HAL Id: tel-01022929
https://tel.archives-ouvertes.fr/tel-01022929
Submitted on 11 Jul 2014
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Synthesis of novel iminogalactitol and epi-isofagomine
derivatives as potential pharmacological chaperones for
Krabbe disease
Anna Biela
To cite this version:
Anna Biela. Synthesis of novel iminogalactitol and epi-isofagomine derivatives as potential phar-macological chaperones for Krabbe disease. Other. Université d’Orléans, 2013. English. �NNT : 2013ORLE2057�. �tel-01022929�
UNIVERSITÉ D’ORLÉANS
ÉCOLE DOCTORALE
SCIENCES BIOLOGIQUES ET CHIMIE DU VIVANTInstitut de Chimie Organique et Analytique, UMR7311
THÈSE
présentée par :Anna BIELA
soutenue le : 9 décembre 2013pour obtenir le grade de : Docteur de l’université d’Orléans Discipline/ Spécialité : Chimie organique
Synthèse de nouveaux dérivés
d’iminogalactitol et d’épi-isofagomine
comme chaperons pharmacologiques
potentiels pour la maladie de Krabbe
THÈSE dirigée par :
Olivier R. MARTIN Professeur, Université d’Orléans - Directeur de thèse
Estelle GALLIENNE Maître de conférences, Université d’Orléans -
Co-encadrant de thèse
RAPPORTEURS :
Tanja WRODNIGG Professeur, Technische Universität, Graz, Autriche
Joanne XIE Professeur, Ecole Normale Supérieure de Cachan
_________________________________________________________________
JURY :
Richard DANIELLOU Professeur, Université d’Orléans, Président du jury
Yves BLERIOT Professeur, Université de Poitiers
Estelle GALLIENNE Maître de conférences, Université d’Orléans
Olivier R. MARTIN Professeur, Université d’Orléans
Tanja WRODNIGG Professeur, Technische Universität, Graz, Autriche
3
Acknowledgements
In this special moment, I would like to thank many people that have supported me the course of my PhD, and without whom this work could not be achieved.
I would like to show gratitude to my supervisor Professor Olivier Martin who three years ago took the risk and welcomed me in his group as a PhD student.He guided me and shared with his experience in sugar chemistry during my stay in his team. I thank him especially for his positive thinking and contestant encouragement even when chemistry was against us.
My second supervisor, Doctor Estelle Gallienne, showed an enormous patience for all my questions, kept correcting my French and helped me to better understand the chemistry I have done. She introduced me into sugar chemistry, lab routine and was not only my superior but also a friend, for what I am really grateful to her.
This thesis could not be done without a fellowship of The Ministry of French Higher Education and Research, for which I am very grateful.
Besides my supervisors, I would like to thank Professor Tanja Wrodnigg (TU Graz, Austria), Professor Joanne XIE (ENS Cachan, France), Professeur Yves Blériot (Université de Poitiers, France) and Professor Richard Daniellou (Université d’Orléans, France) for accepting to read my manuscript, for their time and precious comments to my work.
I would like to express my thanks to Agnès Chartier, Nathalie Percina and their trainee Sakthi who performed HPLC purifications of my final products.
Thank you Marie-Madeleine and Yann for helping me understand and deal with French bureaucracy, for your smile and kindness.
During last four years Orléans and ICOA became my second home, and all Professors, Doctors and Students that I had a pleasure to meet and work with, were a part of my “new” life. I thank you all for your guidelines and positive attitude to a new Polish girl that tried a lot to speak French:) You are unique people that developed my friends.
First of all: thank you Sophie! For your irreplaceable advices concerning laboratory, children, French language… Thank you for listening to all my stories, trying to cheer me up and to find solutions to my (non)chemical problems! Thank you for sharing our morning teas, for helping me with shopping when Artur was not there, for cherries, strawberries, blackberries, a drawer full of sweets and Christophe May! Without you (and your complaining!) lab 7 would never be the same!
4 Thank you Ela! For being my real friend, rescuing me in almost every emergency situation, listening to my problems, sweating together on a gym and the most important: for being the best aunt for my son!
I cannot forget Hélène Blin, who changed rainy Orleans for sunny Marseille, but will always be a part of our lab. You are the most warm-hearted person I met in France. You were so kind that you welcomed me for Christmas at your home. For a long time you were the only French I could understand easily!
There were many Polish students in my lab, and all of them became my dear friends: crazy Ola who loved climbing and was one of the prettiest brides I’ve ever seen; Dominika who loved the order and Scandinavia; Roman who enjoyed my cheesecake and is the tallest person I know; Magda who sang and danced with me in the lab and spread her positive energy all day long; Ania and Oksana: two lovely blonds who seduced my son and both Joannas who survived my welcome dinner:)
In our team we were lucky to always have kind and gifted trainees, who made our lab full of laughter. It was nice to meet you: Hélènes, Asma, Lydie and especially Nathanael, Ludovic and Norhane. I will never forget Garry and his true words about the most beautiful NMR spectrum in my career! I should also mention Julien, who does not mind (or at least not a lot) when I am making fun of him and pays me back with interest.
I would like to thank all of you for my good souvenirs from ICOA: Pascal for the rhubarb and NMR advices, Marie, who appreciated my cookies, Aziz for taking care of wearing suitable clothes, Mathieu for his “Salut Anna!”, Mikael (the cute guy) for constructive comments on my French and English (hope you are going to finally get your “bronzage mythique”!). Thank you Sandrine, Audréy, Stephanie, Magalie, Romain, Nicolas, Mathieu, Katia, Aleksandra, Aurelien, Oskan, Jeremy, Cyril, Chloé!
One of the most trilling experiences during my thesis was the stage in Portland in Doctor Magdalena Petryniak and Greg Potter team in Oregon Health and Science University. I would like to thank you for your very warm reception and guiding me in my adventure with molecular biology! Hope your nights become calm soon!
My deepest thanks go to my family: my Parents Elżbieta and Jerzy, my “little” Sister Agnieszka, my dearest husband Artur and our Son Szymon. They loved me unselfishly and constantly supported me through all my life even if it was not that easy! Thank you for your patience, comprehension and help in every discipline of my life!
5 Table of contents Acknowledgements ... 1 Table of contents ... 5 Glossary ... 9 Abbreviations ... 15 Avant-propos ... 17 Introduction ... 19
I. Lysosomal storage disorders ... 19
I.1 Lysosome ... 19
I.2 Synthesis and transport of lysosomal proteins. ... 20
I.3 Lysosomal storage disorders. ... 21
II. Therapies for LSDs... 27
II.1 General ... 27
II.2 Hematopoietic stem cells therapy ... 28
II.3 Gene therapy ... 30
II.4 Enzyme replacement therapy ... 31
II.5 Substrate reduction therapy ... 34
II.6 Pharmacological chaperone therapy ... 36
III. Krabbe disease ... 41
IV. Galactosidases inhibitors ... 47
IV.1 General ... 47
IV.2 Use of galactosidase inhibitors as PCs for galactosidase-linked LSDs ... 47
IV.3 Additional studies ... 53
Objectives ... 55
I. General comments on GALC-mediated hydrolysis of b-galactosides ... 55
II. Structures of iminosugars of interest ... 57
III. Detailed objectives ... 57
Results and discussion ... 61
I. The synthesis and biological evaluation of 1-C-alkyl-imino-L-arabinitols. ... 61
I.1 Synthetic strategy. ... 61
6
I.3 Synthesis of protected arabinopyranosylamines 4 and 11 ... 63
I.4 C-1 chain elongation of N-protected glycosylamines ... 63
I.5 Intramolecular SN2-type cyclisation ... 66
I.6 Functionalization of double bond and deprotection: preparation of iminosugars 8, 9, 19, 20 and 22. ... 67
I.7 Biological evaluation ... 71
I.8 Conclusions ... 72
II. The synthesis of 1-C-alkyl-imino-D-galactitols. ... 75
II.1 Synthetic strategy ... 75
II.2 Synthesis of N-Z protected galactopyranosylamine 25 and C-1 chain elongation 77 II.3 Synthesis of ketones 41 and 42 ... 79
II.4 Intramolecular reductive amination. ... 80
II.5 Synthesis of 1-C-alkyl-imino-D-galactitols by intramolecular SN2 reaction ... 86
II.6 Conclusions ... 88
III. Alternative synthesis of 1-C-alkyl-imino-D-galactitols from L-sorbose: strategy by way of a C-6 chain elongation. ... 91
III.1 Synthetic strategy ... 91
III.2 Synthesis of 1,2:4,6-di-O-isopropylidene-a-L-sorbofuranose and conversion to a L-tagatofuranose derivative ... 93
III.3 Synthesis of 1,2:4,6-di-O-isopropylidene-a-L-sorbofuranose and inversion of configuration by way of an oxidation with PCC ... 96
III.4 Liberation and oxidation of C-6 hydroxyl group ... 97
III.5 Condensation of aldehyde 66 with sulfinylamines ... 98
III.6 Addition of Grignard reagents ... 98
III.7 Deprotection and reductive amination of amine intermediates ... 101
III.8 Synthesis of azido-derivatives ... 103
III.9 Deprotection and reductive amination of azido-intermediates ... 104
III.10 Synthesis of DGJ ... 106
III.11 Conclusions ... 106
IV. The synthesis of 1-C-alkyl-imino-L-altritols. ... 111
IV.1 Synthetic strategy ... 111
IV.2 Synthesis of protected 1-C-alkyl-imino-L-altritols. ... 111
IV.3 Functionalization of the double bond. ... 112
7
V.1 Synthetic strategy ... 117
V.2 Synthesis of benzyl 2,3-O-isopropylidene-b-L-ribopyranoside 101 ... 119
V.3 Attempted substitution reactions to introduce a CN group ... 120
V.4 Modified synthesis of cyano-derivative ... 121
V.5 Synthesis of nitro-alcohols 112+113 and dehydration assays ... 122
V.6 Reduction of the nitro-intermediates and cyclisation by reductive amination. ... 124
V.7 Conclusions ... 128
VI. Preliminary biological evaluation of the new D-galacto and L-altro-configured iminosugar derivatives ... 131
Conclusion ... 133
Experimental part ... 135
I. General ... 135
II. General treatment with basic ion-exchange resin after hydrogenation ... 135
III. General procedure for purification on Dowex 50WX8 resin (H+, 50-100 mesh) ... 136
IV. General procedure for biological assays... 136
9
Glossary
AglyconeThe compound remaining after replacement of the glycosyl group from a glycoside by a hydrogen atom. For example, for the galactosylceramide the aglycone is a ceramide.
