Letters to Editor
872 Neurology India | Volume 67 | Issue 3 | May‑June 2019
late-onset leukodystrophy phenotype.[5] Genetic testing could not be done in other family members.
In conclusion, special attention should be paid to the radiologic findings of the cases with late‑onset leukodystrophy in which biochemical and metabolic screening tests give unremarkable results. AARS2 gene mutation should then be considered in the differential diagnosis. Definition of further phenotypes will help in developing the clinical algorithms for adulthood leukodystrophies.
Acknowledgement
The authors thank the patient and his family members for their collaboration.
Financial support and sponsorship Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Ahmed RM, Murphy E, Davagnanam I, Parton M, Schott JM, Mummery CJ, et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry 2014;85:770‑81. 2. Dallabona C, Diodato D, Kevelam SH, Haack TB, Wong LJ,
Salomons GS, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014;82:2063‑71.
3. Lynch DS, Zhang WJ, Lakshmanan R, Kinsella JA, Uzun GA, Karbay M, et al. Analysis of mutations in AARS2 in a series of CSF1R‑negative patients with adult‑onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol 2016;73:1433‑9. 4. Rai R, Giri R, Singh P, Verma RK. Leukodystrophy. J Assoc
Physicians India 2016;64:131.
5. Euro L, Konovalova S, Asin‑Cayuela J, Tulinius M, Griffin H, Horvath R, et al. Structural modelling of tissue‑specific mitochondrial alanyl‑tRNAsynthetase (AARS2) defects predicts differential effects on aminoacylation. Front Genet 2015;6:6‑21.
Güneş A Uzun
Department of Neurology, Istanbul Faculty of Medicine, Istanbul University, Millet Cad Capa 34390, Istanbul, TurkeyAddress for correspondence:
Dr. Güneş A Uzun, Department of Neurology, Istanbul Faculty of Medicine, Istanbul University, Millet Cad Capa 34390, Istanbul, Turkey. E‑mail: mavilapina@gmail.com
How to cite this article: Uzun GA. Adult‑onset leukodystrophy with homozygous AARS2 mutation located in the aminoacylation domain. Neurol India 2019;67:871‑2.
© 2019 Neurology India, Neurological Society of India | Published by Wolters Kluwer ‑ Medknow
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution‑NonCommercial‑ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non‑commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Access this article online Website:
www.neurologyindia.com
Quick Response Code:
DOI:
10.4103/0028‑3886.263237
PMID:
xxxx
Primary generalized dystonia due to TOR1A ∆GAG
mutation in an Indian family with intrafamilial
clinical heterogeneity
Sir,
Primary dystonia is a movement disorder characterized by sustained involuntary muscle contraction leading to abnormal postures and twisting movements. The prevalence of dystonia varies worldwide. In India, the prevalence rate is about 49.06 per 100,000 individuals with a higher prevalence of late-onset primary dystonia.[1] There is strong evidence that genetic factors play a significant role in causing dystonia. So far, six genes (TOR1A, CIZ1, THAP1, GNAL, ANO3, and TUBB4) have been implicated for primary torsion dystonia (PTD); however, mutations in TOR1A and THAP1 have been found to be responsible for manifestations in a large number of patients with PTD irrespective of their ethnic background. A trinucleotide deletion (∆GAG) in TOR1A gene has been reported to be the most common cause of PTD (DYT1) in
