Publisher’s version / Version de l'éditeur:
The Journal of Physical Chemistry C, 121, 16, pp. 8832-8840, 2017-04-03
READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. https://nrc-publications.canada.ca/eng/copyright
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la
première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca.
Questions? Contact the NRC Publications Archive team at
PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca. If you wish to email the authors directly, please see the first page of the publication for their contact information.
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
For the publisher’s version, please access the DOI link below./ Pour consulter la version de l’éditeur, utilisez le lien DOI ci-dessous.
https://doi.org/10.1021/acs.jpcc.7b00218
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Molecular dynamics study of guest–host hydrogen bonding in ethylene
oxide, trimethylene oxide, and formaldehyde structure I clathrate
hydrates
Mohammadi-Manesh, Hossein; Ghafari, Hakime; Alavi, Saman
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
NRC Publications Record / Notice d'Archives des publications de CNRC:
https://nrc-publications.canada.ca/eng/view/object/?id=e00f53e5-fbdb-4c06-bc32-524e96ef79d3 https://publications-cnrc.canada.ca/fra/voir/objet/?id=e00f53e5-fbdb-4c06-bc32-524e96ef79d3Molecular Dynamics Study of Guest−Host Hydrogen Bonding in
Ethylene Oxide, Trimethylene Oxide, and Formaldehyde Structure I
Clathrate Hydrates
Hossein Mohammadi-Manesh,
*
,†Hakime Ghafari,
†and Saman Alavi
*
,‡,§†
Department of Chemistry, Yazd University, Yazd, Iran
‡
Department of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, Ontario, Canada K1N 6N5
§
National Research Council of Canada, 100 Sussex Drive, Ottawa, Ontario, Canada K1A 0R6
*
S Supporting InformationABSTRACT: Molecular dynamics simulations are used to study the guest−host hydrogen bonding and guest dynamics in the clathrate hydrate phases of trimethylene oxide (TMO), ethylene oxide (EO), and formaldehyde (FA) as polar guests. Two water models, the SPC/E and TIP4P/ice, were used in the simulations. Binary hydrates are constructed with these guests in the large 14-sided cages and methane placed in the small 12-sided cages of the structure sI clathrate hydrates. The results of these simulations are compared with the nonpolar guests with analogous structures, cyclobutane (CB), cyclopropane (CP), and ethane, respectively, for TMO, EO, and FA. These simulations show that oxygen atoms of the cyclic ethers and carbonyl oxygen of formaldehyde form hydrogen bonds to differing degrees with cage water hydrogen atoms. The TMO size is larger than that of the EO and FA molecules and the closer proximity of the atoms of this large guest to the cage water molecule enhances the probability of guest−host hydrogen bonding in the TMO
clathrate. The TMO oxygen atom is tethered by a lattice water hydrogen atom by hydrogen bonding for longer times, which reduces the range of translational and rotational motions of the TMO guests in the cages compared to EO and FA. The effect of guest−host hydrogen bond formation is the insertion of Bjerrum L-defects in the clathrate water lattice. We consider guest dynamic properties such as velocity autocorrelation function (VACF) and orientational autocorrelation function (OACF) of the different guests in these hydrate phases and compare the hydrogen bonding and non-hydrogen bonding analogues. We discuss some other potential experimentally observable effects of the guest−host hydrogen bonding on the guest and water dynamics in the corresponding clathrate hydrates.
1. INTRODUCTION
Clathrate hydrates are nonstoichiometric crystalline solid solutions composed of a water framework arranged to produce cage structures which encapsulate various gas or liquid species. The guest molecules are trapped in a host framework composed of cavities formed by hydrogen-bonded water molecules.1Clathrate hydrates have been the subject of much study as methane and other natural gas clathrate hydrates have been found in large quantities in ocean sediments and under permafrost regions and can be potentially exploited for a number of practical applications.2 Clathrate hydrate has been
commonly classified according to two criteria: the structure of the water host lattice and the chemical nature of guest molecules. Clathrate hydrates have three common (canonical) framework structures. The clathrate hydrate structure relevant to this work is the cubic structure I (sI or CS-I) which contains two small 12-sided cages (pentagonal dodecahedral, D; 512), six
large 14-sided cages (tetrakaidecahedral, T; 51262), and 48
water molecules per unit cell. The unit cell of the sI phase is shown in Figure S1 of the Supporting Information. Other clathrate hydrate structures are the cubic structure II (sII or
CS-II) which contains 16 D cages, 8 large 16-sided cages (hexakaidecahedral, H; 51264) and 136 water molecules per
unit cell, and hexagonal structure H (sH) hydrates which contain 3 small cages (512), 2 medium cages (435663), 1 large
cage (51268), and 34 water molecules.3
The open structure of the clathrate hydrate is stabilized by the van der Waals interactions of the guest species which occupy the cages and interact with cage walls.
