Development and Applications of Copper(I) Hydride Catalysis in Asymmetric
Reactions and Heterocycle Synthesis
by Yujing Zhou B.S. Chemistry Peking University, 2016
SUBMITTED TO THE DEPARTMENT OF CHEMISTRY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY IN CHEMISTRY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2021
ã2020 Massachusetts Institute of Technology. All rights reserved.
Signature of Author: ____________________________________________________________________ Department of Chemistry October 22, 2020
Certified by: __________________________________________________________________________ Stephen L. Buchwald Camille Dreyfus Professor of Chemistry Thesis Supervisor
Accepted by: __________________________________________________________________________ Adam Willard Associate Professor Graduate Officer
2
This doctoral thesis has been examined by a committee of the Department of Chemistry as follows:
Professor Timothy F. Jamison: ___________________________________________________________ Thesis Committee Chair
Professor Stephen L. Buchwald: __________________________________________________________ Thesis Supervisor
Professor Mohammad Movassaghi: ________________________________________________________ Thesis Committee
3
Development and Applications of Copper(I) Hydride Catalysis in Asymmetric
Reactions and Heterocycle Synthesis
by
Yujing Zhou
Submitted to the Department of Chemistry on October 23, 2020 in Partial Fulfillment of the
Requirements for the Degree of Doctor of Philosophy in Chemistry
Abstract
Chapter 2. Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids as Aldehyde Surrogates
The direct asymmetric copper hydride (CuH)-catalyzed coupling of α,β-unsaturated carboxylic acids to aryl alkenes is developed to access chiral α-aryl dialkyl ketones. A variety of substrate substitution patterns, sensitive functional groups and heterocycles are tolerated in this reaction, which significantly expands the range of accessible products compared to existing hydroacylation methodology. Although mechanistic studies are ongoing, we propose that CuH-catalyzed silylation of unsaturated acids occurs to access a uniquely effective acyl electrophilic coupling partner.
Chapter 3. CuH-Catalyzed Asymmetric Reduction of α,β-Unsaturated Carboxylic Acids to β-Chiral Aldehydes
The copper hydride (CuH)-catalyzed enantioselective reduction of α,β-unsaturated carboxylic acids to saturated aldehydes is reported. This protocol provides a new method to access a variety of β-chiral aldehydes in good yields, with high levels of enantioselectivity and broad functional group tolerance. A reaction pathway involving a ketene intermediate is proposed based on preliminary mechanistic studies and density functional theory calculations.
Chapter 4. CuH-Catalyzed Asymmetric Reductive Amidation of α,β-Unsaturated Carboxylic Acids
The direct enantioselective copper hydride (CuH)-catalyzed synthesis of β-chiral amides from α,β-unsaturated carboxylic acids and secondary amines under mild reaction conditions is reported. The method utilizes readily accessible carboxylic acids, and tolerates a variety of functional groups at β-position including several heteroarenes. A subsequent iridium-catalyzed reduction to γ-chiral amines can be performed in the same flask without purification of the intermediate amides.
Chapter 5. CuH-Catalyzed Asymmetric Hydroamidation of Vinylarenes
A CuH-catalyzed enantioselective hydroamidation reaction of vinylarenes has been developed using readily accessible 1,4,2-dioxazol-5-ones as electrophilic amidating reagents. This method provides a straightforward and efficient approach to synthesize chiral amides in good yields with high levels of enantiopurity under mild conditions. Moreover, this transformation tolerates substrates bearing a broad range of functional groups.
4
Chapter 6. Enantioselective Allylation Using Allene, a Petroleum Cracking Byproduct
Allene (C3H4) gas is produced and separated on million-metric-ton scale per year during petroleum refining
but is rarely employed in organic synthesis. Meanwhile, the addition of an allyl group (C3H5) to ketones is
among the most common and prototypical reactions in synthetic chemistry. Herein, we report that the combination of allene gas with inexpensive and environmentally benign hydrosilanes, such as PMHS, can serve as a replacement for stoichiometric quantities of allylmetal reagents, which are required in most enantioselective ketone allylation reactions. This process is catalyzed by copper catalyst and commercially available ligands, operates without specialized equipment or pressurization, and tolerates a broad range of functional groups. Furthermore, the exceptional chemoselectivity of this catalyst system enables industrially relevant C3 hydrocarbon mixtures of allene with methylacetylene and propylene to be applied directly. Based on our strategy, we anticipate the rapid development of methods that leverage this unexploited feedstock as an allyl anion surrogate.
Chapter 7. Synthesis of Pyrroles through the CuH-Catalyzed Coupling of Enynes and Nitriles
Herein, we describe an efficient method to prepare polysubstituted pyrroles via a copper-hydride (CuH)-catalyzed enyne-nitrile coupling reaction. This protocol accommodates both aromatic and aliphatic substituents and a broad range of functional groups, providing a variety of N-H pyrroles in good yields and with high regioselectivity. We propose that the Cu-based catalyst promotes both the initial reductive coupling and subsequent cyclization steps. Density functional theory (DFT) calculations were performed to help elucidate the reaction mechanism.
Thesis Supervisor: Stephen L. Buchwald Title: Camille Dreyfus Professor of Chemistry
5
Acknowledgements
2020 has been a difficult year for all of us. It was not until recently that I realized how important and precious our freedom and health are, and I am very grateful that my family and friends are all staying safe and doing well during this unprecedented time.
Foremost, I would like to express my deepest thankfulness to my research advisor Prof. Steve Buchwald. I met Steve when he came to Peking University to give an academic talk, and it felt like someone met her long-time idol. I was very fortunate to join Steve’s group afterwards, when I started to realize that he is not only a successful scientist who publishes high-impact papers, but also a practical chemist that solves real problems for industrial manufacturing. His research is elegant, novel, useful and extremely reproducible, and I am significantly influenced by and benefited from his scientific philosophy. He is the one that has the big picture in mind, and always points out the right direction for me. At the same time, I also sincerely appreciate the freedom and flexibility that Steve kindly provides, allowing me to make mistakes, learn lessons, and make better decisions next time. He always “shows off” his dry/self-deprecating (here I quoted Richard’s words) sense of humor during group meetings or lab inspections, and at some point, I hope my English would be better to make good jokes in return.
I want to extend my sincere gratitude to the rest of my thesis committee members, Prof. Tim Jamison and Prof. Mo Movassaghi, for their continuous encouragement, suggestions, and criticism. A special thanks to Prof. Rick Danheiser for teaching us organic chemistry (5.511), attending my oral exam/fourth year proposal presentation, and providing insightful advice. I also want to thank Prof. Neil Garg at UCLA for his long-time help and support. I participated in a summer program and worked in Neil’s lab in 2015, when I had a lot of in person interactions and discussions with him. This two-month training built a solid foundation for my future PhD studies aboard, and I was very grateful to the time, attention, and efforts that he spent.
In addition, I want to acknowledge my undergraduate supervisor Prof. Jianbo Wang, who introduced me to the magic world of organic chemistry. I was inspired by his enthusiasm and passion for chemistry at beginning, and impressed by his caring and support for every student later. Sometimes when I felt very frustrated or self-doubted since I have not obtained any positive results for months, he never lost faith in me. He convinced me that it is important to focus, persist, and be optimistic, which I will be forever grateful.
At the outset of my PhD study, I was very lucky to work with Jeff Bandar, who is now an Assistant Professor at Colorado State University. When he was a postdoc in Steve’s lab, he always worked very efficiently and got tons of interesting results out of minimum experiments. Being a great mentor and friend, Jeff not only taught me basic technical skills and knowledge required for copper hydride chemistry, but also important philosophy to live a happy at the same time successful life in graduate school. Over the past few years I started to get a better understanding of his words, and I feel so lucky to know these things in my early days.
I want to thank my close friends in Steve’s group, Joey Dennis and Richard Liu. I thank them not only for discussing chemistry and science but also offering help and showing support anytime. In lab, they are knowledgeable and trustworthy scientists that I worked with and learned from. Joey is very considerate and understanding, who is the person that I can always count on when I encounter troubles. I remembered the first week I joined the lab, Joey came and said hi to me, which made me feel really welcomed. Richard is a real genius and scientific role model to me, and every day I am so impressed by how much he knows. He also has a great sense of humor, and his jokes or comments always make the pod full of laughter. My best moment in graduate school was dining out with them, and that was a big satisfactory for both my mentation and stomach. Our friendship has eased my toughest time in graduate school, and it will last forever.
I also would like to give my eternal thanks to our best manager Christine Nguyen for taking care of everything for the group, proofreading manuscripts, and providing sincere suggestions to difficult situations we faced. She is always friendly, thoughtful, and responsible, and as Steve said, our group would definitely stop functioning without her.
6
Besides Jeff and Richard, I was privileged to work with another two distinguished postdocs: Oliver Engl and Achim Link. They are both very experienced and knowledgeable in the fields of organic synthesis, and I learned a lot about how to communicate, make plans, divide up the work, and handle conflicts through our collaborations. I also want to thank Prof. Peng Liu, Lin Zhou, and Luke Jesikiewicz at University of Pittsburg for preforming DFT calculations for the pyrrole project. Their professionalism and working efficiency are very impressive to me.