Allele
One member of a pair (or any of the series) of genes occupying a specific spot on a chromosome (called locus) that controls the same trait.
Apoptosis
Programmed cell death as signalled by the nuclei in normally functioning human and animal cells when age or state of cell health and condition dictates.
Astrocytic gliosis
Also known as astrogliosis or astrocytosis, is an abnormal increase in the number of astrocytes due to the destruction of nearby neurons from CNS trauma, infection, stroke, autoimmune responses, and neurodegenerative disease.
Autophagy
Also known as autophagocytosis is the basic catabolic mechanism that involves cell degradation of unnecessary or dysfunctional cellular components through the lysosomal machinery. Autophagy, if regulated, ensures the synthesis, degradation and recycling of cellular components. During this process, targeted cytoplasmic constituents are isolated from the rest of the cell within the autophagosomes, which are then fused with lysosomes and degraded or recycled.
Autosomal
Being encoded by one of the 22 non-sex determining chromosomes.
Cytosol
Also known as an intracellular fluid or cytoplasmic matrix is the liquid found inside cells. It is separated into compartments by membranes.
Cytotoxic
Toxic to cells.
Demyelination
The loss or removal of myelin sheath (e.g. from the nerve fibre).
Dysostosis
10
Endocytosis
A process in which cell takes in materials from the outside by engulfing and fusing them with its plasma membrane.
Endoplasmic reticulum (ER)
A membrane-bounded organelle that occurs as labyrinthine, interconnected flattened sacs or tubules that is connected to the nuclear membrane, runs through the cytoplasm, and may well extend into the cell membrane.
Endosomes
A membrane-bounded compartment inside eukaryotic cells.
ER quality control system
A quality-control system for 'proof-reading' newly synthesized proteins, so that only native conformers reach their final destinations. Non-native conformers and incompletely assembled oligomers are retained, and, if misfolded persistently, they are degraded.
Eukaryotic cell
A cell that contains a nucleus and other organelles enclosed within membranes.
ex vivo
Outside an organism. It refers to experimentation or measurements done in or on tissue in an artificial environment outside the organism with the minimum alteration of natural conditions.
Genotype
The genetic make-up of a cell, an organism, or an individual usually with reference to a specific characteristic under consideration
Hematopoietic stem cells
The blood cells that give rise to all the other blood cells.
Hypertrophic cardiomyopathy
A primary disease of the muscle of the heart in which its portion is thickened without any obvious cause.
Hypotonia
A state of low muscle tone (the amount of tension or resistance to stretch in a muscle), often involving reduced muscle strength.
11
in vitro
Conducted using components of an organism that have been isolated from their usual biological surroundings in order to permit a more detailed or more convenient analysis than can be done with whole organisms.
in vivo
It is experimentation using a whole, living organism.
Lymphoblasts
A different form of a naive lymphocyte that occurs when the lymphocyte is activated by an antigen (from antigen-presenting cells) and increased in volume by nucleus and cytoplasm growth as well as new mRNA and protein synthesis.
Macrophage
The cells produced by the differentiation of monocytes in tissues. Macrophages function in both non-specific defence (innate immunity) as well as help initiate specific defence mechanisms (adaptive immunity) of vertebrate animals. Their roles are to phagocytise, or engulf and then digest, cellular debris and pathogens, either as stationary or as mobile cells.
Missense mutation
A point mutation in which a single nucleotide change results in a codon that codes for a different amino acid.
Monogenic mutation
A single gene mutation.
Necrotic cell death
Also known as necrosis is a form of cell injury that results in the premature death of cells in living tissue. Necrosis is caused by factors external to the cell or tissue, such as infection, toxins, or trauma that result in the unregulated digestion of cell components.
Nonsense mutation
A point mutation in a sequence of DNA that results in a premature stop codon, or a nonsense codon in the transcribed mRNA, and in a truncated, incomplete, and usually nonfunctional protein product. It differs from a missense mutation, which is a point mutation where a single nucleotide is changed to cause substitution of a different amino acid.
Ocular system
An eye and its central visual system.
Oncogenesis
Literally: the creation of cancer. A process by which normal cells are transformed into cancer cells.
12
Phagocytosis
The process of engulfing a solid particle by a phagocyte or a protist to form an internal phagosome. Phagocytosis is a specific form of endocytosis involving the vesicular internalization of solids such as bacteria, and is, therefore, distinct from other forms of endocytosis such as the vesicular internalization of various liquids. Phagocytosis is involved in the acquisition of nutrients for some cells, and, in the immune system, it is a major mechanism used to remove pathogens and cell debris. Bacteria, dead tissue cells, and small mineral particles are all examples of objects that may be phagocytosed.
Phenotype
The composite of an organism's observable characteristics or traits, such as its morphology, development, biochemical or physiological properties, behaviour, and products of behaviour (such as a bird's nest). Phenotypes result from the expression of an organism's genes as well as the influence of environmental factors and the interactions between the two.
Phenylketonuria
An autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase (PAH), rendering it nonfunctional. This enzyme is necessary to metabolize the amino acid phenylalanine to the amino acid tyrosine. When PAH activity is reduced, phenylalanine accumulates and is converted into phenylpyruvate (also known as phenylketone), which can be detected in the urine.
Polysomes
A cluster of ribosomes, bound to a mRNA molecule. Many ribosomes read one mRNA simultaneously, progressing along the mRNA to synthesize the same protein. They may appear as clusters, linear polysomes, or circular rosettes on microscopy, but mainly circular
in vivo.
Recessive
A recessive trait only becomes phenotypically apparent when two copies of a gene (two alleles) are present.
Ribosome
A molecule consisting of two subunits that fit together and work as one to build proteins according to the genetic sequence held within the messenger RNA (mRNA). Some ribosomes occur freely in the cytosol whereas others are attached to the nuclear membrane or to the endoplasmic reticulum (ER)
Rough endoplasmic reticulum (RER)
ER bearing many ribosomes on its outer surface giving it a rough appearance; hence, its name. Since RER has ribosomes attached to its surface it is therefore involved in protein synthesis and secretion. It synthesizes and secretes serum proteins (such as albumin) in the liver, and hormones (such as insulin) and other substances (such as milk) in the glands.
13
Splice-site mutation
A genetic mutation that inserts, deletes or changes a number of nucleotides in the specific site at which splicing of an intron takes place during the processing of precursor messenger RNA into mature messenger RNA.
Trans Golgi network (TGN)
A major secretory pathway sorting station that directs newly synthesized proteins to different subcellular destinations. The TGN also receives extracellular materials and recycled molecules from endocytic compartments.
Transgene expression
Expression of an exogenous gene introduced into the genome of another organism
Vacuole
A membrane-bound organelle which is present in all plant and fungal cells and some protist, animal and bacterial cells. Vacuoles are essentially enclosed compartments which are filled with water containing inorganic and organic molecules including enzymes in solution, though in certain cases they may contain solids which have been engulfed. Vacuoles are formed by the fusion of multiple membrane vesicles and are effectively just larger forms of these. The organelle has no basic shape or size; its structure varies according to the needs of the cell.
