T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Physiopathologie
JURY
Robert SALVAYRE, directeur de thèse Gervaise LOIRAND, rapporteur
Georges NEMOZ, rapporteur Ziad MALLAT, président du jury Nathalie AUGE, co-directeur de thèse Franck PEIRETTI, co-directeur de thèse
Ecole doctorale : Biologie-Santé-Biotechnologies Unité de recherche : U 858, département 3, équipe 10
Directeur(s) de Thèse : Robert SALVAYRE Rapporteurs : Gervaise LOIRANT et Georges NEMOZ
Présentée et soutenue par Edwige TELLIER Le 9 juillet 2008
Titre : Le TNF alpha : maturation par ADAM-17 et activation d’une voie de signalisation
UFR SCIENCES DE LA VIE ET DE LA TERRE
THESE
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III
Discipline : physiopathologie Présentée et soutenue par
EDWIGE TELLIER
Le 9 juillet 2008
Le TNF : maturation par ADAM-17 et activation d’une voie de
signalisation sphingomyéline/céramide
Directeur de thèse : Pr. ROBERT SALVAYRE
Devant le jury JURY : Rapporteurs : Gervaise Loirand
Georges Némoz Membres : Nathalie Augé
Franck Peiretti Robert Salvayre Ziad Mallat
soutenir, me supporter, m’aimer…
A vous, mes très chers parents, sans qui je ne serais pas arrivée jusque là ; j’ai énormément de chance de vous avoir.
A toi, Martine, ton aide et ta générosité ont été plus que précieuses tout au long de ces années.
A toi, Nico, qui partage ma vie, pour tout ce que tu es et tout ce que tu m’offres, merci d’être toujours là et de me rendre si heureuse. Te rencontrer juste avant le début de cette thèse a été la plus belle chose qui me soit arrivée avec cette famille que nous avons fondée.
A vous, Catherine Muller, grâce à qui j’ai réalisé cette thèse, vous m’avez donné confiance en moi et donné le goût de la recherche et je vous en serai toujours reconnaissante.
Je remercie en premier lieu le Pr. Robert Salvayre, mon directeur de thèse, qui m’a acceptéeau sein de son équipe alors qu’il ne me connaissait pas, et qui a également accepté que j’effectue ce travail en collaboration avec l’U626. Merci de la confiance que vous m’avez accordée. Ces quatre années passées pour moitié dans votre laboratoire ont été un plaisir pour moi, et vos conseils et discussions m’ont beaucoup apporté.
Merci également au Pr. Irène Juhan-Vague qui dirigeait l’U626 à Marseille lors du début de ce travail ainsi qu’au Pr. Marie-Christine Alessi d’avoir accepté que je travaille au sein de leur unité alors que je venais de Toulouse et qu’elles non plus ne me connaissaient pas. Travailler dans votre laboratoire a été une expérience très enrichissante pour moi.
Cette thèse ne se serait pas déroulée aussi bien sans l’immense soutien de mes deux encadrants. Nathalie Augé et Franck Peiretti, je vous remercie tous deux pour votre aide au cours de ces quatre ans. Vous avez été là scientifiquement en faisant toujours en sorte que cette thèse se déroule bien et avance, qu’il y ait des résultats, des publications. Vous m’avez également permis de présenter à plusieurs reprises mes travaux lors de congrès. Vos conseils m’ont permis d’apprendre énormément à vos côtés et toutes nos discussions, scientifiques ou pas, ont toujours été un plaisir. Je vous remercie également pour ce soutien moral dont vous avez toujours fait preuve, chacun à votre manière. J’ai beaucoup apprécié de travailler avec vous, vraiment beaucoup.
Je tiens également à remercier Gervaise Loirand et Georges Némoz pour avoir accepté d’évaluer ce travail et pour les corrections que vous y avez apportées.
Merci également à Ziad Mallat d’avoir accepté d’être examinateur et président de mon jury de thèse.
Il y a également bien d’autres personnes de ces deux laboratoires dans lesquels j’ai travaillé et que je voudrais remercier. Merci à Anne Nègre-Salvayre pour son accueil, son aide et ses conseils précieux. Marie, Sylvain, j’ai adoré partagé ce petit bureau avec vous et également tous ces M&M’s (mes hanches s’en souviennent encore d’ailleurs !) avec l’aide de
grande aide sur MT1-MMP ! Et puis vous avez été également là, Gilles, Jean-Claude, Cécile V., Françoise, Catherine que j’aurais aimé connaître plus, Corinne, Stéphanie, Aurélie, Bernadette, Paule, Odile L., et tous les membres de ces deux équipes, je vous remercie.
Et pour finir, je vais également remercier tous ceux qui me sont proches et qui ont été d’un immense soutien dans le bon déroulement de cette thèse. Je vous ai déjà dédié ce travail, mais je vous renouvelle mes remerciements, chers parents, votre exigence a payé ; chère Martine, merci pour toute l’aide que tu nous as apportée dans tes gardes, et ta bonne humeur permanente, j’en ai de la chance que Nico ait une maman si adorable. Merci à toi Nico, de nous avoir offert Maya et Lilas au cours de ces quatre ans. Merci aussi à vous tous qui nous avez entourés toutes ces années, que ce soit à Ponthierry, Champs/Marne, Gap, Laiz, Toulouse, aux Crozets du Haut, à Croydon, Montréal, St-Louis, et j’en oublie probablement…
TNFα : Tumor Necrosis Factor alpha TNF-R : Récepteur au TNFα
ADAM-17 : A Disintegrine and Metalloproteinase-17 HDLs : High Density Lipoproteins
CML : Cellules Musculaires Lisses nSMase : Sphingomyélinase neutre MMP-2 : métalloprotéase 2
MT1-MMP : métalloprotéase de type membranaire 1 SK-1 : sphingosine kinase-1
LTα : lymphotoxine-α
NF-κB : facteur nucléaire kappa B DD : Domaine de mort
LPS : Lipopolysaccharide
MAPK : Protéine Kinase à Activité Mitogène
VCAM-1 : molécule d’adhésion de cellule vasculaire de type 1 ROS : espèces réactives de l’oxygène
SAP 97 : Synapse-Associated Protein 97 FHL2 : Four and a Half Lim Domain
TIMPs : Tissue Inhibitor of Metalloproteinases TRAF : TNF-receptor associated factor
TRADD : TNF Receptor Associated Death Domain RIP : Receptor Interaction Protein
FAN : Factor Associated with Neutral-sphingomyélinase activation NSD : Neutral Sphingomyelinase Domain
JNK : c-Jun N-terminal kinases S1-P : sphingosine-1-phosphate
Le TNFα (Tumor Necrosis Factor alpha) est une cytokine pro-inflammatoire existant sous deux formes moléculaires, une forme transmembranaire étant clivée au niveau de la membrane pour générer une forme soluble. Le TNFα est impliqué dans de nombreux processus physiologiques et physiopathologiques dont l’athérosclérose. Au sein de la plaque, le TNFα soluble semble jouer un rôle majeur : des souris génétiquement modifiées et exprimant exclusivement une forme membranaire non clivable du TNFα développent des plaques d’athérosclérose dont l’état inflammatoire est significativement réduit par rapport à celles des souris sauvages (Canault et al., 2004). Le clivage de la forme membranaire du TNFα est médié par la métalloprotéase ADAM-17 (A Disintegrine and Metalloproteinase-17). A ce jour, la régulation de l’activité de clivage d’ADAM-17 est peu documentée. Nous avons émis l’hypothèse que la localisation membranaire d’ADAM-17 et de ses substrats participait à cette régulation. Nous montrons que la forme mature d’ADAM-17 est contenue dans des domaines particuliers de la membrane, les radeaux lipidiques (Tellier et al., 2006), et que cette localisation conditionne son activité. Nous avons également montré que des modifications de l’homéostasie du cholestérol des cellules par des HDLs conduisaient à une perturbation du clivage des substrats d’ADAM-17 (Tellier et al., 2007).
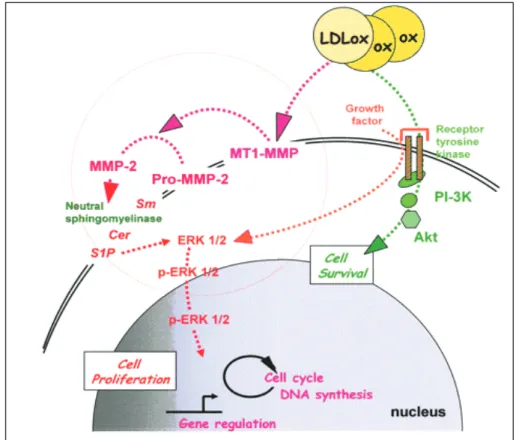
Le TNFα induit de multiples voies de signalisation dans les cellules de la plaque d’athérosclérose, conduisant, entre autres, à la prolifération des CML (cellules musculaires lisses). La littérature montre que le TNFα induit l’activation de sphingomyélinases (SMase) neutres et que le rôle mitogène des SMases implique les métalloprotéinases MMP-2 et MT1-MMP (Auge et al., 2004 ; Nègre-Salvayre et al., 2005). Nous avons posé l’hypothèse que le TNFα pouvait induire la prolifération cellulaire par une voie impliquant l’activation des métalloprotéases et des sphingomyélinases neutres. Nos résultats montrent que le TNFα stimule la prolifération des cellules musculaires lisses et des fibroblastes par activation séquentielle de la pro-protéine convertase furine, des métalloprotéases MT1-MMP et MMP-2, puis de la voie sphingomyéline/céramide (Tellier et al., 2007).
