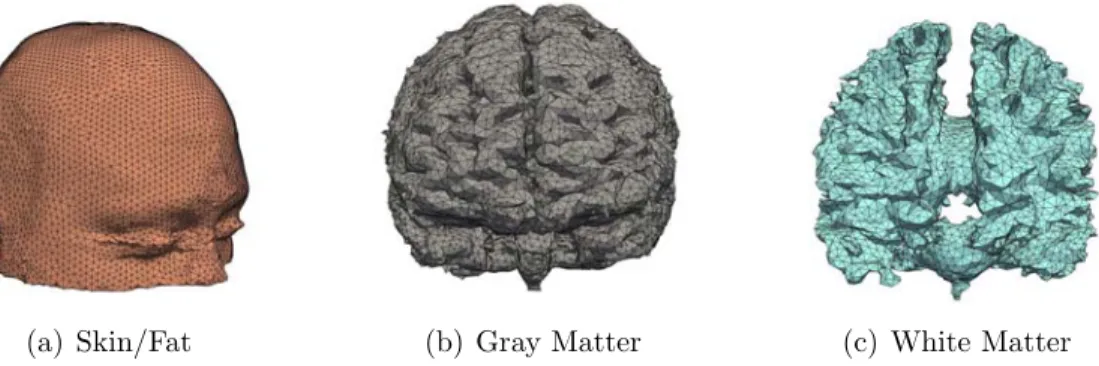
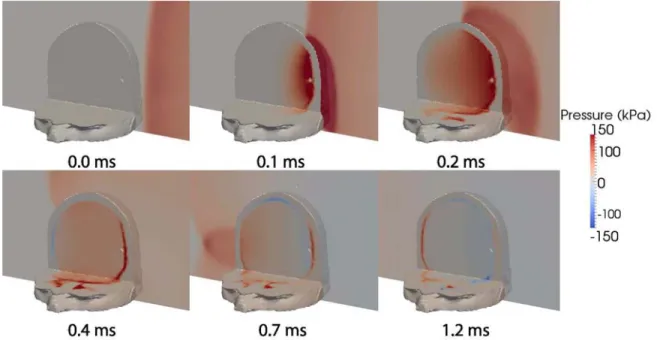
Computational modeling of primary blast effects on the human brain
Texte intégral
Figure






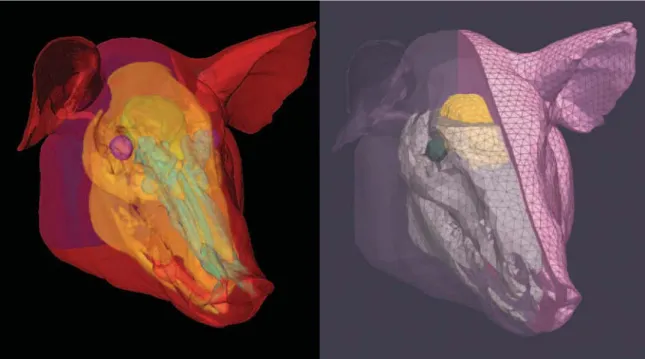
![Figure 4-1: Schematic of the experimental setup (source: [118]). The porcine subjects were positioned such that the shock wave from the shock tube impinged on the right temporal region of the head.](https://thumb-eu.123doks.com/thumbv2/123doknet/14438598.516507/92.918.177.735.111.329/figure-schematic-experimental-porcine-subjects-positioned-impinged-temporal.webp)
Documents relatifs
The thermocouples were located a t the top and bottom surfaces of the insu- lation (inside the polyethylene) above the center of the heat flux transducer.. 2-Hoq/cross srcriotrs
Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of
To assess the effect of REBEL RELIEVER unloading knee brace in conservative treatment of knee osteoarthritis, a randomized controlled trial was conducted in 67 patients
Lead was present both as a small signal in the mass spectra of many particles and as a large signal in a few mass spectra. Figure 4 shows some mass spectra of particles with Pb. 4c),
Er ist respektvoll aber kann auch Klagen ausdrücken: im vierten Buch der Aeneis, wenn Iarbas sich über Aeneas beklagt, stellt er schon Jupiters Macht in Frage 29?.
In analyses restricted to controls only or using concentrations not standardized for sampling conditions, findings concerning phthalates were consistent with those obtained in
Diversity and genetics of nitrogen-induced susceptibility to the blast fungus in rice and wheat.. Increase of Fungal Pathogenicity and Role of Plant Glutamine in
ACE1 / Pi33 : our model system for specific interactions Pi33 pi33 ACE1 ace1 Plant Pathogen. Fudal
