UNIVERSITE ABDELHAMID IBN BADIS MOSTAGANEM
UNIVERSITE FACULTE DES SCIENCES EXACTES ET DE L’INFORMATIQUE
Abdelhamid Ibn Badis DEPARTEMENT DE CHIMIE MOSTAGANEM
MEMOIRE
Présenté pour obtenir le diplôme de
MASTER II EN CHIMIE
Option: ANALYSE SPECTRALE EN CHIMIE
Par
ELKAROUI RABIAA
Soutenu le :15/06/2014 2014 devant la commission d’examen :
Président Mr.BENMALTI Maitre conférence Université de Mostaganem
Examinateur : Mr.BELALIA Maitre conférence Université de Mostaganem
Rapporteur : Mme.RAHMANI Maitre assistante Université de Mostaganem
Etude de la structure SiO
4
par la méthode
Pages
Liste des tableaux
Liste des figures
Introduction générale………...1
Chapitre I : Généralités et Rappels Bibliographiques I.Introduction………...2
I.1 .Silicate………2
I .2.Structure des silicates……….2
I.3.Importance et nature ……….………..3
I.3.1.Rôle géochimique ………..………3
I.3.2.Place particulière en chimie minérale :………..……….3
I .3.3.Structures polymères ………..……..3
I. 4.Classification des minéraux : Silicates………..…...4
I.4.1-Silicates à tétraèdres indépendants ou nésosilicates ………..……4
I.4.2.Silicates à groupes finis de tétraèdres ou Sorosilicates………..…..….…5
I.4.3.Silicates en chaînes ou inosilicates ………..……….5
I.4.3.Silicates en chaînes ou inosilicates ……….….5
I.4.4.Silicates lamellaires ou phyllosilicates ………..6
I.4.5.Les Cyclosilicates………6
I.5.Synthèse bibliographique ………8
Chapitre II : Méthodes de calculs quanto chimique La modélisation en chimie et la chimie assistée par ordinateur………10
II.La chimie quantique ………...…11
II.1.Equation de Schrödinger………..11
Principe de résolution de l’équation de Schrödinger ………12
II.2.Approximation de Born-Oppenheimer……….13
II.3. Approximation orbitalaire………..14
II.4. Expression des OM : théorie LCAO ………..15
Principe et règles générales de la méthode LCAO………....15
II.5. Méthode Hartree-Fock(HF) ………18
II.6.Méthodes Semi-empiriques ……….…….20
II.7.Méthode ab initio ……….21
II.8. Méthode de la fonctionnelle de la densité ………..23
Chapitre III : Calculs et Interprétation des résultats III.1.Introduction………...24
III.2. Interprétations des résultats………..25
III.2.1.Logiciel ………..25
III.2.2.Rappels sur la définition de fréquence ……….…….26
III.2.3.Rappels sur la Spectroscopie infrarouge ………..…….26
III.3.Démarche du travail ………...27
III.4.Résultats et discussion………30
III.4.2.La méthodes de MP2 ………. ………32
III.4.3.La méthodes de DFT………...………34
III-5-Interprétation ………..36
III.5.1.comparaison entre les différentes méthodes et les résultats expérimentaux…...36
III.5.2. comparaison des distances atomiquesentre les différentes méthodes et les résultats expérimentaux………36
Conclusion ……….37
Référence………....38
Introduction Générale
1
Introductiongénérale :
Le silicium est l’élément le plus abondant sur la croûte terrestre.
On le trouve dans de très nombreux silicates. Sur terre, le silicium est le deuxième élément en abondance après l’oxygène. Il se présente principalement sous forme d’oxydes dans les minéraux.
Notre étude c’est porté sur silicates à tétraèdres indépendants ou nésosilicates Pour ces composés, aucun atome d’oxygène n’est lié à deux atomes de silicium. Les tétraèdres 𝑆𝑖𝑂4 sont des anions qui peuvent être associés à des cations. L’intérêt qu’est porté à ce dernier est du principalement à leurs applications industrielles dans lesquelles la stabilité thermique est importante : revêtements des fours, joints haute température, chaudières, systèmes de protection incendie, et ou la limite de température est réduite.
La première partie résume les principes fondamentaux des silicates et les différentes formes de silice. L’importance économique des silicates est considérable et leur rôle géochimique est essentiel ; ils constituent un des chapitres de la chimie minérale les moins bien connus.
Le deuxième chapitre passe en revue les aspects fondamentaux des calculs ab initio ainsi que les différentes méthodes numériques d’optimisation. Cette discussion inclût aussi les
ensembles de fonctions de bases.
Dans le contexte envisagé, nous discuteronsdes avantages et des inconvénients des modèles numériques, ainsi que les limites rencontrées dans ce type de calculs.
Le dernier chapitre abordera les calculs effectués sur la molécule SiO4, nous avons procédé à
l’optimisation de cette dernière suivie d’un calcul de fréquences IR en utilisant 3 méthodes : HF, DFT et MP2 et en les comparant à des résultats expérimentaux pour démontrer laquelle d’entre elles est la plus juste et pertinente.
Chapitre I Généralités et Rappels Bibliographiques
2
I. Introduction :
Les silicates et les différentes formes de silice qui s’y rattachent ont une importante industrielle et géochimique considérable. Ces minéraux, matières premières des industries du bâtiment, de la verrerie, de la céramique et constituants des laitiers métallurgiques, forment la quasi-totalité de l’écorce terrestre. Ce sont, pour la plupart des solutions solides dont la synthèse et l’interprétation des analyses chimiques sont différentes de celles des autres composés inorganiques. C’est la détermination, à partir de la diffraction des rayons X, des structures atomiques de ces composés cristallisés et les synthèses faisant intervenir des minéralisateurs (et en particulier, l’eau agissant à des températures et à des pressions élevées) qui ont résolu les énigmes de ce chapitre de la chimie minérale. On ne considère plus les silicates comme des sels d’acides siliciques, mais comme des assemblages de tétraèdres quasi réguliers (Si, Al) O4, dont les centres sont occupés par des ions
silicium ou aluminium, et les sommets par des ions oxygène.
I.1 .Silicate
Un silicate est un sel dérivant de la silice (SiO2). En chimie, ce sont des composés chimiques. En
minéralogie, les silicates désignent une importante famille de minéraux ; ils constituent 97 % de la croûte terrestre. Il en existe de nombreuses familles.
I .2.Structure des silicates
Figure 1 : Ion orthosilicate tétraédrique
Les silicates sont des minéraux dont le squelette est essentiellement formé par des tétraèdres de silicium et d'oxygène (SiO4) additionnés d’aluminium, magnésium, fer, calcium, potassium,
Chapitre I Généralités et Rappels Bibliographiques
3
I.3.Importance et nature : I.3.1.Rôle géochimique :
Si l’importance économique des silicates est considérable, leur rôle géochimique est essentiel. Ils forment plus de 90% en poids de l’écorce terrestre. Les nombreuses données statistiques sur la composition chimique de la partie superficielle, en particulier celles qui ont été obtenues par F. W. Clarke et H. S. Washington (1924) se rapportant aux roches éruptives, peuvent être présentées sous forme condensée.
I.3.2.Place particulière en chimie minérale :
Jusqu’à une période relativement récente, les silicates ont constitué un des chapitres de la chimie minérale les moins bien connus, car les méthodes classiques d’étude des composés
inorganiques ne leur sont pas en général applicables. Quand ils peuvent être chauffés jusqu’à leur fusion sans se décomposer, ils fournissent souvent, un verre au refroidissement, et beaucoup d’entre eux ne peuvent être reproduits au laboratoire à la pression ordinaire. Leur analyse chimique est difficile, car ce sont presque toujours, des solutions solides dans lesquelles interviennent
différents éléments chimiques par le jeu des remplacements isomorphiques (est la faculté de composés différents de cristalliser avec des mailles de même symétrie et dont les paramètres sont proches, ce qui leur permet de former des cristaux mixtes). Et la traduction des analyses dans une formule chimique a pendant longtemps, fait appel aux hypothèses fausses d’acides siliciques complexes.