En conclusion, nos résultats apportent des données nouvelles sur la régulation de la biodisponibilité du TNFα ainsi que sur son effet pro-athérogène via l’activation d’une voie mitogène de stress.
TNFα (Tumor Necrosis Factor alpha) is a pro-inflammatory cytokine existing in two molecular forms, a transmembrane form cleaved to produced a soluble form. TNFα is implicated in numerous physiological and physiopathologicals processes such as atherosclerosis. Within the lesions, soluble TNFα seems to play a major role : genetically modified mice expressing exclusively a transmembrane form of TNFα develop atherosclerosis lesions with an inflammatory state which is significantly reduced compared to lesions of wild type mice (Canault et al., 2004). Cleavage of TNFα transmembrane form is due to the metalloproteinase ADAM-17 (A Disintegrine and Metalloproteinase-17). Today, regulation of ADAM-17 cleavage activity is poorly documented. We have voiced hypothesis that the membrane localization of ADAM-17 and its substrates was implicated to its regulation. We show that ADAM-17 mature form is localized into membrane particular domains, the lipid rafts (Tellier et al., 2006) and that this localization determined its activity. We also show that cell cholesterol homeostasis modifications by HDLs lead to a ADAM-17 substrates cleavage perturbations (Tellier et al., 2007).
TNFα induce number signaling pathways in atherosclerosis plaques cells, leading to several effects including SMC (smooth muscle cells) proliferation. Literature show that TNFα active neutral sphingomyelinase (nSMase) and that nSMase mitogene effect involve MMP2 and MT1-MMP (Auge et al., 2004 ; Nègre-Salvayre et al., 2005). We have postulated that TNFα could induce cell proliferation by a pathway implicating metalloproteinases and neutral SMase activation. Our results show that TNFα induced mesenchymal cells proliferation is due to pro-protein convertase furin, MT1-MMP ad MMP2 metalloproteinases sequential activation followed by sphingomyeline/ceramide pathway (Tellier et al., 2008).
In conclusion, our results give new data on the TNFα biodisponibility regulation as on pro-atherogene effect via stress mitogene pathway activation.
INTRODUCTION BIBLIOGRAPHIQUE 1
I. Le TNFα 3
I.1. Les membres de la superfamille du TNFα 3
I.2. Le TNFα : synthèse et caractéristiques moléculaires 8
I.3. Les effets biologiques du TNFα 10
I.4. Les effets du TNFα soluble et membranaire 12
II. La régulation du TNFα : le rôle majeur de l’enzyme de conversion du TNF
Alpha (TACE/ADAM-17) 14
II.1. Structure et expression d’ADAM-17 14
II.2. Rôles physiologiques et physiopathologiques d’ADAM-17 15
II.3. Régulation d’ADAM-17 18
II.3.1. Régulation de son expression 18
II.3.2. Régulation de l’activité d’ADAM-17 19
II.3.2.1. La maturation d’ADAM-17 par la furine 19
II.3.2.2. La phosphorylation 20
II.3.2.2. Interaction avec des protéines cytoplasmiques et
localisation cellulaire 21
II.3.2.3. Régulation de l’activation d’ADAM-17 par des
inhibiteurs naturels 21
II.3.2.4. Spécificité des substrats d’ADAM-17 22
III. La signalisation du TNF 23
III.1. Les récepteurs au TNFα 23
III.1.1. Description générale des récepteurs 23
III.1.2. Les molécules adaptatrices associées aux récepteurs du TNFα 25 III.1.2.1. Les protéines adaptatrices associées au TNF-R1 26 III.1.2.2. Les autres protéines adaptatrices 28 III.2. Les voies de signalisation induites par le TNFα 28
III.2.1. L’activation de NF-κB 28
III.2.2. L’activation de JNK, d’AP-1 et de p38-MAPK 30 III.2.3. L’activation de la voie sphingomyéline/céramide par
le TNFα 31
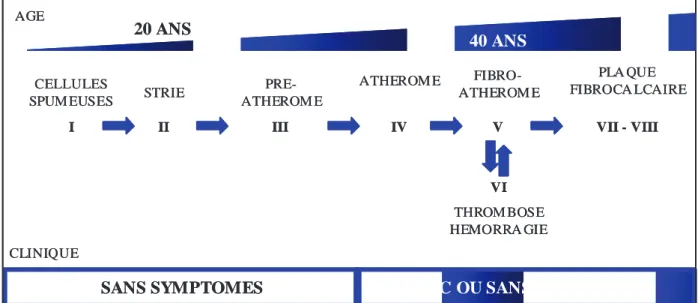
IV. Le TNFα et la physiopathologie de l’athérosclérose 35
IV.1. L’athérosclérose 35
IV.2. Effets du TNFα sur les cellules de la plaque 39
HYPOTHESES DE TRAVAIL 43
I. La régulation du clivage du TNFα par ADAM-17 44 II. Une voie de signalisation prolifératrice du TNFα impliquant métalloprotéases
I. Lignées et cultures cellulaires 47 II. Préparation des microvésicules 47
III. Protéines 48
III.1. Dosage des protéines 48
III.2. Immunoempreintes 48
III.3. Coimmunoprécipitations 48
III.4. Immunocytochimie 48
IV. Activités enzymatiques 49
IV.1. Dosage de l’activité d’ADAM17 sur les microvésicules 49
IV.2. Mesure de l’activité de MT1-MMP et MMP2 49
IV.3. Détermination de l’activité SMase neutre 49
IV.4. Détermination de l’activité sphingosine kinase-1 (SK-1) 50
IV.5. Détermination de l’activité furine 50
V. Analyse de la prolifération cellulaire par étude de l’ADN synthétisé 50
VI. Isolement des HDLs 51
VII. Isolement des radeaux lipidiques 51 VIII. Isolement des fractions enrichies en cytosquelette d’actine 52
IX. SiRNA 52
X. La cytométrie en flux 53
XI. Microscopie à transmission d’électron 53
XII. Animaux 53
XIII. Produits utilisés 54
XIV. Analyses statistiques 55
RESULTATS 56
I. Importance de la localisation membranaire d’ADAM-17 dans le contrôle de
son activité 57
I.1. Les microdomaines membranaires : radeaux lipidiques (lipid raft) 57 I.2. Résultats : « the shedding activity of ADAM-17 is sequestered in
lipid rafts » 58
I.3. Conclusions 71
I.4. Les HDL modifient-elles la localisation membranaire et l’activité
enzymatique d’ADAM-17 ? 74
I.4.1. Les lipoprotéines de forte densité (HDL) 74 I.4.2. Résultats : « HDLs activate ADAM-17-dependant shedding » 75
I.4.3. Conclusions 83
II. La voie de signalisation mitogène métalloprotéase/sphingolipides induite
par le TNFα via la furine 84
II.1 Les travaux précédents 84
II.2. Résultats : « role for furin in tumor necrosis factor alpha-induced
activation of the matrix metalloproteinase/sphingolipid mitogenis pathway » 87
II.3. Conclusions 99
II.4. Importance de la localisation membranaire des métalloprotéases dans
la voie de signalisation TNF/nSMase/prolifération de CML 102
CONCLUSIONS GENERALES 105 ANNEXES
Annexe 1 : “ FHL2 interact with both ADAM-17 and the Cytoskeleton and
regulates ADAM-17 localization and activity” 110
Annexe 2 : “ Proliferation and apoptosis elicited by low density lipoproteins
in vascular cells” 121
Mon travail de thèse est le résultat d’une collaboration développée de ma propre initiative entre mon équipe d’accueil, l’équipe 10 du département 3 de l’unité INSERM U858, dirigée par le Dr. Anne Nègre-Salvayre, et l’U626 du Pr. Marie-Christine Alessi dans laquelle j’ai effectué de nombreux stages. Mes travaux sont basés sur une thématique commune à ces deux laboratoires : la compréhension des mécanismes sous-jacents au TNF .
Les informations nécessaires à la compréhension des différentes voies de signalisation cellulaire induites par le TNF seront apportées dans une introduction bibliographique qui sera focalisée sur :
- La régulation du TNF et le rôle crucial de l’enzyme ADAM-17 qui permet l’obtention de la forme soluble du TNF à partir de sa forme membranaire.
- La signalisation des sphingomyélinases neutres cellulaires activées par le TNF et conduisant à la prolifération cellulaire.
- Les pathologies dans lesquelles le TNF joue un rôle important, notamment l’athérosclérose, pathologie faisant l’objet de nombreux travaux de recherche de la part des équipes dans lesquelles j’ai évolué au cours de cette thèse.
L’introduction bibliographique sera suivie des hypothèses de travail, des méthodologies expérimentales utilisées, puis des résultats expérimentaux que nous avons obtenus ainsi que des conclusions de ce travail.
I. Le TNF !
!