With regard to the nature of the guest species, some soluble guest species form clathrate hydrates, while other water-soluble species are inhibitors of clathrate hydrate formation.4 Jeffrey and McMullan divided clathrate hydrate forming guest molecules into the four groups: hydrophobic compounds, water-soluble acidogenic gases, water-soluble polar compounds, and water-soluble ternary or quaternary alkyl-onium salts.5,6 Guest molecule size and shape are known to affect clathrate formation and decomposition conditions.7 Guest polarity and
Received: January 8, 2017
Revised: April 1, 2017
Published: April 3, 2017
guest−host hydrogen bonding also affect stability ranges of the clathrate hydrate phase, with clathrate hydrates of hydrogen bonding guest molecules decomposing at lower temperatures.8 The nature of the guests in clathrate hydrate phases also affects the dielectric relaxation and host water dynamics.9The hydrate host lattices for dipolar guests have smaller dielectric relaxation times and smaller dielectric relaxation activation energies for water reorientation, but larger guest rotational activation energies. These properties are related to transient hydrogen bonding between the guests and host lattice in the clathrate and the resulting Bjerrum L-defects that are formed and can migrate through the hydrate lattice.10 Hydrogen bonding between guests and host also affects the guest dynamics in the cages which can be investigated by proton and deuterium NMR relaxation times at different temperatures.11−13
Molecular dynamics simulation has been performed on binary sII tetrahydrofuran (THF) + (NH3/CH3OH) and
binary sI CH4+ (NH3/CH3OH) clathrate hydrate phases and
the hydrogen bonding probabilities between ammonia−water and methanol−water lattice were calculated using the radial distribution function.14−16 Simulations show that despite
proton-donating and proton-accepting hydrogen bonding of ammonia and methanol, the presence of hydrophobic guests or moieties in guest molecules in the clathrate hydrate can stabilize them to temperatures near approximately 240 K. In the simulations, water molecules were modeled using TIP4P and the TIP4P/ice force fields and the results show the two different water force fields significantly affect the predicted probability of guest−water hydrogen bonding.14 Molecular dynamics simulations were also used to study hydrogen bonding structure and dynamics of ethanol in binary sI clathrate hydrate with CO2.17 These simulations show that
ethanol forms long-lived (>500 ps) proton-donating and accepting hydrogen bonds with cage water molecules of the large T cages. Ethanol occupancies of greater than 10% in the sI clathrate hydrate large cage of the binary ethanol + CO2
structure sI clathrate hydrate were unstable because of the strong ethanol−water hydrogen bonding. The guest−water host hydrogen bonding was investigated for guests such as tert-butylamine (tBA) sII clathrate with H2S/Xe help gases and the
pinacolone/H2S structure H (sH) clathrate.8 The tBA amine
group has a stronger basicity than the pinacolone carbonyl oxygen, and because of this, tBA guests form stronger hydrogen bonds with water molecules at lower temperature. Molecular dynamics simulations were used to study the formation of guest−host hydrogen bonding for polar guest molecule such as in the sH tert-butylmethyl ether (TBME) and the hydrate phase structure and guest dynamics were compared with hydrates of non-hydrogen bonding analog guest molecules such as neohexane (NH).13 Hydrogen bonding was also compared between five-membered heterocycle ether molecules such as tetrahydrofuran (THF), 1,3-dioxolane and six-membered ring molecules such as tetrahydropyran (THP), and p-dioxane in sII clathrate hydrate.18 To our knowledge, hydrogen bonding in hydrate phases has not been studied in three-membered and four-membered heterocyclic ethers and small molecules with carbonyl functional groups using molecular dynamics simu-lations.
Ethylene oxide (EO) was one of the first water miscible substances discovered to form a clathrate hydrate by Wurtz in 1863.19 The EO hydrate was discussed and characterized in detail using X-ray diffraction by von Stackelberg and Meuthen.20 The single-crystal X-ray crystal structure of this
hydrate was determined by McMullan and Jeffrey21and later refined by Udachin et al.22The IR spectrum of EO hydrate was
measured by Bertie and Othen23,24and the dielectric relaxation by Hawkins and Davidson,25who were also the first to prepare the sI trimethylene oxide (TMO) clathrate hydrate. The powder neutron diffraction of the TMO clathrate hydrate was determined by Rondinone et al.26and the single crystal X-ray
diffraction of this phase with accurate guest positions was determined by Udachin et al.22Gough et al.,27and Udachin et
al.22 detected an ordering transition where the TMO guest molecules align parallel in each row of T cages in the sI clathrate hydrate phase. The IR spectra and rotations of the TMO guests in the sI cages were studied by Bertie and Jacobs28 and the calorimetry of the TMO hydrate phase formation was studied by Handa.29The formaldehyde (FA) clathrate hydrate was synthesized by Ripmeester et al.30and shown to be stable
to temperatures below 250 K.
In this work we use molecular dynamics simulations to identify guest−host hydrogen bonding by the polar guest molecules ethylene oxide, trimethylene oxide, and form-aldehyde in the large T cages of the sI clathrate hydrates with the water lattice. We compare the properties of these hydrate phases with those of the nonpolar structural analogues cyclopropane (CP), cyclobutane (CB), and ethane. Molecular dynamics simulations were performed with the SPC/E three-site and TIP4P/ice four-three-site water potentials to investigate the effect of the water force field on the predicted formation of hydrogen bonding. The TIP4P/ice potential accurately estimates the melting temperature of ice, the slope of the water−ice coexistence curve on the phase diagram, and the melting enthalpy of ice.31These two water models are different in the number of point electrostatic charges they include and each predict a different water dipole moment. The differences in dipole moments for the two models (2.426 D for TIP4P/ice and 2.35 D for SPC/E) affects their ability to form hydrogen bonds with guest molecules (see Figure S2 of theSupporting Information).
In this work, we use the radial distribution function (RDF) and direct observation of the simulation trajectory to detect hydrogen bonding between polar guest molecules, and compare the structure and dynamics of the hydrogen bonding guest hydrates to those of the nonpolar guest molecules with similar shape and mass. We use integration over the RDF plots to obtain the probability of hydrogen bonding between the guests and the cage water molecules and the temperature dependence of the hydrogen bonding probability. Direct observation of the hydrogen bonding in the simulation trajectory allows us to estimate the hydrogen bond lifetime for TMO, EO, and FA sI clathrate hydrates to determine the transient or long-lived nature of guest−host hydrogen bonding. In this study, we show that predicted structural characterizations for the hydrate phases are consistent with the single-crystal X-ray diffraction structures of these materials about symmetry-independent site for TMO and EO molecule.22
The four-membered ether TMO has a larger molecular volume than three-membered ether EO. This places the electronegative oxygen atom of TMO molecule closer to the clathrate hydrate large cage water molecules, and in this work we see that this leads to a greater probability of TMO−water hydrogen bonding as compared to the case of EO. Form-aldehyde guest molecules have a smaller molecular volume compared the other guest molecules, but this guest has a strong dipole moment that facilitates hydrogen bonding.
The Journal of Physical Chemistry C Article
DOI:10.1021/acs.jpcc.7b00218 J. Phys. Chem. C 2017, 121, 8832−8840
To investigate guest dynamics and rotational motion, the velocity autocorrelation function (VACF) and the orientational autocorrelation function (OACF) with unit vectors in two different directions on the guest molecules have been calculated. These quantities are related to NMR relaxation times for the guest molecules. We will discuss this below.