Vector
A DNA molecule used as a vehicle to artificially carry foreign genetic material into another cell, where it can be replicated and/or expressed. A vector containing foreign DNA is termed recombinant DNA. The four major types of vectors are plasmids, viral vectors, cosmids, and artificial chromosomes.
15
Abbreviations
Ac Acetyl
Ac2O acetic anhydride AcOH acetic acid
Atm atmosphere
BBB blood brain barrier
BMT bone-marrow transplantation
Bn Benzyl
Bu Butyl
CNS central nervous system DGJ 1-deoxygalactonojirimycin DMF dimethylformamide
DNJ 1-deoxynojirimycin
ECD electronic circular dichroism
ELSD evaporative light scattering detector
Eq equivalent
ER endoplasmic reticulum ERT enzyme replacement therapy Et2O diethyl ether
EtOAc ethyl acetate
GALC Galactocerebrosidase (= galactoceramidase) GCS glucosylceramide synthase
HPLC high-performance liquid chromatography HSCT hematopoietic stem cell therapy
IC50 half maximal inhibitory concentration IFG isofagomine
iPrOH isopropanol
LSD lysosomal storage disorder MeOH methanol
mM millimolar
MU methylumbelliferyl NEt3 triethylamine
16 NMR nuclear magnetic resonance
PC pharmacological chaperone PCC pyridinium chlorochromate
PCT pharmacological chaperone therapy PNS peripheral nervous system
RER rough endoplasmic reticulum RT room temperature
SM starting material
SRT substrate reduction therapy
t-BuOH tert-butanol
TFA trifluoroacetic acid TGN trans Golgi network THF tetrahydrofuran
TLC thin layer chromatography
TMS trimethylsilane (also trimethylsilyl) TMSOTf trimethylsilyl trifluoromethanesulfonate TS transition state
17
Avant-propos
La maladie de Krabbe est une maladie rare, héréditaire et mortelle, causée par des mutations de la β-galactocérébrosidase (GALC) une glycosidase lysosomale impliquée dans l'hydrolyse de différents galactolipides. Les mutations situées en dehors du domaine catalytique empêchent la GALC d'adopter sa conformation native. L'enzyme mal repliée est ainsi détectée et éliminée de l'organisme. Son substrat non-hydrolysé s'accumule dans le système nerveux central et périphérique entraînant des symptômes mortels avant l’âge de deux ans.
La thérapie chaperon (PCT) est une nouvelle stratégie consistant à administrer, à des concentrations très faibles, une petite molécule ayant des interactions fortes avec l'enzyme. Cela l'aide à adopter une conformation correcte et à restaurer partiellement l’activité au niveau du lysosome, permettant au substrat en excès d’être hydrolysé. La plupart du temps, les chaperons les plus efficaces sont de puissants inhibiteurs de l'enzyme et les iminosucres sont connus pour inhiber fortement les glycosidases. L'efficacité de cette stratégie a été récemment démontrée pour deux maladies lysosomales: les maladies de Fabry et de Gaucher. Dans cette optique, l’objectif de mon projet de thèse consistait à synthétiser de nouveaux iminosucres, inhibiteurs potentiels de b-galactocérébrosidase : des imino-L -arabinitols, imino-D-galactitols et des composés de type galacto-isofagomine (galacto-IFGs), et à évaluer leur potentiel en tant que chaperons pharmacologiques pour la maladie de Krabbe. La conception de ces iminosucres a été dictée par la structure de l'état de transition de la réaction catalysée par la GALC, les données de la littérature sur les inhibiteurs de galactosidases et l'expérience de notre groupe dans la synthèse d’iminosucres en tant que chaperons pharmacologiques pour la maladie de Gaucher.
19
Introduction
I.
Lysosomal storage disorders
I.1 Lysosome
A human organism is a complicated machine, where the organic and inorganic molecules undergo constantly the catabolic and anabolic processes in order to obtain, accumulate and use energy. To preserve the perfect equilibrium, each cell is equipped with a variety of repair and quality control mechanisms, correcting almost every error. Nevertheless, sometimes the cell balance is disturbed and different pathologies can be observed.
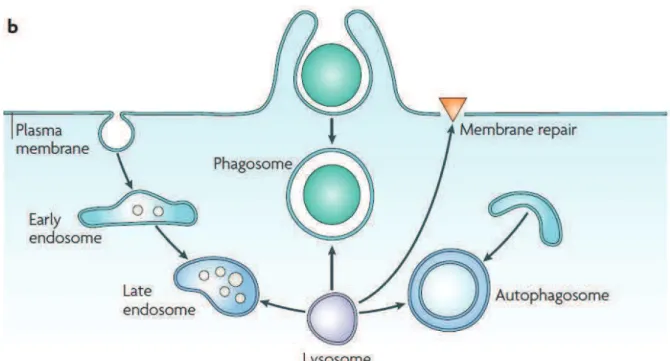
Every cell of our body is composed of diverse organelles, among which the lysosome was originally discovered and described in 1955 by De Duve.1,2 This small, single membrane, bag-like vesicle, also called the cellular digestive machine, contains at least 7 integral membrane proteins3 and about 60 soluble hydrolytic enzymes4 necessary for intracellular digestion. It occurs in almost every eukaryotic cell (common in animals but rare in plants) and its specific functions include digestion of macromolecules (glycosphingolipids, glycogen, oligosaccharides, mucopolysaccharides, glycoproteins and proteins),5 phagocytosis, endocytosis, or autophagy (Figure 1)6 and digestion of any other waste materials. It is also responsible for apoptosis7 and for repair of damage to the plasma membrane by acting as a membrane patch.8 In white blood cells, lysosome content is carefully released into the vacuole around the bacteria and serves to kill and digest it. Lysosomal functions are dependent on lysosomes fusing with target vacuoles and liberation of digestive enzymes.9 Uncontrolled release of lysosome contents into the cytoplasm is also a component of necrotic cell death.10 The pH of lysosome is 4.6-5.0, which is carefully maintained by the proton-pumping ATPases.11 As all enzymes in the lysosome work best at an acidic pH and as the pH of the cytosol is 7.2, the cell is protected from auto-digestion.
1
De Duve, C.; Pressman, B. C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Biochem J 1955, 60, 604. 2
De Duve, C. Nat Cell Biol 2005, 7, 847. 3
Eskelinen, E. L.; Tanaka, Y.; Saftig, P. Trends Cell Biol 2003, 13, 137. 4
Journet, A.; Chapel, A.; Kieffer, S.; Roux, F.; Garin, J. Proteomics 2002, 2, 1026. 5
Futerman, A. H.; van Meer, G. Nat Rev Mol Cell Biol 2004, 5, 554. 6
Luzio, J. P.; Pryor, P. R.; Bright, N. A. Nat Rev Mol Cell Biol 2007, 8, 622. 7
Guicciardi, M. E.; Leist, M.; Gores, G. J. Oncogene 2004, 23, 2881. 8
McNeil, P. L.; Kirchhausen, T. Nat Rev Mol Cell Biol 2005, 6, 499. 9
Hopwood, J. J.; Brooks, D. A. In Organelle Disease; Applegarth, D. A., Dimmick, J. E., Hall, J., Eds.; Chapman and Hall Medical: 1997, p 7.
10
Ferri, K. F.; Kroemer, G. Nat Cell Biol 2001, 3, E255. 11
20 Figure 1.Specific functions of lysosome.6
I.2 Synthesis and transport of lysosomal proteins.
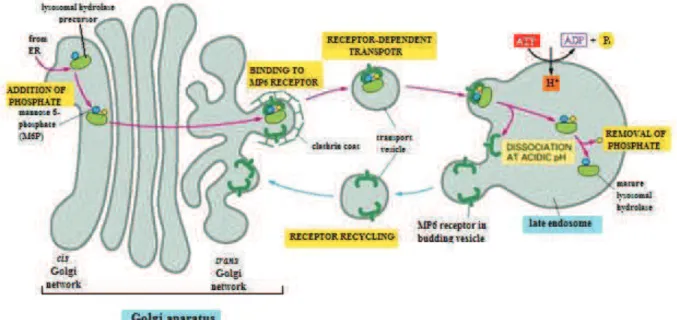
Lysosomal proteins are synthesized on membrane-bound polysomes in the rough endoplasmic reticulum (RER). The soluble lysosomal enzymes contain a hydrophobic N-terminal signal peptide, which directs the ribosome towards the endoplasmic reticulum (ER). After core glycosylation on selected asparagine residues (by transfer of a tetradecasaccharide made of 3 glucose, 9 mannose and 2 N-acetylglucosamine residues, followed by the elaboration of this oligosaccharide by so-called trimming glycosidases),12 the protein is transported to the trans Golgi network (TGN). The next step is the addition of a phosphomannosyl recognition marker (M6P) that mediates enzyme translocation to the lysosomes, which is divided into two phases (Figure 2): the transfer of a N-acetyl-glucosamine-1-phosphate from UDP-GlcNAc (catalyzed by N-acetyl-glucosaminyl-1-phosphotransferase) to one or more mannose residues on the lysosomal protein13 and subsequent removal of the N-acetyl-glucosamine residue to generate a phosphoester intermediate.14,15 Once this active form of an enzyme is created, it forms a complex with one of the two possible receptors: cation dependent or cation independent mannose-6-phosphate receptors (CDM6P-Rc or CIM6P-Rc).16 Clathrin-coated vesicles that contain the complex
6
Luzio, J. P.; Pryor, P. R.; Bright, N. A. Nat Rev Mol Cell Biol 2007, 8, 622. 12
Kornfeld, R.; Kornfeld, S. Annu Rev Biochem 1985, 54, 631. 13
Little, L.; Alcouloumre, M.; Drotar, A. M.; Herman, S.; Robertson, R.; Yeh, R. Y.; Miller, A. L. Biochem J 1987, 248, 151.