Le Tumor Necrosis Factor alpha (TNF ) a été identifié il y a plus de 30 ans (Old LJ, 1985) comme étant produit lors d’une activation du système immunitaire. Cette molécule a été décrite comme exerçant un effet cytotoxique sur diverses cellules tumorales et causant une nécrose tumorale dans certains systèmes de modèles animaux (Gaur et Aggarwal, 2003). L’ADNc du TNF a été cloné et son homologie de structure et de fonction avec la lymphotoxine- (LT ) définie. Deux récepteurs membranaires ont été identifiés, chacun étant capable de lier les deux cytokines. Le TNF s’inscrit dans une large famille de cytokines, la superfamille des ligands TNF .
Aujourd’hui, la recherche d’articles sur la base de données PubMed nous donne accès à près de 80 000 articles sur le TNF et les membres de sa famille. Il va donc de soi que cette revue générale ne traitera du TNF que dans ses aspects les plus généraux et sera volontairement orientée vers la compréhension du travail que nous avons réalisé.
I.1. Les membres de la superfamille du TNF !
La superfamille du TNF !se compose d’au moins 19 membres possédant de 15 à 18 % d’homologie de séquence entre eux. La plupart de ces protéines sont principalement produites par les cellules du système immunitaire : macrophages, monocytes et lymphocytes, mais également par d’autres types cellulaires comme les fibroblastes (tableau 1).
Toutes ces molécules partagent une voie de signalisation qui induit l’activation du facteur nucléaire kappa B (NF"B) et de protéines kinases à activité mitogène (par exemple, c-jun N-terminal kinase) (tableau 2). A l’exception de la lymphotoxine- (LT ) et de l’inhibiteur de croissance de cellules endothéliales vasculaires (VEGI) qui sont sécrétés, les membres de la superfamille du TNF sont tous des protéines transmembranaires de type II avec un domaine extracellulaire carboxy-terminal, un domaine amino-terminal intracellulaire et un domaine unique transmembranaire. Le domaine extracellulaire, connu pour être le domaine d’homologie du TNF , montre 20 à 30 % d’identité entre les membres de la superfamille et est responsable de la liaison à leur récepteur respectif. Le ligand induisant l’apoptose relié au TNF (TRAIL) et le ligand CD95 (CD95L) sont les membres ayant la plus forte homologie avec le TNF .
Cytokine Cellules Récepteurs Cellules LT TNF- LT FasL TRAIL TWEAK 4-IBBL OX40L CD40L CD27L CD30L APRIL Blys LIGHT VEGI GITRL RANKL EDA-1 EDA-2 ? ? ? Cellules NK, T, et B Cellules NK, T, et B Cellules T, B, NK, DC, macrophages Cellules T activées et cellules non-T Lymphocytes, cellules DC
Monocytes
Cellules B, cellules dentritiques, macrophages
Cellules T et B Cellules T et B Cellules NK, B et T Cellules T, monocytes
Organes lymphoïdes secondaires Cellules T, cellules DC
Monocytes, macrophages Cellules T, granulophages, monocytes, cellules DC
Cellules endothéliales
Cellules T activées, ostéoblastes
Peau Peau TNF-R1 TNF-R2 TNF-R1, TNF-R2 LT- R Fas DcR3 DR4, DR5, DcR1, DcR2 Fn 14 4-IBB OX-40 CD40 CD27 CD30 BCMA, TACI TACI, BCMA BAFF-R HVEM LT- R DR3 GITR RANK OPG EDAR XEDAR DR6 RELT TROY
Toutes les cellules Cellules endothéliales et immunitaires
Voir au-dessus
Cellules NK, CD4+ CD8+ T Cellules nuclées
Cellules de poumon et de colon La plupart des cellules
Cellules endothéliales, fibroblastes Cellules T activées, monocytes et cellules NK Cellules T Cellules Reed-Sternberg Cellules T CD4+ ,CD8+ Cellules Reed-Sternberg Cellules T et B Voir au-dessus Cellules B Cellules T lymphoïdes Cellules non lymphoïdes hématopoiétiques, et cellules stromales
Cellules T activées Cellules T CD25+ CD4+ Précurseurs des ostéoclastes Précurseurs des ostéoclastes, cellules endothéliales, autres Dérivées ectodermales Dérivées ectodermales Cellules T restantes Tissus lymphoïdes
Peau embryonnaire, épithélium, follicule de cheveux et cerveau
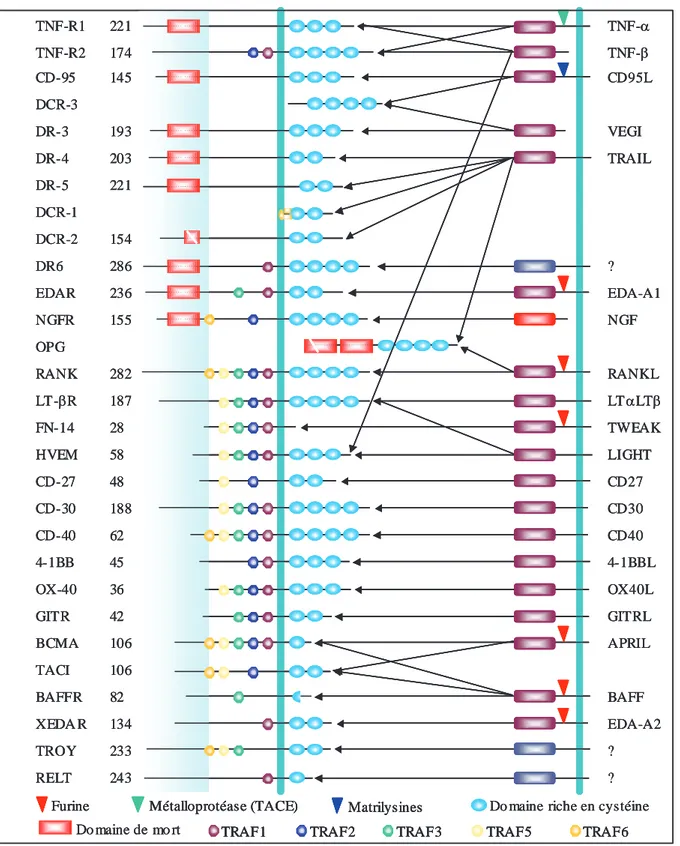
Dans leur majorité, les membres de la superfamille du TNF sont libérés de la surface cellulaire par un clivage protéolytique engageant des protéases distinctes (figure 1). Le TNF et le RANKL (Ligand Activateur de Récepteur au Nuclear Factor-kappa B) sont clivés par des métalloprotéases de la famille ADAM (A Disintegrine And Metalloprotease) (Black et al., 1997 ; Lum et al., 1999),et le CD95L par des matrilysines (Powell et al., 1999). Le facteur d’activation des cellules B (BAFF) à dysplasine ectodermale (EDA), le faible inducteur d’apoptose associé au TNF (TWEAK) et le ligand inducteur de prolifération (APRIL) sont
Tableau 1 : expression cellulaire des ligands et des récepteurs de la superfamille du TNF (selon Gaur et Aggarwal, 2003)
clivés par des membres de la famille des proprotéines convertases (Schneider et al., 1999 ; Chen et al., 2001).
Les membres de la superfamille du TNF ont des récepteurs distincts (tableau 2) qui présentent des homologies au niveau de leur domaine extracellulaire (figure 1) et ne présentent pas d’activité enzymatique. Certains possèdent un domaine de mort (Death Domain : DD) dans leur partie cytoplasmique. La majorité desmembres de la superfamille du TNF avec fonction de récepteurs sont des protéines transmembranaires de type I ayant un domaine extracellulaire N-terminal et un domaine intracellulaire C-terminal.
Ligand Récepteur Apoptose Prolifération NF-"B JNK p42-MAPK p38-MAPK
TNF Lt FasL VEGI TRAIL LT# CD27L CD30L 4-IBBL TWEAK LIGHT CD40L OX40L RANKL APRIL BAFF GITRL EDA-A1 EDA-A2 NGF ? ? ? ? TNF-R1,R2 TNF-R1,R2 Fas, DCR-3 DR3 DR4, DR5,DCR-2, DCR-1 LT-#R CD27 CD30 4-1BB Fn 14 LT-#R, HVEM, DCR-3 CD40 OX40 RANK TACI TACI, BCMA GITR EDAR XEDAR NGFR TROY DR6 RELT OPG + + + + + + + + + + + - - - - - + - - - - ? - + + - - - + + + - + + + + + + + + - - + + - + + + + + + + + + + + + + + + + + + + + + + + + + + + + + - + - + + + + - + - + + + - + - + - + ? + + - - - - + - - - - - - + - - - - - + - - ? + + + - - - - - - - - - - - + - - - - - + - - ? + Tableau 2 : régulation de l’apoptose et de la prolifération des cellules par les membres de la superfamille du TNF (selon Gaur et Aggarwal, 2003).