various populations.[2-5] This mutation was found to be more common in Ashkenazi Jew patients though it was found in most of the world dystonia cohorts. DYT1 dystonia is an autosomal dominant primary dystonia with a reduced penetrance (30%– 40%). Phenotypic characteristics of patients include early disease onset (<26 years) and the disease begins with the involvement of limbs which gradually progresses to other body parts except the craniocervical and oromandibular regions, leading to generalized dystonia. In this study, we report a family of four members from West Bengal, India, affected with DYT1 dystonia due to ∆GAG mutation in TOR1A gene with atypical phenotype and intrafamilial phenotypic variability. The patients were diagnosed in the Movement Disorders Clinic at Bangur Institute of Neurosciences, Kolkata, India, and have [Downloaded free from http://www.neurologyindia.com on Friday, July 26, 2019, IP: 47.15.218.131]
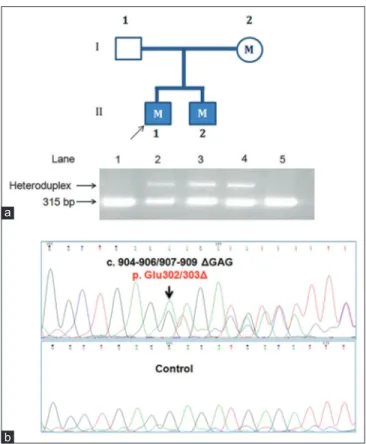
Figure 1: Identification of TOR1A ∆GAG mutation in an Indian family. (a) Upper panel: the pedigree with four family members. Shaded squares: affected individuals, shaded square with arrow: proband, M: presence of TOR1A ∆GAG variant. Lower panel: 7% PAGE of TOR1A exon 5 PCR products for all the family
members (lanes 1, 2, 3, and 4 for I‑1, II‑1, II‑2, and I‑2, respectively) and one unrelated control (lane 5). The top gel band shows the heteroduplex formed due
to the deletion. (b) The sequencing chromatogram of TOR1A exon 5 showing deletion (top panel) in affected individuals compared with the control (bottom panel)
b a Letters to Editor
Neurology India | Volume 67 | Issue 3 | May‑June 2019 873
negative history for developmental complications, malignancy, or any brain injury. Electroencephalogram, nerve conduction velocity (NCV) test with H‑reflex, neuroimaging (computed tomography, magnetic resonance imaging, etc.) studies for any brain lesions, and measurement of biochemical parameters (e.g., serum ceruloplasmin, uric acid) were found to be normal. None of them had a Kayser–Fleischer (KF) ring, as verified by routine slit‑lamp eye examination.
Case I
A 26-year old male patient [Figure 1a, proband, II: 1] was diagnosed as primary generalized dystonia with disease onset at 10 years. The proband was suffering from writing problem and holding objects in both the upper limbs. He felt an abnormal sensation with cramps in the right upper limb during writing and subsequently developed problem in holding objects. Gradually, the disease progressed to the lower limbs with abnormal posture during walking with a pattern of twisting gait. Later on, postural and action tremors developed in both the upper limbs along with a flexed posture in the right upper limb. The patient also developed truncal dystonia, an atypical feature for generalized dystonia with asymmetric distribution more towards the right side of the body (laterocorpus). The neck and shoulder position tilted slightly to the left side. At that time, the patient was treated with levodopa–carbidopa (55 mg TDS), clonazepam (0.5 mg BDS), tetrabenazine (25 mg BDS), and trihexyphenidyl (4 mg BDS).
Case II
The brother of proband [Figure 1a, II: 2], a 21-year old male, was also affected with primary generalized dystonia. He started having problem with dragging and lifting of toes during walking at 10 years of age. Gradually, the disease progressed to the upper body parts leading to generalized dystonia. He developed writer’s cramp with overflow of muscle contraction and action tremor during writing with the right upper limb. Whole-body tremor, both postural and action, was also present. Asymmetric twisting posture with stiffness developed on the right side of the body. Twisting in the right toe was found during walking, though the tandem walking was normal. The tone and activation in both the upper limbs were normal. The patient was treated with the medicines including levodopa–carbidopa (110 mg TDS), clonazepam (0.5 mg BDS), trihexyphenidyl (2 mg TDS), and propranolol (40 mg OD). The personalized medications prescribed for both the patients were dependent on variable biological responses.