Il en a résulté un divorce entre chimistes et minéralogistes; pour ces derniers, la parenté des propriétés cristallographiques et physiques rapproche dans une même famille, comme celles des micas et des pyroxènes, des minéraux qui, du point de vue chimique, apparaissaient très différents. C’est la diffraction cristalline des rayons X, entreprise d’abord par W. L. Bragg et ses élèves, qui ont réconcilié chimistes et minéralogistes; et la détermination des structures atomiques a eu pour conséquence de faire du chapitre des silicates l’un des plus clairs de la chimie inorganique.
I .3.3.Structures polymères :
Les tétraèdres SiO4 peuvent s'associer pour former des structures de taille très grande (structures
polymères). Ces structures sont caractérisées par de longs enchaînements covalents -Si-O-Si-O-Si-O- qui peuvent être unidimensionnels ou former des réseaux bi- ou tri-dimensionnels.
Les macroanions, comme les anions, sont associés à des cations qui assurent la neutralité électrique de l'ensemble.
Chapitre I Généralités et Rappels Bibliographiques
4
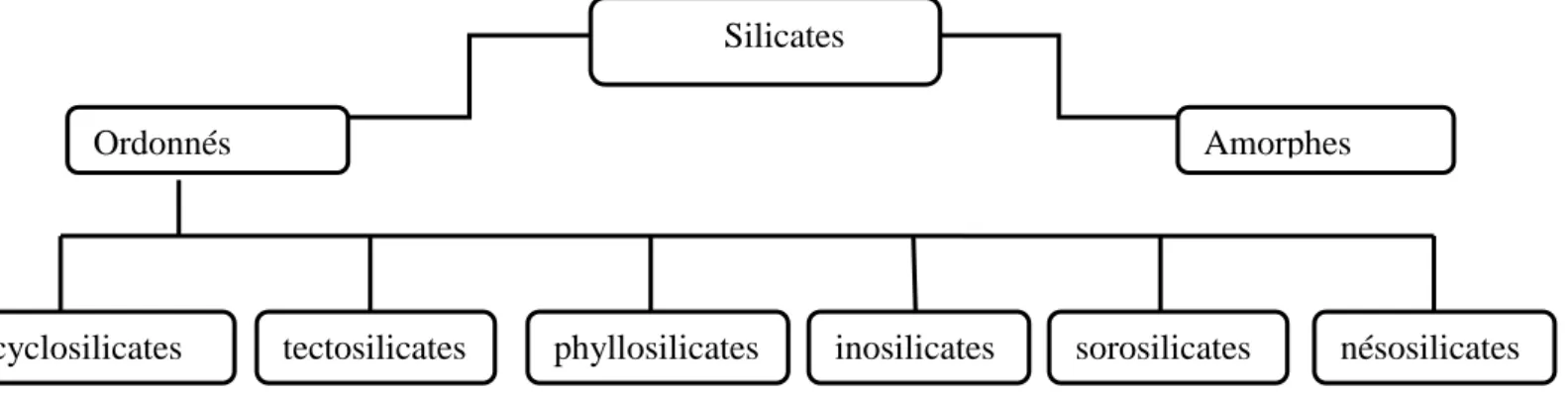
I. 4.Classification des minéraux : Silicates
Les silicates peuvent être classés selon plusieurs critères, mais les deux classifications les plus employées en minéralogie sont basées sur l’enchaînement des tétraèdres :
la classification est basée sur l’enchaînement des tétraèdres; dans la classification cristallochimique, les silicates se répartissent dans les familles suivantes :
Figure 2 : Classification des silicates
I.4.1-Silicates à tétraèdres indépendants : ou nésosilicates
Pour ces composés, aucun atome d’oxygène n’est lié à deux atomes de silicium. Les tétraèdres SiO4 sont des anions associés par l’intermédiaire des cations. Un exemple est la forstérite Mg2SiO4;
Dans cette famille, le rapport atomique Si/O est, au plus, égal à 1/4.
Figure 3 : nésosilicate
Les différents types structuraux de cette famille sont distingués en fonction de la nature des cations :
Gros cations= Zr
Moyens cations= Fe, Mg Petits cations= Be, Zn
Silicates
Ordonnés Amorphes
cyclosilicates seses
Chapitre I Généralités et Rappels Bibliographiques
5
I.4.2.Silicates à groupes finis de tétraèdres : ou Sorosilicates
Si les Nesosilicates sont à tétraèdres indépendants c'est-à-dire sans O commun, les Sorosilicates en revanche sont caractérisés par des groupes de 2 tétraèdres reliés par un O commun.
Figure 4 :sorosilicate
Il existe très peu de Sorosilicates, En fait les minéraux courant de ce groupe ont, à côté des radicaux [Si2 O7], des radicaux [Si O4].
I.4.3.Silicates en chaînes ,ou inosilicates :
Les tétraèdres s’associent dans ces composés pour former des chaînes linéaires infinies et les silicates correspondants ont souvent une texture fibreuse. Si la chaîne est simple, sa composition chimique est SiO3, et la composition chimique en même temps que les propriétés
cristallographiques sont bien mises en évidence en faisant apparaître la formule chimique du maillon de la chaîne.
Chapitre I Généralités et Rappels Bibliographiques
6
I.4.4.Silicates lamellaires, ou phyllosilicates :
Ce sont des silicates en feuillets, les tétraèdres ont en commun 3 O qui forment une face (base), On réalise ainsi des couches de tétraèdres, disposés selon un motif hexagonal, avec des pointes d'un même côté. La base de la couche correspondant aux bases triangulaires est
électriquement neutre. Dans la famille des argiles, on observe dans les tétraèdres des substitutions de l'atome Si4+ par Al, K, Na, Mg ou Fe. Ainsi les minéraux argileux sont constitués par une trame silicatée, qui s'étend d'une façon infinie dans un plan (001) et qui est obtenue par la condensation latérale d'une infinité de chaînes de type amphibole rubané ou de type Pyroxène. Ces plans se superposent et on abouti suivant les cas à des motifs à 2 couches, 3 couches, 4 couches.
Figure 6 :phyllosilicate I.4.5.Les Cyclosilicates :
Ces minéraux sont constitués par des tétraèdres disposés en « chaine fermée » comme le montre la figure ci-dessous, pour constituer ces anneaux il peut y avoir 3 ou 4ou 6 tétraèdres. Ce nombre de tétraèdre conditionne le système cristallin des minéraux formés :
- 3 tétraèdres : système rhomboédrique - 4 tétraèdres : système quadratique - 6tétraèdres : système hexagonale
Chapitre I Généralités et Rappels Bibliographiques
7
Figure 7 :cyclosilicate
I.4.6.Silicates à charpente tridimensionnelle : Ou tectosilicates :
Ce sont des silicates construits à l’aide de tétraèdre disposés en édifice à trois dimension, en fait les tétraèdres de base SiO4 que l’on retrouve dans tous les silicates sont ici soudés les uns aux autres par leurs quatre sommets ; donc chaque atome d’oxygène appartient à deux tétraèdres voisins.
Toutes les formes de silice à l’exception de la stishovite peuvent être considérées comme des tectosilicates, car leur structure atomique est un assemblage des mêmes tétraèdres quasi réguliers SiO4 liés entre eux par leurs quatre sommets ; comme chacun des atomes d’oxygène se trouve lié à deux atomes de silicium.
La composition chimique correspond à la formule SiO2. Les formes les plus importantes de silice
cristallisée, des points de vue géochimique et industriel, sont le quartz, la tridymite et la cristobalite
Chapitre I Généralités et Rappels Bibliographiques
8
Le graphe suivant représente les polymorphes de silice existant à différentes températures et pressions
Figure 9: Diagramme de phase de SiO2 [1]
I.5.Synthèse bibliographique :
Le silicium et l’oxygène sont des éléments clés dans la technologie microélectronique moderne. L’intérêt qu’est porté aux silicatesne cesse de croitre ; cet intérêt est du principalement à leurs applications industrielles dans lesquelles la stabilité thermique est importante : revêtements des fours, joints haute température, chaudières, systèmes de protection incendie, et ou la limite de température est réduite.
L’etude qu’on a efféctué se porte sur le calcul de l’energie d’optimisation ainsi que les fréquences de vibrations IR de la molécule SiO4, que nous avons comparé avec des résultats expérimentaux.