2. COMPUTATIONAL METHODS
In the simulations of the sI clathrate hydrates, we assume all large T cages (51262) are filled with the TMO, EO, and FA
guests, and all the small D cages (512) are fully occupied by
methane gas. All simulations are performed on 3 × 3 × 3 replicas of the sI unit cell at temperatures between 50 and 250 K and 1 bar pressure, which constitutes the stability region of these clathrates. The sum of Lennard-Jones and electrostatic point charges is used to represent the intermolecular van der Waals potentials between atoms i and j on different molecules
∑
ε σ σ πε = − + ⎧ ⎨ ⎪ ⎩ ⎪ ⎡ ⎣ ⎢ ⎢ ⎛ ⎝ ⎜⎜ ⎞ ⎠ ⎟⎟ ⎛ ⎝ ⎜⎜ ⎞ ⎠ ⎟⎟ ⎤ ⎦ ⎥ ⎥ ⎫ ⎬ ⎪ ⎭ ⎪ V r r r q q r ( ) 4 4 ij i j ij ij ij ij ij i j ij , 12 6 0 (1)where εijand σij are the Lennard-Jones potential energy and
distance parameters of the ij pair separated by a distance of rij
and qi and qj are the electrostatic point charges on the atoms.
The Lorentz−Berthelot combination rules are used to calculate Lennard-Jones parameters in potentials between unlike atoms. We used the extended simple point charge (SPC/E)32 and TIP4P/ice30 potentials for water (see Figure S1 of the Supporting Information), the General Amber Force Field (GAFF) for the ether, formaldehyde, and cycloalkane guests,33 the OPLS force field for the ethane molecules,34 and the Murad−Gubbins potential for methane.35Initial structures of large cage guests are optimized with the Gaussian 09 (G09) suite of programs at the B3LYP/6-311++G(d,p) level of theory.36Point charges for the guest molecules are determined
from the “charges from electrostatic potential using a grid based method” (CHELPG) calculations37 on the optimized
struc-tures. The Lennard-Jones interaction parameters and atomic point charges of the guests and water are reported in Table S1 of theSupporting Information. Constant pressure−temperature (NPT) molecular dynamics simulations with periodic boundary conditions are performed using the DL_POLY 2.18 software program.38The NPT simulations were performed for a total
time of 500 ps with the first 50 ps used for temperature scaled equilibration. The modified Nosé−Hoover barostat algorithm with 0.1 and 1 ps relaxation times for the thermostat and barostat, respectively, and Verlet leapfrog algorithm39−41 are
used with a time step of 1 fs for integrating the equations of motion. Final NPT configurations are used for NVE dynamics calculations at each temperature. The NVE simulations for the dynamics were performed for a total time of 250 ps with the first 50 ps used for equilibration. All interactions in the simulation box were computed within a cutoff distance of Rcutoff
= 15.0 Å.
3. RESULTS AND DISCUSSION
3.1. Structure and Guest−Host Hydrogen Bond Formation. Snapshots of the formation of guest−host hydrogen bonding between EO, TMO, and FA guest molecules and the sI large cage waters at 200 K from simulations with the SPC/E potential are shown in Figure 1. The three guest molecules can act as hydrogen bond acceptors from water. To
form a hydrogen bond with the guest, a water molecule rotates from its position in the hydrate lattice to orient an OH bond toward the guest electronegative O atom (the “OS” atom). Accompanying the formation of guest−host hydrogen bonding, a water−water hydrogen bond must be broken, causing formation a Bjerrum L-defect in the water lattice which is shown inFigure 2. These defects are defined as the absence of a covalently bonded hydrogen between two adjacent water oxygen atoms in the lattice. The formation of guest-induced Bjerrum L-defects in clathrate hydrates, unlike the case ice structures, is not accompanied by the formation of Bjerrum D-defects. The D-defects form where there are two hydrogen atoms between adjacent water oxygen atoms in a lattice.
If the guest−host hydrogen bond is short-lived, hydrogen bond between guest OS atom and the water breaks and water OH group can rotate back into the lattice framework position, leading to annihilation of the Bjerrum L-defect. If the guest− host hydrogen bond is stable, further rotations of the water molecules in the lattice can lead to the migration of the L-defect to adjacent water sites in the lattice. These phenomena are shown inFigure 2. When migration of the L-defect occurs, the guest−water hydrogen bond is “locked in” by a process requiring the rotation of a minimum of two water molecules to eliminate the L-defect. This increases the lifetime of the guest host hydrogen bond.
In this work, the presence of guest−host hydrogen bonds is quantitatively characterized by the −O···H−OH radial distribution function (RDF). The RDF plots for the guest
Figure 1.Snapshots of TMO, EO, and FA guests in the sI large cages
without hydrogen bonding (left) and with hydrogen bonding (right) 0.2 ps later. The cages are extracted from simulations of the hydrate supercell with periodic boundary conditions. The guest−water hydrogen bond leads to the formation of a Bjerrum L-defect between neighboring waters which can migrate within the water lattice. Simulations are at 200 K using the SPC/E water model.
OS atom with cage water hydrogen (HW) and oxygen (OW) atoms at 200 K are shown for the two water potentials in Figures 3and4, respectively. Analogous RDF plots between a
selected carbon atom of cyclopropane, cyclobutane, and ethane with the HW and OW atoms are given in these figures for comparison. InFigure 3, the first peaks in the OS···HW RDF plots of TMO and FA for both SPC/E and TIP4P/ice water potential occur at short distances (<2 Å) which are in the range of normal hydrogen bonding. The large and sharp peak in the TMO clathrate RDF plot is indicative of strong hydrogen bonding between TMO OS atom and water HW atom. The TMO hydrogen bonding is predicted to be stronger for TIP4P/ ice than the SPC/E simulation and generally the range of hydrogen bonding distances predicted by the SPC/E potential is broader. The absence of a short-range peak for cyclopropane, cyclobutane, and ethane shows that there are no short spatial correlations between a selected carbon atom and cage water HW atoms for guest molecules of this size. In Figure 4, hydrogen bonding is primarily shown by a peak in the OS··· OW RDF at distances of ∼3 Å or shorter.
