HAL Id: cea-01477621
https://hal-cea.archives-ouvertes.fr/cea-01477621
Submitted on 27 Feb 2017
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Time-resolved photoemission spectroscopy on a
metal/ferroelectric heterostructure
J.E. Rault, G. Agnus, T. Maroutian, V. Pillard, Ph. Lecoeur, G. Niu, B.
Vilquin, M.G. Silly, A. Bendounan, F. Sirotti, et al.
To cite this version:
J.E. Rault, G. Agnus, T. Maroutian, V. Pillard, Ph. Lecoeur, et al.. Time-resolved
photoemis-sion spectroscopy on a metal/ferroelectric heterostructure. Physical Review B: Condensed Matter
and Materials Physics (1998-2015), American Physical Society, 2013, 88, pp.155107.
�10.1103/Phys-RevB.88.155107�. �cea-01477621�
J. E. Rault,1, ∗ G. Agnus,2 T. Maroutian,2 V. Pillard,2 Ph. Lecoeur,2 G.
Niu,3, † B. Vilquin,3 M. G. Silly,4 A. Bendounan,4 F. Sirotti,4 and N. Barrett1, ‡
1CEA, DSM/IRAMIS/SPCSI, F-91191 Gif-sur-Yvette Cedex, France
2
Institut d’Electronique Fondamentale, Univ Paris-Sud, CNRS UMR 8622, 91405 Orsay Cedex, France
3
Universit´e de Lyon, Ecole Centrale de Lyon, Institut des Nanotechnologies de Lyon, F-69134 Ecully cedex, France
4Synchrotron-SOLEIL, BP 48, Saint-Aubin, F91192 Gif sur Yvette CEDEX, France
In thin film ferroelectric capacitor the chemical and electronic structure of the electrode/FE interface can play a crucial role in determining the kinetics of polarization switching. We investigate
the electronic structure of a Pt/BaTiO3/SrTiO3:Nb capacitor using time-resolved photoemission
spectroscopy. The chemical, electronic and depth sensitivity of core level photoemission is used to probe the transient response of different parts of the upper electrode/ferroelectric interface to voltage pulse induced polarization reversal. The linear response of the electronic structure agrees quantitatively with a simple RC circuit model. The non-linear response due to the polarization switch is demonstrated by the time-resolved response of the characteristic core levels of the electrode and the ferroelectric. Adjustment of the RC circuit model allows a first estimation of the Pt/BTO interface capacitance. The experiment shows the interface capacitance is at least 100 times higher than the bulk capacitance of the BTO film, in qualitative agreement with theoretical predictions from the literature.
PACS numbers: 77.80.-e 73.21.Ac 73.40.-c 77.84.-s
I. INTRODUCTION
Epitaxially strained oxide thin films make ferroelec-tric (FE) based heterostructures of real interest for new
electronic devices1,2. High read and write speeds are of
prime importance for implementing ferroelectric based electronics. The dynamic process of polarization reversal is crucial to understand the application limits in terms of
maximum frequency and long-term fatigue3. It is
there-fore essential to understand the polarization switching kinetics in thin film ferroelectric capacitor structures.
Polarization reversal in the prototypical BaTiO3
ferro-electric singles crystals was first studied by W.J. Merz4.
The switching dynamics have been described by classical nucleation theory of Kolmogorov, Avrami and Ishibashi in which the polarization is reversed first by domain
nu-cleation then domain wall motion5–7. In smaller
capac-itive stuctures, nucleation limited switching have been
found to provide a more accurate description8–10.
Fol-lowing the model of Merz, the switching time depends
exponentially on the activation field4. Gruverman et al.
have shown that there are high and low field regimes for polarization reversal. Small capacitive structures under a high field switch more rapidly whereas larger area ca-pacitors switch more quickly under low fields because the
kinetics are nucleation limited9.
Most characterization of FE switching has been done using electrical methods but accurate switching measure-ments are often limited by the RC circuit load on the device and parasitic resistances. Despite this problem, it has been shown that the activation field can be de-termined independently of the RC circuit characteristics,
the effect of the latter being a time dilation of the FE switch11.
The properties of the electrode/FE interface may also determine the switching kinetics. For example, it has been suggested that defect related local fields determine
the switching time in BiFeO3 capacitors12. At the
elec-trode/FE interface, chemistry, strain and electronic or-dering combine in a complex way to screen the inter-face polarization charge of the ferroelectric layer. An ac-curate description of the dynamical electronic structure may help to better understand the switching process and the phenomena underlying the results of electrical mea-surements. However, experimental data on the electronic properties of such systems are scarce, principally due to the intrinsic difficulty of measuring a buried interface.