Chapitre I Généralités et Rappels Bibliographiques
9
BHASKAR J SAIKIA et all [2,3] ont trouvé les fréquences IR de la molécule SiO4suivantes :
Une vibration antisymétrique d’élongation Si-O correspondant à une fréquence de 1080-1175cm-1.
Une vibration symétrique d’élongation Si-O correspondant à une fréquence variant de 780 à 800 cm-1.
Ainsi qu’une vibration symétrique et antisymétrique de l’angle de liaison entre O-Si-O respectivement de 695cm-1 et 464cm-1.
D’autres résultats concernant la distance entre les atomes ainsi que l’angle de liaison ont été trouvé [4,5], la silice est constituée d’un arrangement de tétraèdres SiO4 reliés entre eux par
L’intermédiaire des sommets oxygènes.
Ces tétraèdres sont caractérisés par la distance atomique Si-O (de 1,6 à 1,63 Å). La valeur de l’angle θ entre les liaisons O-Si-O θ varie de 110° à 180°.
Dans notre travail nous allons tenter de procéder aux calculs des fréquences IR de la molécule optimisée à travers différentes méthodes et les comparer avec ceux de la littérature pour connaitre quelle méthode se rapproche le plus de l’expérience dans le cas de la molécule SiO4.
Chapitre II Méthodes de calculs quanto chimique
10
La modélisation en chimie et la chimie assistée par ordinateur :
L’utilisation d’un modèle implique une représentation simplifiée d’un système ou d’un processus pour une meilleure compréhension. Les chimistes conçoivent, transforment et étudient des objets bien réels qu’ils ne voient pas. Ils doivent donc continuellement chercher à améliorer les représentations de concepts qu’ils se font des atomes ou des molécules. Les modèles qui en découlent sont l’œuvre d’une patiente accumulation de faits expérimentaux et d’une rigoureuse confrontation de ces observations qui permettent de donner à la réalité invisible une image de plus en plus riche [6].
L’utilisation de méthodes théoriques pour l’obtention de modèles qui puissent prédire et comprendre les structures, les propriétés et les interactions moléculaires est connue sous le nom de «Modélisation Moléculaire». Celle-ci permet de fournir des informations qui ne sont pas disponibles par l’expérience et joue donc un rôle complémentaire à celui de la chimie expérimentale. Ainsi, la modélisation moléculaire peut par exemple permettre de se faire une idée précise de la structure de l’état de transition pour une réaction donnée, ce qui est difficile, voire impossible, pour la chimie expérimentale.
La chimie théorique [7] permet l’étude de diverses entités moléculaires existantes ou non, afin de déterminer les différentes propriétés physico-chimique de celles-ci, leur réactivité ou encore les différentes conformations qu’elles peuvent adopter en solution ou en phase gazeuse. Ces études permettent à la fois de reproduire la réalité, de mieux comprendre les phénomènes expérimentaux, mais aussi de prévoir des réactions ou des structures non encore élucidées.
Le nombre d’études théoriques a fortement augmenté avec le développement des outils informatiques dans les 20 dernières années ; des procédures de calculs numériques ainsi que des ordinateurs toujours plus puissants ont été mis au point, rendant ainsi possible l’étude de systèmes de plus en plus compliqués, et permettant l’utilisation de techniques de calculs et de niveaux de théorie de plus en plus poussées.[8]
Cependant, les domaines de la chimie théorique sont très nombreux : chimie quantique, mécanique moléculaire et la dynamique moléculaire.
Chapitre II Méthodes de calculs quanto chimique
11
II.La chimie quantique :
Les méthodes de chimie quantique permettent le calcul de la structure électronique de systèmes tels que les atomes, les molécules neutres, les espèces radicalaires, les ions, les clusters d’atomes, les surfaces de solides, etc.
Des algorithmes de calculs très précis sont utilisés pour minimiser l’énergie totale en fonction des paramètres structuraux et pour prédire la structure la plus stable des composés étudiés.
Les fonctions d’onde ainsi obtenues permettent de calculer des propriétés électroniques et magnétiques, de même qu’elles conduisent à l’obtention d’indices de réactivité ou encore à d’autres caractéristiques. Ces méthodes permettent donc aussi bien l’interprétation de résultats expérimentaux, que la prédiction de propriétés pour lesquelles aucune expérience n’a pu encore fournir d’information.
Dans ce chapitre, il a été choisi de décrire dans le détail la théorie dite de Hartree-Fock et celle de la fonctionnelle de densité parce qu’elles sont à la base des méthodes qui ont été utilisées dans les études qui vont être présentées.
La chimie quantique tient compte de la structure électronique d’un système et consiste en l’utilisation de méthodes basées sur la résolution de l’équation de Schrödinger indépendante du temps. En résolvant l’équation aux valeurs propres et vecteurs propres ĤΨ=EΨ, ou Ĥ est l’hamiltonien non relativiste, E l’énergie totale et Ψ la fonction d’onde du système à ce stade, il sera alors possible de déterminer toutes les informations du système étudié.
II.1.Equation de Schrödinger[9] :
Toute l’information que l’on peut obtenir sur un système constitué d’un ensemble de
particules est contenue dans la fonction d’onde Ψ du système. Pour un système composé de n électrons la fonction d’onde est obtenue en résolvant l’équation de Schrödinger indépendante du temps
ĤΨ=EΨ (1)
Dans cette équation E est l’énergie du système et Ĥ est l’opérateur hamiltonien du système. Ψ est la fonction d’onde du système, fonction des coordonnées des coordonnées des noyaux, des électrons et contient toute l’information du système, E est l’énergie associée à la
Chapitre II Méthodes de calculs quanto chimique
12
distribution électronique correspondante. Les valeurs propres de H sont les valeurs observables de cette énergie et les fonctions d’onde correspondantes sont les fonctions propres associées.
Ψ est écrite sous la forme ;
Ψ (r, R)= χn (R).φe (r , R) (2)
En remplaçant dans l’expression classique de l’énergie totale les grandeurs physiques par les opérateurs qui leurs sont associées en mécaniques quantique ; l’hamiltonien pour un système de N noyaux et n électrons, devient comme suit :
H=TN+ Te+ VeN +Vee +VNN ou (3) TN=∑
𝑃𝑗2 2𝑀𝑗 𝑁
𝑗=1 énergie cinétique du noyau
Te=∑ 𝑃𝑖2 2𝑚𝑖
𝑛
𝑖=1 énergie cinétique des électrons
Ve N= - ∑ ∑ 𝑍𝑗е² 𝑅𝑖𝑗 𝑁 𝑗=1 𝑛
𝑖=1 énergie d’attraction des électrons avec les noyaux
Ve e= + ∑ ∑ е² 𝑟𝑖𝑖′ 𝑛 𝑖′>𝑖 𝑛
𝑖=1 énergie de répulsion des électrons
VNN=∑ ∑ 𝑍𝑗𝑍𝑗′е² 𝑅𝑗𝑗′ 𝑁 𝑗′>𝑗 𝑁
𝑗=1 énergie de répulsion des noyaux
Les variables de ce système : j et j ’ représentent les noyaux et i et i’ les électrons.
Principe de résolution de l’équation de Schrödinger :
Du fait de la complexité de cet hamiltonien Ĥ, à la fois fonction des coordonnées
électroniques et nucléaires. La fonction d’onde totale du système doit comporter à la fois les coordonnées électroniques et nucléaires que nous désignons par r et R.
La résolution de l’équation de Schrödinger pour la recherche de la fonction d’onde totale Ψ(r, R) permettent la description de l’état d’un tel système moléculaire s’avère être une tache extrêmement complexe dans ces conditions. Il est donc nécessaire d’effectuer certaines approximations afin de calculer les fonctions d’ondes approchées du système.
Chapitre II Méthodes de calculs quanto chimique
13
Trois approximation sont le plus souvent utilisées pour calcules la fonction d’onde d’une molécule.
Il s’agit de l’approximation de Born-Oppenheimer, orbitalaire, et de la théorie LCAO (Linear Combination of Atomic Orbitals).