This study was approved by “Bioethical Committee for Animal and Human Research Studies, University of Calcutta” following the guidelines of Indian Council for Medical Research. For genetic study, blood samples were collected from the patients and their parents [Figure 1]. All exons, promoter and 3'UTR of TOR1A and THAP1 genes were screened by polymerase chain reaction and sequencing, as described elsewhere.[6,7] A heterozygous trinucleotide deletion mutation (c.904-906/907-909∆GAG; p.302/303∆E) in TOR1A gene was detected in both the patients and their asymptomatic mother [Figure 1, I: 2], but not in their father [Figure 1, I: 1]. To understand the lack of penetrance in mother, we also analyzed the presence
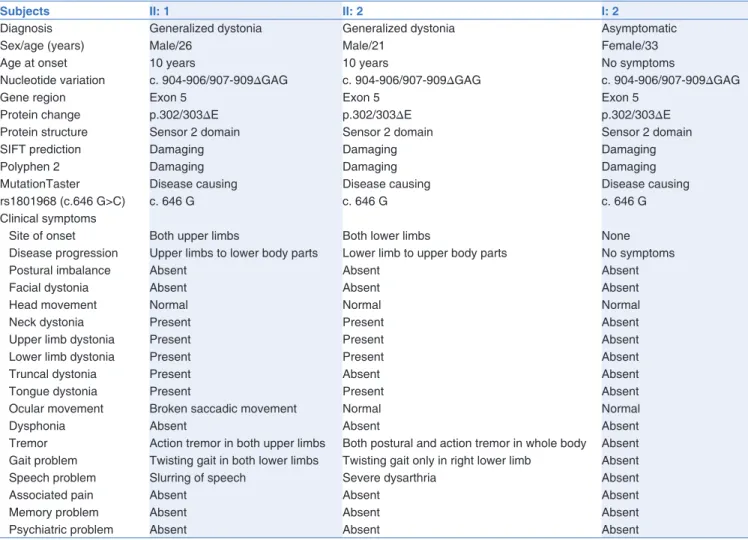
and orientation of c. 646C allele of rs1801968 (c.646G>C, p.D216E), as the presence of this allele in trans-orientation to the ∆GAG can greatly suppress the disease expression.[8] However, none of the family members harbored the c. 646C allele in exon 4 of TOR1A gene. The clinical features of the individuals harboring ∆GAG mutations are presented in Table 1. Interestingly, while there are similarities in clinical features, several significantly different phenotypes were also observed among them.
Among the clinical features of primary dystonia, two phenotypes are relatively common: (1) early onset of symptoms and (2) onset of dystonia in a limb. However, the intrafamilial phenotypic variability of dystonia patients having ∆GAG deletion is relatively rare.[3,9] The occurrence of ∆GAG mutation in Indian primary dystonia patients is rare.[6] In this study, we are reporting two generalized dystonia patients having TOR1A ∆GAG mutation from a single family with atypical intrafamilial phenotypic variability. The anatomical sites for disease onset were different for the two patients; while the proband had upper limb onset, his brother had lower limb onset. The direction of disease progression to the generalized form was exactly opposite for both the patients. In addition, as the disease progressed, the proband developed truncal dystonia, whereas the sibling developed a tremor dominant generalized dystonia. So far, there are a few reports where [Downloaded free from http://www.neurologyindia.com on Friday, July 26, 2019, IP: 47.15.218.131]
Letters to Editor
874 Neurology India | Volume 67 | Issue 3 | May‑June 2019
Table 1: Clinical characteristics of individuals harboring TOR1A ∆GAG mutation
Subjects II: 1 II: 2 I: 2
Diagnosis Generalized dystonia Generalized dystonia Asymptomatic
Sex/age (years) Male/26 Male/21 Female/33
Age at onset 10 years 10 years No symptoms
Nucleotide variation c. 904-906/907-909∆GAG c. 904-906/907-909∆GAG c. 904-906/907-909∆GAG
Gene region Exon 5 Exon 5 Exon 5
Protein change p.302/303∆E p.302/303∆E p.302/303∆E
Protein structure Sensor 2 domain Sensor 2 domain Sensor 2 domain
SIFT prediction Damaging Damaging Damaging
Polyphen 2 Damaging Damaging Damaging
MutationTaster Disease causing Disease causing Disease causing
rs1801968 (c.646 G>C) c. 646 G c. 646 G c. 646 G
Clinical symptoms
Site of onset Both upper limbs Both lower limbs None
Disease progression Upper limbs to lower body parts Lower limb to upper body parts No symptoms
Postural imbalance Absent Absent Absent
Facial dystonia Absent Absent Absent
Head movement Normal Normal Normal
Neck dystonia Present Present Absent
Upper limb dystonia Present Present Absent
Lower limb dystonia Present Present Absent
Truncal dystonia Present Absent Absent