14
Reitman, M. L.; Kornfeld, S. J Biol Chem 1981, 256, 4275. 15
Reitman, M. L.; Kornfeld, S. J Biol Chem 1981, 256, 11977. 16
21 fuse with late endosomes, where due to the acidic pH the enzyme dissociates from the receptor. The released M6P-Rc recycles back to the TGN or move to the plasma membrane to integrate the exogenous ligands.17 Unlike soluble hydrolases, lysosomal membrane proteins do not require mediation by MP6 receptors. They are transported via the plasma membrane or by a direct intracellular route.18
Figure 2. Transport from the Trans Golgi Network to lysosomes.19
I.3 Lysosomal storage disorders.
The lysosomal storage diseases (LSDs) are a group of genetic, inherited disorders, resulting from impaired activity of one of the lysosomal proteins. There are over 5020 such diseases described so far (Table 1). Individually, LSDs have incidence rate of less than 1:100 000 births but overall they occur with incidence rate between 1:5 000-1:8 000 births,21 which makes them relatively common and represents an important health problem. Moreover, some populations show higher risk for certain LSDs. Among them we can list Ashkenazi Jewish population with greater prevalence of Gaucher,22 Tay-Sachs and Niemann-Pick diseases,23 or the Finnish population more susceptible to aspartylglucosaminuria24 and to
17
Lodish, A.; Berk, A.; Zipursky, S. Molecular Cell Biology. 4th edition; W. H. Freeman;: New York, 2000. 18
Linder, M. E.; Deschenes, R. J. Biochemistry 2003, 42, 4311. 19
Alberts, B.; Johnson, A.; Lewis, J. In Molecular Biology of the Cell. 4th edition; Garland Science: New York, 2002.
20
Parkinson-Lawrence, E. J.; Shandala, T.; Prodoehl, M.; Plew, R.; Borlace, G. N.; Brooks, D. A. Physiology (Bethesda) 2010, 25, 102.
21
Meikle, P. J.; Hopwood, J. J.; Clague, A. E.; Carey, W. F. JAMA 1999, 281, 249. 22
Beutler, E.; Grabowski, G. In The Metabolic and Molecular Bases of Inherited Disease. 8th ed.; McGraw-Hill: New York, 2001, p 3635.
23
Vallance, H.; Ford, J. Crit Rev Clin Lab Sci 2003, 40, 473. 24
22 infantile/juvenile neuronal ceroid lipofuscinosis (NCLs).25 LSDs are generally monogenic disorders, however, for most of them different mutations have been detected in the same gene in different patients. The mutations (missense, nonsense and splice-site) lead to complete or reduced loss of activity of mutant proteins. Although some enzymes can be synthesized at a normal level, and some of them are also functionally competent, most of them are not correctly folded and as a consequence they are not properly processed and trafficked to the lysosome. Many of them are detected and discarded by the quality control system in the ER. Most of the dysfunctions observed in LSDs result from a hydrolase deficiency (and subsequent substrate accumulation), but mutations in genes coding for any of the lysosomal proteins or even proteins processing them can lead to those illnesses (Figure 3)5.
Figure 3. Different basis of LSDs5
For example, the deficiency of two lysosomal membrane transporter proteins: sialin and cystinosin causes sialic-acid-storage disease and cystinosis respectively.26,27 It is also known that Danon disease results from mutations in abundant lysosome-associated membrane protein-2 (LAMP2).28 Moreover, any perturbation of lysosomal enzymes transport through TGN can lead to LSD5 such as I Cell or pseudo-Hurler disease, where mutations occur in genes coding for N-acetyl-glucosaminyl-1-phosphotransferase (see I.2). In some cases (multiple sulfatase deficiency,29 galactosialidosis30) the mutation affects only one enzyme, but
25
Santavuori, P. Brain Dev 1988, 10, 80. 26
Verheijen, F. W.; Verbeek, E.; Aula, N.; Beerens, C. E.; Havelaar, A. C.; Joosse, M.; Peltonen, L.; Aula, P.; Galjaard, H.; van der Spek, P. J.; Mancini, G. M. Nat Genet 1999, 23, 462.
27
Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S. A.; Callen, D. F.; Gribouval, O.; Broyer, M.; Bates, G. P.; van't Hoff, W.; Antignac, C. Nat Genet 1998, 18, 319.
28
Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J. E.; Oh, S. J.; Koga, Y.; Sue, C. M.; Yamamoto, A.; Murakami, N.; Shanske, S.; Byrne, E.; Bonilla, E.; Nonaka, I.; DiMauro, S.; Hirano, M. Nature 2000, 406, 906.
5
Futerman, A. H.; van Meer, G. Nat Rev Mol Cell Biol 2004, 5, 554. 29
Schmidt, B.; Selmer, T.; Ingendoh, A.; von Figura, K. Cell 1995, 82, 271. 30
23 as it is necessary for the stability or activity of numerous proteins, the deficiency of many enzymes is observed. On the other hand, in illnesses such as NCLs or Batten Disease31 it was observed that defects in up to 8 distinct genes cause similar pathology by accumulation of similar substances in lysosomes. There are also mutations severe enough to be lethal in early stages of development and because of that they will never be associated with any lysosomal storage diseases.
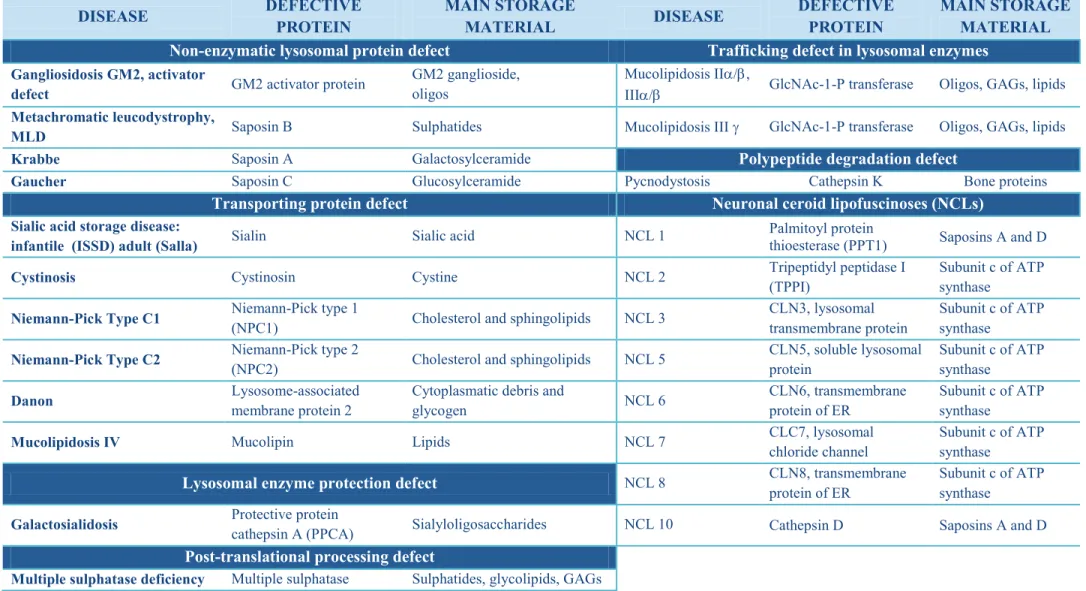
Being aware of the complexity and the diversity of LSDs it is difficult to find an appropriate classification model. They can be grouped according to the type of material accumulated or on the basis of the deficient enzyme. Table 1 represents all known LSDs classified according to the molecular defect and provides details on both the defective enzyme and the main substrate accumulated.32
The severity and the type of the disorder depend not only on the enzyme implied but also on the degree to which its activity is compromised. Therefore, a complete or almost complete loss of enzymatic activity leads to the most severe pathology, with an early onset and death. Interestingly, residual enzymatic activity is often sufficient for late onset (juvenile or adult form) and milder symptoms to be observed.33 The same enzyme defect can lead to different symptoms. In Pompe disease a severe infantile onset of the disorder is expressed by hypotonia, hypertrophic cardiomyopathy and failure to thrive, where the adult onset has no cardiac findings, but proximal muscle weakness and respiratory insufficiency.34 The nature of the undegraded storage material and the location of its accumulation play also a crucial role by changing the architecture and function of cells, tissues and organs. The cells with a high turnover of enzyme’s substrate are usually more concerned than other cells. For example, mucopolysaccharidose type IVA (MPS IVA), called also Morquio A syndrome, results from the impaired activity of N-acetyl-galactosamine-6-sulphatase. This enzyme breakdowns keratan sulfate, mostly found in the skeletal system, which thus implies severe skeletal growth problems, dysostosis and joint disease.35 Alternatively, the accumulation in the central nervous system (CNS) leads to much more severe disorders than when other tissues are affected. This is the case in Krabbe disease36 characterized by the deficiency of galactocerebrosidase: the enzyme degrading galactosylceramide. This sphingolipid is highly
31
Cooper, J. D. Curr Opin Neurol 2003, 16, 121. 32
Filocamo, M.; Morrone, A. Hum Genomics 2011, 5, 156. 33
Boyd, R. E.; Lee, G.; Rybczynski, P.; Benjamin, E. R.; Khanna, R.; Wustman, B. A.; Valenzano, K. J. J Med Chem 2013, 56, 2705.
