, A
β42 is degraded by NEP
both in vitro and in vivo at a 6-fold lower rate, (
Shirotani
of reducing AICD and thus preventing NEP-mediated
degradation of A
β.
et al., 2001
). As a result, the upregulation of AICD (and
thus, NEP expression), which would be anticipated in
subjects with presenilin mutations, would not
com-Experimental Procedures
pletely abolish the accumulation of A
β42 in these
cases. It should be noted that, in agreement with the
Cell Culture and Transfectionsabove hypothesis, neprilysin expression and activity
Primary cultured neurons, HEK293 cells, telencephalon murine (TSM1) cell lines, blastocyst-derived cells, PS-, βAPP-, andwere higher only in brain tissues with familial
Alzheim-p97Fe65-deficient fibroblasts were obtained and cultured as
pre-er’s disease linked to various presenilin-1 mutations,
viously described (Vincent et al., 1996; De Strooper et al., 1999;
while sporadic AD cases displayed neprilysin levels
Zhang et al., 2000; Herreman et al., 2000 Armogida et al., 2001;
similar to those exhibited by normal brain tissues (
Fig-
Leissring et al., 2002; Wang et al., 2004). Mouse embryonicfibro-ures 6
G–6I). Interestingly, PS1 mutations selectively af-
blasts derived from APLP2 or APP/APLP-deficient mouse embryosfect neprilysin and do not alter insulin-degrading en-
and their littermates (Heber et al., 2000) were immortalized with the large T antigen of SV40. Several clonal lines were established andzyme expression.
cultured in DMEM/10% fetal calf serum containing 2 mM glutamine
The above observations are also of direct practical
and 50M β-mercaptoethanol. Transient transfections were carried
interest because they indicate the possibility of new
av-out with DAC 30 (Eurogentec).
enues for controlling A
β levels without directly affecting
γ-secretase. This latter concept is important because of
the various developmental and postnatal side-effects
Fluorimetric Assays of Enzymatic ActivitiesNEP activity was measured on intact cells or in cell homogenates
associated with the inhibition of
γ-secretase-mediated
with Suc-Ala-Ala-Phe-7AMC in the absence or presence of
phos-cleavage of other signaling molecules, including Notch
phoramidon as described previously (Checler, 1993). When NEP
(
Sisodia and St. George-Hyslop, 2002; Haass and De
activity was measured in brain tissues, brains were homogenized