II.2.Approximation de Born-Oppenheimer[10] :
en 1927, Born et Oppenheimer ont proposé de simplifier la résolution de l’équation(1) en séparant la partie électronique de la partie nucléaire dans la fonction d’onde Ψ.
Cette approximation appelée aussi approximation adiabatique est basée sur le fait que les électrons se déplacent beaucoup plus rapidement que les noyaux, ceci est dû à la masse beaucoup plus faible des électrons (environ 1836 fois moindre de celle du proto). Par conséquent, les électrons réagissent quasi instantanément à une modification de la position des noyaux [7], ainsi l’hamiltonien dans l’équation (3) s’écrit :
H=Te +VeN +Vee+VNN (4)
Cela mène à la séparation de l’équation de Schrödinger en deux équations indépendantes : L’équation de Schrödinger relative aux électrons dans le champ des noyaux supposés fixes : H0ψe (ri, Ri)=E0ψ0 (ri, Ri) (5)
ri: les coordonnées de n électrons.
Ri : les coordonnées des noyaux fixes.
L’autre équation décrit le mouvement des noyaux : les vibrations, rotations et translations de la molécule.
L’énergie adiabatique est alors la somme de l’énergie E0 solution de l’équation électronique et
l’énergie de répulsion entre les noyaux.
Eadiabatique=E0+VNN (6)
L’énergie totale du systéme est la somme de l’énergie adiabatique, vibrationnelle, ainsi que l’énergie de rotation et de translation qui correspondent au mouvement nucléaires :
Chapitre II Méthodes de calculs quanto chimique
14
La fonction d’onde du Système, solution de l’équation de Schrödinger dans l’approximation de Born et Oppenheimer, peut donc s’écrire sous la forme d’un produit de deux fonctions :
Ψ(r, R)=χn (R).φe (r, R) (8)
e : réfère à un état électronique
n : réfère à un état vibrationnel
la fonction d’onde totale ψ(r, R ) , solution de l’équation de Schrödinger dans l’approximation de Born et Oppenheimer, peut donc s’écrire sous la forme d’un produit d’une fonction d’onde électronique φe (r, R) et une fonction d’onde nucléaire χn(R).
En tenant compte des mouvements de translation ou des mouvements de vibration des noyaux dans une molécule, l’équation de Schrödinger avec l’hamiltonien donné par l’équation (4) et la fonction d’onde (8) s’écrit :
[{∑ 𝑃𝑖 2 2𝑚𝑖 𝑛 𝑖=1 }-{∑ ∑ 𝑍𝑗е² 𝑅𝑖𝑗 𝑁 𝑗=1 𝑛 𝑖=1 } + {∑ ∑ 𝑟е² 𝑖𝑖′ 𝑛 𝑖′>𝑖 𝑛 𝑖=1 } + {∑ ∑ 𝑍𝑗𝑍𝑗′е² 𝑅𝑗𝑗′ 𝑁 𝑗′>𝑗 𝑁 𝑗=1 }] χn (R).φe (r, R)= εn χn (R). φe (r, R) (9)
Les électrons sont ainsi considérés comme se déplaçant dans un champ moyen créé par des noyaux immobiles, et sont donc sujets à un potentiel nucléaire statique.
L’équation de Schrödinger à n électrons et à N noyaux peut ainsi être séparée en une partie nucléaire et une partie électronique. Puisque la fonction d’onde nucléaire dépend uniquement des coordonnées des noyaux, la fonction d’onde électronique est alors calculée pour une position donnée des noyaux et dépendra de paramètres liés aux coordonnées nucléaires.
II.3. Approximation orbitalaire[11] :
L’approximation orbitalaire introduite par Hartree en 1928 [3], suggère d’écrire la fonction d’onde poly électronique qui dépend des coordonnées d’un seul électron ; c’est
l’approximation de Hartree.
φe (е1 ,е2,…. , еi,….,еn)= φ1(е1 ) φ(е2)…φi(еi)…φn (еn)
Pour un système à n électrons la fonction d’onde poly électronique Ψ la plus simple s’écrira donc sous la forme d’un produit d’orbitales supposées normalisées :
Chapitre II Méthodes de calculs quanto chimique
15
φe (е1, е2, …, еi,…, еn)= Xi=1n φi(еi)
Chacune d’elle satisfait une équation aux valeurs propres séparées. hi (еi) φi (еi) = εi φi (еi)
Où hi est l’opérateur associé à l’énergie d’un électron dans le champ des noyaux.
Les fonctions d’onde mono électroniques φi (еi) sont appelées orbitales moléculaires selon le cas.
L’énergie totale sera la somme des énergies individuelles εi
E ≈ ∑𝑛𝑖 𝜀𝑖
Les valeurs propres εi de hi sont appelées énergies orbitalaires.
Si l’on joint à l’orbitale φi (еi) une fonction de spin α(i) ou β(i) pour le i ème électron, alors on obtient une spin-orbitale φi (еi) α(i) par exemple. Il est important de bien distinguer les orbitales, qui est une fonction d’onde pour un et un seul électron du système de la fonction d’onde de l’atome ou de la molécule en entier, qui est un produit de n orbitales ou spin-orbitales.
II.4. Expression des OM : théorie LCAO :
Si la résolution de l’équation de Schrödinger sous forme analytique directe est simple pour les Cas mono électroniques élémentaires (H, H2+) [12], les solutions exactes de cette équation ne
peuvent être généralisables à des systèmes poly électroniques plus complexes.
Toutes approche théorique dans laquelle les orbitales moléculaires sont développées en termes de combinaisons linéaires des orbitales atomiques est désignée par la méthode LCAO (Linear Combination of Atomic Orbitas).
Principe et règles générales de la méthode LCAO :
Afin d’exposer le principe de la méthode LCAO, on considère l’approche la plus élémentaire pour une molécule diatomique[13] dans cette approche, on écrit les orbitales d’une molécule AB sous la forme suivante :
Chapitre II Méthodes de calculs quanto chimique
16
φ =Caφa+Cbφb (1-3-1)
Où φa et φb sont des orbitales atomiques centrées sur le noyau A et B respectivement sont supposées connues, contrairement aux coefficients inconnus Ca et Cb qui sont déterminés
variationnellement.
L’expose suivant utilise l’hypothèse que les orbitales atomiques φa et φb ainsi que les coefficients Ca et Cb sont choisis réels.
L’application du principe variationnel requière la minimisation de l’énergie totale ε par
rapport aux coefficients du développement et l’on obtient alors les équations : 〈E〉φ Définie par : 〈E〉φ= ∫ φ∗hφ.dr
∫ φ∗φ.dr (1-3-2)
Par rapport à Ca et Cb (le dénominateur, représente la constante de normalisation de la fonction φ)
L’équation (1-3-2) représente dans l’approximation des électrons indépendants, l’énergie mono-électronique moyenne dans l’orbitale d’essais φ.
Avec φ=Caφa+ Cbφb On a : ε=〈𝐸〉φ=Ca2Haa+C2bHbb+2CaCbHab Ca2+Cb2+2CaCbSab (1-3-3) Haa =∫ φa ∗ hφ adr , Hbb=∫ φb ∗ hφbdr (1-3-4) Hab=∫ φa ∗ hφ bdr = ∫ φb ∗ hφadr (1-3-5) Et Sab=∫ φa ∗ φbdr=∫ φb ∗ φadr (1-3-6)
L’intégrale Sab est communément appelée intégrale de recouvrement des orbitales φa et φb.
Notons que cette intégrale n’est pas nulle : deux orbitales atomiques centrées sur deux noyaux différents ne sont pas orthogonales.
Haa , Hbb et Sab sont des intégrales connues puisqu’elles ne dépendent que des fonctions
Chapitre II Méthodes de calculs quanto chimique
17
En minimisant 〈E〉φ par rapport aux coefficients du développement suivant le théorème de Ritz [12], on obtient alors les équations :
Ca(Haa-ε) + Cb(Hab-εSab)=0
Ca(Hab-εSab) + Cb(Hbb-ε)=0 (1-3-7)
La résolution de ce système d’équations linéaires passe par l’annulation d’un déterminant construit sur les équations inconnues(les coefficients Ca et Cb et les εi relatives).