34
Heese, B. A. Semin Pediatr Neurol 2008, 15, 119. 35
Tomatsu, S.; Montano, A. M.; Oikawa, H.; Smith, M.; Barrera, L.; Chinen, Y.; Thacker, M. M.; Mackenzie, W. G.; Suzuki, Y.; Orii, T. Curr Pharm Biotechnol 2011, 12, 931.
36
24 abundant in neurons and its buildup results in progressive neuroregression. Only in some cases (e.g. Krabbe disease) the accumulated substrate is cytotoxic itself. Usually, its accumulation activates secondary and tertiary biochemical pathways, which have not been well understood until now. Genetic defects that lay at the basis of all LSDs can change cellular processes, alternate signaling pathways and modify gene expression, which leads finally to tissue damage and death.5 However, even if the pathology development strongly relies on all the factors above and even if the gene mutations are well documented, prediction of illness seriousness and progression is almost never accurate.
5
25 DISEASE DEFECTIVE PROTEIN MAIN STORAGE MATERIAL DISEASE DEFECTIVE PROTEIN MAIN STORAGE MATERIAL Mucopolysaccharidoses (MPSs) Gangliosidosis GM2, Tay-sachs b-Hexosaminidase A GM2 ganglioside, oligos, glycolipids MPS I (Hurler, Scheie, Hurler/Scheie) a-Iduronidase
Dermatan sulfate, heparan sulfate
Gangliosidosis GM2,
Sandhoff b-Hexosaminidase A+B GM2 ganglioside, oligos
MPS II (Hunter) Iduronate sulphatase Dermatan sulfate, heparan sulfate
Gaucher
(Types I, II, III) Glucosylceramidase Glucosylceramide
MPS III A (Sanfilippo A) Heparan sulphamidase Heparan sulfate Krabbe b-Galactocerebrosidase Galactosylceramide
MPS III B (Sanfilippo B) Acetyl
a-glucosaminidase Heparan sulfate
Metachromatic
leukodystrophy Arylsulphatase A Sulphatides
MPS III C (Sanfilippo C) Acetyl CoA:
a-glucosaminidase Heparan sulfate
Niemann-Pick
(type A, type B) Sphingolyelinase Sphingomyelin
MPS III D (Sanfilippo D) N-acetyl
glucosamine-6-sulphatase Heparan sulfate Oligosaccharidoses (glycoproteinoses)
MPS IV A (Morquio A) Acetyl
glucosamine-6-sulphatase
Keratan sulfate,
chondroiotin 6-sulfate Aspartylglycosaminuria Glycosylasparaginase Aspartylglucosamine
MPS IV B (Morquio B) b-Galactosidase Keratan sulfate Fucosidosis a-Fucosidase Glycoproteins,
glycolipids, oligos
MPS VI (Morteaux-Lamy) Acetyl galactosamine Dermatan suplhate a-Mannosidosis a-Mannosidase Mannose-rich oligos
MPS VII (Sly) b-Glucuronidase
Dermatan suplhate, heparan sulfate, chondroiotin 6-sulfate Schindler N-Acetylgalactosaminidase Sialylated/ asialoglycopeptides, glycolipids
MPS IX (Natowicz) Hyaluronidase Hyluronan b-Mannosidosis b-Mannosidase Man(b1-4)GlnNAc
Sphingolipidoses Sialidosis Neuraminidase glycopeptides, oligos
Fabry a-Galactosidase A Globotriasylceramide Glycogenoses
Farber Acid ceramidase Ceramide Wolman/CESD Acid lipase Cholesterol esters
Gangliosidosis GM1
(Types I, II, III) GM1-b-galactosidase
GM1 ganglioside, keratan
sulfate, oligos, glycolipids Glycogenosis II/Pompe
a 1,4-glucosidase
26 DISEASE DEFECTIVE PROTEIN MAIN STORAGE MATERIAL DISEASE DEFECTIVE PROTEIN MAIN STORAGE MATERIAL
Non-enzymatic lysosomal protein defect Trafficking defect in lysosomal enzymes
Gangliosidosis GM2, activator
defect GM2 activator protein
GM2 ganglioside, oligos
Mucolipidosis IIa/b,
IIIa/b GlcNAc-1-P transferase Oligos, GAGs, lipids
Metachromatic leucodystrophy,
MLD Saposin B Sulphatides Mucolipidosis III g GlcNAc-1-P transferase Oligos, GAGs, lipids
Krabbe Saposin A Galactosylceramide Polypeptide degradation defect
Gaucher Saposin C Glucosylceramide Pycnodystosis Cathepsin K Bone proteins
Transporting protein defect Neuronal ceroid lipofuscinoses (NCLs)
Sialic acid storage disease:
infantile (ISSD) adult (Salla) Sialin Sialic acid NCL 1
Palmitoyl protein
thioesterase (PPT1) Saposins A and D
Cystinosis Cystinosin Cystine NCL 2 Tripeptidyl peptidase I
(TPPI)
Subunit c of ATP synthase
Niemann-Pick Type C1 Niemann-Pick type 1
(NPC1) Cholesterol and sphingolipids NCL 3
CLN3, lysosomal transmembrane protein
Subunit c of ATP synthase
Niemann-Pick Type C2 Niemann-Pick type 2
(NPC2) Cholesterol and sphingolipids NCL 5
CLN5, soluble lysosomal protein Subunit c of ATP synthase Danon Lysosome-associated membrane protein 2
Cytoplasmatic debris and
glycogen NCL 6
CLN6, transmembrane protein of ER
Subunit c of ATP synthase
Mucolipidosis IV Mucolipin Lipids NCL 7 CLC7, lysosomal
chloride channel
Subunit c of ATP synthase
Lysosomal enzyme protection defect NCL 8 CLN8, transmembrane
protein of ER
Subunit c of ATP synthase
Galactosialidosis Protective protein
cathepsin A (PPCA) Sialyloligosaccharides NCL 10 Cathepsin D Saposins A and D
Post-translational processing defect
Multiple sulphatase deficiency Multiple sulphatase Sulphatides, glycolipids, GAGs
Table 1. List of lysosomal storage disorders with the deficient enzyme and main storage material.32
32
27
II.
Therapies for LSDs
II.1 General
The clinical course of LSDs is chronic, progressive, and frequently lethal in the late childhood. Although the symptoms associated with those illnesses are often neurological, they can also be multisystemic, with skeletal, CNS, cardiovascular, and ocular system involvement. As mentioned before, there is no clear correlation between the mutation and its phenotypic consequences. This is why next to genetic and molecular analysis, the preliminary (urine and serum) tests and clinical interview are indispensable. Furthermore, it was proved that earlier diagnosis and presymptomatically treatment provide the best outcome. This highlights the need of newborn screening (NBS) for LSDs. The NBS concept was originally developed and used in the early 1960s37 predominantly for patients suffering from phenylketonuria. Presently, by using tandem mass spectrometry identification of enzyme reaction’s products or immunologic protein assays, it is possible to screen several LSDs at once.38 Although this technique still needs optimization, it gives a hope for early diagnosis and a life-saving therapy. It is already applied in the New York state where the NBS for Krabbe disease has been performed since 2006.34 The newborn screening has another advantage: it can reveal an illness in parents of sick children, giving them opportunity of early treatment. However, there is also the dark side of the NBS: they are often done without parental consent, which raises psychosocial and ethical concerns.