The temperature dependence of the first peak in the OS··· HW RDF plots in distances of r < 2.5 Å, which represent guest−water hydrogen bonding, are shown for TMO, EO, and FA using TIP4P/ice and SPC/E water potentials in Figures S3−S5 of theSupporting Information.
The average hydrogen bonding probability ⟨P(T)⟩ can be calculated from the first peak of OS···OW RDF, g(r)
∫
ρ π ⟨P T( )⟩ = r g r( )4 r dr 0 2 min (2)where rmin= 3 Å is the cutoff distance criterion for hydrogen
bonding interactions between the guest and cage water molecules based on OS···OW distance in Figure 4. The probability of hydrogen bond formation for various guests at different temperature from simulations using the SPC/E and TIP4P/ice force field is given inTable 1and shown inFigure 5. As seen inFigure 5, at each temperature the TMO molecules have the largest probability of hydrogen bonding with cage waters, with the TIP4P/ice potential predicting two water molecules meeting the OS···OW distance criterion. This may be related to the larger TMO molecule size which positions the
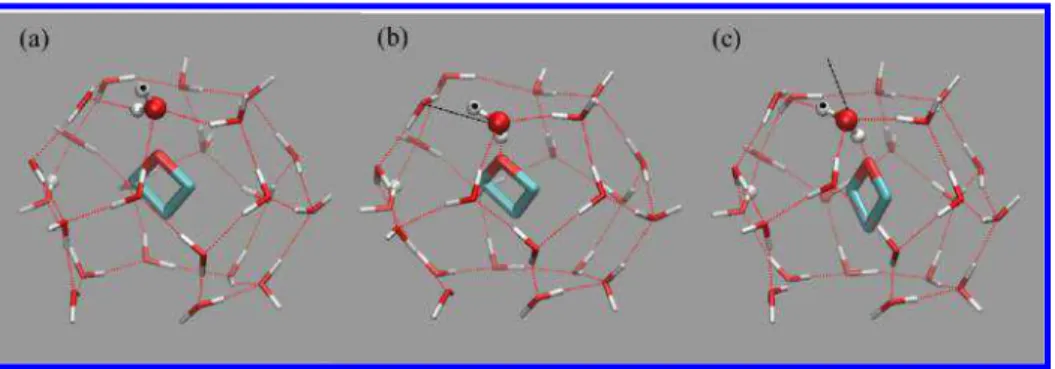
Figure 2.Consecutive snapshots of a selected TMO molecule in the large cage sI clathrate hydrate. (a) Large cage with a non-hydrogen-bonded
TMO molecule. (b) Formation of hydrogen bond between TMO and one hydrogen atom of the specially marked water molecule and formation of a Bjerrum L-defect (marked by the black dashed line) between the special water and adjacent water molecule in the hexagonal face. (c) Rotation of the specially marked water while maintaining the hydrogen bond to TMO to form a hydrogen bond between hydrogen (marked by a black dot) and the neighboring water in the hexagonal face of the large sI cage. The Bjerrum L-defect has now migrated outside of the cage (see the location of the black dashed line) and the hydrogen bond of the water with TMO is “locked in”.
Figure 3.OS···HW RDF plot for TMO, EO, and FA molecules at 200
K for (a) SPC/E, and (b) TIP4P/ice water model for different guest molecules. The RDFs for hydrocarbons with similar carbon backbones are given for comparison to emphasize the effects of hydrogen bonding.
Figure 4.OS···OW RDF plot for TMO, EO, and FA molecules at 200
K for (a) SPC/E, and (b) TIP4P/ice water model for different guest molecules. The RDFs for hydrocarbons with similar carbon backbones are given for comparison to emphasize the effects of hydrogen bonding.
The Journal of Physical Chemistry C Article
DOI:10.1021/acs.jpcc.7b00218 J. Phys. Chem. C 2017, 121, 8832−8840
oxygen atom in TMO closer to the cage water molecules and the larger partial electrostatic charge of the ether O atom on TMO (seeTable S1).
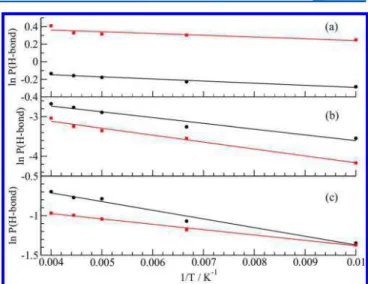
The guest−water hydrogen bonding probability increases with temperature in all cases. The van’t Hoff plots for the change in the logarithm of probability of hydrogen bonding with inverse temperature are given in Figure 6. Between the temperature range of 100 and 250 K, the van’t Hoff plots are linear and the enthalpies of hydrogen bond formation for the three guests extracted from the slopes of these lines are given in Table 2. In these hydrates, the guest−water hydrogen bond formation is endothermic which implies that the hydrogen bonds between water molecules of clathrate lattice are stronger than the TMO−water, EO−water, and FA−water hydrogen bonds. Thermal vibrations of the guest molecules and water molecules in the lattice can provide the activation energy required to break lattice water−water hydrogen bonds to form guest−water hydrogen bonds. The smaller value of the hydrogen bond formation enthalpy for TMO molecule shows that the hydrogen bond between the TMO molecule and water is stronger than the other guest molecules.