To my fellow labmates, nothing would be accomplished without their help and suggestions. I knew Sheng Feng since my sophomore year, and we established our special and lasting friendships since then. Although we have very different values, interests, perspectives, and career aspirations, we talked about everything and supported each other over these years. Further, I want to thank Alex Schuppe, who seems pretty intimidating to me at first (probably just because he is very knowledgeable) but actually very fun and friendly. He has rich experiences in total synthesis, methodology, lab safety, cooking, coffee brewing, and cat raising. He is always willing to help, and I constantly learned a lot from him. I would like to extend my gratitude to Heemal Dhanjee for upgrading and optimizing our inventory system, Veronika Kottisch for organizing group meetings, and Elaine Reichert for taking great care of the gloveboxes. Many thanks to Martin Gazvoda, Chuan-Jin Hou, Hojoon Park, Jason Tao, Ryan King, Levi Knippel, Aaron Mallek, Jacob Rodriguez, Azin Saebi, Erica Tsai, and Jess Xu. It is a great honor to work with you all, and I thank you sincerely.
My appreciation also goes to a lot of our former group members that I overlapped during these years. Scott McCann is a very knowledgeable and warmhearted postdoc. He kindly taught me a lot of useful knowledge about palladium chemistry, kinetics, and workout, as well as helped me proofread my manuscripts. Yuxuan Ye was a senior graduate student in Steve’s group, who helped me get familiarized with MIT and the group when I first started. He always has many unique opinions and ideas about science, news, and games, and I constantly enjoyed and benefited from our conversations. I also want to thank Jeff Yang and Frieda Zhang for always being helpful and supportive. I still miss our monthly trip to Costco for big-pack food and delicious pizza, which never happened again since Jeff graduated. I want to express my thankfulness to other alumni who offered generous help to me since I joined the group: Mike Gribble, Bryan Ingoglia, Saki Ichikawa, Anthony Rojas, Sheng Guo, Chengxi Li, Zhaohong Lu, Haoxuan Wang, Gary Zhang, Liang Zhang, Yiming Wang, Xueqiang Wang, Yang Zhao, Nick White, Klaus Speck, Andy Thomas, Kwangmin Shin, Liela Bayeh, Mycah Uehling, Aaron Sather, Stig Friis, Mike Pirnot, and Yang Yang. It is always a great privilege to work with so many outstanding postdocs and graduate students from different research background.
Outside the lab, I have been blessed with friendships with Kristen Flynn, Vickie Wang, Yifeng Qi, Chenyue Sun, Luming Yang, Ke Qin, Gen Li, and Wencong Wang. Thanks for celebrating my successes and easing my stresses throughout these years. I also want to thank my long-time friends whom I know from high school or college: Aixi Zhang, Qianyue Wang, Chunyi Liu, Jiaxin Qiao, Qingyu Ye, Honghong Lin, Yan Cong, Chang Liu, Shizhong Dai, Xianyuan Zhao and Mei Lin. Thanks for listening to my complaints and showing support when I felt depressed, even if we were only able to communicate on the internet.
I want to thank the Department of Chemistry at MIT, Bristol Myers Squibb, and Merck for kindly providing fellowships to support my graduate study. In addition, several corporations, including Sigma–Aldrich, Takasago, Solvias, Nippon Chemical, and Strem, have generously donated phosphine ligands used in my PhD research. Last but most importantly, I want to thank my parents for their unconditional love, caring, understanding, and support throughout my entire life. Graduate school is a big adventure, and I am so grateful that they are always here for me.
7
Preface
Portions of this dissertation have been adapted from the following published articles co-written by the author and are reproduced with permission from the American Chemical Society or Wiley-VCH.
Zhou, Y.; Bandar, J. S.; Buchwald, S. L. Enantioselective CuH-Catalyzed Hydroacylation Employing
Unsaturated Carboxylic Acids as Aldehyde Surrogates. J. Am. Chem. Soc. 2017, 139, 8126–8129.
Zhou, Y.; Bandar, J. S.; Liu, R. Y.; Buchwald, S. L. CuH-Catalyzed Asymmetric Reduction of
α,β-Unsaturated Carboxylic Acids to β-Chiral Aldehydes. J. Am. Chem. Soc. 2018, 140, 606–609.
Zhou, Y.*; Engl, O. D.*; Bandar, J. S.; Chant, E. D.; Buchwald, S. L. CuH-Catalyzed Asymmetric
Hydroamidation of Vinylarenes. Angew. Chem. Int. Ed. 2018, 57, 6672–6675. (* denotes equal contribution.)
Liu, R. Y.*; Zhou Y.*; Yang, Y.; Buchwald, S. L. Enantioselective Allylation from Allene, a Petroleum Cracking Byproduct. J. Am. Chem. Soc. 2019, 141, 2251–2256.
(* denotes equal contribution.)
Zhou, Y.; Zhou, L.; Jesikiewicz, L. T.; Liu, P.; Buchwald, S. L. Synthesis of Pyrroles through the
CuH-Catalyzed Coupling of Enynes and Nitriles. J. Am. Chem. Soc. 2020, 142, 9908–9914.
Link, A.; Zhou, Y.; Buchwald, S. L. CuH-Catalyzed Asymmetric Reductive Amidation of α,β-Unsaturated Carboxylic Acids. Org. Lett. 2020, 22, 5666–5670.
8
Respective Contributions
This thesis contains work that is the result of collaborative efforts between the author and other colleagues at MIT. The specific contributions of the author are detailed below.
The work in Chapter 2 was a collaborative effort between Dr. Jeffrey Bandar and the author. Dr. Bandar performed initial experiments. The experiments presented in Chapter 2 were conducted by the author. The work in Chapter 3 was a collaborate effort between Dr. Jeffrey Bandar, Dr. Richard Liu, and the author. The author conducted the experiments, and the DFT calculations were performed by Dr. Liu.
The work in Chapter 4 was a collaborate effort between Dr. Achim Link and the author. The author discovered the reaction and performed initial optimizations. Dr. Link and the author conducted the experiments.
The work in Chapter 5 was a collaborative effort shared equally between Dr. Oliver Engl and the author. Dr. Jeffrey Bandar and Emma Chant carried out initial experiments.
The work in Chapter 6 involving allene gas was a collaborative effort shared equally between Dr. Richard Liu and the author. The reported DFT calculations were performed by Dr. Liu.
The work in Chapter 7 was a collaborate effort between Dr. Lin Zhou, Luke Jesikiewicz, Prof. Peng Liu, and the author. The author conducted the experiments, and the DFT calculations were performed by Dr. Zhou, Luke, and Prof. Liu.
9
Table of Contents
Chapter 1. Introduction to Copper Hydride Chemistry in Organic Synthesis
1.1 Discovery, Synthesis, and Reactivity of Copper(I) Hydride Complex………...12
1.2 CuH-Catalyzed Asymmetric 1,4- and 1,2-Reduction Reactions……….13
1.3 CuH-Catalyzed Enantioselective Olefin Hydrofunctionalization Reactions………. 16
1.4 References………. 20
Chapter 2. Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids as Aldehyde Surrogates 2.1 Introduction………24
2.2 Results and Discussion……….. 25
2.3 Conclusion………. 30
2.4 Experimental……….. 31
2.5 References and Notes………. 54
2.6 Spectra and Chromatograms..……… 58
Chapter 3. CuH-Catalyzed Asymmetric Reduction of α,β-Unsaturated Carboxylic Acids to β-Chiral Aldehydes 3.1 Introduction.……….105
3.2 Results and Discussion……….106
3.3 Conclusion………110
3.4 Experimental……….112
3.5 References and Notes………147
3.6 Spectra and Chromatograms.………152
Chapter 4. CuH-Catalyzed Asymmetric Reductive Amidation of α,β-Unsaturated Carboxylic Acids 4.1 Introduction.……….222
4.2 Results and Discussion……….223
4.3 Conclusion………229
4.4 Experimental……… 230
4.5 References and Notes………282
4.6 Spectra and Chromatograms.………289
Chapter 5. CuH-Catalyzed Asymmetric Hydroamidation of Vinylarenes 5.1 Introduction.……….354
5.2 Results and Discussion……….355
10
5.4 Experimental……… 361
5.5 References and Notes………381
5.6 Spectra and Chromatograms.………384
Chapter 6. Enantioselective Allylation Using Allene, a Petroleum Cracking Byproduct 6.1 Introduction.……….437
6.2 Results and Discussion……….438
6.3 Conclusion………446
6.4 Experimental……… 447
6.5 References and Notes………472
6.6 Spectra and Chromatograms.………480
Chapter 7. Synthesis of Pyrroles through the CuH-Catalyzed Coupling of Enynes and Nitriles 7.1 Introduction.……….524
7.2 Results and Discussion……….526
7.3 Conclusion………532
7.4 Experimental……… 533
7.5 References and Notes………558
11
12
1.1 Discovery, Synthesis, and Reactivity of Copper(I) Hydride Complex
The first report on copper hydride (CuH) chemistry described a ligand-free CuH complex prepared from the reduction of copper (II) sulfate (CuSO4) with phosphinic acid (H3PO2) by Würtz in 1844 (Scheme
1a).1 This red, pyrophoric solid was isolated in a polymeric form, and readily decomposed to form Cu (0)
and H2 at ambient temperature. Despite being the first metal hydride compound discovered, the reactivity
of this species was not well understood until over a century later by the pioneering work of Whitesides2 and
Stryker3. They disclosed the use of phosphine-ligated CuH complexes for chemoselective reductions. In
particular, Stryker’s work demonstrated that [(Ph3P)CuH]6, a hexameric ligated CuH species, could be
employed as a mild metal hydride reducing agent and facilitate conjugate reductions of α,β-unsaturated carbonyl compounds (Scheme 1b).