For most of the LSDs, as soon as they are well diagnosed, immediate therapy is initiated. The vital need of developing existing and finding new therapies for LSDs is due to two facts. Firstly, for many of the lysosomal storage disorders, even if well diagnosed, no specific or definitive treatment is available. Secondly, due to the high regulatory and research costs and lack of competition on LSDs’ medicines market, the therapies are very expensive (Table 2).39
37
Marsden, D.; Larson, C.; Levy, H. L. J Pediatr 2006, 148, 577. 38
Li, Y.; Scott, C. R.; Chamoles, N. A.; Ghavami, A.; Pinto, B. M.; Turecek, F.; Gelb, M. H. Clin Chem 2004, 50, 1785.
34
Heese, B. A. Semin Pediatr Neurol 2008, 15, 119. 39
Beutler, E. Mol Genet Metab 2006, 88, 208.
Disease Treatment Annual Cost (per patient) Gaucher ERT $ 145,000 - $ 290,000
Gaucher SRT $ 91,000
Fabry ERT $ 156,000
MPS I ERT $ 340,000
MPS IV ERT $ 377,000
28 Classically, the treatment consists in symptomatic care of disease manifestations. The therapies applied in LSDs can be roughly divided into those that act on the symptoms and those that act on the cause (Figure 4).40
1 4 2 5 3 SUBSTRATE X Biosynthetic enzyme Substrate-reduction therapy
Determination of the secondary cellular and biochemical pathways that are affected by substrate accumulation and intervention in
these pathways
Hematopoitetic stem cell therapy
Gene therapy Defective lysosomal hydrolase Enzyme-replacement therapy
Design of small molecules to reactivate defective enzymes (chemical chaperones)
PRODUCT
Figure 4. LSDs therapy strategies. 1- stem cell therapy, 2-gene therapy, 3-ERT, 4-SRT, 5-PCT.40
General treatment includes bone-marrow transplantation41 (BMT) or splenectomy (partial or complete surgical removal of the spleen) applied in Gaucher disease42. The latter was recommended earlier only in life-saving cases and is no longer used. More important therapeutic approaches are those which can act on accumulating substrate (substrate reduction therapy SRT), on deficient enzyme (enzyme replacement therapy ERT and the novel pharmacological chaperone therapy PCT) or which use directly gene therapy.
II.2 Hematopoietic stem cells therapy
Although BMT was successful in some cases, it has two major drawbacks: the requirement of matched donors and a high mortality rate. A more recent version of this method uses hematopoietic stem cells from placental cord blood.43 The objective of hematopoietic stem cells therapy (HSCT) is to engraft healthy cells into a patient without functional enzyme.44 The donor leukocytes in the host tissue produce and secrete the enzyme, which is then taken up by enzyme-deficient host cells (Figure 5).45 This principle is called cross-correlation and is a basis of ERT (II.4).
40
van Gelder, C. M.; Vollebregt, A. A.; Plug, I.; van der Ploeg, A. T.; Reuser, A. J. Expert Opin Pharmacother 2012, 13, 2281.
41
Hoogerbrugge, P. M.; Brouwer, O. F.; Bordigoni, P.; Ringden, O.; Kapaun, P.; Ortega, J. J.; O'Meara, A.; Cornu, G.; Souillet, G.; Frappaz, D.; et al. Lancet 1995, 345, 1398.
42
Salky, B.; Kreel, I.; Gelernt, I.; Bauer, J.; Aufses, A. H., Jr. Ann surg 1979, 190, 592. 43
Martin, P. L.; Carter, S. L.; Kernan, N. A.; Sahdev, I.; Wall, D.; Pietryga, D.; Wagner, J. E.; Kurtzberg, J. Biol Blood Marrow Transplant 2006, 12, 184.
44
Peters, C.; Steward, C. G. Bone Marrow Transplant 2003, 31, 229. 45
29 Figure 5. The concept of cross-correlation in HSCT.
Hematopoietic stem cells therapy can be applied in two ways: by transplanting donor healthy cells to a patient or by associating it with ex-vivo gene therapy, where after genetic modifications, the patient own cells are re-grafted to its organism (II.3).46 The first HSCT for LSDs was performed on Hurler syndrome (MPS I) patient in 198047 resulting in biochemical and clinical improvements up to 13 months after. Since then over 900 HSCTs 48 were done (Figure 6). Most of them concern Hurler disease, metachromatic leukodystrophy (MLD) and Krabbe disease.49
Figure 6. Estimated frequencies of HSCTs for various inborn errors in metabolism since 1980. * Other leukodystrophies/white-matter diseases.50
Even though HSCT is a very promising option for treating LSDs at the source of the problem (it is easier to prevent the pathology than to correct it) it has serious limitations. First of all, HSCT is most successful in the neonatal and presymptomatic stage of disease, especially when CNS dysfunction is involved.51 This requires early diagnosis and transplantation in a young age. Furthermore, hematopoietic stem cells migrate only to several
46
Priller, J.; Flugel, A.; Wehner, T.; Boentert, M.; Haas, C. A.; Prinz, M.; Fernandez-Klett, F.; Prass, K.; Bechmann, I.; de Boer, B. A.; Frotscher, M.; Kreutzberg, G. W.; Persons, D. A.; Dirnagl, U. Nat Med 2001, 7, 1356.
47
Hobbs, J. R.; Hugh-Jones, K.; Barrett, A. J.; Byrom, N.; Chambers, D.; Henry, K.; James, D. C.; Lucas, C. F.; Rogers, T. R.; Benson, P. F.; Tansley, L. R.; Patrick, A. D.; Mossman, J.; Young, E. P. Lancet 1981, 2, 709. 48
Rovelli, A. M.; Steward, C. G. Bone Marrow Transplant 2005, 35 Suppl 1, S23. 49
Boelens, J. J.; Prasad, V. K.; Tolar, J.; Wynn, R. F.; Peters, C. Pediatr Clin North Am 2010, 57, 123. 50
Boelens, J. J. J Inherit Metab Dis 2006, 29, 413. 51
Escolar, M. L.; Poe, M. D.; Provenzale, J. M.; Richards, K. C.; Allison, J.; Wood, S.; Wenger, D. A.; Pietryga, D.; Wall, D.; Champagne, M.; Morse, R.; Krivit, W.; Kurtzberg, J. N Engl J Med 2005, 352, 2069.
30 organs (shown in mice)52 such as bone marrow, liver, spleen, lungs and CNS, and barely reach the skeletal muscles and cartilage (shown in rats53) which results in no therapeutic improvement in skeletal diseases.54 Finally, like for BMT, the transplant-related mortality rate is about 10-25%41 and the surgery delays are too long. To limit the risk, careful selection of donor graft source is required. Moreover, the application of cord blood stem cells has reduced the time to transplantation and the danger of the graft-versus-host disease.
II.3 Gene therapy
As the majority of therapies applied to LSDs treat secondary complications of the disease, the somatic gene therapy would be the most attractive and effective approach.55 It consists in inserting a functional gene to produce a correctly working enzyme and replace the deficient one. The LSDs are monogenic, for many of them the mutation has been identified and a relatively small amount of active lysosomal enzyme can resolve the disease, which make them perfect candidates for such type of therapy. Thanks to a phenomenon known as cross correlation phenomenon genes do not have to be transferred to all cells of the organism, which means that only a small percentage of transducted cells can have a therapeutic effect. Gene transfer can be applied in vivo and ex vivo. The first method requires a vector (based on disabled adenovirus, adeno-associated virus, lentivirus or herpes virus)56 carrying the transgene which is injected into circulation or directly into a target tissue. The second one implies in vitro genetic correction of patients cells (often hematopoietic stem cells) followed by re-implantation (Figure 7).40 Despite a number of animal models57 and biochemical and clinical progress,58 very little development has been made in applying this method to humans. Three human clinical trials of gene therapy for Gaucher disease did not show significant improvements.59 One of the drawbacks of applying gene therapy to humans is the potential
52
Kennedy, D. W.; Abkowitz, J. L. Blood 1997, 90, 986. 53
Odorfer, K. I.; Egerbacher, M.; Unger, N. J.; Weber, K.; Jamnig, A.; Lepperdinger, G.; Kleiter, M.; Sandgren, E. P.; Erben, R. G. J Cell Mol Med 2011, 15, 2232.
54
Aldenhoven, M.; Boelens, J. J.; de Koning, T. J. Biol Blood Marrow Transplant 2008, 14, 485. 41
Hoogerbrugge, P. M.; Brouwer, O. F.; Bordigoni, P.; Ringden, O.; Kapaun, P.; Ortega, J. J.; O'Meara, A.; Cornu, G.; Souillet, G.; Frappaz, D.; et al. Lancet 1995, 345, 1398.
55
Barranger, J. M.; Novelli, E. A. Expert Opin Biol Ther 2001, 1, 857. 56
Sands, M. S.; Haskins, M. E. Acta Paediatr Suppl 2008, 97, 22. 40
van Gelder, C. M.; Vollebregt, A. A.; Plug, I.; van der Ploeg, A. T.; Reuser, A. J. Expert Opin Pharmacother 2012, 13, 2281.