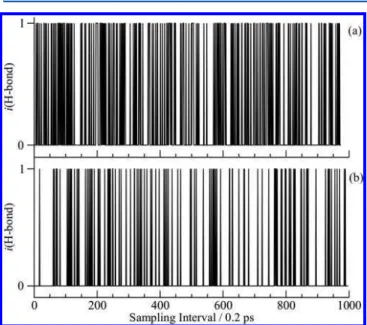
The guest−host hydrogen bond formation probability can also be directly studied by examining simulation trajectories probed at set intervals and applying the distance criteria for hydrogen bonding directly for the guest and cage water
molecules. For each guest, if the distance between the oxygen atom (OS) and a water hydrogen atom (HW) in the large cages is less than 2.1 Å, the guest is considered to form a hydrogen bond and assigned a hydrogen bond index value i(HB) = 1. Non-hydrogen-bonded guest configurations are assigned a value i(HB) = 0. This hydrogen bonding distance criterion has previously been used in hydrate studies.42 The occurrence of hydrogen bonding over time between a sample FA guest and large cage water molecules from sample trajectories at 150 and 250 K is shown inFigure 7. Hydrogen bonds of this guest with the cage water molecules are seen to form and break numerous times in the 200 ps duration of the simulation. InFigure 7, the probability of hydrogen bonding (i.e., occurrence of i(HB) = 1 values) is greater at the higher temperature. The average lifetimes of the hydrogen bonds are short at both temperatures and the hydrogen bonds form and break at short time intervals. Similar figures are given for EO, FA, and TMO in Figures S6− S8 of theSupporting Informationin the range of 50 to 250 K. The lifetimes of the hydrogen bonding events for TMO are the longest among the three guests studied. The hydrogen bonding of the guests with the water molecules is stochastic and guests in different cages may show slightly different hydrogen bonding probabilities and lifetimes. The hydrogen bonding probabilities from the RDF are an average of the hydrogen bonding for all guests.
As discussed below, guest−host hydrogen bonding affects the guest rotational dynamics.
3.2. Dynamic Properties. Hydrogen bonding tethers guests to cage water molecules and greatly reduces their range of translational and rotational motions in the cage. The dynamic motion of the guest in the cage can be characterized by the velocity autocorrelation function (VACF) defined by Table 1. Probability of Hydrogen Bond Formation for TMO,
EO, and FA Guests at Different Temperatures from Simulations with the SPC/E and TIP4P/Ice Water
Potentials Using the OS···OW Distance of 3 Å as the Cutoff inEquation 2
TMO EO FA
T/K SPC/E TIP4P/ice SPC/E TIP4P/ice SPC/E TIP4P/ice
50 0.6937 1.1893 0.0238 0.0181 0.1530 0.1770 100 0.7525 1.2836 0.0288 0.0155 0.2600 0.2540 150 0.7960 1.3547 0.0385 0.0288 0.3432 0.3077 200 0.8367 1.3711 0.0555 0.0350 0.4551 0.3525 225 0.8537 1.3887 0.0629 0.0389 0.4623 0.3698 250 0.8743 1.5057 0.0689 0.0479 0.4982 0.3795
Figure 5. Temperature dependence of the ensemble average
probability of hydrogen bonding for (a) TMO, (b) FA, and (c) EO guests from simulations using the SPC/E (black lines) and TIP4P/ice (red lines) water force fields using integration of the RDF.
Figure 6.Van’t Hoff plot for the logarithm of the probability of the (a)
TMO, (b) FA, and (c) EO guest−water hydrogen bond formation as a function of the inverse temperature in the sI clathrate using the SPC/E (black lines) and TIP4P/ice (red lines).
Table 2. Enthalpy of Hydrogen Bond Formation (kJ·mol−1)
for the TMO, EO, and FA Guests with Water
guest molecule ΔHH‑bond(SPC/E) ΔHH‑bond(TIP4P/ice)
TMO 0.2001 0.1671
EO 1.2103 1.4554
= ⟨ · ⟩ ⟨ · ⟩ C t t v v v v ( ) ( ) (0) (0) (0) i i i i (3)
where vi(t) is velocity of atom i of the guest at time t. The
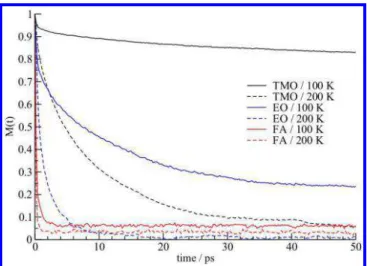
VACF functions of the OS atoms of TMO, EO, and FA guests at 150 and 250 K from NVE calculations are shown inFigure 8.
The VACFs decay quickly to zero at about 0.2 ps, after which they have a negative region and a minimum between 0.25 and 0.4 ps. After the minimum, the VACFs decay to zero a second time. This second zero in time is called the velocity randomization time and is ∼0.5 ps for the EO guests, which is faster this time for TMO at ∼0.7 ps and FA at ∼1 ps. The VACF of TMO shows short time oscillations with a period of about 0.05 s which are related to oscillatory motion of the
TMO guest as it is tethered to the water by strong hydrogen bonding. The initial decay and reversal of the VACF are related to the motion of the guest in the cage. The guest approaches the cage wall and slows down and ultimately collides with the cage wall, and subsequently reverses its direction of motion. The decay of the VACF to zero is related to the randomization of the direction of the guest velocity relative to its initial direction of motion.
The orientation autocorrelation function (OACF) character-izes the rotational dynamics of the guest molecules in the large cages by the change in orientation of a unit vector μ(t) defined for the guest molecules. The OACF, M(t), is defined by
μ μ μ μ θ = ⟨ · ⟩ ⟨ · ⟩ = ⟨ ⟩ M t( ) ( ) (0)t t (0) (0) cos ( ) (4)
where μ(0) gives the guest orientation at time 0, μ(t) is the orientation at a later time t. and θ(t) is the rotational angle of the μ between times 0 and t. Two unit vectors can be defined to characterize the guest rotations in the cages and these are shown in Figure S9 of theSupporting Informationfor FA, EO, and TMO. The unit vectors along the molecule symmetry axis going through the O atom are labeled as μ∥. This unit vector
lies along the CO axis in FA and along the C2symmetry axes
in EO and TMO. The other unit vector characterizing the molecular rotation is called μ⊥and is along the H···H direction
in FA, along the C···C axis in EO, and along the C1···C3axis in
TMO.