Furine Métalloprotéase (TACE) Matrilysines Do maine riche en cystéine Do maine de mo rt TRAF1 TRAF2 TRAF3 TRAF5 TRAF6 TNF-R1 TNF-R2 CD-95 DCR-3 DR-3 DR-4 DR-5 DCR-1 DCR-2 DR6 EDAR NGFR OPG RANK LT-#R FN-14 HVEM CD-27 CD-30 CD-40 4-1BB OX-40 GITR BCMA TACI BAFFR XEDA R TROY RELT 221 174 145 193 203 221 154 286 236 155 282 187 28 58 48 188 62 45 36 42 106 106 82 134 233 243 TNF- TNF-# CD95L VEGI TRAIL ? EDA-A1 NGF RANKL LT LT# TWEAK LIGHT CD27 CD30 CD40 4-1BBL OX40L GITRL APRIL BAFF EDA-A2 ? ? Furine Métalloprotéase (TACE) Matrilysines Do maine riche en cystéine
Do maine de mo rt TRAF1 TRAF2 TRAF3 TRAF5 TRAF6 Furine Métalloprotéase (TACE) Matrilysines
Furine Métalloprotéase (TACE) Matrilysines Do maine riche en cystéine Do maine de mo rt TRAF1TRAF1 TRAF2TRAF2 TRAF3TRAF3 TRAF5TRAF5 TRAF6TRAF6 TNF-R1 TNF-R2 CD-95 DCR-3 DR-3 DR-4 DR-5 DCR-1 DCR-2 DR6 EDAR NGFR OPG RANK LT-#R FN-14 HVEM CD-27 CD-30 CD-40 4-1BB OX-40 GITR BCMA TACI BAFFR XEDA R TROY RELT 221 174 145 193 203 221 154 286 236 155 282 187 28 58 48 188 62 45 36 42 106 106 82 134 233 243 TNF- TNF-# CD95L VEGI TRAIL ? EDA-A1 NGF RANKL LT LT# TWEAK LIGHT CD27 CD30 CD40 4-1BBL OX40L GITRL APRIL BAFF EDA-A2 ? ? TNF-R1 TNF-R2 CD-95 DCR-3 DR-3 DR-4 DR-5 DCR-1 DCR-2 DR6 EDAR NGFR OPG RANK LT-#R FN-14 HVEM CD-27 CD-30 CD-40 4-1BB OX-40 GITR BCMA TACI BAFFR XEDA R TROY RELT 221 174 145 193 203 221 154 286 236 155 282 187 28 58 48 188 62 45 36 42 106 106 82 134 233 243 TNF- TNF-# CD95L VEGI TRAIL ? EDA-A1 NGF RANKL LT LT# TWEAK LIGHT CD27 CD30 CD40 4-1BBL OX40L GITRL APRIL BAFF EDA-A2 ? ?
Figure 1 : les différents membres de la superfamille du TNF et leurs récepteurs. NF-"B : facteur nucléaire kappa B, RANKL : ligand activateur du récepteur au NF-"B, ADAM : A Disintegrine And Metalloprotease, BAFF : facteur d’activation des cellules B, EDA : dysplasine ectodermale, TWEAK : faible inducteur d’apoptose ressemblant au TNF, APRIL : ligand d’induction de prolifération, BCMA : antigène de maturation des cellules B, TACI : ligand d’interaction à la cyclophyline. BAFFR : récepteur au BAFF, XEDAR : récepteur à l’EDA lié à X, DCR1 : decoy récepteur 1, OPG : ostéoprotégérine, DR : récepteur de mort, NGFR : récepteur de facteur de croissance nerveuse, GITR : récepteur de la famille des TNF-R induit par les glucorticoïdes, HVEM : médiateur d’entrée du virus herpès, RELT : récepteur exprimé dans les tissus lymphoïdes, TRAF :
Toutefois, certains récepteurs comme BCMA (antigène de maturation des cellules B), TACI (ligand d’interaction à la cyclophyline), BAFFR (récepteur au BAFF), XEDAR (récepteur à l’EDA lié à X), sont dépourvus de séquence de peptide signal et sont définis comme des protéines transmembranaires de type III.
…
Après liaison à leurs récepteurs respectifs, les membres de la superfamille du TNF ont la capacité d’induire à la fois l’apoptose, la survie et/ou la prolifération cellulaire dans la même cellule en fonction de l’environnement (tableau 2). Ils sont, de ce fait, associés à un large éventail de rôles physiologiques. Citons comme exemple la régulation du système immunitaire. Le TNF , la LT , la LT#, RANKL et!l’interaction CD95/CD95L induisent des signaux essentiels dans la morphogénèse des organes lymphoïdes secondaires (Locksley et al., 2001) et dans la régulation du système immunitaire (Nagata et Golstein, 1995 ; Gaur et
Hématopoièse SIDA Maladie d’Alzheimer Maladie de Crohn
Immunité innée Régression tumorale Rejet de greffe Diabète (type II) Scléroses multiples Protection contre les infections bactériennes Arthrite rhumatoïde Problème cardiaque
Athérosclérose Maladie du foie
Asthme allergique Fièvre Surveillance immunitaire Lupus érythémateux systémique Ostéoporose / résorption osseuse Choc septique Maladies lymphoprolifératives Fibrose pulmonaire Métastase tumorale
Superfamille
du TNF
Figure 2 : les principaux effets physiologiques et pathologiques des membres de la superfamille du TNF . Bien que le TNF et les membres de sa famille soient essentiels à l’hématopoièse, à la protection contre des infections bactériennes, à la surveillance immunitaire et à la régression tumorale (comme schématisé en vert), la modification de leur régulation conduit à un certain nombre de maladies (en bleu). Ce type de rôles à « double tranchant » est typique de la plupart des membres de la superfamille du TNF (selon Aggarwall BB, 2003)
dans des processus pathologiques : le choc septique, la réplication virale, la résorption osseuse, l’arthrite rhumatoïde, le diabète et d’autres maladies inflammatoires (figure 2).
I.2. Le TNF !$!synthèse et caractéristiques moléculaires!
Le gène du TNF fait environ 3 kb et est porté par le chromosome 6 humain. Ce gène contient trois introns et quatre exons. Le promoteur du gène du TNF !possède des sites de fixation aux facteurs de transcription tels que NF-"B et c-Jun susceptibles de moduler l’expression de gènes à réponse précoce inflammatoire (Drouet et al., 1991). Les ARNm du TNF sont traduits en une protéine transmembranaire de 233 acides aminés, soit 26 kDa (Kriegel et al., 1988). Durant leur transport à la membrane plasmique, les monomères de TNF s’assemblent en homotrimères stables par des liaisons non covalentes (Tang et al., 1996) et deviennent biologiquement actifs.
La protéine TNF !est formée de trois domaines : un domaine cytoplasmique qui contient un site de phosphorylation unique sur le résidu sérine 2, un site de palmitoylation sur une cystéine du domaine cytoplasmique et deux sites de myristoylation sur les lysines 19 et 20 ; un domaine transmembranaire caractéristique des protéines transmembranaires de type II grâce auquel le TNF !s’intègre dans la membrane plasmique et un domaine extracellulaire qui lie les récepteurs au TNF (figure 3).
La forme soluble du TNF est obtenue par un clivage protéolytique entre les résidus alanine 76 et valine 77 de l’ectodomaine de la forme membranaire (figure 4). Ce clivage fait majoritairement intervenir ADAM-17 (A Disintegrin and Metalloproteinase 17) (Black et al., 1997 ; Moss et al., 1997 ; Zheng et al., 2004) et, minoritairement, d’autres protéines comme ADAM-10 (Rosendalh et al., 1997), ADAM-19 (Zheng et al., 2004) et autres (Gearing et al., 1995).
Figure 3 : structure du TNF !monomérique. Cyt : domaine cytoplasmique ; TM : domaine transmembranaire ; aa : acides aminés. KK : lysines, sites de myristoylation ; C : cystéine, site de palmitoylation. Cyt (35 aa) TM (21 aa) Domaine extracellulaire (177 aa) A76V77 Site de clivage C KK N-ter C-ter
Plusieurs types cellulaires sont capables de synthétiser du TNF (figure 5). Dans le cadre de notre travail, soulignons que la plupart des cellules qui composent la plaque d’athérosclérose sont susceptibles de produire du TNF . Le TNF est en effet exprimé dans les plaques d’athérosclérose(Barath et al., 1990 ; Arbustini et al., 1989 ; Rus et al., 1991 ; Tipping et al., 1993), notamment par les cellules de l’immunité innée dont les macrophages qui sont la source majeure de TNF . Les cellules musculaires lisses (Warner et al., 1989) et, dans certaines conditions de stimulation, des cellules endothéliales (Axel et al., 1997 ; Imaizumi et al., 2000) produisent également du TNF . Les cellules mammaires sont impliquées dans les événements athérosclérotiques et constituent une source supplémentaire de TNF puisqu’elles sont capables de produire des taux importants de TNF présynthétisé au sein de leurs granules, en plus du TNF sécrété de novo lors de stimulations (Gordon et Galli, 1990). Le TNF !est également produit par les cellules T et B, et les cellules NK au sein de la plaque.