57
Haskins, M. ILAR J 2009, 50, 112. 58
Ioannou, Y. A.; Enriquez, A.; Benjamin, C. Expert Opin Biol Ther 2003, 3, 789. 59
Dunbar, C. E.; Kohn, D. B.; Schiffmann, R.; Barton, N. W.; Nolta, J. A.; Esplin, J. A.; Pensiero, M.; Long, Z.; Lockey, C.; Emmons, R. V.; Csik, S.; Leitman, S.; Krebs, C. B.; Carter, C.; Brady, R. O.; Karlsson, S. Hum Gene Ther 1998, 9, 2629.
31 risk of oncogenesis.60 Additionally, viral vectors can activate inflammatory and immunological responses.61 The success of gene therapy depends on the level of transgene expression, as well as on the ability of the treatment to cross the blood-brain barrier (BBB) to reach the CNS. Overall, to become a real option to treat LSDs, gene therapy still needs a great deal of optimization on the vectors and procedures safety, specificity and efficiency.
Figure 7. In vivo (direct) and ex vivo (cell-based) gene therapy. Extracted from http://stemcells.nih.gov/info/2001report/pages/chapter11.aspx II.4 Enzyme replacement therapy
Pompe disease was discovered in 1963 by Hers,62 which one year later brought in the idea of treatment by replacing defective enzyme by a healthy one.63 In 1968, Neufeld and coworkers discovered that culturing together cells of patients suffering from Hurler disease (a-iduronidase) and Hunter disease also called MPS II (iduronate sulphatase) leads to correcting of both enzymes’ defects.64 The earliest attempts of ERT were unsuccessful65 (Pompe disease) owing to the low doses, short treatment duration, the lack of knowledge about receptor-mediated endocytosis and insufficient sources of highly purified enzymes with appropriate markers for targeted uptake. The first victorious ERT was achieved in 199166 for type I Gaucher disease, using human placental glucocerebrosidase. The enzyme was
60
Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G. P.; Soulier, J.; Lim, et al. J Clin Invest 2008, 118, 3132. 61
Cheng, S. H.; Smith, A. E. Gene Ther 2003, 10, 1275. 62
Hers, H. G. Biochem J 1963, 86, 11. 63
De Duve, C. Fed Proc 1964, 23, 1045. 64
Fratantoni, J. C.; Hall, C. W.; Neufeld, E. F. Science 1968, 162, 570. 65
Valayannopoulos, V. Handb Clin Neurol 2013, 113, 1851. 66
Barton, N. W.; Brady, R. O.; Dambrosia, J. M.; Di Bisceglie, A. M.; Doppelt, S. H.; Hill, S. C.; Mankin, H. J.; Murray, G. J.; Parker, R. I.; Argoff, C. E.; et al. N Engl J Med 1991, 324, 1464.
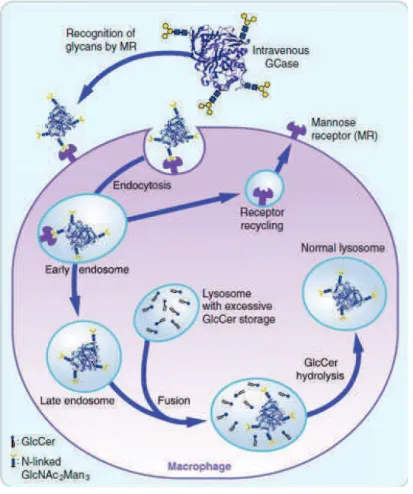
32 previously partially deglycosylated, to enhance its uptake by mannose receptor of macrophages (Figure 8).67 Since then, this therapy was applied to five other 34 LSDs: Fabry, Hunter, Hurler, Maroteaux-Lamy (MPS VI) and Pompe diseases. ERTs for metachromatic leukodystrophy, Morquio A disease and lysosomal acid lipase deficiency are in clinical trials.40
Figure 8. Macrophage uptake of intravenously administered recombinant GCase.67
Although studies during the last decade have demonstrated the benefits of using ERT, the overall outcome varies from disease to disease. The success of ERT strongly depends on the following factors: accessibility of affected cells,68 molecular composition and turn-over of the tissue, antibody formation69 against the therapeutic enzymes and finally, irreversible cell damages impairing intracellular trafficking.70 As a consequence, ERT has little effect on the
67
Phenix, C. P.; Rempel, B. P.; Colobong, K.; Doudet, D. J.; Adam, M. J.; Clarke, L. A.; Withers, S. G. Proc Natl Acad Sci U S A 2010, 107, 10842.
34
Heese, B. A. Semin Pediatr Neurol 2008, 15, 119. 40
van Gelder, C. M.; Vollebregt, A. A.; Plug, I.; van der Ploeg, A. T.; Reuser, A. J. Expert Opin Pharmacother 2012, 13, 2281.
68
Brands, M. M.; Frohn-Mulder, I. M.; Hagemans, M. L.; Hop, W. C.; Oussoren, E.; Helbing, W. A.; van der Ploeg, A. T. J Inherit Metab Dis 2013, 36, 227.
69
Brooks, D. A. Mol Genet Metab 1999, 68, 268. 70
Fukuda, T.; Ahearn, M.; Roberts, A.; Mattaliano, R. J.; Zaal, K.; Ralston, E.; Plotz, P. H.; Raben, N. Mol Ther 2006, 14, 831.
33 brain (due to the BBB), skeletal tissue (slow turn-over of the bones and slow diffusion to cartilage through the matrix), and valvular heart disease in LSD. Animal studies have shown though, that some of the problems associated with cell and tissue accessibility may be overcome by increasing the doses of ERT.71 However, this approach can increase the immunological response and the therapy cost. Concerning the mild allergic reactions observed during intravenous ERT, they are neutralized with antihistamine and non-steroidal anti-inflammatory drugs.
Despite the development mentioned above, low recombinant enzyme uptake by the cells is still the biggest challenge in ERT. It can be resolved by several strategies that will improve enzyme trafficking and recognition by both cells and lysosomes. The first consist in conjugating the enzyme with synthetic oligosaccharides having M6P (see I.2). This idea was tested on a mouse model of Pompe disease (enzyme used: oxime-neo-recombinant human a-glucosidase) and has shown decrease of glycogen accumulated during illness in muscles.72 Another approach is so called glycosylation independent lysosomal targeting, which consist in replacing the enzyme’s N-terminal peptide by an N-terminal fragment of insulin-like growth factor 2. Thanks to the high affinity of this signal peptide to M6PR, the modified enzyme is better recognized and more efficiently binds to cell and/or lysosome membrane.73 Two more solutions for increased enzyme uptake are either to modulate the expression of M6PR on the target cells74 or to inject enzyme directly to the tissue of interest (e.g. to intracerebrospinal fluid75). However, the latter procedure remains controversial from ethical and practical point of view. Completely different problem lies in passing BBB. No receptor recognition enhancement is successful so far in this case. To overcome this problem, one of the possibilities is to bind the recombinant protein to a monoclonal antibody against an endogenous BBB receptor.76 This strategy proved to be working in Hunter disease animal models.
Finally, regardless of how optimized and adapted ERT will be, it remains an expensive treatment (see Table 2) and a long-life burden to the patients.
71
Ioannou, Y. A.; Zeidner, K. M.; Gordon, R. E.; Desnick, R. J. Am J Hum Genet 2001, 68, 14. 72
Maga, J. A.; Zhou, J.; Kambampati, R.; Peng, S.; Wang, X.; Bohnsack, R. N.; Thomm, A.; Golata, S.; Tom, P.; Dahms, N. M.; Byrne, B. J.; LeBowitz, J. H. J Biol Chem 2013, 288, 1428.