Tongue dystonia Present Present Absent
Ocular movement Broken saccadic movement Normal Normal
Dysphonia Absent Absent Absent
Tremor Action tremor in both upper limbs Both postural and action tremor in whole body Absent
Gait problem Twisting gait in both lower limbs Twisting gait only in right lower limb Absent
Speech problem Slurring of speech Severe dysarthria Absent
Associated pain Absent Absent Absent
Memory problem Absent Absent Absent
Psychiatric problem Absent Absent Absent
generalized dystonia patients harboring TOR1A ∆GAG have truncal involvement.[3-5,10] Interestingly, the age of onset for both the patients was the same, that is, 10 years. This may be due to the age-related spatial expression of certain modifier genes in the cerebellum or could be triggered by environmental factors. Genetic analysis reveals that the mutant allele was inherited from their asymptomatic mother. Thus, in this family, the disease penetrance and phenotypic variation could be governed by some other genetic and/or environmental factors. This finding was previously presented in a conference as the first report of TOR1A ∆GAG mutation among Indian primary dystonia patients and published as a meeting proceeding.[11]
Acknowledgement
The authors are thankful to the participating patients and their family members for their cooperation toward this study. Financial support and sponsorship
This study was supported by a grant from the Department of Science and Technology [(DST-PURSE) and (DST-CSI to JR)], Govt. of India. SG was supported by DST, INSPIRE fellowship.
Conflicts of interest
There are no conflicts of interest.
References
1. Das SK, Banerjee TK, Biswas A, Roy T, Raut DK, Chaudhuri A,
et al. Community survey of primary dystonia in the city of Kolkata,
India. Mov Disord 2007;22:2031‑6.
2. Grundmann K, Laubis‑Herrmann U, Bauer I, Dressler D, Vollmer‑Haase J, Bauer P, et al. Frequency and phenotypic variability of the GAG deletion of the DYT1 gene in an unselected group of patients with dystonia. Arch Neurol 2003;60:1266‑70. 3. Szczaluba K, Jurek M, Milewski M, Friedman A, Kadziolka B,
Szolna A, et al. Clinical characteristics of carriers of a GAG deletion in the DYT1 gene amongst Polish patients with primary dystonia. Eur J Neurol 2007;14:659‑62.
4. O’Riordan S, Cockburn D, Barton D, Lynch T, Hutchinson M. Primary torsion dystonia due to the Tor1A GAG deletion in an Irish family. Ir J Med Sci 2002;171:31‑2.
5. Akbari MT, Zand Z, Shahidi GA, Hamid M. Clinical features, DYT1 mutation screening and genotype‑phenotype correlation in patients with dystonia from Iran. Med Princ Pract 2012;21:462‑6. 6. Naiya T, Biswas A, Neogi R, Datta S, Misra A, Das S, et al. Clinical [Downloaded free from http://www.neurologyindia.com on Friday, July 26, 2019, IP: 47.15.218.131]
Letters to Editor
Neurology India | Volume 67 | Issue 3 | May‑June 2019 875
characterization and evaluation of DYT1 gene in Indian primary dystonia patients. Acta Neurol Scand 2006;114:210‑5.
7. Giri S, Naiya T, Equbal Z, Sankhla CS, Das SK, Ray K, et al. Genetic screening of THAP1 in primary dystonia patients of India. Neurosci Lett 2017;637:31‑7.
8. Risch NJ, Bressman SB, Senthil G, Ozelius LJ. Intragenic Cis and Trans modification of genetic susceptibility in DYT1 torsion dystonia. Am J Hum Genet 2007;80:1188‑93.
9. Opal P, Tintner R, Jankovic J, Leung J, Breakefield XO, Friedman J,
et al. Intrafamilial phenotypic variability of the DYT1 dystonia:
From asymptomatic TOR1A gene carrier status to dystonic storm. Mov Disord 2002;17:339‑45.