To compare guest rotational dynamics, the OACFs of three guest molecules at 100 and 200 K for μ∥and μ⊥unit vectors are
shown inFigures 9 and 10, respectively, for simulations with
the SPC/E water potential. When a guest molecule is hydrogen bonded to a cage water, the range of rotation of the μ∥vector
becomes limited. InFigure 9, at 100 K, the M∥(t) for μ∥for the
largest TMO guests initially decays quickly, but stabilizes at a value of ∼0.65. The OACF of the smaller EO and FA molecules decay at similar rates, but the EO OACF goes to a limit of ∼0.55, while the FA OACF goes to ∼0.35. The nonzero asymptotic limits of the M∥(t) and therefore ⟨cos θ(t)⟩ show
that the guest molecules do not attain isotropic distribution in the equatorial plane of the oblate large sI cages within the 50 ps
Figure 7. Hydrogen bond formation for a selected FA in an sI
clathrate large cage from simulation trajectories at temperature (a) 250 K and (b) 150 K. A i(HB) = 1 shows the formation of a guest−host hydrogen bond in the cage and configurations with no hydrogen bonding have i(HB) = 0. Results are for simulations performed with the SPC/E water potential.
Figure 8.Velocity autocorrelation function for the oxygen atoms for
(a) FA, (b) EO, and (c) TMO at 150 K (black lines) and 250 K (red lines) and ambient pressure. Simulations were performed using the SPC/E water potential.
Figure 9.Decay of the orientational autocorrelation functions for the
unit vector in direction of the symmetry axis of the guest molecules,
μ∥, of TMO, EO, and FA at 100 and 200 K and ambient pressure.
Simulations were performed using the SPC/E water potential.
The Journal of Physical Chemistry C Article
DOI:10.1021/acs.jpcc.7b00218 J. Phys. Chem. C 2017, 121, 8832−8840
time frame of the simulation trajectory. At 200 K, all M∥(t)
decay more rapidly as the guest molecules move more quickly in the cages and have shorter-lived hydrogen bonds with the cage water molecules. The asymptotic distribution of the guests in the equatorial plane of the cages becomes more isotropic at the higher temperature.
Even when a guest molecule is tethered to a water molecule by hydrogen bonding, the μ⊥vector can rotate.Figure 10shows
that the decay of M⊥(t) for TMO is slower than for EO and FA.
The smaller radius of rotation of the H atoms in FA allows for the fast isotropic rotation of H atoms about the CO axis. For the other two guests, the smaller EO reaches more uniform distribution of the CH2groups around the C2axis direction due
to its smaller size.
The M(t) functions for CP, CB, and ethane are compared with the analogous hydrogen bonding guests in Figure S10 of the Supporting Information. As expected, the OACFs of the non-hydrogen-bonding guests decay much faster than the tethered hydrogen bonded guests.
Bertie and Jacobs have measured the infrared spectra of ethylene oxide, cyclopropane, and trimethylene oxide clathrate hydrates at 4.3 K.28 They are able to observe rotational
vibrations for the guests which they assign to rotations of the guests about different inertial axes. These different rotational frequencies are implied by the different decay times of the
M∥(t) and M⊥(t) functions inFigures 9and10. For EO they
obtain the rotational force constants of 487 and 264 ferg·rad−2
and for TMO the rotational force constants of 1190 and 1130 ferg·rad−2. The larger rotational force constants of the TMO
are shown in the present simulations to be related to the greater hydrogen bonding and larger size of the TMO molecule, both of which are manifested in the slower decay of the M∥(t) and M⊥(t) functions.
The NMR 13C T
1 and 1H T1ρ relaxation times of guest
molecules in clathrate hydrate cages correlate with guest rotation.13An NMR measurement of these quantities should be
able to detect the consequences of guest−host hydrogen bonding on the relaxation times.
The guest−host hydrogen bonding which introduces Bjerrum L-defects into the water lattice also effects of the dynamics of the water molecules in the lattice.18,25,43 Measurements of the dielectric relaxation of the sI hydrate
phase was summarized by Davidson who gives the dielectric relaxation time and water reorientation activation energies of the sI clathrate hydrates of cyclopropane, EO, and TMO; see Table 3.42 The dielectric relaxation time and water rotation
activation energy43 for water in the clathrate hydrate of the non-hydrogen-bonding cyclopropane guest are larger than the clathrate hydrates of the hydrogen bonding EO and TMO guests. The hydrogen bonding in the clathrate hydrate lattice of cyclopropane are not disrupted by the guest-induced Bjerrum L-defects and this leads to significantly larger water dielectric relaxation times. The dielectric relaxation time and water rotation activation energy for water for EO are larger than TMO. This can be understood on the basis of the greater probability of hydrogen bonding of the TMO guest with the cage water which induces a greater number of L-defects into the clathrate hydrate water network, facilitating water rotational reorientation and dielectric relaxation.
4. CONCLUSIONS
We performed molecular dynamics simulations of guest−host hydrogen bonding in the large cage sI clathrate hydrate with the guest molecules, formaldehyde, trimethylene oxide, and ethylene oxide, and their alkane analogs ethane, cyclopropane, and cyclobutane with the TIP4P/ice and SPC/E water potentials. We observe that the choice of water force field quantitatively affects the predicted hydrogen bonding of guests with water in clathrate hydrates, but does not affect the qualitative trends observed for hydrogen bonding probability between the three guests.
The formaldehyde carbonyl group oxygen atom and the ether oxygen atoms of the TMO and EO molecules can form hydrogen bonds with sI large cage water molecules. The dipole moments of the EO, TMO, and FA guest molecules are 2.10, 2.49, and 2.79 D (Debye), respectively. To enhance hydrogen bond formation, in addition to large dipole moment, the guest molecule must also have a relatively large molecular volume that positions the electronegative oxygen atom in close proximity to the large cage water molecule. In this study TMO molecules are larger than the FA and EO molecules; therefore, TMO oxygen atoms are positioned at closer distances to the cage water. This increases the guest−host hydrogen bonding.
The water molecules with TIP4P/ice potential have a stronger electrostatic interaction with the guests and other neighbor water molecules than the SPC/E model, based on atomic partial charges (see Table S1 of the Supporting Information). The TIP4P/ice model, therefore, often predicts greater guest−host hydrogen bonding probability than SPC/E potential. The effect of the water force field on the probability of hydrogen bonding, however, is smaller for FA and EO guests. The probability of hydrogen bonding for these guests increases with temperature which implies that the guest−water hydrogen bonds are weaker than water−water hydrogen bonds
Figure 10.Decay of the orientational autocorrelation functions for the
μ⊥unit vector of TMO, EO, and FA at 100 and 200 K and ambient
pressure. Simulations were performed using the SPC/E water potential.