73
Koeberl, D. D.; Li, S.; Dai, J.; Thurberg, B. L.; Bali, D.; Kishnani, P. S. Mol Genet Metab 2012, 105, 221. 74
Hemsley, K. M.; Hopwood, J. J. Int J Clin Pharmacol Ther 2009, 47 Suppl 1, S118. 75
Pardridge, W. M.; Boado, R. J. Methods Enzymol 2012, 503, 269. 76
34 II.5 Substrate reduction therapy
Substrate reduction therapy (SRT) is a drug-based treatment with the objective to diminish lysosomal storage by inhibition of the substrate biosynthesis.77 As an applied treatment, SRT is used only for glycosphingolipidoses (see Table 1) so far.78 Those disorders result from accumulation of various glycosphingolipids (GLS), which all have the same first step of biosynthesis (Scheme 1): the addition of a glucose unit to ceramide by ceramide-specific glucosyltransferase (also termed glucosylceramide synthase, GCS).79
Scheme 1. Biosynthesis of glycosphingolipids in humans.80
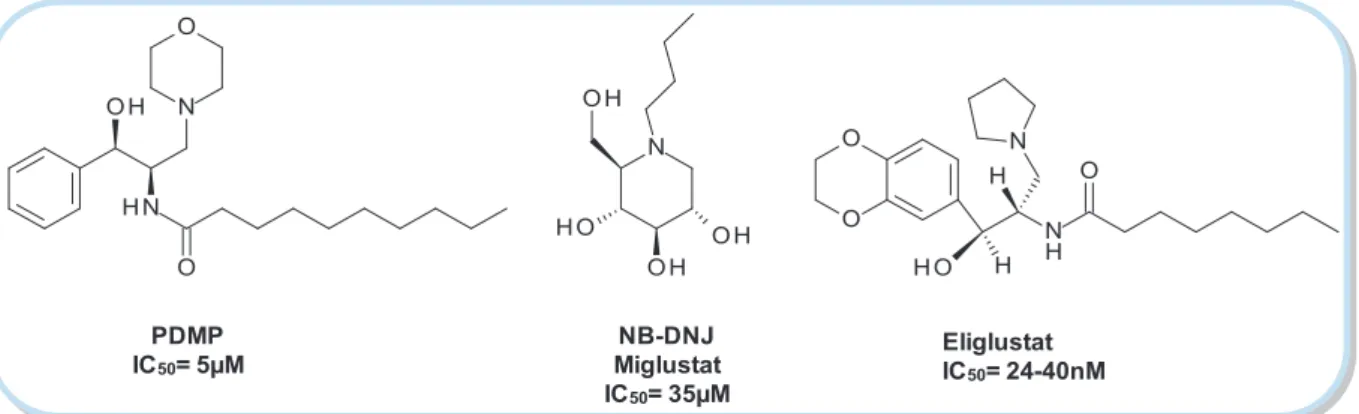
This approach was introduced in the 1970s by Radin81 and coworkers who identified the first type of glucosyltransferase inhibitors. PDMP (D,L -threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol) (Figure 9) and its analogues are powerful inhibitors of GCS, but relatively cytotoxic due to their hydrophobicity. Additionally, they provoke accumulation of free ceramide. New generations of PDMP derivatives proved to be efficient glucosylceramide synthase inhibitors;82 nevertheless none of them was tested on humans so far.
77
Cox, T. M. Acta Paediatr Suppl 2005, 94, 69. 78
Aerts, J. M.; Hollak, C. E.; Boot, R. G.; Groener, J. E.; Maas, M. J Inherit Metab Dis 2006, 29, 449. 79
Lachmann, R. H.; Platt, F. M. Expert Opin Investig Drugs 2001, 10, 455. 80
Platt, F. M.; Butters, T. D. Expert Rev Mol Med 2000, 2, 1. 81
Vunnam, R. R.; Radin, N. S. Chem Phys Lipids 1980, 26, 265. 82
35 Eliglustat IC50= 24-40nM NB-DNJ Miglustat IC50= 35µM PDMP IC50= 5µM
Figure 9. GCS inhibitors.83, 84, 85 NB-DNJ is used in SRT.
Another group of GCS inhibitors includes the N-alkylated iminosugars. The leading compound NB-DNJ (N-butyldeoxynojirimycin, miglustat) was initially designed to be an antiviral agent:86 by being a good inhibitor of intracellular a-glucosidases I and II, this compound blocks glucosylation of viral proteins and ends virus life cycle. Unfortunately, the 6-month clinical trial87 has shown no important efficacy of the drug. In 1994, while working on an in vitro model of Gaucher disease, Platt and Butters84 found that NB-DNJ reduced significantly the endogenous glucosylceramide synthesis. Although this iminosugar is not GCS-specific (it inhibits also a-glucosidase I and II as well as digestive a-glucosidases), the therapy is more efficient than in the HIV case80 (thanks to cytosolic orientation of ceramide-specific UDP-glucosyltransferase the enzyme is more accessible for the drug). After a series of encouraging assays on animals,88 the first clinical test was performed on mild-to-moderate type 1 (non-neuronopathic) Gaucher patients.89 The results of this experiment and follow-up studies lead to registration of miglustat (Zavesca®, 2003) as a drug for Gaucher type I patients, for whom ERT cannot be applied. In 2009, NB-DNJ was also approved as a drug for Niemann-Pick disease, as it proved to stabilize one of the neurological symptoms of the disorder.90 On the other hand, even though NB-DNJ was shown to cross BBB in some mouse
83
Inokuchi, J.; Radin, N. S. J Lipid Res 1987, 28, 565. 84
Platt, F. M.; Neises, G. R.; Dwek, R. A.; Butters, T. D. J Biol Chem 1994, 269, 8362. 85
McEachern, K. A.; Fung, J.; Komarnitsky, S.; Siegel, C. S.; Chuang, W. L.; Hutto, E.; Shayman, J. A.; Grabowski, G. A.; Aerts, J. M.; Cheng, S. H.; Copeland, D. P.; Marshall, J. Mol Genet Metab 2007, 91, 259. 86
Fenouillet, E.; Gluckman, J. C. J Gen Virol 1991, 72 ( Pt 8), 1919. 87
Fischl, M. A.; Resnick, L.; Coombs, R.; Kremer, A. B.; Pottage, J. C., Jr.; Fass, R. J.; Fife, K. H.; Powderly, W. G.; Collier, A. C.; Aspinall, R. L.; et al. J Acquir Immune Defic Syndr 1994, 7, 139.
80
Platt, F. M.; Butters, T. D. Expert Rev Mol Med 2000, 2, 1. 88
Jeyakumar, M.; Butters, T. D.; Cortina-Borja, M.; Hunnam, V.; Proia, R. L.; Perry, V. H.; Dwek, R. A.; Platt, F. M. Proc Natl Acad Sci U S A 1999, 96, 6388.
89
Cox, T.; Lachmann, R.; Hollak, C.; Aerts, J.; van Weely, S.; Hrebicek, M.; Platt, F.; Butters, T.; Dwek, R.; Moyses, C.; Gow, I.; Elstein, D.; Zimran, A. Lancet 2000, 355, 1481.
90
36 models, it didn’t demonstrate any effect on Gaucher type III patients over a 2-year period of treatment.91
Despite its benefits to LSDs patients, miglustat has gastrointestinal side-effects92 and remains an expensive therapy. An alternative to this drug may be eliglustat (Phase III trial in Gaucher type I patients in progress), which due to a higher specificity for ceramide-specific UDP-glucosyltransferase shows minimal side-effects.93 Other trials of SRT for LSDs are pending: e.g. for MPSs using a soybean isoflavone genistein.94
To summarize, SRT seems to be a promising approach to treat some LSDs. The advantages include lower cost, oral administration and possibility to treat the disorders with neurological manifestations. However, further investigation is required to find novel compound with higher specificity to be used at lower dosage and to avoid side-effects.
II.6 Pharmacological chaperone therapy
In biology, a chaperone is a protein which helps in the folding of nascent polypeptide chains, refolding of denatured proteins. It also prevents aggregation of surface-exposed hydrophobic parts of the proteins that have problems with folding. Apart from that, chaperones can play an important role in signal transduction, in the maintenance of the organized state of the cytoplasm and other intracellular compartments, in the motions inside the cell, and some other vital functions of the cells.95 Pharmacological chaperone therapy (PCT) arises from the same principle, with the difference of using low-molecular-weight molecules (PCs) instead of proteins as chaperones. As mentioned previously, LSDs are the genetic disorders resulting from deficiencies of lysosomal proteins. In some cases, the mutant enzymes retain their activity or it is slightly compromised,96 but as they are often misfolded, they are recognized by the ER quality control system of the cell.97 In consequence, mutants are polyubiquitinated and translocated to the cytosol, where they are degraded by proteases.98 However, even small improvement in protein stability or conformation may prevent their
91
Tylki-Szymanska, A.; Groener, J. E.; Kaminski, M. L.; Lugowska, A.; Jurkiewicz, E.; Czartoryska, B. Mol Genet Metab 2011, 104, 627.
92
Reuser, A. J.; Wisselaar, H. A. Eur J Clin Invest 1994, 24 Suppl 3, 19. 93
Lukina, E.; Watman, N.; Arreguin, E. A.; Dragosky, M.; Iastrebner, M.; Rosenbaum, H.; Phillips, M.; Pastores, G. M.; Kamath, R. S.; Rosenthal, D. I.; Kaper, M.; Singh, T.; Puga, A. C.; Peterschmitt, M. J. Blood 2010, 116, 4095.
94
Piotrowska, E.; Jakóbkiewicz-Banecka, J.; Tylki-Szymanska, A.; Liberek, A.; Maryniak, A.; Malinowska, M.; Czartoryska, B.; Puk, E.; Kloska, A.; Liberek, T.; Baranska, S.; Wegrzyn, A.; Wegrzyn, G. Current Therapeutic Research 2008, 69, 166.
95
Ellis, R. J.; Hemmingsen, S. M. Trends Biochem Sci 1989, 14, 339. 96
Fan, J. Q. Biol Chem 2008, 389, 1. 97
Ellgaard, L.; Helenius, A. Curr Opin Cell Biol 2001, 13, 431. 98