Concernant les inducteurs de la libération du TNF , des cytokines comme l’Il-1, l’INF-%& l’Il-2 (Economou et al., 1989), le GM-CSF (Granulocyte-Macrophage Colony-Stimulating Factor) (Cannistra et al., 1987), et le CSF-1 (Colony-Colony-Stimulating Factor) (Warren et al., 1986) induisent la libération de TNF . Le TNF peut induire sa propre synthèse et sa libération par les monocytes. Dans les macrophages péritonéaux inflammatoires, il induit une augmentation de la quantité de ses ARNm (Kindler et al., 1989). Des extraits membranaires
ADAM-17 TNFs (17 kDa ) N H2 N H2 N H2 TNFm (26 kDa ) Milieu e xtracellula ire Membrane p lasmique Milieu intracellula ire N H2 N H2 N H2 ADAM-17 TNFs (17 kDa ) N H2 N H2 N H2 N H2 N H2 N H2 TNFm (26 kDa ) Milieu e xtracellula ire Membrane p lasmique Milieu intracellula ire N H2 N H2 N H2 N H2 N H2 N H2
Figure 4 : le clivage du TNF par ADAM-17 (TNFm : TNF membranaire, TNFs : TNF soluble)
de germes, de virus ou de champignons ainsi que la membrane de certaines cellules tumorales stimulent également la production de TNF .
Le LPS est l’inducteur le plus puissant de la production de TNF ' Il est actif sur les monocytes et les macrophages provenant de différents tissus, que ce soient les cellules de Kuppfer ou les macrophages pulmonaires. Les macrophages péritonéaux de souris activés par le LPS libèrent 10 fois plus de TNF que ceux non activés. Un traitement simultané à l’interféron gamma (INF-%) rend les macrophages encore plus sensibles à de faibles taux de LPS et constitue un activateur de la réaction au LPS (Stein et al., 1991).
I.3. Les effets biologiques du TNF !
Tous les types de cellules nucléées sont potentiellement capables de répondre au TNF . Le TNF exerce différents effets in vivo via ses deux récepteurs membranaires (Smith et al., 1990 ; Loetscher et al., 1990). Certaines cellules expriment les deux types de récepteurs comme les cellules NK (Shalaby et al., 1990) et les cellules endothéliales (Naume et al., 1991)), d’autres l’un ou l’autre des récepteurs comme les monocytes (Rossol et al., 2007). Les
TNF
Cellules musculaires lisses MACROPHAGES Lymphocytes Mastocystes Leucocytes polynucléaires Kératinocytes Astrocytes et cellules microgliales Cellules intestinales Cellules tumoraleseffets du TNF sur les tissus dépendent de la nature du récepteur, mais aussi de la nature du tissu, du contexte cellulaire ainsi que de la durée d’exposition à cette cytokine.
Le TNF exerce un large panel d’effets biologiques : mort cellulaire par nécrose ou apoptose, différenciation cellulaire, prolifération, inflammation, tumorigénèse et réplication virale (figure 6).
Le principal rôle du TNF concerne la régulation des cellules immunitaires, mais on lui connaît aussi un rôle important dans les désordres inflammatoires pathogènes tels que l’arthrite rhumatoïde et la maladie de Crohn (Sack M, 2002 ; Kooloos et al., 2007), l’asthme, le choc septique, les colites ulcératives, les fièvres hémorragiques et la cachexie (Locksley et al., 2001) (figure 2).
Le TNF a d’abord été considéré comme un agent tumoricide utilisable. Cependant, il apparaît évident que l’effet anti-tumoral observé in vivo est dépendant de sa capacité à modifier la réponse immunitaire et non pas via l’induction de l’apoptose des cellules
Figure 6 : les réponses cellulaires médiées par le TNF (selon McEwan et al., 2002)
Protéases P ro li fé ra ti o n Inflammation Récepteurs au TNF MAPK p38MAPK NF-"B Céramide
Protéases Espècesréactives de l’oxygène JNK
AP-1
Apoptose Nécrose Oncose Paraptose
Mort cellulaire Prolifération Différenciation Réplicationvirale Inflammation Réarrangements du cytosquelette
tumorales cibles (Hock et al., 1993). Le TNF a donc une fonction essentielle dans le développement des maladies immunitaires (Baugh et al., 2001 ; Weinberg et al., 2005). Il est impliqué dans les processus inflammatoires de la maladie de Parkinson (Sawada et al., 2006), de la maladie de Pages, une maladie conduisant à la formation excessive de tissu osseux (Kurihara et al., 2006), dans le psoriasis (Tan et al., 2007), l’asthme (Cazzola et al., 2006), … Cette cytokine intervient également dans certaines pathologies rénales, toujours en rapport avec son implication dans l’inflammation et le système immunitaire (Vielhauer et al., 2007), au cours du développement de maladies inflammatoires des intestins, comme les colites ulcérantes (Cottone et al., 2006) (figure 2), dans diverses maladies dégénératives comme la sclérose multiple et est responsable des effets délétères observés lors des pancréatites aiguës, représentant un déterminant majeur de la progression systémique de cette pathologie, en particulier des dommages au niveau pulmonaire et hépatique (Malleo et al., 2007). Le rôle du TNF dans les maladies cardiovasculaires sera décrit dans le dernier chapitre de cette revue.
I.4. Les effets du TNF soluble et membranaire
Nous avons mentionné que le TNF existait sous deux formes moléculaires, le TNF transmembranaire (tmTNF ( et le TNF soluble (sTNF (& le passage d'une forme à l'autre faisant intervenir l’activité protéolytique d’ADAM-17.
La majorité des effets biologiques documentés du TNF concerne le TNF soluble et pendant longtemps, l’étude des effets du tmTNF a été négligée. La création des modèles murins génétiquement modifiés dans lesquels le tmTNF est rendu non clivable a ensuite permis d’analyser l’effet de l’expression exclusive du tmTNF sur les fonctions physiologiques. Dans les neurones et les astrocytes murins (Akassoglou et al., 1997), des désordres neurologiques ressemblant à des manifestations neuro-inflammatoires ont été constatés. L’expression exclusive d’un transgène non clivable du tmTNF chez la souris déficiente en TNF protège du choc septique endotoxémique, mais n’empêche pas l’atteinte hépatique de se développer, à des degrés toutefois moins importants que chez les souris sauvages (Mueller et al., 1999). L’expression du transgène induit aussi l’augmentation de la concentration plasmatique d’IL-12, indiquant que la nature de la forme moléculaire du TNF régule la production d’IL-12, une cytokine centrale dans le déterminisme de la différenciation fonctionnelle des lymphocytes T «naïfs» (Trinchieri G, 1998), ce qui montre que le tmTNF
1999 ; Canault et al., 2004). En outre, le tmTNF module l’expression de cytokines centrales dans la réaction immuno-inflammatoire comme RANTES, MIP1-!, IL-10, et participe à l’organisation structurelle du tissu des organes lymphoïdes secondaires (Ruuls et al., 2001). Dans le cœur, la surexpression du tmTNF provoque une hypertrophie cardiaque de type concentrique conférant aux souris le phénotype de la cardiomyopathie dilatée (Dibbs et al., 2003). Le tmTNF induit la résistance à l’infection bactérienne par Mycobacterium bovis bacillus Calmette-Guérin (Olleros et al., 2002) et contrôle la réaction inflammatoire induite par Mycobacterium tuberculosis (Saunders et al., 2005). Enfin, un modèle murin d’expression du tmTNF ciblé spécifiquement dans le tissu adipeux (Xu et al., 2002a) a permis de mettre en évidence un défaut de signalisation insulinique responsable d’une réduction du transport du glucose dans l’adipocyte et d’une perte de la masse adipeuse. Cette même équipe montre qu’au cours de l’obésité, l’expression du tmTNF à la membrane des adipocytes est augmentée par inhibition de son clivage (Xu et al., 2002b), suggérant un rôle du tmTNF dans la différenciation adipocytaire.
Il apparaît donc clairement que la forme moléculaire sous laquelle le TNF !existe peut conditionner ses effets biologiques. ADAM-17 étant responsable du clivage du tmTNF à la surface cellulaire, cette enzyme se retrouve à une étape clé de la régulation de la production du TNF .
II. La régulation du TNF : le rôle majeur de l’enzyme de
conversion du TNF Alpha (TACE/ADAM-17)
La partie extracellulaire du TNF membranaire est clivée par une enzyme, l’ADAM-17, libérant le TNF soluble. Initialement appelée TACE pour TNF Alpha Converting Enzyme, elle appartient à la superfamille des protéases membranaires à zinc ADAM (A Disintegrin And Metalloproteinase) comprenant au moins trente-cinq membres. Cette protéine a été purifiée et clonée en 1997 par deux groupes indépendants (Black et al., 1997 ; Moss et al., 1997).
Pour comprendre les travaux que nous avons réalisés en vue de déterminer les mécanismes de régulation du clivage du TNF !par ADAM-17, il convient de décrire cette protéine et de détailler certains mécanismes de sa régulation fonctionnelle.
II.1. Structure et expression d’ADAM-17
La protéine ADAM-17 est codée par un gène d’environ 50 kb constitué de 19 exons et situé sur le chromosome 2 chez l’homme (12 chez la souris). Un seul transcrit mature est observé dans de nombreux tissus humains dont le cœur, le placenta, … (Black et al., 1997) et deux transcrits obtenus par épissage alternatif ont été mis en évidence chez la souris (Cerretti et al., 1999). L’expression d’ADAM-17 est ubiquitaire, les ARNm d’ADAM-17 étant présents dans tous les tissus étudiés (cœur, foie, cerveau, …) (Black et al., 1997).