Table 3. Dielectric Relaxation Time τD and the Activation
Energy of Water Reorientation (kJ·mol−1) for the
Cyclopropane, EO, and TMO Clathrate Hydrates
guest molecule τD(−40 °C)/μs ΔEact(kJ·mol−1)
Cyclopropane 280 42
Ethylene oxide 0.33 32
in the lattice. The guest−host hydrogen bonding leads to the appearance of Bjerrum L-defects in the water lattice which can migrate through the lattice away from the cage holding the guest, effectively locking in the guest−host hydrogen bond. The guest−host hydrogen bond and guest-induced defects in the water lattice have limited lifetimes that depend on the temperature.
The next cyclic ether in the series is tetrahydrofuran (THF). Due to the size of the THF guest molecule, it cannot be incorporated in the 14-sided 51262cages of the sI clathrate, and
this guest forms a structure II clathrate hydrate and fits in large 16-sided 51264cages of this phase. The dipole moment of THF
is smaller than TMO and the larger 16-sided cage sizes of the sII clathrate hydrate contribute to THF having weak hydrogen bond probability with the cage water molecules.18,44
The velocity and orientational autocorrelation function are used to investigate guest dynamical properties. The decay of the orientational autocorrelation function, M(t), depends on the direction of the unit vector μ(t) vector in the guest molecule. When hydrogen bonding occurs, the μ∥(t) vector is in the
direction pointing from the guest molecule toward the host lattice, and the M∥(t) function decays more slowly, which is
observed for EO, FA, and TMO molecules. The rotation of the μ⊥(t) vector, which is roughly parallel to the cage wall during
hydrogen bonding, will depend on the size of the guest molecule.
■
ASSOCIATED CONTENT*
S Supporting InformationThe Supporting Information is available free of charge on the ACS Publications websiteat DOI:10.1021/acs.jpcc.7b00218.
Table of force field parameters used in the simulations and the following figures: structure I clathrate hydrate unit cell; two water models used in the simulations; OS··· HX RDF and hydrogen bonding at different temper-atures for the guests using the SCP/E and TIP4P/ice water potentials; orientational autocorrelation function for the hydrogen and non-hydrogen bonding guests (PDF)
■
AUTHOR INFORMATION Corresponding Authors *E-mail: mohammadimanesh@yazd.ac.ir. *E-mail: saman.alavi@nrc-cnrc.gc.ca. ORCID Saman Alavi: 0000-0001-9463-8766 NotesThe authors declare no competing financial interest.
■
ACKNOWLEDGMENTSThe authors would like to thank John A. Ripmeester for discussions on hydrogen bonding in clathrate hydrates and providing an unpublished chapter on the history of clathrate hydrates from which some of the references of the introduction were taken. H. M. and H. G. thank the University of Yazd for computational support.
■
REFERENCES(1) Sloan, E. D., Jr; Koh, C. Clathrate Hydrates of Natural Gases; CRC press: 2007.
(2) Susilo, R.; Alavi, S.; Ripmeester, J. A.; Englezos, P. Molecular Dynamics Study of Structure H Clathrate Hydrates of Methane and Large Guest Molecules. J. Chem. Phys. 2008, 128, 194505.
(3) Sloan, E. D. Fundamental Principles and Applications of Natural Gas Hydrates. Nature 2003, 426, 353−363.
(4) Gough, S.; Hawkins, R.; Morris, B.; Davidson, D. Dielectric Properties of Some Clathrate Hydrates of Structure II. J. Phys. Chem. 1973, 77, 2969−2976.
(5) Jeffrey, G. A.; McMullan, R. K. In Progress in Inorganic Chemistry; John Wiley & Sons: Inc., 2007; p 43.
(6) Jeffrey, G. A. In Inclusion Compounds, Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D., Eds.; Academic Press: London, 1984; Vol. 3, pp 135−190.
(7) Takeya, S.; Kida, M.; Minami, H.; Sakagami, H.; Hachikubo, A.; Takahashi, N.; Shoji, H.; Soloviev, V.; Wallmann, K.; Biebow, N.; et al. Structure and Thermal Expansion of Natural Gas Clathrate Hydrates. Chem. Eng. Sci. 2006, 61, 2670−2674.
(8) Alavi, S.; Udachin, K.; Ripmeester, J. A. Effect of Guest−Host Hydrogen Bonding on the Structures and Properties of Clathrate Hydrates. Chem. - Eur. J. 2010, 16, 1017−1025.
(9) Davidson, D. W.; Ripmeester, J. A. In Inclusion Compounds, Atwood, J. L.; J. Davies, E. D.; MacNicol, D. D., Eds; Academic Press: London, 1984; Vol. 1, pp 69−128.
(10) Bjerrum, N. Structure and Properties of Ice. Science 1952, 115, 385−390.
(11) Garg, S.; Davidson, D.; Ripmeester, J. NMR Behavior of the Clathrate Hydrate of Tetrahydrofuran. I. Proton Measurements. J. Magn. Reson. 1974, 15, 295−309.
(12) Davidson, D.; Garg, S.; Ripmeester, J. NMR behavior of the Clathrate Hydrate of Tetrahydrofuran. II. Deuterium Measurements. J. Magn. Reson. 1978, 31, 399−410.
(13) Susilo, R.; Alavi, S.; Moudrakovski, I. L.; Englezos, P.; Ripmeester, J. A. Guest−Host Hydrogen Bonding in Structure H Clathrate Hydrates. ChemPhysChem 2009, 10, 824−829.
(14) Shin, K.; Kumar, R.; Udachin, K. A.; Alavi, S.; Ripmeester, J. A. Ammonia Clathrate Hydrates as New Solid Phases for Titan, Enceladus, and Other Planetary Systems. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14785−14790.
(15) Shin, K.; Udachin, K. A.; Moudrakovski, I. L.; Leek, D. M.; Alavi, S.; Ratcliffe, C. I.; Ripmeester, J. A. Methanol Incorportation in Clathrate Hydrates and the Implications for Oil and Gas Pipeline Flow Assurance and Icey Planetary Bodies. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8437−8442.