Scheme 1. (a) Discovery and (b) application of CuH complexes in chemeoselective reductions of α,β-unsaturated
carbonyl compounds.
Subsequently, considerable effort has been devoted to improving the generality and scope of CuH-catalyzed reduction reactions. Breakthroughs were made in two directions4: catalytic transformations
realized by using stoichiometric hydride sources, and enantioselective reductions enabled by employing chiral bisphosphine ligands.
Hydrogen gas was originally used as the terminal reductant to prepare Stryker’s reagent from a mixture of phosphine, CuCl, and NaOt-Bu (Scheme 2a).3a,5a However, initial protocols for the preparation of the
CuH complex necessitated high pressure (1500 psi) of H2,5b which poses significant challenges for operation
and safety. Shortly thereafter, stoichiometric quantities of hydride reagents, including silanes (R3SiH), tin
(a) Synthesis of ligand-free polymeric copper hydride species
(b) Conjugate reduction of unsaturated carbonyl compounds using [(Ph3P)CuH]6
CuSO4 + H3PO2 (CuH)n (decomposes to form Cu and H2) O O [(Ph3P)CuH]6 C6D6, rt
13
hydrides (R3SnH), and boranes (R2BH), were employed as convenient alternatives to H2 for the in-situ
regeneration of ligated CuH complex.6 Among these, silanes, which are generally inexpensive,
environmentally benign, and have readily tunable properties, became the most common choice of reducing agent (Scheme 2b).
Scheme 2. Synthesis of Stryker’s reagent [(Ph3P)CuH]6 using (a) hydrogen gas and (b) hydrosilane.
Additionally, a variety of copper salts have been demonstrated to be effective precursors for accessing reactive CuH species. Initially, CuOt-Bu, either independently synthesized or prepared in-situ by mixing CuCl and NaOt-Bu was used to access various LCuH complexes in the presence of a proper ligand and a reductant.5a,7 However, CuOt-Bu is a highly air- and moisture-sensitive compound and requires cautious
manipulation. To solve this problem, Yun and co-workers described the use of Cu(OAc)2 as a more stable
and commercially available alternative to CuOt-Bu in the synthesis of Stryker’s reagent.8 In the intervening
years, many copper(II) salts have been shown to efficiently used to generate LCuH catalysts and modulate the reactivity of these species in a variety of synthetic transformations.
1.2 CuH-Catalyzed Asymmetric 1,4- and 1,2-Reduction Reactions
The CuH-catalyzed asymmetric 1,4-reduction of α,β-unsaturated carbonyl compounds has been extensively developed as a general method to deliver optically active β-chiral ketones and esters. In 1999, Buchwald first disclosed a CuH-catalyzed enantioselective conjugate reduction of α,β-unsaturated esters using a CuH catalyst ligated by a chiral bisphosphine ligand, (S)-p-tol-BINAP (Scheme 3a).9 This reaction
was soon be further expanded to the asymmetric 1,4-reduction of a broad range of Michael acceptors, including α,β-unsaturated ketones,10 lactones11,12 lactams11, and sulfones13, affording enantioenriched
1,4-CuCl + NaOt-Bu
(a) Synthesis of Stryker’s reagent using hydrogen gas
+ PPh3 H2 (1 atm) toluene/benzene, rt [(Ph3P)CuH]6 CuCl + KOt-Bu + PPh3 PhMe2SiH benzene, rt [(Ph3P)CuH]6
(b) Synthesis of Stryker’s reagent using hydrosilane
50–65% yield
14
reduction products in high yields and enantioselectivities with excellent functional group tolerance. The addition of a hindered alcohol, such as t-BuOH, formed a copper alkoxide species from intermediary copper enolate, which led to a more rapid catalyst regeneration through s-bond metathesis with a hydrosilane. Further studies showed that improvements in reactivity and stereoselectivity could be achieved by employing SEGPHOS (Scheme 3b)14 and JOSIPHOS (Scheme 3c)15 ligated CuH catalysts. The use of these
bisphosphine ligands resulted in increased stability of LCuH complex, thus enabling efficient conjugate reductions with lower catalyst loading.
Scheme 3. Representative examples of CuH-catalyzed asymmetric 1,4-reduction reactions. PMHS =
Polymethylhydrosiloxane (HSiMeO)n.
The enolate intermediates derived from conjugate reduction of α,β-unsaturated carbonyl compounds have been utilized as carbon-based nucleophiles in cascade reactions to enable the preparation of structurally complexed compounds with consecutive stereogenic centers (Scheme 4a-b).16 Both inter- and
intramolecular trapping transformations have been developed, providing C–C bond forming products in processes that displayed good enantio- and diastereoselectivity. Meanwhile, a conceptually similar strategy involves two-step one-pot protocols, where silyl enol ether intermediates from a CuH-catalyzed reduction
Me OEt O CuCl, NaOt-Bu (S)-p-tol-BINAP PMHS, toluene, rt OEt O Me (a) 84% yield, 95:5 er PAr2 PAr2 O O O O P(p-tol)2 P(p-tol)2 Ar = 4-MeO-3,5-t-Bu-C6H2 (R)-DTBM-SEGPHOS (S)-p-tol-BINAP O Me [(Ph3P)CuH]6 (R)-DTBM-SEGPHOS PMHS, toluene, 0 °C O Me 90% yield, 98:2 er (b) (c) n-Hex Me Me O CuCl, NaOt-Bu (R,S)-PPF-Pt-Bu2 PMHS, toluene, –78 °C 95% yield, 99.5:0.5 er n-Hex Me O (R,S)-PPF-Pt-Bu2 Fe Pt-Bu2 P Ph Ph Me Me
15
reaction were telescoped in further coupling reactions with a wide range of electrophiles, including carbonyl compounds (Scheme 4c),17 alkyl halides (Scheme 4d),18 and aryl halides (Scheme 4e)19.
Scheme 4. CuH-catalyzed sequential reduction and C–C bond formation reactions.
CuH catalysis has also been widely used for asymmetric 1,2-reduction of ketones, aldehydes, and imines. Stryker, early on, demonstrated that the supporting ligands on LCuH species have a significant effect on the chemoselectivity (1,2- vs 1,4-reduction) (Scheme 5a). When a combination of dialkylarylphosphines and Stryker’s reagent was employed, 1,2-reduction of an α,β-unsaturated ketone was observed as the predominant product.7a,20 Shortly after the initial report, Lipshutz developed an improved
procedure to supplant the use of H2 with hydrosilane.21 The use of chiral bisphosphine ligands in this
catalytic system, including Roche’s MeO-BIPHEP (Scheme 5b) and Takasago’s SEGPHOS (Scheme 5c),
(a) O Me CO2Et Me O [(Ph3P)CuH]6 toluene, –23 °C Me HO CO2Et O Me 82% yield, 6:1 dr Ph Me O OMe O + CuF(PPh3)3 PhSiH3 toluene, –50 °C (R,S)-TANIAPHOS Ph OMe O Me Me OH 98% yield, 11:1 dr 97.5:2.5 er (b) Fe PCy2PCy2 Me2N (R,S)-TANIAPHOS (c) CuCl, NaOt-Bu (S)-p-tol-BINAP Ph2SiH2, toluene, 0 °C TBAT, BnBr DCM:toluene = 1:1, rt O Bn O Bn Bn 67% yield, 94:6 dr 97:3 er (d) CuCl, NaOt-Bu (S)-p-tol-BINAP
Ph2SiH2, toluene, 0 °C THF/pentane, rt O Bn O Bn 75% yield 97.5:2.5 er Pd(OAc)2, Johnphos, CsF t-Bu Br t-Bu (e) O
Me (Metoluene, rt2SiH)2O TiCl4 [(Ph3P)CuH]6 Me CHO O Me OH Me 72% yield
16
has enabled CuH chemistry to become a general method to deliver optically active alcohols by enantioselective reduction.22
The catalytic asymmetric reduction of imines is more challenging compared to the analogous transformation of carbonyl compounds, presumably due to the formation of a strong Cu–N bond following imine reduction and the fact that many imines exist as E/Z mixutures (and the rate of their interconversion is slow relative to the rate of their reduction). To address these issues, an electron-withdrawing di-(3,5-xylyl)phosphinyl group was installed on the nitrogen atom to facilitate the rapid reduction (Scheme 5d).23 Scheme 5. Representative examples of CuH-catalyzed asymmetric 1,2-reduction reactions.