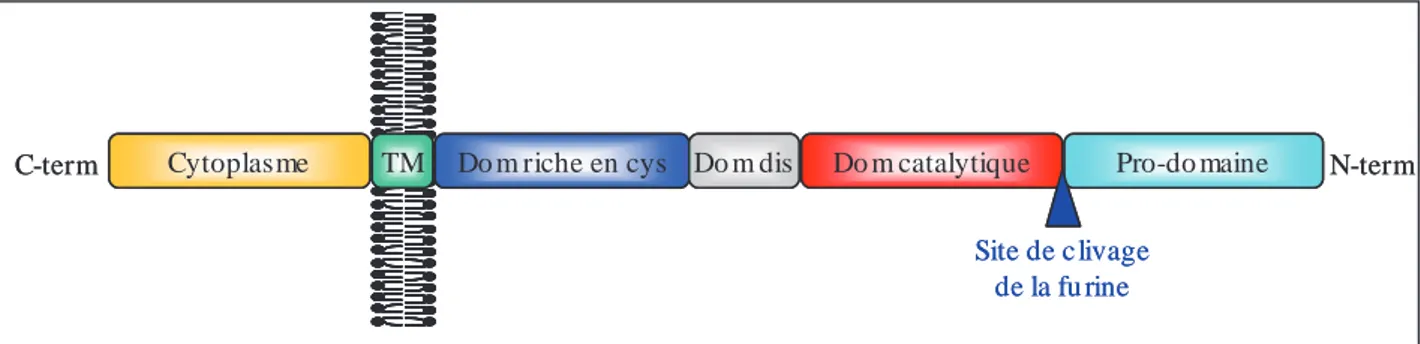
ADAM-17 est une glycoprotéine transmembranaire de type I composée de plusieurs domaines, existant sous une pro-forme de 120 kDa et une forme mature de 90 kDa (figure 7).
Figure 7 : structure des domaines d’ADAM-17. Cytoplasme : domaine cytoplasmique ; TM : domaine transmembranaire ; Dom riche en cys : domaine disintégrine riche en cystéines ; Dom dis : domaine disintégrine ; Dom catalytique : domaine catalytique (selon Black et al., 2003 ; Peiretti et al., 2003).
Cytoplasme TM Do m riche en cys Do m catalytique Pro-do maine
C-term N-term
Site de c livage de la fu rine
Do m dis
Cytoplasme TM Do m riche en cys Do m catalytique Pro-do maine
C-term N-term
Site de c livage de la fu rine
Le peptide signal est situé à l’extrémité N-terminale de la protéine. Suivent le pro-domaine qui possède un rôle de chaperon moléculaire et qui inhibe l’activité de l’enzyme, le domaine catalytique qui contient un atome de zinc nécessaire à l’activité métalloprotéase et un domaine disintégrine homologue à celui retrouvé dans les SVMP (métalloprotéases contenues dans le venin de serpent) qui suggère une possibilité d’interaction avec des intégrines (Bax et al., 2004). Des arguments expérimentaux sont, par ailleurs, en faveur d’un rôle d’ADAM17 dans l’adhésion cellulaire (Tsakadze et al., 2006). Se trouve ensuite un domaine riche en cystéine dont le rôle n’est pas défini avec certitude, mais qui pourrait participer à la reconnaissance des substrats (Reddy et al., 2000) et être impliqué dans la maturation de l’enzyme (Milla et al., 1999). Le domaine transmembranaire semble également important dans le contrôle de l’activité et de la localisation membranaire de l’enzyme (Li et al., 2007). Enfin, la partie cytoplasmique contient des sites potentiels d’interaction avec des protéines cytoplasmiques et des sites de phosphorylation.
II.2. Rôles physiologiques et physiopathologiques d’ADAM-17
L’étude des rôles physiologiques et physiopathologiques d’ADAM-17 est compliquée par le fait que l’invalidation du gène chez la souris conduit à une mortalité post-natale précoce (Peschon et al., 1998). Toutefois, l’activité métalloprotéase d’ADAM-17 a été très étudiée et plus de quarante protéines transmembranaires de natures très diverses ont été décrites comme étant des substrats d’ADAM-17 (Smalley et Ley, 2005) (tableau 3), ce qui suggère l’implication de cette enzyme dans de nombreux processus physiologiques.
Des facteurs de croissance et leurs récepteurs sont substrats d’ADAM-17. Cette enzyme serait donc impliquée dans la régulation du développement. Elle joue également un rôle dans la régulation des signaux générés par les récepteurs Notch qui influencent la prolifération, la différenciation et l’apoptose (Artavanis-Tsakonas et al., 1999) à tous les stades du développement des organismes pluricellulaires. La mortalité précoce des souris déficientes en ADAM-17 est en partie expliquée par un défaut de maturation des protéines impliquées dans le développement. L’importance d’ADAM-17 dans le contrôle du développement en fait une cible activement étudiée dans le cancer (Borrell-Pages et al., 2003 ; Fabre-Lafay et al., 2005). Certaines cytokines et leurs récepteurs sont des substrats d’ADAM-17. Parmi celles-ci, nous ne citerons que le TNF ' Comme nous l’avons décrit précédemment, son activité biologique peut différer selon qu’il est ou pas clivé par
ADAM-17. Les récepteurs du TNF sont aussi substrats d’ADAM-17, ce qui permet une régulation plus fine de la signalisation TNF . En effet, la perte des récepteurs à la surface cellulaire réduirait la sensibilité des cellules au TNF . En outre, les formes solubles des récepteurs au TNF conservent leur capacité à fixer le TNF !et agissent comme des compétiteurs des récepteurs membranaires (Aderka et al., 1996).
ADAM-17 est aussi responsable du clivage de certaines molécules d’adhésion et serait impliquée dans le contrôle de l’adhésion leucocytaire. Les travaux de différentes équipes ont montré, sur des modèles cellulaires en culture, qu’ADAM-17 clive la fractalkine (Garton et al., 2001; Tsou et al., 2001), la L-sélectine (Peschon et al., 1998) et la molécule d’adhésion de cellule vasculaire de type 1 (VCAM-1) (Garton et al., 2003). En régulant l’action de cytokines et l’adhésion leucocytaire, ADAM-17 contrôlerait ainsi la réaction immuno-inflammatoire.
ADAM-17 serait impliquée dans les maladies à forte dominance inflammatoire avec production exagérée de TNF . C’est notamment le cas des pathologies inflammatoires de l’intestin (Brynskov et al., 2002) ou de l’arthrite rhumatoïde (Ohta et al., 2001). En physiopathologie cardio-vasculaire, domaine d’intérêt des laboratoires dans lesquels j’ai travaillé, peu de données existent sur le rôle d’ADAM-17. Deux travaux montrent l’implication de ADAM-17 dans les cardiomyopathies dilatées avec excès de TNF libéré (Satoh et al., 1999 ; Fedak et al., 2006). Un autre groupe s’est intéressé à l’activité d’ADAM-17 dans les leucocytes humains chez des coronariens ayant eu un infarctus du myocarde (Shimoda et al., 2005). Ils constatent que le niveau d’expression et l’activité d’ADAM-17 en terme de libération du TNF par les monocytes ex vivo sont associés aux complications cardiaques ultérieures de ces patients.
Les travaux portant sur le rôle d’ADAM-17 dans le développement de l’athérosclérose chez l’animal (Canault et al., 2004) montrent qu’ADAM-17 est exprimé au sein des lésions athéromateuses et que cette expression est corrélée à la progression de ces lésions au cours du temps. D’autre part, l’augmentation d’ADAM-17 dans les lésions est reliée à une capacité plus grande de l’aorte à libérer ex vivo les récepteurs au TNF . Ces données suggèrent donc que, dans la lésion athéroscléreuse, ADAM-17 participe à la réaction inflammatoire et contribue à l’élévation des taux circulants des récepteurs au TNF . Ce travail, qui montre également qu’ADAM-17 est exprimée dans les plaques humaines, a été récemment confirmé par une étude (Satoh et al., 2008).
ADAM-17 exerce également un rôle dans la physiologie plaquettaire à travers le clivage de la GPIb! (Bergmeier et al., 2003; Bergmeier et al., 2004) et de la GPV (Rabie et
al., 2005).Ces glycoprotéines membranaires présentes à la surface des plaquettes forment, en association avec la glycoprotéine IX (GPIX), les complexes (GP)2-Ib-V-IX, récepteurs du
facteur von Willebrand, qui initient la formation du thrombus. L’implication d’ADAM-17 dans le clivage de la GPIb et de la GPV plaquettaires lui confèrerait ainsi un rôle dans la régulation de la thrombogénèse via le contrôle de l’agrégation plaquettaire (Aktas et al., 2005).
Substrats d’ADAM-17
Amphiregulin
Amyloid-beta precursor protein Vetrivel et al., 2005 AXL Receptor Tyrosine Kinase
CD30 Von Tresckow et al., 2004 CD40 Vidalain et al., 2000 CD117 (cKIT) Jahn et al., 2002 Cellular prion protein PrP Baron et al., 2002 Collagen XVII Zimina et al., 2005 Epiregulin
Fractaline (CX3CL1) Muehlhoefer et al., 2000 Glycoprotein GP of Ebola virus Bavari et al., 2002 Glycoprotein IB !!!!!!!!!!!! Dorahy et al., 1996 Glycoprotein V Shrimpton et al., 2002 Growth hormone receptor Yang et al., 2004 Heparin binding epidermal growth factor
Human epidermal growth factor receptor 4 Mineo et al., 1999 Human Meprin
Il-6 receptor Matthews et al., 2003 Interleukin-1 receptor 2
Interleukin-15 receptor
Low-density lipoprotein receptor Ness et al., 2003 L-selectin Sitrin et al., 2004 Macrophage colony stimulating fector receptor
Mucin 1 Handa et al., 2001 Nectin 4
Neuregulins Frenzel et al., 2001 Neurogenic locus notch homolog protein
P75 neurotrophin receptor Bilderback et al., 1997 Transforming growth factor- !