Le système ne possède de solutions non triviales que seulement si
|(𝐻(𝐻𝐻𝐻− 𝐻) (𝐻𝐻𝐻− 𝐻𝐻𝐻𝑏)
𝐻𝐻− 𝐻𝐻𝐻𝐻) (𝐻𝐻𝐻− 𝐻) | =0 (1-3-8)
On obtient à la fin du développement un système d’équations algébriques du second ordre pour l’énergie ε qui possède deux solutions,ε+ et ε…
Une fois que les coefficients sont tirés du système (1-3-7), on obtient deux orbitales moléculaires avec une combinaison linéaire de la forme limitée de (1-3-1).
Le principe de la méthode LCAO a été expliqué pour le cas d’une molécule diatomique, or cette méthode ne se limite pas à un développement limité à deux termes ; et peut donc être généralisée pour donner lieu aux règles suivantes généralisant tout développement LCAO :
Deux orbitales atomiques ne se combinent (ne se mélange) de façon appréciable que si :
1- La combinaison de deux orbitales atomiques ayant des énergies proches exige un maximum de recouvrement de celle-ci.
2- Le mélange de n orbitales atomiques donnes n orbitales moléculaires.
3- La symétrie du système peut éventuellement imposer certaines formes de combinaison linéaire des orbitales atomiques ; ces combinaisons linéaires sont adaptées à la
symétrie. Par exemple, dans le cas d’une molécule diatomique homo nucléaire commeH2+, toutes les orbitales σ1, σ2, etc. Décrites précédemment sont adaptées à la
symétrie de cette molécule.
Face aux complexités de la fonctionnelle d’énergie H de l’ensemble des fonctions d’ondes admissibles on doit simplifier ce problème. Pour cela deux approches sont utilisées :
Chapitre II Méthodes de calculs quanto chimique
18
La première est celle de Hartree-Fock (HF) qui est l’application du principe variationnel à la minimisation de l’énergie et de la simplification de l’espace des fonctions d’ondes construites sur la base d’un déterminant de Slater.
Quand aux deuxièmes méthodes, elle consiste à simplifier la fonctionnelle d’énergie du système en utilisant la théorie de la fonctionnelle de densité(DFT).
II.5. Méthode Hartree-Fock(HF) :
L’utilisation de l’équation de Schrödinger, basée sur l’hamiltonien Ĥ et la fonction d’onde Ψ, nous permet de trouver l’énergie effective électronique en utilisant la méthode des variations.[14]
Le problème consiste dés lors à recherche la meilleure fonction d’onde en minimisant l’énergie effective électronique par rapport aux paramètres de la fonction d’onde. Fock et Slater ont utilisé cette idée en développant de façon simultanée et indépendante ce qui est connue comme les équations de Hartree-Fock.
Soit un système à couche fermée « Closed shells », c'est-à-dire sans électron célibataire, le système d’équation de Hartree-Fock va se simplifier sous la forme qui suit qui ne tiendra compte que des orbitales spatiales φ.
Fi(1) φi(1)= εiφi(1) (II-1)
εi est l’énergie de l’orbitale I et Fi est l’opérateur de Fock donné par :
Fi = h(1)+∑ [2𝐻𝐻 𝐻(1) − 𝐻𝐻(1)] (II-2)
Dans l’équation (2), le Terme h(1) est l’opérateur pour un électron H(1)= -1 2⁄ ∇² − ∑ Zα
ηα N
α=1 (II-3)
H(1) est l’opérateur qui prend en compte l’énergie cinétique de l’électron 1 et son énergie potentielle d’interaction avec le noyau α.
Le terme Ji est l’opérateur d’échange auquel correspond l’intégrale d’échange suivante :
Jij=∫ φi∗(1) φj∗(2)1
Chapitre II Méthodes de calculs quanto chimique
19
Kij est l’opérateur d’échange auquel correspond l’intégrale d’échange suivante:
Kij =∫ φi∗(1) φj∗(2)1
rijφj(1)φi(2).dτ12 (II-5)
Les intégrales Jij et Kij sont respectivement appelées intégrales de coulomb et intégrales
d’échange ; l’intégrale de coulomb a un équivalent en mécanique classique, alors que l’intégrale d’échange provient de la nécessité d’antis symétriser la fonction d’onde. Les intégrales de coulomb et d’échange décrivent les interactions entre électrons. Jij
Représente l’interaction coulombienne moyenne entre deux électrons situés dans les orbitales φi etφj, sans tenir compte de leur spin.
L’intégrale d’échange Kij réduit l’interaction coulombienne entre deux électrons situés dans les orbitales φi et φj ayant des spins parallèles. Ce terme est une conséquence directe du principe de Pauli et conduit à une valeur d’énergie Ee plus basse, donc à une
stabilisation.
Par l’intermédiaire de l’intégrale d’échange on introduit ainsi une corrélation électronique entre électrons ayant des spins parallèles, c’est-à-dire que ces deux électrons ne peuvent pas se mouvoir indépendamment l’un de l’autre.
L’énergie électronique totale est obtenue par la sommation sur toutes les orbitales occupées et s’écrit dans l’hypothèse de couches fermées.
E= 2∑nj=1Ij+∑Ni=1∑ (2Jnj=1 ij− Kij) (II-6) Ou Ij= ∫ φj(1) (−12∇j2−
Z
rj) φj(1)dτ12 (II-7)
Les équations de Hartree-Fock sont un jeu d’équation intégro-différentielles couplées, et ne peuvent être résolues que par processus itératif. Le couplage se constate par le fait que les intégrales Jij et Kij sont définies en fonction des orbitales φi et φj.
Ce qui veut dire que pour déterminer l’opérateur de Hartree F(1) dans (1) on a besoin de connaître les autres orbitalesφj.
Chapitre II Méthodes de calculs quanto chimique
20
L’existence de plusieurs méthodes de calculs quanto chimique, est due aux divers niveaux d’approximations pour le calcul des intégrales d’interactions biélectroniques. Parmi ces méthodes on citera brièvement trois grandes classes.
-les méthodes semi-empiriques
-la méthode ab-initio
-la méthode de la fonctionnelle de densité(DFT).
II.6.Méthodes Semi-empiriques :
L’ensemble de ces méthodes utilise le formalisme Hartree-Fock dans le cadre de la théorie LCAO. Elles conduisent en conséquence aux équations de Roothaan. Parmi ces méthodes, les plus connues sont dénommées : CNDO, INDO, MINDO et MNDOO. Elles ont toutes en commun un point essentiel qui est de négliger un certain nombre de termes
biélectronique contenus dans l’opérateur de Fock, ces termes (intégrales) sont négligés, simplifiés ou approchés en corrélant les résultats à des données expérimentales. Cette approximation est dite d’annulation, partielle ou totale du recouvrement diatomique différentiel [18].
CNDO [19], [20] Complete Neglect of Differentiel Overlap, comme son nom l’indique,
toutes les intégral possèd ent un recouvrement differential sont négligés.
INDO [21], [22]Neglect of Differential Diatomic Overlap ; ou l’intégral d’échange est
conservée.
MINDO [23], [24],[25] Minimum Neglect of Differential Overlap ; la para métrisation est
faite de façon à reproduire les enthalpies de formation expérimentales, mais elle ne tiend pas compte du recouvrement différentiel mono centrique.
MNDO [26] corrige ce défaut en ne négligeant que le recouvrement différentiel
diatomique, cependant elle présente un défaut au niveau de la reproduction de la liaison hydrogène, une autre méthode de calcul à été développée dans laquelle toutes ces erreurs sont corrigées ; c’est la méthode AMI [27],[28](Austin Model 1).
D’autres méthodes semi-empiriques existent telles que : SINDOI, INDO/S, CNDO/S (paramètre pour des spectres électroniques) et la ZINDOR.
Chapitre II Méthodes de calculs quanto chimique
21
Les méthodes semi-empiriques offrent la possibilité de traiter des systèmes relativement gros et un temps de calcul moindre comparé avec celui de la méthode ab-initio.