1.3 CuH-Catalyzed Enantioselective Olefin Hydrofunctionalization Reactions
In recent years, the development of CuH-catalyzed transformations has expanded to include the asymmetric functionalization of olefins. The asymmetric hydroamination of alkenes has unveiled new
(a) Me Me Me Me O [(Ph3P)CuH]6, Me2PPh 500 psi H2 benzene, t-BuOH, rt Me Me Me Me OH 75% yield 1,2:1,4-selectivity = 49:1 (b) CuCl, NaOt-Bu (R)-3,5-xyl-MeO-BIPHEP PMHS, toluene, –50 or –78 °C Et O Et OH 95% yield, 97.5:2.5 er PAr2 PAr2 Ar = 3,5-Me-C6H3 (R)-3,5-xyl-MeO-BIPHEP MeO MeO (c) CuCl, NaOt-Bu (R)-DTBM-SEGPHOS PMHS, toluene, –50 °C Me O Me OH 85% yield, 97.5:2.5 er O O PAr2 PAr2 O O O O Ar = 4-MeO-3,5-t-Bu-C6H2 (R)-DTBM-SEGPHOS (d) Me N P(xylyl)2 O CuCl, NaOMe (R)-DTBM-SEGPHOS TMDS, t-BuOH, toluene, rt Me HN P(xylyl)2 O 99% yield, 98:2 er
17
reactivity of CuH catalysis. In 2013, the Miura24 and Buchwald25 group independently reported enantio-
and regioselective hydroamination reaction of styrenes (Scheme 6a). The key step in these transformations involved stereoselective Markovnikov insertion of a bisphosphine-ligated CuH species into the C–C double bond of styrenes (olefin hydrocupration), followed by the interception of the putative benzyl copper species by hydroxylamine esters (Scheme 6b). This formal hydroamination process allows for the generation of chiral trialkyl amines in a highly regio- and enantioselective manner under the mild conditions.
Scheme 6. CuH-catalyzed enantioselective olefin hydroamination reactions.
Further work was carried out to expand the types of electrophilic aminating reagents as well as the scope of alkene coupling partners. By carefully tuning the structure of hydroxylamine ester electrophiles and modifying reaction conditions, the successfully development of efficient procedures for the synthesis of tertiary aryl dialkyl26 and secondary amines was accomplished.27 Moreover, 1,2-benzisoxazole could
also be employed as an electrophilic nitrogen source for the preparation of enantioenriched primary amines (Scheme 6c).28 In addition to styrenes, a variety of olefin classes, including terminal olefins,25
1,1-disubstituted olefins,29 vinyl silanes,30 vinyl boronates,31 alkynes,32 strained,33 and unactivated internal + Bn2N OBz L*CuH Me NBn2 Conditions A (Miura) CuCl, (S,S)-Me-DuPhos PMHS, LiOt-Bu, THF, rt 65% yield, 87:13 er Conditions B (Buchwald) Cu(OAc)2, (R)-DTBM-SEGPHOS DEMS, THF, 40 °C 91% yield, 2:98 er (a) L*CuH L*CuOBz Ph olefin insertion (hydrocupration) electrophile interception R3SiH R3SiOBz transmetalation (catalyst regeneration) catalytic cycle Ph Me CuL* Bn2N OBz Ph Me NBn2 (b) Proposed mechanism (c) Ar N O R O NEt2 for primary amines: for secondary amines:
R N O H O NMe2 for aryldialkylamines: N O
18
olefins,34 could all be successfully converted to products in the hydroamination process and afford chiral
amines with high levels of enantiopurity.
Concurrently, this strategy was also applied to many additional asymmetric olefin hydrofunctionalization reactions by intercepting the in-situ generated alkyl copper intermediates with a variety of electrophiles (Scheme 7).35,36 In this context, readily available and stable olefins could be
employed as surrogates for relatively unstable and/or highly reactive carbon-based nucleophiles in the subsequent coupling reactions. The mild conditions of the CuH catalytic system usually lead to better functional group compatibility as compared to the corresponding reactions performed with stochiometric organometallic reagents. Moreover, the use of chiral ligands provides opportunities for the installation of stereogenic centers with high efficiency.
Scheme 7. CuH-catalyzed asymmetric olefin hydrofunctionalization reactions.
This dissertation focuses on the further development and applications of CuH catalysis in asymmetric transformations and heterocycle synthesis. Chapters 2–4 describe the development of CuH-catalyzed enantioselective transformations of α,β-unsaturated carboxylic acids for the synthesis of chiral ketones, aldehydes, and amides. Chapter 5 discusses an asymmetric hydroamidation reaction of vinylarenes using readily accessible 1,4,2-dioxazol-5-ones as the reactive electrophilic amidating reagents. In addition, Chapter 6 describes CuH-catalyzed stereo- and regioselective addition of allylcopper species to ketones by
Ph R R R2 R1 Ph Me R Me R R2 R1 CuL* CuL* CuL* CuL* Me CuL* L*CuH E X Ph Me R Me R R2 R1 E E E E Me E R3SiH
19
using allene gas as a surrogate for traditional allyl organometallic reagents. Finally, Chapter 7 discloses an efficient method to prepare polysubstituted pyrroles via a CuH-catalyzed enyne-nitrile coupling reaction.
20
1.4 References
(1) (a) Würtz, A. Compt. Rend. Hebd. Séances Acad. Sci. 1844, 18, 702. (b) Würtz, A. Compt. Rend. Hebd. Séances Acad. Sci. 1879, 89, 1066. (c) Würtz, A. Compt. Rend. Hebd. Séances Acad. Sci. 1880, 90, 22. (2) Whitesides, G. M.; San Filippo, J.; Stredronsky, E. R.; Casey, C. P. J. Am. Chem. Soc. 1969, 91, 6542. (3) (a) Brestensky, D. M.; Huseland, D. E.; McGettigan, C.; Stryker, J. M. Tetrahedron Lett. 1988, 29, 3749. (b) Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc. 1988, 110, 291. (c) Brestensky, D. M.; Stryker, J. M. Tetrahedron Lett. 1989, 30, 5677. For the first report of Stryker’s reagent synthesis, see: (d) Bezman, S. A.; Churchill, M. R.; Osborn, J. A.; Wormald, J. J. Am. Chem. Soc. 1971, 93, 2063. (4) For selected reviews, see: (a) Jordan, A. J.; Lalic, G.; Sadighi, J. P. Chem. Rev. 2016, 116, 8318. (b) Pirnot, M. T.; Wang, Y.-M.; Buchwald, S. L. Angew. Chem. Int. Ed. 2016, 55, 48. (c) Suess, A. M.; Lalic, G. Synlett 2016, 27, 1165. (d) Deutsch, C.; Krause, N.; Lipshutz, B. H. Chem. Rev. 2008, 108, 2916. (e) Rendler, S.; Oestreich, M. Angew. Chem. Int. Ed. 2007, 46, 498.
(5) (a) Chiu, P.; Li, Z.; Fung, K. C. M. Tetrahedron Lett. 2003, 44, 455. (b) Goeden, G. V.; Caulton, K. G. J. Am. Chem. Soc. 1981, 103, 7354.
(6) Lawrence, N. J.; Drew, M. D.; Bushell, S. M. J. Chem. Soc., Perkin Trans. 1 1999, 3381.
(7) (a) Chen, J.-X.; Daeuble, J. F.; Stryker, J. M. Tetrahedron 2000, 56, 2789. (b) Leung, L. T.; Leung, S. K.; Chiu, P. Org. Lett. 2005, 7, 5249.
(8) Lee, D.-W.; Yun, J. Tetrahedron Lett. 2005, 46, 2037.
(9) Appella, D. H.; Moritani, Y.; Shintani, R.; Ferreira, E. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9473.
(10) (a) Moritani, Y.; Appella, D. H.; Jurkauskas, V.; Buchwald, S. L. J. Am. Chem. Soc. 2000, 122, 6797. (b) Lipshutz, B. H.; Servesko, J. M. Angew. Chem. Int. Ed. 2003, 42, 4789. (b) Lipshutz, B. H.; Servesko, J. M.; Petersen, T. B.; Papa, P. P.; Lover, A. A. Org. Lett. 2004, 6, 1273. (d) Lipshutz, B. H.; Frieman, B. A.; Unger, J. B.; Nihan, D. M. Can. J. Chem. 2005, 83, 606.
(11) (a) Hughes, G.; Kimura, M.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 11253. (12) Rainka, M. P.; Milne, J. E.; Buchwald, S. L. Angew. Chem. Int. Ed. 2005, 44, 6177.
21
(13) (a) Llamas, T.; Arrayás, R. G.; Carretero, J. C. Angew. Chem. Int. Ed. 2007, 46, 3329. (b) Desrosiers, J.-N.; Charette, A. B. Angew. Chem. Int. Ed. 2007, 46, 5955.
