Croissance et étude des propriétés des nanostructures de cobalt obtenues par électrodéposition
Texte intégral
Figure
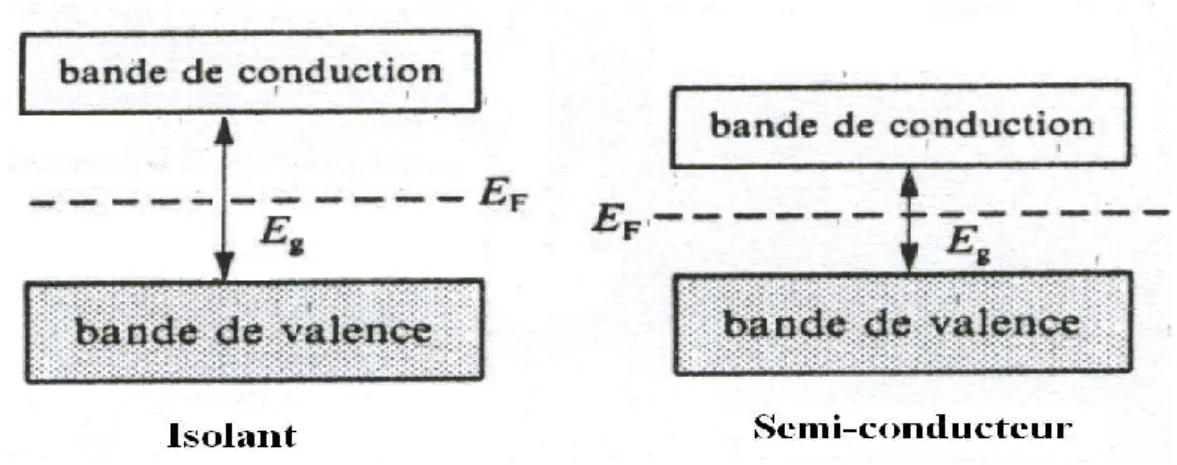
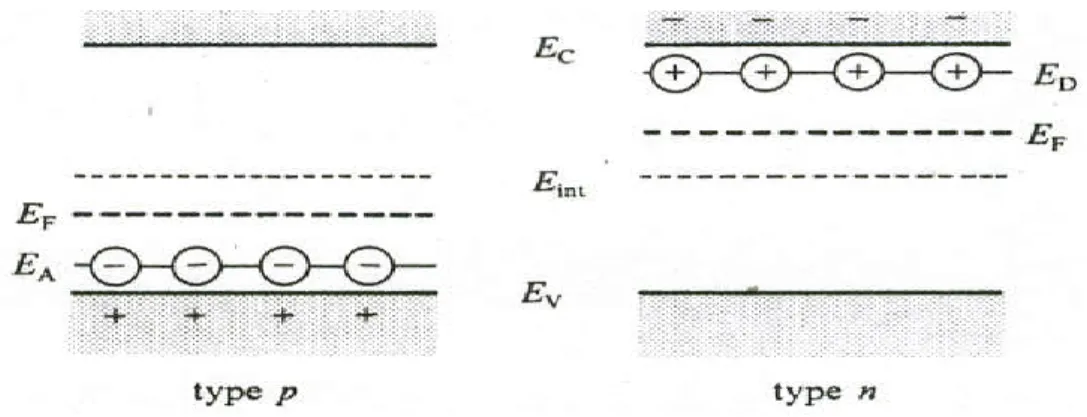

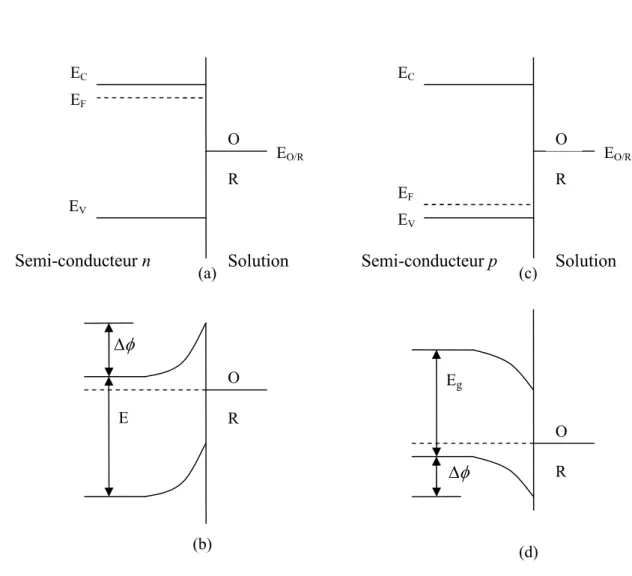

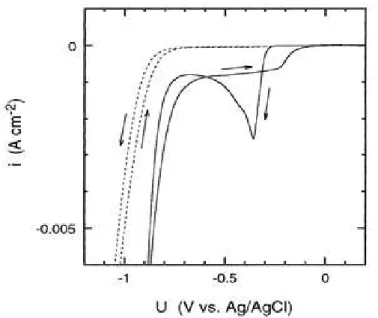
![Figure 1.9: Représentation schématique des différentes étapes pouvant intervenir au cours d’une réaction électrochimique [17]](https://thumb-eu.123doks.com/thumbv2/123doknet/3404861.98629/27.892.199.730.290.527/figure-représentation-schématique-étapes-pouvant-intervenir-réaction-électrochimique.webp)


Documents relatifs
déformation optimale dans le canal est obtenue pour 0. L’originalité de cette étude par rapport à celles de la littérature est la mesure de la déformation en
Le moment magnétique de l’atome de phosphore s’oriente dans la direction opposée à celle des moments magnétiques des atomes de cobalt et avec une faible magnitude de -0.04 µ
The first mechanism of catalyst deactivation is relevant to carbon deposition on the most active cobalt sites presumably located on the steps and edges of cobalt nanoparticles
généralisé la méthode classique de Neumann-Fredholm qui consiste à représenter la solution sous forme d'un potentiel de double couche et à ramener le problème à la résolution
De ce fait, le microscope à force atomique nous a permis d'étudier la morphologie (aspect qualitatif) et la topographie (aspect quantitatif) de surface descouches de nickel
Les alliages à base de cobalt sont largement utilisés pour la fabrication de divers dispositifs soit implantés dans le corps par la chirurgie (leurs applications y compris la
Le présent travail consiste en l'élaboration et la caractérisation des couches minces de ZnO non dopées et dopées par différentes concentrations de cobalt (Co) (0.5, 1,
Dans cette approche, un espace de formes d'ordre réduit des courbes d'indentation et des empreintes issues d'un ensemble de simulations numériques, appelées