10. Ikeuchi T, Shimohata T, Nakano R, Koide R, Takano H, Tsuji S. A case of primary torsion dystonia in Japan with the 3‑bp (GAG) deletion in the DYT1 gene with a unique clinical presentation. Neurogenetics 1999;2:189‑90.
11. Giri S, Biswas A, Das SK, Ray K, Ray J. Molecular basis of DYT1 and DYT6 primary dystonia in Indian patients. Mol Cytogenet 2014;7(Suppl 1):P121.
Subhajit Giri, Arindam Biswas,
Shyamal Kumar Das
1, Kunal Ray
2, Jharna Ray
S. N. Pradhan Centre for Neurosciences, University of Calcutta, 1Bangur Institute of Neurosciences, Kolkata, West Bengal, 2Academy of Scientific and Innovative Research (AcSIR), New Delhi, India
How to cite this article: Giri S, Biswas A, Das SK, Ray K, Ray J. Primary generalized dystonia due to TOR1A ΔGAG mutation in an Indian family with intrafamilial clinical heterogeneity. Neurol India 2019;67:872‑5.
© 2019 Neurology India, Neurological Society of India | Published by Wolters Kluwer ‑ Medknow
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution‑NonCommercial‑ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non‑commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Access this article online Website:
www.neurologyindia.com
Quick Response Code:
DOI:
10.4103/0028‑3886.263172
PMID:
xxxx
Address for correspondence:
Dr. Jharna Ray, S. N. Pradhan Centre for Neurosciences, University of Calcutta, Kolkata, West Bengal, India. E‑mail: jharnaray@gmail.com
Thalamic abscess caused by a rare
pathogen ‑ streptococcus sanguinis ‑ A report and
a review on thalamic abscess
Sir,
Thalamic location of a brain abscess is very unusual. Only 41 cases of thalamic abscess have been reported so far in the literature. The reported cases have been usually secondary to hematogenous spread from cyanotic heart disease, dental caries, etc. Streptococcus sanguinis is a normal oral commensal found in dental plaques and is a very unusual cause of intracranial infection. Only 7 cases of brain abscess caused by S. sanguinis have been reported so far. Thalamic abscess caused by S. sanguinis has not been reported so far.
A 42-year old man presented with headache, vomiting, and diminished sensation of right half of the body including face. He had no trauma to head, or any other source of infection elsewhere in the body including the oral cavity. Neurological examination was unremarkable except for the subjective sensory impairment of the right half of the body including face. Computed tomographic (CT) scan and magnetic resonance imaging (MRI) of the brain showed a well‑defined ring lesion with surrounding edema in the left thalamus, extending into the body and occipital horn of the left lateral ventricle, with mass effect and mild obstructive hydrocephalus
[Figures 1 and 2]. The diagnosis of a high grade glioma and abscess were considered. Investigations did not reveal any immunocompromised state.
A left parietal craniotomy was performed. The cortical incision was made in the left posterior parietal cortex, and the occipital horn of lateral ventricle was entered. The lesion was found projecting into the lateral ventricle. Tapping revealed creamy yellow pus, suggesting an abscess. Taking adequate precautions to prevent spillage of pus into rest of the ventricular system, the abscess capsule, which was thin, was excised. The abscess bed and ventricle was thoroughly irrigated with antibiotic solution and the craniotomy was closed with external ventricular drain in the occipital horn [Figure 3].
The culture of the pus revealed Streptococcus sanguinis, which was confirmed by VITEK 2 system (Version: 06.01 MIC interpretation guideline. bioMérieux Inc.). The organism was sensitive to amipicillin, linezolid, ofloxacin, penicillin and vancomycin. The patient was treated with appropriate antibiotics with intravenous antibiotics for 3 weeks and oral antibiotics for another 3 weeks. The patient made an uneventful recovery. At the time of discharge and at a [Downloaded free from http://www.neurologyindia.com on Friday, July 26, 2019, IP: 47.15.218.131]