(14) Saito, T.; Yokozawa, T.; Ishizaki, T.; Moroi, T.; Sayo, N.; Miura, T.; Kumbayashi, T. Adv. Synth. Catal. 2001, 343, 264.
(15) Blaser, H.-U.; Brieden, W.; Pugin, B.; Spindler, F.; Studer, M.; Togni, A. Top. Catal. 2002, 19, 3. (16) (a) Chiu, P.; Chen, B.; Cheng, K. F. Tetrahedron Lett. 1998, 39, 9229. (b) Deschamp, J.; Chuzel, O.; Hannedopuche, J.; Riant, O. Angew. Chem. Int. Ed. 2006, 45, 1292. (c) Chuzel, O.; Deschamp, J.; Chausteur, C.; Riant, O. Org. Lett. 2006, 8, 5943.
(17) Lipshutz, B. H.; Chrisman, W.; Noson, K.; Papa, P.; Scalfani, J. A.; Vivian, R. W.; Keith, J. M. Tetrahedron 2000, 56, 2779.
(18) Yun, J.; Buchwald, S. L. Org. Lett. 2001, 3, 1129.
(19) Chae, J.; Yun, J.; Buchwald, S. L. Org. Lett. 2004, 6, 4809.
(20) Chen, J.-X.; Daeuble, J. F.; Brestensky, D. M.; Stryker, J. M. Tetrahedron 2000, 56, 2153. (21) Lipshutz, B. H.; Chrisman, W.; Noson, K. J. Organomet. Chem. 2001, 624, 367.
(22) (a) Lipshutz, B. H.; Noson, K.; Chrisman, W. J. Am. Chem. Soc. 2001, 123, 12917. (b) Lipshutz, B. H.; Noson, K.; Chrisman, W.; Lower, A. J. Am. Chem. Soc. 2003, 125, 8779. (c) Lipshutz, B. H.; Lower, A.; Noson, K. Org. Lett. 2002, 4, 4045. (d) Moser, R.; Bošković, Ž. V.; Crowe, C. S.; Lipshutz, B. H. J. Am. Chem. Soc. 2010, 132, 7852. (e) Lee, D.-W.; Yun, J. Tetrahedron Lett. 2004, 45, 5415. (f) Sirol, S.; Courmarcel, J.; Mostefai, N.; Riant, O. Org. Lett. 2001, 3, 4111. (g) Albright, A.; Gawley, R. E. J. Am. Chem. Soc. 2011, 133, 19680.
(23) Lipshutz, B. H.; Shimizu, H. Angew. Chem. Int. Ed. 2004, 43, 2228.
(24) Miki, Y.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem. Int. Ed. 2013, 52, 10830. (25) Zhu, S.; Niljianskul, N.; Buchwald, S. L. J. Am. Chem. Soc. 2013, 135, 15746. (26) Ichikawa, S.; Zhu, S.; Buchwald, S. L. Angew. Chem. Int. Ed. 2018, 57, 8714. (27) Niu, D.; Buchwald, S. L. J. Am. Chem. Soc. 2015, 137, 9716.
22
(29) Zhu, S.; Buchwald, S. L. J. Am. Chem. Soc. 2014, 136, 15913.
(30) Niljianskul, N.; Zhu, S.; Buchwald, S. L. Angew. Chem. Int. Ed. 2015, 54, 1638. (31) Nishikawa, D.; Hirano, K.; Miura, M. J. Am. Chem. Soc. 2015, 137, 15620. (32) Shi, S.‐L.; Buchwald, S. L. Nat. Chem. 2015, 7, 38.
(33) Feng, S.; Hao, H.; Liu, P.; Buchwald, S. L. ACS Catal. 2020, 10, 282. (34) Yang, Y.; Shi, S.‐L.; Niu, D.; Liu, P.; Buchwald, S. L. Science 2015, 349, 62. (35) Liu, R. Y.; Buchwald, S. L. Acc. Chem. Res. 2020, 53, 1229.
(36) For selected examples, see: (a) Saxena, A.; Choi, B.; Lam, H. W. J. Am. Chem. Soc. 2012, 134, 8428. (b) Yang, Y.; Perry, I. B.; Lu, G.; Liu, P.; Buchwald, S. L. Science 2016, 353, 144. (c) Wang, Y.-M.; Buchwald, S. L. J. Am. Chem. Soc. 2016, 138, 5024. (d) Bandar, J. S.; Ascic, E.; Buchwald, S. L. J. Am. Chem. Soc. 2016, 138, 5821. (e) Friis, S. D.; Pirnot, M. T.; Buchwald, S. L. J. Am. Chem. Soc. 2016, 138, 8372. (f) Lee, J.; Torker, S.; Hoveyda, A. H. Angew. Chem. Int. Ed. 2017, 56, 821. (g) Xu, G.; Zhao, H.; Fu, B.; Cang, A.; Zhang, G.; Zhang, Q.; Xiong, T.; Zhang, Q. Angew. Chem. Int. Ed. 2017, 56, 13130. (h) Gui, Y.-Y.; Hu, N.; Chen, X.-W.; Liao, L.-L.; Ju, T.; Ye, J.-H.; Zhang, Z.; Li, J.; Yu, D.-G. J. Am. Chem. Soc. 2017, 139, 17011. (i) Gribble, M. W., Jr.; Guo, S.; Buchwald, S. L. J. Am. Chem. Soc. 2018, 140, 5057. (j) Ye, Y.; Kim, S.-T.; Jeong, J.; Baik, M.-H.; Buchwald, S. L. J. Am. Chem. Soc. 2019, 141, 3901. (k) Schuppe, A. W.; Borrajo-Calleja, G. M.; Buchwald, S. L. J. Am. Chem. Soc. 2019, 141, 18668.
23
Chapter 2. Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated
Carboxylic Acids as Aldehyde Surrogates
24
2.1 Introduction
Chiral α-aryl ketones represent an important functional group due to their synthetic utility and common
occurrence in molecules of broad interest.1 While a number of catalytic methods exist for preparing enantioenriched quaternary α-aryl ketones2, catalytic assembly of acyclic tertiary variants via C–C bond formation is made challenging by the acidic nature of the stereocenter.3,4 This has recently been addressed
via transition metal-catalyzed asymmetric coupling reactions of α-bromo ketones to aryl organometallic
reagents5a,b, benzylic bromides to acyl chlorides5c, and benzylic zinc reagents to thioesters5d; additionally, chiral Lewis acid-catalyzed insertion of aryldiazoalkanes into aldehyde C–H bonds has also been described.5e Despite this progress, complementary catalytic methods for preparing chiral tertiary α-aryl dialkyl ketones from abundant and stable functional groups remain in high demand.
Hydroacylation, typically achieved by the addition of an aldehyde C–H bond across an alkene π-bond,
is a streamlined approach for the construction of ketones from readily available functional groups.6 While numerous enantioselective intramolecular hydroacylation processes have been reported, intermolecular variants remain less developed.7 The primary challenge associated with this transformation is suppressing aldehyde decarbonylation, which often occurs readily for substrates without a coordinating substituent.8 Nonetheless, impressive examples of enantioselective intermolecular hydroacylation have been reported using substrates of broad value that feature a coordinating substituent (e.g. salicylaldehydes,
2-(methylthio)benzaldehydes and α-substituted acrylamides).9 Meanwhile, the branch-selective addition of
simple aliphatic aldehydes to styrenes provides direct access to dialkyl ketones bearing an α-aryl
stereocenter, although only racemic methods for this transformation have been reported (Scheme 1a).10 To avoid the problems associated with aldehyde decarbonylation and stereocenter epimerization, we reasoned that a complementary approach toward hydroacylation could potentially provide rapid access to enantioenriched tertiary α-aryl dialkyl ketones.
25
Scheme 1. Previous work in hydroacylation using (a) aldehydes, (b) aryl anhydrides, and (c, this work) α,β-unsaturated
carboxylic acids to access chiral α-aryl ketones.
As part of a broader program using chiral copper hydride (CuH) species as catalysts for enantioselective hydrofunctionalization reactions11, we sought to utilize acyl electrophiles as surrogates for aldehydes in order to address limitations of existing hydroacylation methodology. Inspired by prior work by Miura12a and Krische12b, we recently reported a CuH-catalyzed method for coupling styrenes to symmetrical aryl anhydrides to afford chiral α-aryl ketones or, after concomitant 1,2-reduction, chiral
alcohols (Scheme 1b).13 In order to expand the synthetic utility of this hydroacylation strategy, we recognized the need to develop an improved method that avoids use of symmetrical anhydrides and, importantly, employs aliphatic aldehyde surrogates. We herein report the use of α,β-unsaturated carboxylic
acids as direct coupling partners in a CuH-catalyzed dual hydroacylation and reduction process that provides access to highly enantioenriched tertiary α-aryl dialkyl ketones (Scheme 1c).