(16) Alavi, S.; Shin, K.; Ripmeester, J. A. Molecular Dynamics Simulations of Hydrogen Bonding in Clathrate Hydrates with Ammonia and Methanol Guest Molecules. J. Chem. Eng. Data 2015, 60, 389−397.
(17) Alavi, S.; Ohmura, R.; Ripmeester, J. A. A Molecular Dynamics Study of Ethanol−Water Hydrogen Bonding in Binary Structure I
Clathrate Hydrate with CO2. J. Chem. Phys. 2011, 134, 054702.
(18) Alavi, S.; Susilo, R.; Ripmeester, J. A. Linking Microscopic Guest Properties to Macroscopic Observables in Clathrate Hydrates: Guest-Host Hydrogen Bonding. J. Chem. Phys. 2009, 130, 174501.
(19) Wurtz, A. Mémoire sur l’Oxyde d’éthylène et les Alcools Polyéthyléniques. Ann. Chim. Phys. Ser. 1863, 369, 317.
(20) von Stackelberg, M.; Meuthen, B.; Feste Gashydrate. VII. Hydrate wasserlöslicher Äther. Z. Elektrochem. 1958, 62, 130−131.
(21) McMullan, R. K.; Jeffrey, G. Polyhedral Clathrate Hydrates. IX. Structure of Ethylene Oxide Hydrate. J. Chem. Phys. 1965, 42, 2725− 2732.
(22) Udachin, K. A.; Ratcliffe, C. I.; Ripmeester, J. A. Structure, Dynamics and Ordering in Structure I Ether Clathrate Hydrates from Single-Crystal X-ray Diffraction and 2H NMR Spectroscopy. J. Phys. Chem. B 2007, 111, 11366−11372.
(23) Bertie, J. E.; Othen, D. A. The Infrared Spectrum of Ethylene
Oxide Clathrate Hydrate at 100 K Between 4000 and 360 cm−1. Can. J.
Chem. 1973, 51, 1159−116.
The Journal of Physical Chemistry C Article
DOI:10.1021/acs.jpcc.7b00218 J. Phys. Chem. C 2017, 121, 8832−8840
(24) Bertie, J.; Othen, D. The Infrared Spectrum of Ethylene Oxide
Clathrate Hydrate Between 360 and 20 cm−1, at 100° K. Can. J. Chem.
1972, 50, 3443−3449.
(25) Hawkins, R.; Davidson, D. Dielectric Relaxation in the Clathrate Hydrates of Some Cyclic Ethers. J. Phys. Chem. 1966, 70, 1889−1894. (26) Rondinone, A. J.; Chakoumakos, B. C.; Rawn, C. J.; Ishii, Y. Neutron Diffraction Study of Structure I and Structure II Tri-methylene Oxide Clathrate Deuterate. J. Phys. Chem. B 2003, 107, 6046−6050.
(27) Gough, S.; Garg, S.; Davidson, D. Ordering of Guest-Molecule Dipoles in the Structure I Clathrate Hydrate of Trimethylene Oxides. Chem. Phys. 1974, 3, 239−247.
(28) Bertie, J. E.; Jacobs, S. M. Far Infrared Absorption and Rotational Vibrations of the Guest Molecules in Structure I Clathrate Hydrates Between 4.3 and 100 K. Can. J. Chem. 1977, 55, 1777−1785. (29) Handa, Y. A Calorimetric Study of Trimethylene Oxide and its Structure I and Structure II Clathrate Hydrates in the Temperature Range 85 to 270 K. Can. J. Chem. 1985, 63, 68−70.
(30) Ripmeester, J.; Ding, L.; Klug, D. A Clathrate Hydrate of Formaldehyde. J. Phys. Chem. 1996, 100, 13330−13332.
(31) Abascal, J.; Sanz, E.; Fernández, R. G.; Vega, C. A Potential Model for the Study of Ices and Amorphous Water: TIP4P/Ice. J. Chem. Phys. 2005, 122, 234511.
(32) Berendsen, H.; Grigera, J.; Straatsma, T. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269−6271.
(33) Cornell, W. D.; Cieplak, P.; Bayly, C. I.; Gould, I. R.; Merz, K. M.; Ferguson, D. M.; Spellmeyer, D. C.; Fox, T.; Caldwell, J. W.; Kollman, P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179−5197.
(34) Jorgensen, W. L.; Madura, J. D.; Swenson, C. J. Optimized Intermolecular Potential Functions for Liquid Hydrocarbons. J. Am. Chem. Soc. 1984, 106, 6638−6646.
(35) Murad, S.; Gubbins, K. In Computer Modelling of Matter, Lykos, P.; Ed.; ACS Symposium Series Vol. 86; American Chemical Society: Washington, DC, 1978; p 62; DOI: 10.1021/bk-1978-0086.ch005
(36) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.; Wallingford, CT, 2009.
(37) Breneman, C. M.; Wiberg, K. B. Determining Atom-Centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11, 361−373.
(38) Smith, W.; Forester, T.; Todorov, I. The DL_POLY Molecular Simulation Package, v 2.18; Daresbury Laboratory: Daresbury, UK, 2007.
(39) Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511−519.
(40) Hoover, W. G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A: At., Mol., Opt. Phys. 1985, 31, 1695.
(41) Melchionna, S.; Ciccotti, G.; Lee Holian, B. Hoover NPT Dynamics for Systems Varying in Shape and Size. Mol. Phys. 1993, 78, 533−544.
(42) Davidson, D. W. Clathrate hydrates. In Water. A Comprehensive Treatise, Franks, F., Ed.; Plenum: New York, 1973; p 180.
(43) Petrenko, V. F.; Whitworth, R. W. Physics of Ice; Oxford University Press: Oxford, 1999.
(44) Alavi, S.; Ripmeester, J. A. Effect of Small Cage Guests on Hydrogen Bonding of THF in Binary Structure II Clathrate Hydrates. J. Chem. Phys. 2012, 137, 054712.