The use of complementary in-operando, i.e. submit-ted to a realistic voltage excitation, structural character-ization techniques is therefore desirable. Ideally, these techniques should be independent of the electrical cir-cuit required to switch the polarization. For instance, Gorfman et al. used time-resolved x-ray diffraction to determine the lattice dynamics and atomic positions in piezoelectrics13.
X-ray photoemission spectroscopy (XPS) is a power-ful, direct tool for characterizing the chemical and
elec-tronic interface structure. It has been used with
in-situ bias to demonstrate the migration of oxygen
va-cancies in Pt/HfO214or to conduct impedance-like
mea-surements on Rb deposited on silicon15. Chen and
Klein used in-situ photoemission spectroscopy on a BTO monocristal to deduce the barriers properties as a
func-tion of ferroelectric polarizafunc-tion16. Our group studied
a Pt/BaTiO3/Nb:SrTiO3(Pt/BTO/NSTO)
2 ture with bias application and found polarization
de-pendent barriers and a non-trivial behavior of the
core-levels as a function of applied bias17. All of these studies
show the potential of photoemission experiments for in-operando work.
An exciting route for a fundamental understanding of the interface electronic structure of FE devices would therefore be time-resolved photoelectron spectroscopy. State-of-the-art delay-line detectors allow detection of photoemitted electrons with a time-resolution down to
5 to 10 ns18. Although switching times can be of the
or-der of a nanosecond19, in many cases it may be sufficient
to resolve the transient response of the electronic struc-ture to the applied voltage pulse. The aim is to detail the transient response of the electronic structure to the switching pulse and the role of the interface and bulk film capacitance in defining ultimate device speed.
We investigate the electronic structure of an elec-trode/FE interface using time-resolved photoemission
spectroscopy. The static properties of the
ferroelec-tric capacitance have been studied by photoemission in
Ref. 17. Bias pulses on a Pt/BTO/NSTO capacitor
probe the time-resolved chemical and electronic structure of the Pt/BTO interface. An equivalent circuit model is used predicting a behavior which compares well with the photoemission results on the real system. This shows the potential of time-resolved photoemission spectroscopy to follow the chemical/electronic changes in working model FE microelectronic devices.
II. EXPERIMENT
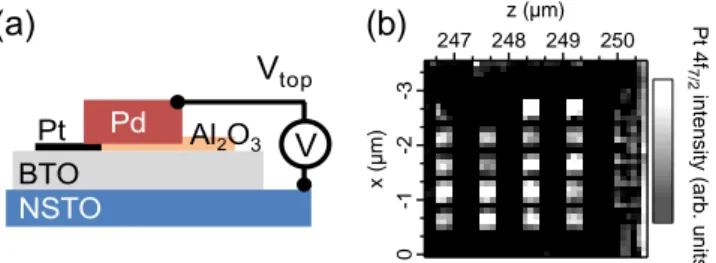
The sample is a Pt (2.8 nm)/BTO (64 nm)/NSTO
het-erostructure grown by Molecular Beam Epitaxy. The
growth conditions and heterostructure properties are
de-scribed in Ref. 17. 300 × 300 µm2 thick Pt electrodes
were patterned by ionic beam etching. Thicker (300
nm) Palladium (Pd) pads overlapping part of the Pt electrodes have been deposited by evaporation to en-able wire-bonding of the top electrodes to the sample
holder. A highly insulating layer of Al2O3was deposited
by evaporation onto bare BTO to suppress interference
of the Pd pads with the capacitance. The device
ar-chitecture is fully described in Ref. 17 and is shown in Fig. 1a. When the polarization is pointing from the bot-tom to top electrode (P+) the leakage current is limited by Schottky emission at the upper, Pt/BTO interface whereas in the P- state the current flow is determined by the quasi-ohmic BTO/NSTO interface. The device
was then introduced in ultrahigh vacuum (10−8 Pa) in
the XPS set-up of the TEMPO Beamline at the SOLEIL
synchrotron radiation source20. The 100×100 µm2beam
could be directed onto a single top electrode located by a map of the whole sample using the Pt absorption edge (see Fig. 1b). A photon energy of 1100 eV was used to optimize the signal from the BTO close to the interface (estimated probing depth of ≈3-4 nm). The overall
en-ergy resolution was 220 meV. The sample-holder allows in-situ bias application and electrical measurements via high quality coax wires to limit parasitic behavior due to the electrical environment.
Pt BTO NSTO Pd
V
V
top Al2O3(a)
-3 -2 -1 0 x (µm) 250 249 248 247 z (µm) Pt 4f 7/2intensity (arb. units)
(b)
FIG. 1. (a) Schematic of the capacitor; (b) Pt 4f
inten-sity map for the Pt/BTO/NSTO sample showing 20 identical
Pt/BTO/NSTO capacitors (300 × 300 µm2) on the 5 × 5 mm2
surface, allowing location of the wired capacitor.