2.2 Results and Discussion
In CuH-catalyzed hydrofunctionalization reactions, chemoselective hydrocupration of an olefin in the presence of an electrophilic coupling partner is required in order to obtain high product yields.14 This requisite is realized when using aryl anhydride coupling partners (Scheme 1b); however, no hydroacylation of 4-fluorostyrene was seen when butyric anhydride, an aliphatic anhydride (1a), was subjected to the
Ar +
(a) Typical approach to hydroacylation using aliphatic aldehydes
O H R metal catalyst O R Me Ar
(b) Our previous work using aryl anhydrides as aldehyde surrogates
Ar2 Ar1 Me O Ar2 R3SiH L*CuH + Ar1 O O Ar1 O Ar1 Me OH Ar2 or
(c) α,β-Unsaturated acid electrophiles for hydroacylation (this work)
R1 OH O + Ar R 3SiH L*CuH O R1 Ar R2 R2
hydroacylation and 1,2-reduction
hydroacylation and 1,4-reduction
26
previously reported reaction conditions (Scheme 2a). Instead, only direct reduction of butyric anhydride to
n-butanol was observed. We hypothesized that reactions of α,β-unsaturated anhydrides may display similar
chemoselectivity as with aryl anhydrides to allow for styrenyl hydroacylation, and with concomitant
1,4-reduction of the unsaturated carbonyl, would produce chiral α-aryl dialkyl ketones. This hypothesis was
validated when crotonic anhydride (1b) was subjected to the reaction conditions, which provided chiral ketone 3a in 70% yield and 97:3 er after treatment with NH4F. In the course of searching for acyl electrophile alternatives to anhydrides, we discovered that α,β-unsaturated carboxylic acids directly engage
in this hydroacylation process. Thus, the use of crotonic acid (1c) and 8.0 equiv of silane provided ketone
3a in 85% yield and 94:6 er.
Scheme 2. (a) Discovery and (b) SAR of CuH-catalyzed tandem hydroacylation and 1,4-reduction with unsaturated
acyl electrophiles.a
a Yields determined by 1H NMR analysis of crude reaction mixture. (S,S)-Ph-BPE = 1,2-Bis((2S,5S)-2,5-diphenylphospholano)ethane.
This finding prompted a structure-activity relationship (SAR) study of the electrophilic acyl species (Scheme 2b). First, no coupling was observed when butanoic acid (1d) or its silyl ester 1e were used, clearly
O O O Me Me R X O O Me Me F 1, acyl electrophile Cu(OAc)2 (4.0 mol %) (S,S)-Ph-BPE (4.4 mol %) THF, rt, 24 h (NH4F workup) + 2, Ar = 4-FC6H4 O O O Me Me OH O Me 0% yield 1a 70% yield, 97:3 er 1b
a) Initial discovery of asymmetric hydroacylation using enoic acids
85% yield, 94:6 er
1c b) Structure-activity relationship study of acyl electrophiles
Cl O Me OR O Me H O Me OH O Me OSiMe(OMe)2 O Me 0% yield 1d 0% yield 1g 0% yield R = Me, 1h 0% yield 1f 1j 20% yield, 90:10 er Ar 3a
Me(OMe)2SiH (4.0 equiv)
(1.5 equiv) OSiMe(OMe)2 O Me 1e 0% yield 0% yield R = tBu, 1i
27
indicating the requirement for α,β-unsaturation in this process. Crotonaldehyde (1f) also provided no
product, which suggests reduction of the acid to an aldehyde does not occur prior to hydroacylation. Additionally, other crotonoyl-based electrophiles, such as crotonoyl chloride (1g) or alkyl crotonates (1h and 1i), provided no hydroacylation product. On the other hand, the use of methyl(dimethoxy)silyl crotonate (1j) led to 20% yield of ketone 3a in 90:10 er, suggesting silylated crotonic acid may be an active intermediate in the direct coupling of acids.
Based on the above observations and previous studies on CuH chemistry, we are currently able to propose the reaction pathway outlined in Scheme 3.15 First, carboxylic acid deprotonation and silylation is catalyzed by CuH in the presence of a hydrosilane, generating activated acyl electrophile 4 (Step A).16 Additionally, enantioselective hydrocupration of a styrene generates a chiral copper(I) benzylic intermediate (5, Step B), which represents the active nucleophilic partner. The reaction ultimately produces silyl enol ether 6, which is observed in high yield as judged by 1H NMR spectroscopy of the crude reaction mixture and allows access to chiral ketone 3 upon treatment with NH4F.17 At this time, the mechanism by which chiral benzyl copper 5 and the silylated acid 4 couple (Step C), and if other reaction intermediates are involved, is unknown and is the subject of ongoing investigations.18,19
Scheme 3. Currently proposed pathway for hydroacylation.
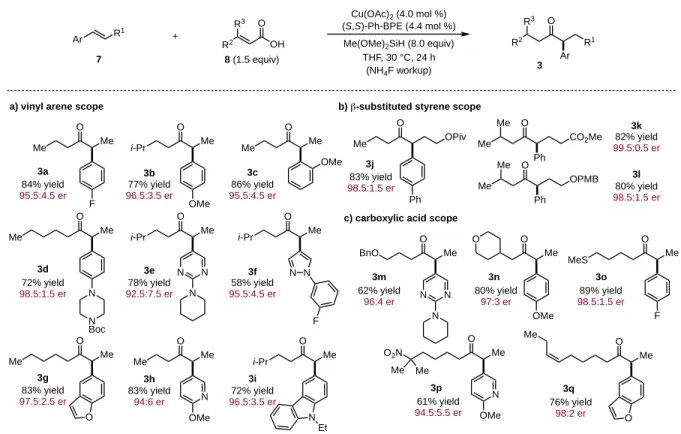
The current scope of accessible chiral α-aryl ketones through the CuH-catalyzed coupling of
α,β-unsaturated acids to aryl alkenes is shown in Table 1.20a First, the vinyl arene scope was explored using simple unsaturated carboxylic acid coupling partners (Table 1a). Electron deficient and electron rich, as well as ortho-substituted styrenes, provide chiral ketones in excellent yield and enantioselectivity (3a-c). A variety of alkenes containing coordinating atoms or consisting of heterocycles, such as a piperizine,
Ar L*Cu Me Ar OH O R O[Si] O R L*CuH L*CuH [Si]–H R O[Si] Ar Me Step B Step A Step C Step A: Step B: Step C: enantioselective hydrocupration acid silylation (activation)
hydroacylation and 1,4-reduction (mechanistic studies ongoing)
4
5
28
pyrimidine, pyrazole, pyridine, benzofuran and carbazole, engage in this hydroacylation process delivering ketones in high enantiopurity (3d-i). In contrast to our previous hydroacylation method utilizing aryl anhydride reagents, we found that β-substituted styrenes couple to unsaturated acids in both high yield and
enantioselectivity (Table 1b).20bβ-substituted styrenes containing functional groups that could potentially be reduced by reactive CuH intermediates, such as a t-butyl ester (3j) and methyl ester (3k), are well tolerated, as is a PMB-protected alcohol (3l). Carboxylic acid substrates containing ethers (3m and 3n), a thioether (3o) and potentially-reducible functional groups, such as a nitro group (3p) and a cis-alkene (3q), couple to styrenes in excellent yield and stereoselectivity (Table 1c).
Table 1. Ketone substrate scope for the direct coupling of aryl alkenes to α,β-unsaturated carboxylic acids.a
a All yields represent average isolated yields of two runs, performed with 1 mmol of alkene.
The substrates shown in Table 1 demonstrate that β-alkyl- and symmetrical β,β-dialkyl-substituted unsaturated carboxylic acids are competent hydroacylation coupling partners for a variety of styrene types.20c α-Alkyl substituted unsaturated acids, such as tiglic or angelic acid, do not engage in
7 O R2 Ar Cu(OAc)2 (4.0 mol %) (S,S)-Ph-BPE (4.4 mol %) Me(OMe)2SiH (8.0 equiv)
THF, 30 °C, 24 h (NH4F workup) 3 R1 R2 OH O + Ar R 1 R 3 R3 Me O Me F Me O Me O Me OMe OMe 3a 3b 3c 84% yield 95.5:4.5 er 86% yield 95.5:4.5 er 77% yield 96.5:3.5 er O Me i-Pr O Me i-Pr O Me N N N 3d 3e 3f 72% yield 98.5:1.5 er 58% yield 95.5:4.5 er 78% yield 92.5:7.5 er N N Boc Me N N F Me O Me N i-Pr O Me 3h 3i 83% yield 94:6 er 72% yield 96.5:3.5 er OMe O Me 3g 83% yield 97.5:2.5 er Me O N Et O Ph 3l 80% yield 98.5:1.5 er OPMB Me O Ph 3k 82% yield 99.5:0.5 er CO2Me Me O 3j 83% yield 98.5:1.5 er OPiv Ph i-Pr 3n O O Me OMe 80% yield 97:3 er O Me F 89% yield 98.5:1.5 er MeS O Me N N N 62% yield 96:4 er BnO 3o 3m O Me O O2N Me O 3q 76% yield 98:2 er Me Me Me N OMe 3p 61% yield 94.5:5.5 er 8 (1.5 equiv)
a) vinyl arene scope b) β-substituted styrene scope
c) carboxylic acid scope
Me Me Me
29
hydroacylation under the current reaction conditions. Similarly, acrylic acid, an unsubstituted unsaturated carboxylic acid that would provide access to ethyl ketones, does not participate in this transformation (Scheme 4a). To address this limitation, we found that β-ethoxyacrylic acid (9) acts as an acrylic acid surrogate to deliver chiral ethyl ketones in good yield and enantiopurity (Scheme 4b). This approach, which we propose proceeds via a 1,4-reduction/β-ethoxy elimination pathway, is general as shown for ortho-substituted substrate 10a and β-ortho-substituted styrene-derived ketone 10b.