II.7.Méthode ab initio :
Les méthodes de chimie quantique peuvent être classées sur la base du traitement des intégrales de répulsion inter électronique, cependant le calcul des intégrales dépend considérablement des fonctions utilisées. De toute évidence, les meilleurs fonctions qu’on puisse utiliser sont les orbitales atomiques de Hartree-fock. Ces fonctions sont
malheureusement difficiles à manipuler. Toutes, il est possible d’utiliser les orbitales atomiques de Slater : des fonctions analytiques du type :
φsf =N
n(r a⁄ )0 n−1 exp(− ξr a⁄ 0)
Où ξ : une constante déterminant la taille de l’orbitale.
Bien que les fonctions de Slater présentent l’avantage de décrire raisonnablement les orbitales atomiques, elles sont peu commodes d’utilisation pour les calculs des intégrales. Pour contourner ce problème, chaque orbitale atomique de Slater est approchés par plusieurs fonctions gaussiennes de type :
φgf = N
n(α) 𝐻𝐻−1exp (−𝐻𝐻2)
Où α : une constante déterminant l’expression radiale de la fonction
C’est ainsi que la base minimale STO-NG a été définie, N est le nombre des fonctions gaussiennes.
Les bases gaussiennes ont, par contre, une assez mauvaise représentation des orbitales atomiques de Hartree-Fock à l’origine (dérivée non nulle) ni aux grandes distances (décroissance trop rapide avec r). Pour compenser la représentation incomplète des orbitales atomiques des fonctions gaussiennes, on utilise donc des fonctions bi
exponentielles. La base construite sur ce tupe de fonction est dite base Double Zeta (DZ).
Afin de simplifier le calcul des intégrales bi électroniques, on développe les essentielles е−ξr sur une base de fonction gaussienne de la forme е−ξr2.
Qui sont des fonctions largement répondues en chimie quantique, car le produit de deux gaussiennes centrées en deux points différents peut s’écrire à l’aide d’une seule
Chapitre II Méthodes de calculs quanto chimique
22
gaussienne centré en un point situé sur le segment de ces deux points. Ainsi, toutes les intégrales électroniques se ramènent à une somme d’intégrales mono centriques qui se calculent aisément. On utilise souvent une base où chaque orbitale de Slater (STO) est développée en utilisant trois gaussiennes d’où sa dénomination de STO-3G appelée base minimale.
Il existe bon nombre de base de gaussiens possibles. Les plus communément utilisées sont celles qui ont été développées par Pople et collaborateurs [18]. Le niveau suivant développé par Pople comprend les bases split-valence telles que 3-21G, 4-31G et 6-31G, où le premier chiffre représente le nombre de gaussiennes utilisées pour représenter les orbitales de
coeur.les orbitales de valences y sont représentées par deux fonctions qui sont composées du nombre de gaussiennes dans la seconde partie de la dénomination de la base. Ainsi la base 6-31G du carbone, par exemple, utilisera six gaussiennes pour représenter l’orbitale 1s, trois gaussiennes pour l’orbitale 2s et 1 gaussienne pour représenter les orbitales 2p.
Pour une plus grande flexibilité on peut encore rajouter des fonctions de polarisation. La
dénomination la plus ancienne est l’ajout d’un astérisque sur la base en question (par exemple 6-31G∗), et dans une désignation plus récente, le caractère de la fonction ajoutée est
explicitement donné : 6-31G(d). la base 6-31G∗ ou 6-31G(d) signifie ainsi qu’un jeu de fonctions d a été ajouté à tous les atomes (sauf H) dans la molécule, alors que 6-31G∗∗ ou 6-31G(p, d) signifie qu’un jeu de fonction p a été ajouté aux hydrogènes et que des fonctions d ont été ajoutées aux autres atomes. Ces fonctions sont généralement indispensables.
Quant aux fonctions diffuses, celles-ci sont utile pour décrire des régions très éloignées du noyau, on ajoute un jeu de fonctions (généralement p ou d) avec un faible exposant α. Elles sont par exemple importantes pour décrire les interactions faibles inter moléculaires (ex : liaisons hydrogènes entre deux molécules).
L’utilisation des calculs ab initio nous permet d’augmenter la qualité des résultats en augmentant le niveau des calculs ou la qualité de la base de fonctions. La seule limitation dans le cas ab initio étant naturellement liée au temps requis pour effectuer de tels calculs.
Chapitre II Méthodes de calculs quanto chimique
23
II.7. Méthode de la fonctionnelle de la densité :
La théorie de la fonctionnelle de la densité, ou DFT, est basée sur le théorème de Hohenberg-Kohn[14] qui établit que l’énergie d’un système de n électrons dans son étant fondamental est une fonctionnelle de la densité électronique au point de l’espace r de ce système, ρ (R).
ρ (R)=φ∗(r)φ(r) (V-1)
Ainsi, contrairement aux méthodes décrites précédemment, la théorie de la fonctionnelle de la densité ne consiste pas à chercher une fonction d’onde complexe, Ψ, à 3 N dimensions
décrivant le système à étudier, mais plutôt une simple fonction à trois dimensions : la densité électronique totale ρ[21,22,23]
Chapitre III Calculs et Interprétation des résultats
24
III.1.Introduction :
La chimie numérique ou chimie informatique, parfois aussi chimie computationnelle, est une branche de la chimie qui utilise les lois de la chimie théorique exploitées dans des
programmes informatiques spécifiques afin de calculer structures et propriétés des molécules, les solides, les agrégats atomiques (ou clusters), les surfaces, etc…
En appliquant autant que possible ces programmes à des problèmes chimiques réels. La frontière entre la simulation effectuée et le système réel est définie par le niveau de précision requis et/ou la complexité des systèmes étudiés et les théories employées lors de la
modélisation. Les propriétés recherchées peuvent être la structure (géométrie, relations entre constituants), l'énergie totale, l'énergie d'interaction, les charges, dipôles et moments
multipolaires, fréquences vibrationnelles, réactivité ou autres quantités spectroscopiques, etc. Le domaine le plus représenté de la discipline est le traitement des configurations
électroniques des systèmes.
L'expression chimie numérique est parfois également utilisée pour désigner tous les champs scientifiques qui recouvrent à la fois la chimie et l'informatique.
Les codes utilisés en chimie numérique moléculaire sont basés sur de nombreuses et différentes méthodes de chimie quantique qui permettent la résolution de l'équation de Schrödinger associée à l’hamiltonien moléculaire. Les méthodes qui n'incluent aucun paramètre empirique ou semi-empirique dans leurs équations, c'est-à-dire qui dérivent
directement des principes théoriques, sans inclusion de données expérimentales, sont appelées méthodes ab initio. Cela n'implique pas que la solution obtenue est la solution exacte; elles consistent toutes en des approximations à divers degrés des calculs de mécanique quantique. Cela signifie qu'une approximation particulière est définie de manière rigoureuse sur les premiers principes (théorie quantique) puis résolue avec une marge d'erreur qui est connue de manière qualitative à l'avance. Si des méthodes par itérations numériques sont utilisées, le but est d'itérer jusqu'à atteindre la précision machine.
Dans notre travail nous avons utilisé 3 méthodes ab-initio différentes puis nous avons comparé les résultats avec ceux de l’expérience, les méthodes utilisées sont :
1-La méthode HF : La méthode de Hartree-Fock est une méthode de résolution approchée de
Chapitre III Calculs et Interprétation des résultats
25
comme un champ moyen. C'est à dire que chaque électron ressent l'effet des autres électrons de façon moyenne (comme si les autres électrons formaient un nuage homogène d'électrons).
On peut alors choisir différentes fonctions mathématiques pour décrire les orbitales (STO 3G, 3-21G, 6-31G, 6-31G(d,p), 6-311++G(2d,p)).
L'ensemble de ces fonctions constitue la base.
2-La méthode DFT : Les méthodes post-HF étant très coûteuses en temps de calcul, les
méthodes DFT (DensityFunctionalTheory) ont été développées pour prendre en compte ces effets.
Les méthodes DFT sont relativement récentes mais sont de plus en plus utilisées, notamment en biologie. Elles sont moins coûteuses en temps de calcul que les méthodes post-HF et peuvent apporter une très bonne fiabilité pour certains systèmes moléculaires.