TNF-related activation-induced cytokine
Transforming tyrosine kinase protein Nishio et al., 2004 Tumor necrosis factor-
Tumor necrosis factor receptor I Cottin et al., 1999 Tumor necrosis factor receptor II Feng et al., 2001 Vascular cell adhesion molecule 1
Tableau 3 : les différents substrats d’ADAM-17, protéines pour lesquelles il existe une évidence qu’ADAM-17 est responsable du clivage ou y participe (selon Smalley et Ley, 2005).
II.3. Régulation d’ADAM-17
II.3.1. Régulation de son expression
Les ARNm d’ADAM-17 sont présents de façon constitutive dans toutes les cellules et tissus étudiés. Des études in vitro ont montré une augmentation des ARNm d’ADAM-17 dans les cellules endothéliales stimulées par des cytokines pro-inflammatoires ou des facteurs de croissance (Bzowska et al., 2004) et dans des lignées de cellules monocytaires traitées par des LDLs de patients diabétiques de type 2 (Worley et al., 2007). Une augmentation des ARNm d’ADAM-17 a été observée in vivo dans des tissus pathologiques inflammatoires dans des cas d’ostéoarthrite, d’arthrite rhumatoïde (Patel et al., 1998), de cardiomyopathie dilatée (Satoh et al., 1999) et de myocardite (Satoh et al., 2000), ainsi que dans des tissus cancéreux rénaux (Roemer et al., 2004) et hépatiques (Ding et al., 2004) ou hypoxiques (Zheng et al., 2007). Toutefois, les variations des niveaux d’ARNm décrits dans la littérature ne sont jamais drastiques.
L’analyse de la région 5’ du promoteur du gène d’ADAM-17 murin révèle l’existence de plusieurs sites de fixation de facteurs de transcription tels qu’AP2 et Sp1, mais aucun site pour les facteurs transcriptionnels classiquement activés au cours des réactions inflammatoires, tels que NF"B ou AP1 (Mizui et al., 1999). Une analyse du promoteur humain d’ADAM-17 a mis en évidence des éléments de réponse fonctionnels à l’hypoxie (H3 et H4 situés sur les régions -991 et -410 du promoteur) qui lient le facteur de transcription HIF-1 (Hypoxia-Inducible Factor-1) (Charbonneau et al., 2007).
L’expression protéique d’ADAM-17 est augmentée dans certaines conditions pathologiques, notamment dans les lésions athérosclérotiques (Canault et al., 2006). Certains travaux suggèrent que la régulation du clivage des substrats d’ADAM-17 implique directement la modulation de l’activité plutôt que la synthèse de l’enzyme ou de ses substrats (Kiessling et Gordon, 1998).
II.3.2. Régulation de l’activité d’ADAM-17
Le rôle important d’ADAM-17 dans le développement embryonnaire et dans la réponse immunitaire suppose une régulation fine de son activité. Cependant, les mécanismes conduisant à l’activation d’ADAM-17 demeurent mal connus.
II.3.2.1 La maturation d’ADAM-17 par la furine
Le passage de la pro-forme à la forme mature d’ADAM-17 se réalise par clivage du pro-domaine lors du transport de la protéine dans les compartiments médians de l’appareil de Golgi. Cette étape est indispensable à l’obtention de la forme active d’ADAM-17 (Milla et al., 1999). La pro-protéine convertase furine est à l’origine de la maturation d’ADAM-17 (Peiretti et al., 2003b). Toutefois, la participation d’autres pro-protéines convertases n’est pas exclue (Endres et al., 2003 ; Srour et al., 2003). Les mécanismes de régulation de l’activation de la furine sont mal connus (Thomas G, 2002) et peuvent être indirectement impliqués dans la régulation d’ADAM-17.
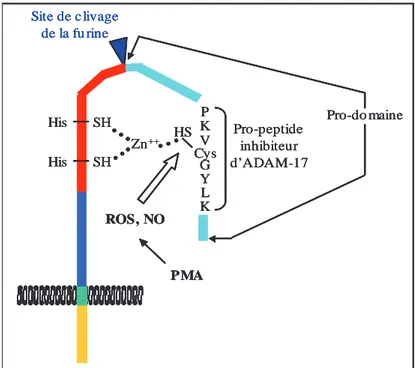
Figure 8 : mécanisme possible de l’activation d’ADAM-17 par les espèces réactives de l’oxygène (ROS) et le monoxyde d’azote (NO) (selon Zhang et al., 2001).
Site de c livage de la fu rine P K V Cys G Y L K Pro-do maine Pro-peptide inhibiteur d’ADAM-17 His His SH SH
...
. ..
Zn++ HS. . .
PMA ROS, NO Site de c livage de la fu rine P K V Cys G Y L K Pro-do maine Pro-peptide inhibiteur d’ADAM-17 His His SH SH...
. ..
Zn++ HS. . .
PMA ROS, NO Site de c livage de la fu rine P K V Cys G Y L K Pro-do maine Pro-peptide inhibiteur d’ADAM-17 His His SH SH...
...
. ..
. ..
Zn++ HS. . .
. . .
PMA ROS, NOD’autre part, il a été proposé que le clivage du pro-domaine d’ADAM-17 était nécessaire, mais non suffisant, à l’activation d’ADAM-17. En effet, après son clivage, le pro-domaine resterait associé à l’enzyme et l’activité d’ADAM-17 aurait lieu après dissociation du pro-domaine, permettant au site actif dépendant du Zn2+ d’adopter la bonne conformation pour l’activité d’ADAM-17.
Un mécanisme possible suggère que des espèces réactives de l’oxygène (ROS) ou du monoxyde d’azote (NO) oxydent le groupe thiol (SH) de la cystéine du pro-domaine (cystéine switch), ce qui conduit à la dissociation complète entre le pro-domaine et le site catalytique, libérant ainsi la forme active d’ADAM-17 (Zhang et al., 2000 ; Zhang et al., 2001) (figure 8). Ces résultats ont, cependant, été remis en question par une équipe américaine (Gonzales et al., 2004).
II.3.2.2. La phosphorylation
La partie cytoplasmique d’ADAM-17 possède quatre sites potentiels de phosphorylation : la tyrosine 702 (Moss et al., 1997), la thréonine 735 (Diaz-Rodriguez et al., 2002) en réponse aux facteurs de croissance et les sérines 791 et 819 (Fan et al., 2003).
La phosphorylation de résidus précis de la partie cytoplasmique d’ADAM-17 serait un élément de contrôle de son activité. Lors d’une stimulation par le PMA (phorbol 12-myristate 13-acetate), la protéine Erk (Extracellular signal-regulated kinase) associée à ADAM-17 phosphoryle le résidu thréonine 735 (Diaz-Rodriguez et al., 2002). Cette phosphorylation d’ADAM-17 sur la thréonine 735 serait nécessaire à l’adressage ainsi qu’à la maturation de l’enzyme (Soond et al., 2005). Cependant, l’intervention directe de Erk dans la régulation de l’activité d’ADAM-17 par le PMA n’a pas été démontrée (Montero et al., 2002). Un autre travail a montré qu’ADAM-17 phosphorylée par la voie PI3 Kinase activée par une phosphoinositide kinase 1 coupe l’amphiréguline ligand de l’EGFR (Zhang et al., 2006). L’ensemble de ces données suggère une régulation de l’activité d’ADAM-17 par la phosphorylation de sa partie cytoplasmique. Toutefois, des résultats déjà anciens montrent que les esters de phorbols stimulent de façon comparable la libération des substrats d’ADAM-17, que celle-ci possède ou pas sa région cytoplasmique, suggérant que l’effet activateur des esters de phorbol est indépendant de la phosphorylation d’ADAM-17.
II.3.2.3 Interaction avec des protéines cytoplasmiques et localisation cellulaire
Des protéines intracellulaires peuvent interagir avec la partie cytoplasmique d’ADAM-17 et modifier ainsi son activité. Le domaine cytoplasmique d’ADAM-17 comporte des motifs homologues à ceux intervenant dans la fixation des protéines possédant des séquences de type SH2 et SH3. D’autre part, l’association d’ADAM-17 à une kinase (Diaz-Rodriguez et al., 2002) ou à une phosphatase (Zheng et al., 2002) telle que PTPH1 suggère l’importance des mécanismes de phosphorylation ou de déphosphorylation dans sa régulation.