Scheme 4. The use of (a) acrylic acid and (b) β-ethoxyacrylic acid to access chiral ethyl ketones.
To demonstrate the scalability of this methodology, a 10 mmol scale reaction using just 1 mol % catalyst loading is shown in Eq (1) to provide 1.5 g of chiral ketone 3a with 96:4 er.
EtO OH O + conditions as above Me O Ph OPiv 60% yield 97.5:2.5 er 10b 10 OH O Ar Me O Me Ar + Cu(OAc)2 (4.0 mol %) (S,S)-Ph-BPE (4.4 mol %) Me(OMe)2SiH (8.0 equiv)
THF, 30 °C, 24 h (NH4F workup)
acrylic acid
no product
a) No coupling observed using acrylic acid electrophile
b) Utilizing β-ethoxyacrylic acid to access chiral ethyl ketones
Ar O Me Ar R R Me O Me 66% yield 97.5:2.5 er 10a OMe 9 (1.5 equiv) 7 O Me Me F Cu(OAc)2 (1.0 mol %) (S,S)-Ph-BPE (1.1 mol %) Me(OMe)2SiH (8.0 equiv)
THF, 30 °C, 48 h (NH4F workup) 1.5 g 76% yield 96:4 er OH O Me + F 2, 10 mmol (1) 3a (1.5 equiv)
30
2.3 Conclusion
Given the ready accessibility of α,β-unsaturated acids and aryl alkenes, this method represents a highly
practical approach to chiral α-aryl dialkyl ketones containing a range of functional groups and substitution
patterns.21 Furthermore, by utilizing in situ activated acyl electrophiles as surrogates for aldehydes, this work addresses current substrate limitations of existing hydroacylation methodogy. The mechanism of this unprecedented coupling reaction is currently under investigation, as are additional CuH-catalyzed transformations involving unsaturated carboxylic acid substrates.
31
2.4 Experimental
General reagent information. All reactions were performed under a nitrogen atmosphere using the
indicated method in the general procedures. Tetrahydrofuran (THF) was dried and deoxygenated by passage through two packed columns of neutral alumina and copper(II) oxide under a positive pressure of argon. Copper(II) acetate was purchased from Strem and was used as received. 1,2-Bis((2S,5S)2,5-diphenylphospholano)ethane and 1,2-Bis((2R,5R)2,5-1,2-Bis((2S,5S)2,5-diphenylphospholano)ethane (Ph-BPE) ligands were purchased from Strem Chemicals Inc. and Sigma Aldrich Co. and stored in a nitrogen-filled glove box. Dimethoxy(methyl)silane (DMMS) was purchased from Tokyo Chemical Industry Co. (TCI) and stored in a nitrogen-filled glove box at −20 °C for long term storage. All other solvents and commercial reagents were used as received from Sigma Aldrich, Alfa Aesar, Acros Organics, TCI and Combi-Blocks, unless otherwise noted. Flash column chromatography was performed using 40-63 µm silica gel (SiliaFlash® F60 from Silicycle), or with the aid of a Biotage Isolera Automated Flash Chromatography System using prepacked SNAP silica cartridges (10-100 g). Organic solutions were concentrated using a Buchi rotary evaporator.
General analytical information. All new compounds were characterized by NMR spectroscopy, IR
spectroscopy, elemental analysis, optical rotation and melting point analysis (if solids). 1H, 13C and 19F
NMR spectra were recorded in CDCl3 on a Bruker AMX-400 spectrometer. Chemical shifts for 1H NMR
are reported as follows: chemical shift in reference to residual CHCl3 at 7.26 ppm (δ ppm), multiplicity (s
= singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, td = triplet of doublets, m = multiplet), coupling constant (Hz), and integration. Chemical shifts for 13C NMR are reported
in terms of chemical shift in reference to the CDCl3 solvent signal (77.16 ppm). Chemical shifts for 19
F-NMR are reported in terms of chemical shift in reference to an external standard (α,α,α-trifluorotoluene set to d −63.7 ppm). IR spectra were recorded on a Thermo Scientific Nicolet iS5 spectrometer (iD5 ATR, diamond) and are reported in terms of frequency of absorption (cm-1). Melting points were
32
P-1010 digital polarimeter. Elemental analyses were performed by Atlantic Microlabs Inc., Norcross, GA. ESI- and DART-MS spectrometric data were recorded on a Bruker Daltonics APEXIV 4.7 Tesla Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR-MS). Enantiomeric ratios (er’s) were determined either by chiral HPLC analysis using Agilent 1200 Series chromatographs or by chiral SFC analysis using a Waters Acquity UPC2 instrument; specific columns and analytical methods are provided in the experimental details for individual compounds; the wavelengths of light used for chiral analyses are provided with the associated chromatograms. Thin-layer chromatography (TLC) was performed on silica gel 60Å F254 plates (SiliaPlate from Silicycle) and visualized with UV light or potassium permanganate
stain. Preparatory thin-layer chromatography (Prep-TLC) was performed on silica gel GF with UV 254 (20 x 20 cm, 1000 microns, catalog # 02013 from Analtech) and visualized with UV light. Isolated yields reported in Tables 2 and 3 reflect the average values from two independent runs.
2.4.1 Acyl electrophile study for hydroacylation reactions
General procedure. In a nitrogen-filled glove box, a catalyst stock solution was prepared by adding
Cu(OAc)2 (19.8 mg, 0.11 mmol), (S,S)-Ph-BPE (61.6 mg, 0.12 mmol), and THF (5.5 mL) to an oven-dried
25 mL round bottom flask containing a magnetic stir bar. This mixture was stirred for 10 min as a homogenous blue solution formed. At this time, dimethoxy(methyl)silane (1.35 mL, 11 mmol) was added and the solution was stirred for another 5 min, during which time the solution developed a bright yellow color. An aliquot of this solution (623 μL, corresponds to 4 mol% catalyst loading) was then transferred to a separate reaction vial (Fisherbrand, 13 x 100 mm, catalog no. 1495925A) containing 4-fluorostyrene (30 μL, 0.25 mmol, 1.0 equiv) and acyl electrophile (0.375 mmol, 1.5 equiv). The reaction tube was capped (Thermo Scientific 13 mm screw cap with TEF/SIL septa, catalog no. C4015-66A), removed from the glove
X R O + Cu(OAc)2 (4.0 mol %) (S,S)-Ph-BPE (4.4 mol %) DMMS (4.0 equiv) THF (0.50 M), rt, 24 h (1.5 equiv) 3a (NH4F workup) 1 2 F Me O Me F Me OSiMe(OMe)2 Me F 1H NMR observed product before NH
33
box, and stirred for 24 h at rt. Water (3 mL) was added to quench the reaction and the mixture was extracted with Et2O (3 x 2 mL). The combined extracted organic solution was dried with anhydrous Na2SO4, filtered
and concentrated in vacuo. A stock solution (1 mL) of 1,1,2,2-tetrachloroethane in CDCl3 (0.25 mmol, 1.0
equiv per mL of solution) was added to dissolve the crude residue. 1H NMR analysis of the solution was
used to determine the product yield by comparison to 1,1,2,2-tetrachloroethane standard using the chemical shift of silyl enol ether observed at 4.60 ppm.
For ee analysis, saturated NH4F in methanol was added to the crude reaction residue and the mixture
was stirred for 30 min. The mixture was concentrated in vacuo. The resulting residue was redissolved in EtOAc, and filtered through a plug of silica gel with washing additional EtOAc. The collected EtOAc solution was concentrated in vacuo, and the crude material was purified by preparative thin layer chromatography to yield the title ketone product as a colorless oil (10% EtOAc in hexane). HPLC analysis: Chiralpak OJ-H (Hex/IPA = 99/1, 1 mL/min, 220 nm, 23°C), 6.69 min (minor), 7.47 min (major).
Electrophile Amount used Product yield Selectivity (er)
1a 49 μL 0% - 1b 56 μL 70% 97:3 1c* 32 mg 85% 94:6 1d 28 μL 0% - 1e** 72 mg 0% - 1f 31 μL 0% - 1g 36 μL 0% - 1h 40 μL 0% - 1i 59 μL 0% - 1j** 71 mg 20% 90:10
*8 equiv of DMMS used; ** see below for preparation of 1e and 1j.