The coercive voltage Vc− needed to switch from P+
to P− is 0.80 V and V
c+, to switch from P− to P+, is
0.40 V. The loop shows a 0.6 V offset with a P+remnant
state at zero voltage. In order to ensure fully switched polarization states we apply voltages beyond the
coer-cive values, Vtop = 0.35 V (0.85 V) for the P+ (P−
state). To investigate the time-resolved properties, we use a train of voltages pulses. One is a non-switching pulse, the other is a switching pulse. This design allows to discriminate phenomena due to ferroelectric polariza-tion (non-linear ferroelectric behavior, switching pulse) from those due to the whole device (linear dielectric
be-havior, non-switching pulse)21.
At t = 0, we set Vtop = 0.85 V, the idle state, which
corresponds to the P− polarization. At t = 1.9 µs, an
upward pulse Vtop = 1.35 V is applied with a rise (fall)
time of 2.5 ns and a width of 2 µs, then Vtopgoes back to
the idle state. Thus, during this non-switching pulse, the
system stays in the P− state. At t = 5.8 µs, a downward
pulse Vtop = 0.35 V is applied and the system switches
from P− to P+. At t = 7.8 µs, V
top returns to the idle
state and the system switches from P+ to P− (see
Fig-ure 2). Snapshot spectra of the photoemitted electrons are acquired with a time step of 45 ns over 9 µs. The pulse train is repeated a thousand times in order to ac-quire a sufficient photoemission signal/noise ratio.
III. RESULTS
A. Photoemission Data
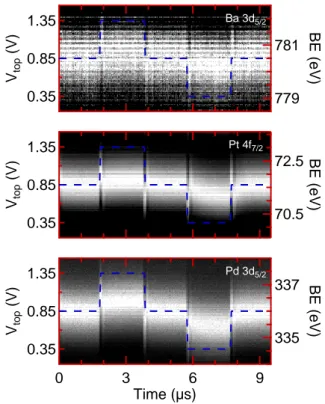
Figure 3 shows photoelectron intensity as a function
of time and binding energy for Pd 3d5/2, Pt 4f7/2 and
Ba 3d5/2 core levels corresponding to the pad, electrode
and ferroelectric responses to the switching. The shift of the binding energy scale due to the voltage pulses is clearly visible on the core-level spectrum, but this
rep-1.6 1.2 0.8 0.4 0.0 Vtop (V) 9 6 3 0 Time (µs) BTO NSTO Pt BTO NSTO Pt P -P+ Vc+ V
c-FIG. 2. Vtoppulse train as a function of time. The first pulse
is non-switching P−to P−, the second is switching P−to P+
then back to P−. The BTO film is in the P+ state between
t = 5.8 µs and t = 7.8 µs. The coercive voltages Vc+and Vc−
are indicated by dotted lines.
1.35 0.85 0.35 Vtop (V) 781 779 BE (eV) Ba 3d5/2 1.35 0.85 0.35 Vtop (V) 72.5 70.5 BE (eV) Pt 4f7/2 9 6 3 0 Time (µs) 1.35 0.85 0.35 Vtop (V) 337 335 BE (eV) Pd 3d5/2
FIG. 3. Photoemitted electron intensity as a function of time
and binding energy (BE) for the three core-levels Pd 3d5/2,
Pt 4f7/2and Ba 3d5/2. The blue dotted curve shows the pulse
train applied on the top electrode.
resentation is not well-suited for further analysis. We used a fitting procedure which subtracts the secondary electron background and fits the instantaneous spectrum to a Gaussian shape for each time step. It returns the binding energy position of the peak as a function of time and allows a proper comparison of core-level response to the electrical excitation. The width of each peak is kept constant after a preliminary fit procedure using a high-resolution reference spectrum taken with both elec-trodes grounded. The shape for all core-levels did not change with time except for the spectra close to the rise
and fall of the bias pulses. At these moments, the peaks are highly distorted leading to spikes (typical width of 150 ns) in the time-dependent binding-energy curves. A typical spectrum far from the spikes is shown in Fig-ure 4 together with the fit. Doniach-Sunjic lineshapes
are more suited to metallic peaks22, however, since we
ex-tract binding energy differences between identical peaks (in shape) at different time, the use of Gaussian shape is a good approximation and leads to a less time-consuming
procedure. We used one component to fit the Ba 3d
spectrum though it is known that two components are
observed at the Pt/BTO interface17. However, the
sig-nal/noise ratio of the Ba core-level from the buried in-terface was not sufficient to properly fit two components at every time step (Fig. 4b).