3-La méthode MP2 : La théorie de la perturbation de Møller-Plesset (MP) est une des
nombreuses méthodes post-Hartree-Fock ab initio en chimie quantique appliquée dans le cadre de la chimie numérique. Elle améliore la méthode de Hartree-Fock en y apportant les effets de corrélation électronique au moyen de la théorie de la perturbation de Rayleigh-Schrödinger (RS-PT) au deuxième (MP2), troisième (MP3) ou quatrième (MP4) ordre habituellement.
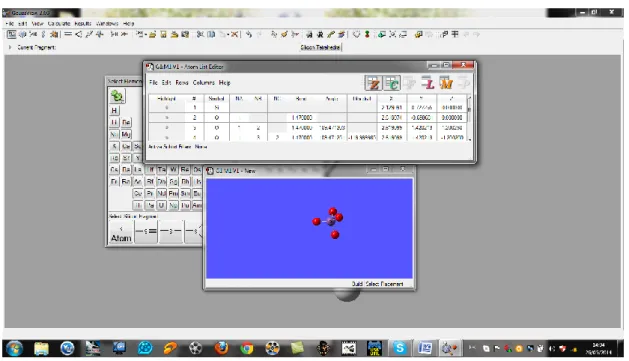
III.2. Interprétations des résultats III.2.1.Logiciel :
Dans notre travail nous avons utilisés un logiciel pour calculer l’énergie d’optimisation de la molécule SiO4 ainsi que les fréquences IR à partir de trois méthodes :
Chapitre III Calculs et Interprétation des résultats
26
Figure 10 :Création de la molécule
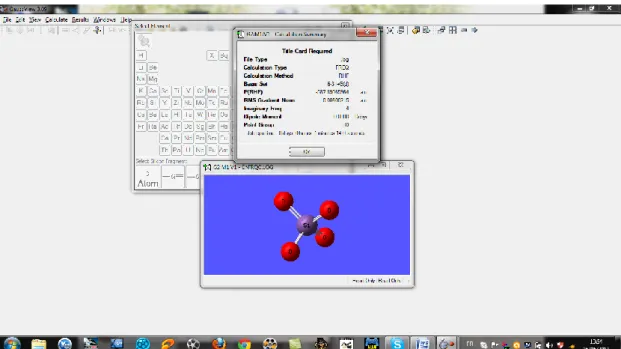
On utilise quelques précisions sur le choix des bases et leurs significations, pour les fonctions de polarisations :
6-31G(d) ou 6-31G* c’est une base avec des fonctions de polarisations ajoutées aux atomes lourds.
6-31G+(d) est une base 6-31G(d) avec des fonctions diffuses ajoutés aux atomes lourds.
Après les calculs on obtient un résultat contenant l’énergie d’optimisation, la distance des liaisons, l’angle et les fréquences IR ainsi que les spectres.
III.2.2.Rappels sur la définition de fréquence :
La fréquence représente le nombre de fois que se reproduit un processus à l'identique dans un temps donné. Le temps que mets le processus pour se réaliser une fois est appelé période. Ainsi les systèmes qui suivent un processus répété régulièrement ont une fréquence exprimée en Hertz (Hz) ou en nombre de période par seconde.
III.2.3.Rappels sur la Spectroscopie infrarouge :
Quand on soumet une molécule à une radiation infrarouge, la structure moléculaire se met à vibrer. Ceci à pour effet de modifier les distance interatomique (vibration de valence ou d’élongation) ainsi que les angles de valence(vibrations de déformation).lorsque la longueur d’onde (l’énergie)apportée par le faisceau lumineux est voisine de l’énergie de vibration de la
Chapitre III Calculs et Interprétation des résultats
27
molécule ,cette dernière va absorber le rayonnement et on enregistre une diminution de l’intensité réfléchie ou transmise. le domaine infrarouge est compris entre 4000cm-1 et
400 cm-1 correspond au domaine d’énergie de vibration des molécules.
La spectroscopie infrarouge est fréquemment utilisée pour la caractérisation des silicates, et plus encore dans l’étude des transformations qui s’y rapportant. [29]
III.3.Démarche du travail :
Notre travail c’est porté sur la molécule SiO4en utilisant trois méthodes :HF,DFT et
MP2.Avec une base (6-31G , + , *), on a effectué une optimisation de la molécule suivie d’un calcul de fréquence .
Chapitre III Calculs et Interprétation des résultats
28
Figure 12:représente les bases de calculs.
Les résultats des calculs s’affichent sur une autre fenêtre :
Figure 13 : représente le calcul en cours.
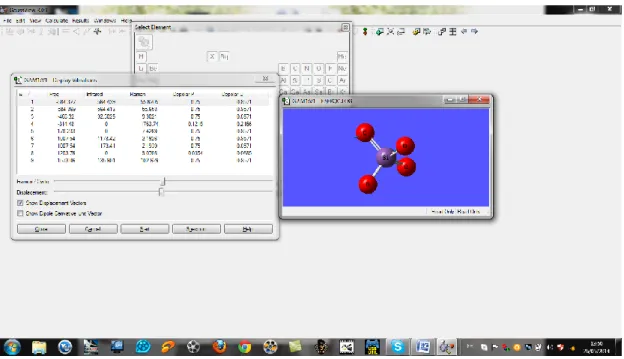
A la fin on obtient des résultats concernant l’énergie, les charges et les fréquences IR notés sur les figures suivantes :
Chapitre III Calculs et Interprétation des résultats
29
Figure 14: représentant les résultats obtenus.
Chapitre III Calculs et Interprétation des résultats
30
Figure 16:représentant les fréquences de vibration de la molécule SiO4.
Chapitre III Calculs et Interprétation des résultats
31
III.4.Résultats et discussion:
III.4.1.la méthode d’Hartree Fock (HF):
Nous avons procédé à l’optimisation de notre molécule nous avons trouvé l’énergie suivante -587.47491060 a.u.
Par la suite nous avons procédé au calcul des fréquences IR, les résultats sont notés ci-dessous :
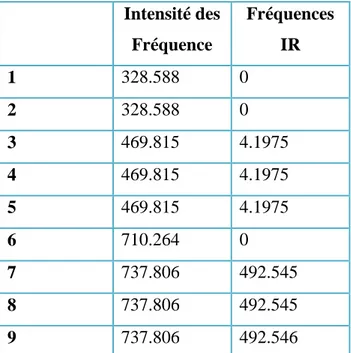
Tableau 1: représente les fréquences de la molécule optimisée pour la méthode HF
Figure 18: represente le spectre d’IR de la méthodes HF 0 200 400 600 800 1000 1200 1400 0 200 400 600 800 1000 inte ns it é fréquence
spectre IR de HF
Intensité des Fréquence Fréquences IR 1 328.588 0 2 328.588 0 3 469.815 4.1975 4 469.815 4.1975 5 469.815 4.1975 6 710.264 0 7 737.806 492.545 8 737.806 492.545 9 737.806 492.546Chapitre III Calculs et Interprétation des résultats
32
Les charges des différents atomes de la molécule SiO4 sont mentionnées ci- après :
Figure 19:represente les charges des atomes de SiO4. III.4.2.La méthodes de MP2 :
Nous avons procédé à l’optimisation de notre molécule nous avons trouvé l’énergie
d’optimisation suivante -589.03787946 a.u ; par la suite nous avons procédé à un calcul de fréquences sur cette dernière les résultats sont mentionnés ci-après :
Tableau 2: représente les fréquences de la molécule optimisée pour la méthode MP2. Intensité des Fréquence Fréquences IR 1 126.872 0 2 290.48 64.658 3 290.723 64.6862 4 489.727 0.0054 5 577.87 43.7908 6 810.372 0.0005 7 890.46 81.0592 8 892.091 81.5299 9 1224.07 85.587
Chapitre III Calculs et Interprétation des résultats
33
Figure 20: represente le spectre d’IR de la méthodes MP2
Les charges des différents atomes de la molécule SiO4 sont mentionnées ci- après :
Figure 21:représente les charges des atomes pour la molécule SiO4. 0 20 40 60 80 100 120 140 160 180 0 200 400 600 800 1000 1200 1400 1600 in te n sité fréuence
spectre IR de MP2
Chapitre III Calculs et Interprétation des résultats
34
III.4.3.La méthodes de DFT :
Nous avons procédé à l’optimisation de notre molécule nous avons trouvé l’énergie
d’optimisation suivante -587.91663534 a.u ; par la suite nous avons effectué un calcul de fréquence sur cette dernière, les résultats sont les suivants :
Tableau 3: représente les fréquences de la molécule optimisée pour la méthode DFT.