2.4.2 Procedures for CuH-catalyzed hydroacylation reactions General Procedure for 1 mmol Scale Reaction.
O O O Me Me O O O Me Me OH O Me 1a 1b 1c Cl O Me OMe O Me OtBu O Me H O Me OH O Me OSiMe(OMe)2 O Me 1d 1g 1h 1i 1f 1j OSiMe(OMe)2 O Me 1e
34
An oven-dried reaction tube (Fisherbrand, 20 x 125 mm, catalog no. 1495925C) containing a magnetic stir bar was charged with alkene (if a solid, 1.0 mmol, 1.0 equiv) and unsaturated carboxylic acid (if a solid, 1.5 mmol, 1.5 equiv). The reaction tube was loosely capped (Cap: Kimble Chase Open Top S/T Closure catalog no. 73804-15425; Septum: Thermo Scientific 1.3 mm silicone/PTFE catalog no. B7995-18) and then brought into a nitrogen-filled glove box. If the alkene or the acid was a liquid, they were added via pipette at this time. To another reaction tube (same type as above) in the glove box was added Cu(OAc)2
(7.3 mg, 0.04 mmol, 0.04 equiv) and (S,S)-Ph-BPE (22.3 mg, 0.044 mmol, 0.044 equiv), followed by the addition of THF (2 mL) via syringe. The resulting mixture was stirred at rt for 15 min until a deep blue-colored homogenous solution formed. Dimethoxymethylsilane (DMMS, 0.99 mL, 8.0 mmol, 8.0 equiv) was then added via a 1 mL syringe and the resulting solution was stirred at rt for an additional 2 min as the solution turned to a yellow color. The resulting yellow solution was then transferred in one portion to the reaction tube containing the alkene and acid via a syringe (Caution: gas evolution observed). The reaction tube was capped after the bubbling stopped, and then was removed from the glove box. The cap was wrapped in parafilm, and the reaction tube was inserted into an oil bath preheated to 30 °C. The reaction solution was stirred for 24 h, and then removed from the oil bath and allowed to cool to rt, at which time 15 mL sat. NH4F in MeOH was slowly added to quench the reaction (Caution: gas evolution observed).
The mixture was stirred for 30 min, transferred to a 100 mL round bottom flask, and concentrated in vacuo. The resulting residue was redissolved in EtOAc, and filtered through a plug of silica gel with washing additional EtOAc. The collected EtOAc solution was concentrated in vacuo, and the crude material was immediately added to a chromatography column and purified by silica gel column chromatography or Biotage Isolera Automated Flash Chromatography System. (Note: If not purified immediately, the crude material will form an insoluble gel upon standing making purification more difficult.)
Large-scale synthesis of 3a. This example was conducted according to general procedure on 10 mmol O
R2
Ar Cu(OAc)2 (4.0 mol %)
(S,S)-Ph-BPE (4.4 mol %) Me(OMe)2SiH (8.0 equiv)
THF, 30 °C, 24 h R1 R2 OH O + Ar R 1 R3 R3 (1.5 equiv)
35
scale with a reduced catalyst loading. An oven-dried 100 mL round bottom flask containing a magnetic stir bar was charged with crotonic acid (1.29 g, 15.0 mmol, 1.5 equiv). The reaction flask was then brought into a nitrogen-filled glove box. 4-Fluorostyrene (1.22 g, 10.0 mmol, 1.0 equiv) was added via pipette at this time. To another 50 mL round bottom flask in the glove box was added Cu(OAc)2 (18.2 mg, 0.10 mmol,
0.010 equiv) and (S,S)-Ph-BPE (55.8 mg, 0.11 mmol, 0.011 equiv), followed by the addition of THF (20 mL) via syringe. The resulting mixture was stirred at rt for 15 min until a deep blue-colored homogenous solution formed. Dimethoxymethylsilane (DMMS, 9.84 mL, 80 mmol, 8.0 equiv) was then added via a 12 mL syringe and the resulting solution was stirred at rt for an additional 5 min as the solution turned to a yellow color. The resulting yellow solution was then transferred in one portion to the flask containing the alkene and acid via a syringe (Caution: gas evolution observed). The reaction flask was capped after the bubbling stopped, and then was removed from the glove box. The cap was wrapped in parafilm, and the reaction tube was inserted into an oil bath preheated to 30 °C. The reaction solution was stirred for 48 h, and then removed from the oil bath and allowed to cool to rt, at which time 50 mL sat. NH4F in MeOH was
slowly added to quench the reaction (Caution: gas evolution observed). The mixture was stirred for 30 min, transferred to a 250 mL round bottom flask, and concentrated in vacuo. The resulting residue was redissolved in EtOAc, and filtered through a plug of silica gel with washing additional EtOAc. The collected EtOAc solution was concentrated in vacuo, and the crude material was immediately added to a chromatography column and purified by silica gel column chromatography. (Note: If not purified immediately, the crude material will form an insoluble gel upon standing making purification more difficult.)
(S)-2-(4-fluorophenyl)hexan-3-one (3a)
The general procedure was followed using 4-fluorostyrene (122 mg, 1.0 mmol, 1.0 equiv) and crotonic acid (129 mg, 1.5 mmol, 1.5 equiv). Silica gel column chromatography (0 to 10% Et2O in hexanes) yielded the title product as a colorless oil (Run 1: 166 mg, 85%
yield, 95.5:4.5 er; Run 2: 162 mg, 83% yield, 95:5 er; 10 mmol scale: 1.48 g, 76% yield, 96:4 er). 1H NMR (400 MHz, CDCl 3) δ 7.16–7.19 (m, 2H), 6.99–7.03 (m, 2H), 3.74 (q, J = 6.8 Hz, 1H), Me O Me F
36
2.32 (t, J = 7.2 Hz, 2H), 1.46–1.57 (m, 2H), 1.37 (d, J = 7.2 Hz, 3H), 0.79 (t, J = 7.2 Hz, 3H); 13C NMR
(101 MHz, CDCl3) δ 210.7, 162.0 (d, J = 244.0 Hz), 136.6 (d, J = 3.0 Hz), 129.5 (d, J = 7.0 Hz), 115.8 (d,
J = 21.0 Hz), 52.2, 43.1, 17.7, 17.3, 13.7; 19F NMR (376 MHz, CDCl
3) δ –116.7. EA Calcd. for C12H15FO:
C, 74.20; H, 7.78. Found: C, 74.47; H, 7.79. IR (neat): 1712, 1508, 1222, 1159, 1014, 836 cm-1. Specific rotation [α]D23: +221 (c = 1.0, CHCl3). HPLC analysis: Chiralpak OJ-H (Hex/IPA = 99/1, 1.0 mL/min, 220
nm, 23°C), 6.69 min (minor), 7.47 min (major), 95.5:4.5 er.
(S)-2-(4-methoxyphenyl)-6-methylheptan-3-one (3b)
The general procedure was followed using 4-methoxystyrene (134 mg, 1.0 mmol, 1.0 equiv) and 4-methyl-2-pentenoic acid (171 mg, 1.5 mmol, 1.5 equiv). Silica gel column chromatography (0 to 10% Et2O in hexanes) yielded the title product as a colorless oil (Run 1: 178 mg, 76%
yield, 96.5:3.5 er; Run 2: 181 mg, 77% yield, 96:4 er). 1H NMR (400 MHz, CDCl 3) δ
7.10–7.15 (m, 2H), 6.84–6.88 (m, 2H), 3.79 (s, 3H), 3.71 (q, J = 6.8 Hz, 1H), 2.31–2.36 (m, 2H), 1.33–1.43 (m, 6H),0.76-0.80 (two doublets, 6H); 13C NMR (101 MHz, CDCl
3) δ 211.6, 158.8, 133.0, 129.0, 114.4,
55.4, 52.2, 39.1, 32.9, 27.7, 22.5, 22.3, 17.7. EA Calcd. for C15H22O2: C, 76.88; H, 9.46. Found: C, 76.78;
H, 9.39. IR (neat): 1710, 1510, 1246, 1177, 1034, 834 cm-1. Specific rotation [α]
D23: +208 (c = 1.0, CHCl3). SFC analysis: AD-H (5:95 IPA: scCO2 to 10:90 IPA: scCO2 linear gradient over 6 min with 1 min hold
time), 2.44 min (major), 2.71 min (minor), 96:4 er.
(S)-2-(2-methoxyphenyl)hexan-3-one (3c)
The general procedure was followed using 2-methoxystyrene (134 mg, 1.0 mmol, 1.0 equiv) and crotonic acid (129 mg, 1.5 mmol, 1.5 equiv). Silica gel column chromatography (0 to 10% Et2O in hexanes) yielded the title product as a colorless oil
(Run 1: 182 mg, 88% yield, 95:5 er; Run 2: 174 mg, 84% yield, 95.5:4.5 er). 1H NMR (400 MHz, CDCl 3) δ 7.24 (ddd, J = 8.4 Hz, J = 7.6 Hz, J = 2.0 Hz, 1H), 7.11 (dd, J = 7.6 Hz, J = 1.6 Hz, 1H), 6.93 (td, J = 7.6 Hz, J = 1.2 Hz, 1H), 6.88 (dd, J = 8.0 Hz, J = 0.8 Hz, 1H), 4.08 (q, J = 7.2 Hz, 1H), 3.83 (s, 3H), 2.30 (t, J O Me OMe i-Pr Me O Me OMe