Par une approche en double hybride, des travaux montrent que l’extrémité C-terminale d’ADAM-17 peut interagir avec SAP 97 (Synapse-Associated Protein 97) (Peiretti et al., 2003a). Cette protéine jouerait un rôle dans le processus de localisation membranaire de certains récepteurs et canaux membranaires (Fujita et Kurachi, 2000). L’importance de cette interaction dans le contrôle de l’activité de clivage d’ADAM-17 est démontrée par le fait que la surexpression de SAP 97 réduit fortement la libération des substrats d’ADAM-17 (Peiretti et al., 2003a). Une partie plus interne du domaine cytoplasmique d’ADAM-17 lie FHL2 (Four and a Half Lim domain 2) (annexe 1 : Canault et al., 2006). FHL2 serait impliquée dans l’association de la forme mature d’17 au cytosquelette. L’interaction ADAM-17/FHL2 semble fonctionnelle puisque des cellules FHL2-/- présentent une augmentation d’ADAM-17 à leur surface et une diminution de la libération des substrats d’ADAM-17.
Les mécanismes exacts par lesquels FHL2 et SAP97 régulent l’activité d’ADAM-17 ne sont pas encore établis. Toutefois ces deux protéines modulent la localisation cellulaire d’ADAM-17 et il peut être suggéré que c’est par cette action qu’elles modifient son activité (Peiretti et al., 2003a ; Canault et al., 2006).
II.3.2.3. Régulation de l’activation d’ADAM-17 par des inhibiteurs naturels
Parmi les quatre membres de la famille des inhibiteurs naturels de métalloprotéases, les TIMPs (Tissue Inhibitor of Metalloproteinase), seul TIMP3 inhibe l’activité d’ADAM-17 (Amour et al., 1998). L’expression de TIMP3 est induite par le PMA et par des cytokines anti-inflammatoires comme le TGF- (Edwards et al., 1996), mais est diminuée par les
modulation de l’expression de TIMP3 ait des répercussions sur l’activité d’ADAM-17. L’importance de TIMP3 dans la régulation de l’inflammation est démontrée in vivo chez des souris déficientes en TIMP3 chez qui on note une forte inflammation hépatique (Mohammed et al., 2004) et, après injection de LPS, une inflammation systémique plus marquée que dans les souris sauvages (Smookler et al., 2006).D’autre part, parmi des souris hétérozygotes pour le récepteur à l’insuline, celles développant un diabète présentent une déficience constitutive en TIMP3, responsable d’une activité exacerbée d’ADAM-17 et d’un excès de TNF circulant. L’inflammation vasculaire observée qui en résulte apporte un argument expérimental aux complications vasculaires associées à la résistance à l’insuline et au syndrome métabolique (Federici et al., 2005).
II.3.2.4. Spécificité des substrats d’ADAM-17
Même si un résidu valine apparaît préférentiellement impliqué dans la liaison peptidique clivée par ADAM-17, l’alignement des séquences correspondant aux régions de clivage des substrats d’ADAM-17 ne permet pas de mettre en évidence de site consensus de coupure, indiquant que la séquence primaire des substrats n’intervient pas de manière prépondérante dans la spécificité de coupure. Cette observation a poussé une équipe américaine à proposer un modèle dans lequel ADAM-17 pourrait cliver ses substrats à condition que le site de clivage soit à une certaine distance de la membrane plasmique (Mohan et al., 2002).
Une façon d’envisager la spécificité de coupure d’ADAM-17 pour ses substrats serait une régulation précise de l’expression d’ADAM-17 et de ses substrats dans le temps et dans l’espace. C’est vers cette voie que nous avons orienté notre travail et émis une hypothèse de travail qui sera développée plus loin.
III. La signalisation du TNF
III.1. Les récepteurs au TNF !
!
III.1.1. Description générale des récepteurs
Le TNF exerce ses activités biologiques par interaction avec deux récepteurs membranaires : TNF-R1 (TNF récepteur 1, CD120a, p55/60) et TNF-R2 (TNF récepteur 2, CD120b, p75/80) de 55 et 75 kDa respectivement (Hohmann et al., 1989 ; Engelmann et al., 1990a ; Engelmann et al., 1990b ; Brockhaus et al., 1990) (figure 9). Ces récepteurs peuvent lier le TNF membranaire et le TNF soluble, ainsi qu’une molécule homotrimérique sécrétée, la lymphotoxine- (LT- ) (Wajant et al., 2003).
TNF-R1 et TNF-R2, glycoprotéines transmembranaires de type I et substrats d’ADAM-17, contiennent chacun quatre répétitions riches en cystéine dans leur domaine extracellulaire. La première, très conservée, permet l’assemblage des récepteurs en trimères (Chan et al., 2000 ; Siegel et al., 2000). Le TNF-R1 et le TNF-R2 possèdent 28 % d’homologie de séquence dans le domaine extracellulaire de fixation au TNF , mais aucune
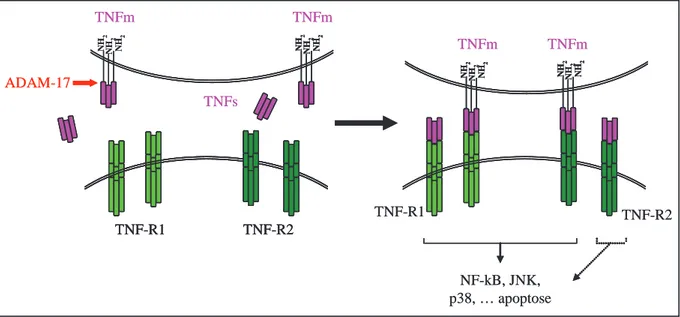
Figure 9 : le TNF lié à la membrane (TNFm) et le TNF soluble (TNFs) lient tous les deux le TNF-R1 et le TNF-R2. Cependant, alors que le TNF membranaire active les deux récepteurs, le TNF !soluble stimule essentiellement le TNF-R1 et a une signalisation limitée sur le TNF-R2 (selon Wajant et al., 2003).
TNF-R1 TNF-R2 ADAM-17 TNFm TNFs N H2 N H2 NH 2 TNFm TNF-R1 TNF-R2 N H2 N H2 NH 2 N H2 N H2 NH 2 TNFm N H2 N H2 NH 2 TNFm NF-kB, JNK, p38, … apoptose TNF-R1 TNF-R2 TNF-R1 TNF-R2 ADAM-17 TNFm TNFs N H2 N H2 NH 2 TNFm TNF-R1 TNF-R2 N H2 N H2 NH 2 N H2 N H2 NH 2 N H2 N H2 NH 2 N H2 N H2 NH 2 TNFm N H2 N H2 NH 2 N H2 N H2 NH 2 TNFm NF-kB, JNK, p38, … apoptose
dans le domaine intracellulaire, suggérant qu’ils pourraient activer des voies de signalisation différentes (Grell et al., 1994). Il semble essentiel que les récepteurs s’homotrimérisent pour initier les voies de signalisation. Des travaux montrent en effet que l’initiation de l’activation du récepteur est un processus complexe : en absence de ligand, les domaines distaux riches en cystéine de TNF-R1 et TNF-R2 permettent des interactions homophiliques entre les molécules de récepteurs (Chan et al., 2000). Dans ce statut multimérisé, ces « preligand binding assembly domain » (PLAD) peuvent aboutir à la formation d’homotrimères de récepteurs inactifs et non à une autoactivation spontanée. Ceci est souvent observé lors d’une surexpression de ces récepteurs dans des systèmes in vitro. La liaison du ligand sur le TNF-R préformé induit l’activation d’un changement conformationnel du complexe « réceptif » et permet la formation de complexes récepteurs très ordonnés qui acquièrent alors leur compétence de signalisation.
Le TNF-R1 est exprimé constitutivement dans la plupart des tissus alors que l’expression de TNF-R2 est fortement régulée et spécifique des cellules du système immunitaire. Dans la plupart des cellules, le TNF-R1 est stocké dans des pools Golgiens et est, soit exporté directement à la membrane plasmique, soit libéré e étant associé à des exosomes dans le milieu extracellulaire sans clivage préalable (Hawari et al., 2004). Il apparaît comme le médiateur de la signalisation du TNF alors que, dans le système lymphoïde, c’est le TNF-R2 qui semble jouer un rôle majeur. Des études basées sur l’utilisation d’anticorps agonistes ou antagonistes spécifiques des TNF-R1 ou TNF-R2, ainsi que l’invalidation de gènes codant pour les deux types de récepteurs, ont montré que la majorité des effets biologiques induits par le TNF sont médiés par le TNF-R1. Par exemple, des études in vivo ont montré que les souris KO pour le TNF-R1 sont résistantes à la mort consécutive à l’injection intrapéritonéale de LPS qui s’accompagne de la production de TNF et d’IL-1.
Cependant, l’importance du TNF-R2 est souvent mésestimée. En effet, le TNF membranaire, en plus du TNF soluble, peut activer ce récepteur (Grell et al., 1995). Nous retiendrons que les deux récepteurs au TNF fonctionnent en collaboration l’un avec l’autre. Le concept de « ligand passing » (Tartaglia et al., 1993b) décrit que la liaison du TNF soluble au TNF-R1 est potentialisée par le TNF-R2. Dans ce système, le TNF soluble lierait le TNF-R2, puis serait transféré sur le TNF-R1 sur lequel il agirait. D’autres modèles de coopération ont également été décrits (Grell et al., 1995 ; Grell et al., 1999) ; tous montrent que les voies de signalisation induites par le TNF soluble ou membranaire se potentialisent