Figure 22: représente le spectre IR pour la méthode la DFT 0 20 40 60 80 100 120 140 160 180 0 200 400 600 800 1000 1200 1400 1600 in ten si té fréquence
spectre IR de DFT
Intensité des fréquence Fréquences IR 1 126.537 0.0001 2 277.393 52.2135 3 277.443 52.2059 4 533.014 0 5 664.097 47.1136 6 847.573 26.3864 7 848 26.3621 8 877.242 0.0002 9 1261.5 166.903Chapitre III Calculs et Interprétation des résultats
35
Les charges des différents atomes de la molécule SiO4 sont mentionnées ci- après :
Chapitre III Calculs et Interprétation des résultats
36
III-5-Interprétation :
III.5.1.comparaison entre les différentes méthodes et les résultats expérimentaux: Méthode HF Méthode MP2 Méthode DFT Résultats expériment aux [2,3]
vibration antisymétrique d’élongation Si-O Vibration symétrique d’élongation Si-O Vibration symetrique de l’angle de liaison entre O-Si-O
Vibration antisymetrique de l’angle de liaison entre O-Si-O
737,806 710,264 328,558 469.815 891 810,285 577,741 290.676 1085,54 877,242 664,097 277.393 1080-1175 (cm-1) 780-800 695 464
Tableau 4: représentant les résultats de fréquence de vibration de déférentes méthodes
III.5.2. comparaison des distances atomiques entre les différentes méthodes et les résultats expérimentaux:
Liaison (Si-O)
Distance théorique Distance expérimentaux HF DFT MP2 Si-O(1) Si-O(2) Si-O(3) Si-O(4) 1.704941 1.704941 1.704941 1.704941 1.631559 1.631733 1.631794 1.631658 1.646895 1.646980 1.646950 1.647177 1.6-1.63A°
pages
Figure 1 :Ion orthosilicate tétraédrique………2
Figure 2 : Classification des silicates………..….4
Figure 3 : nésosilicate……….………….4 Figure 4 :sorosilicate………5 Figure 5 :inosilicate………..5 Figure 6 :phyllosilicate………...6 Figure 7 :cyclosilicate………...…………...…7 Figure 8 :tectosilicate………...………...7
Figure 9: Diagramme de phase de SiO2…...8
Figure 10 : Création de la molécule………....26
Figure 11:représente la fenêtre du logiciel pour modifier et lancer le calcul…………..27
Figure 12:représente les bases de calculs………....28
Figure 13 : représente le calcul en cours………...28
Figure 14: représentant les résultats obtenus………...29
Figure 15: représentant la charge de la molécule SiO4………...29
Figure 16:représentant les fréquences de vibration de la molécule SiO4…………...30
Figure 17: représentant le spectre IR………...…...30
Figure 18: represente le spectre d’IR de la méthodes HF………..…...31
Figure 19:represente les charges des atomes de SiO4………..……..32
Figure 20: represente le spectre d’IR de la méthodes MP2………..………..33
Figure 21:représente les charges des atomes pour la molécule SiO4………..………...33
Figure 22: représente le spectre IR pour la méthode la DFT………...34
pages
Tableau 1: représente les fréquences de la molécule optimisée pour la méthode HF……..31 Tableau 2: représente les fréquences de la molécule optimisée pour la méthode MP2…...32 Tableau 3: représente les fréquences de la molécule optimisée pour la méthode DFT……34
Conclusion
37
:
A partir des tableaux nous pouvons conclure que les valeurs des fréquences de la molécule SiO4 optimisées et les distances interatomiques que nous avons trouvés
pour la méthode DFT se rapprochent de ceux de l’expérience donc nous pouvons dire que cette méthode donne de très bons résultats vu qu’ils sont en accord avec ceux de l’expérience.
Ce qui nous confirme le fait que la méthode de la DFT est une méthode performante et très fiable.
Le choix de la base a joué un rôle déterminant dans la représentation fidèle des propriétés structurales.
Au final nous pouvons conclure que la simulation et l’utilisation de la méthode de la DFT nous a permis d’obtenir des résultats très satisfaisants.
38
[1] http://jcrystal.com/steffenweber/gallery/StructureTypes/st1.html
[2]Ramakrishnan M and Vaidyanadhan R 2008 Geology of India(Bangalore: Geological Society of India) p 556
[3]Saikia B J, Parthasarathy G, Sarmah N C and Baruah G D 2008 Bull. Mater. Sci. 31 155.
[4] M. J. S. Dewar, E. G. Zoebisch, E. F. Healy, J. J. P. Stewart, J. Am. Chem. Soc, 107, 3902-3909. 1985.
[5] R.L. Mozzi, B.E.Warren, The Structure of Vitreous silica, J. Appl. Cryst.,vol.2, p.164, 1969 .
[6] Doucet, J.P. ; Weber, J. Compture-aided molecular design : Theory and application, Academic Press, London, 266, 1996.
[7] F .Jansen, "Introduction to Computational Chemistry", John Wiley and sons,Inc.,New York. 1999.
[8] Rivail, J.L. Eléments de chimie quantique à l’usage des chimistes, Deuxième édition, Savoirs Actuels, I nter-Edition/CNRS Edition, 1994.
[9] E. Schrodinger, Ann. Phys, 79,361, 1926.
[10] M. Born,J .R. Oppenheimer . Ann. Physiks. 84. 457, (1927).
[11] Y. Jean F. Volatron, « les orbitales moléculaires en chimie ».49.80(1991). [12] O. Burrau, Klg. Danske,Videnskab. Selekab. 7.1(1927).
[13]C.Cohen-Tanoudji, B.Diu , F.Laloe, Mécanique quantique. Hermann Ed.p1374(1973).
[14] M .J.S.Dewar the molecular Orbital theory of Organic Chemistry.Mc .Graw Hill.Ed. P36(1969).
[15] J.C.Slater, « The Self Consistent Field for Molecules and Solids » Mc Graw Hill.N.Y.1974.
[16] V. Minkine,B .Simkine,R.Miniaev, « Théorie de la structure Moleculaire »Ed Mir,Moscou .
[17] C.C.J.Roothaan, Rev.Mod. Phys.23.69(1951).
[18] J.A.Pople,D. L. Beveredge, Approximate Molecular Orbital Theory Mc . Graw Hill.New .York .N.Y 1970.
39
[19] J.A. Pople, G.A.Segal, J. Chem. Phys, 43, S136 , 1965;44,389, 1996. [20] J.A. Pople, D.L Beveredge, P.A. Dobosh,J. Chem. Phys .47.2026. 1967. [21] M.J.S. Dewar, D.M.Stroch,J .Am. Chem. Soc , 107, 3898-3902. 1985. [22] M.J.S. Dewar, W. Thiel,Ibid,99,4899. 1977.
[23] N.C. Baird, M. J. S. Dewar,J. Chem .Phys , 50, 1262. 1969. [24] M.J.S. Dewar, E. Haselbach, J. Am. Chem. Soc , 92, 590. 1970.
[25] R.C. Bingham, M.J.S.Dewar, D.H. Lo, J. Am. Chem. Soc, 97, 1285, 1975. [26] M.J.S. Dewar,W. Thiel,J . Am. Chem. Soc . 99.4899.4907. 1977.
[27] J.A.Pople,D.P.Santry,G.A.Segal,J . Chem . Phys , 43, S129.1965.
[28] Dr. Walt Volland, Bellevue Community College all rights reserved, Bellevue, Washington, 1999.
![Figure 9: Diagramme de phase de SiO 2 [1]](https://thumb-eu.123doks.com/thumbv2/123doknet/13143941.388823/12.892.85.733.193.646/figure-diagramme-de-phase-de-sio.webp)