Intensity (arb. units)
1040 1035
1030
Kinetic Energy (eV)
Pt 4f (a)
Intensity (arb. units)
338 334 330 326 322
Kinetic Energy (eV)
Ba 3d5/2 (b)
FIG. 4. Example of spectrum of (a) Pt 4f and (b) Ba 3d5/2
core-level taken at one time step (black circles) after subtrac-tion of a linear background. The Gaussian fitting lineshape is shown in plain red.
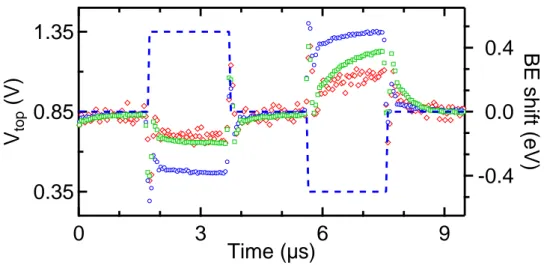
Figure 5 shows the time dependence of the binding energies obtained from the fitting procedure. All spec-tra are referenced relative to their binding energy in the
idle state at Vtop= 0.85 V. As expected, for the upward
(downward) pulse, every core-level shows a decrease (in-crease) in binding energy due to the more positive (nega-tive) voltage applied on the top electrode in comparison to the idle state. At every pulse rise and fall, spikes are observed in the binding energy. This is likely to be due to a parasitic capacitance with a very small time-constant although we cannot single out its location in the circuit. A capacitance with a time-constant below the rise time of the generator will act as a differentiator, i.e. will differen-tiate the signal with respect to time. The time derivative of a step pulse is the delta function we observe as binding
4 energy spikes here. Within the framework of the domain
nucleation-domain switching model, Merz distinguished the lead current due to rapid domain nucleation from the current due to domain wall motion and domain switch-ing. Rapid domain nucleation should therefore give rise to a current spike, whereas the response due to domain switching should be broader. Therefore, the core level binding energy spike may also be tentatively correlated with the onset of domain nucleation.
For the non-switching pulse, the core-levels reach a steady state value before the end of the pulse. While Pd follows quite closely the amplitude of the source pulse (∆BE = 0.4 eV compared to a pulse ∆V of 0.5 V), Pt
and Ba only shift by 0.20 eV. For the P− to P+ pulse,
the core-level shifts have a higher time constant and each core-level shift is quantitatively different. The ∆BE val-ues for the Pd, Pt and Ba core levels after 2 µs are 0.25, 0.37 and 0.50 eV respectively. It is therefore clear that a simple electrostatic model is not sufficient to describe the Pt/BTO interface and that the response of the electronic structure to switches in polarization is non-linear.
B. Simulations
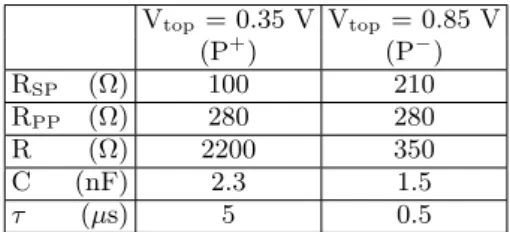
We conducted numerical simulations of the time-dependent linear behavior of the capacitor using PSpice
software23. This software can simulate the electrical
re-sponse of a circuit under a pulse excitation. An upward (downward) pulse identical to the experimental one was used and the circuit response in terms of the potential drop across each component calculated. We assume the resistances between the pulsed source and the palladium
pad (source impedance, RSP) and between the palladium
pad and the platinum electrode (imperfect metal/metal
contact, RPP) are ohmic. The resistance values should
not change with ferroelectric switching and are therefore kept constant. The Pt/BTO/NSTO stack is modeled by a RC circuit of resistance R and capacitance C and of
time constant τ = RC. The Pt 4f7/2core-level is
there-fore a direct probe of the voltage across the capacitor. An RC model accounts for the linear dielectric proper-ties but cannot of course directly model the FE switching, intrinsically non-linear. We have therefore used R and C as fitting parameters that change in response to polariza-tion switch. Figure 6 displays the equivalent circuit used in the simulations. The values which give the best fit to experimental data are given in Table I for each polariza-tion state and the results of the simulapolariza-tion are shown in Figure 7.
We first discuss the behavior of the Pt and Pd core-levels. It was not possible to properly fit the
photoemis-sion data without changing the resistance RSP between
the palladium pad and the pulse generator. Indeed, when the load (here the Pd/Pt/BTO/NSTO capacitor) on the pulse generator is too small (this is the case during the non-switching pulse), the source is not able to deliver the full pulse magnitude. In the framework of our simple
Vtop= 0.35 V Vtop= 0.85 V (P+) (P−) RSP (Ω) 100 210 RPP (Ω) 280 280 R (Ω) 2200 350 C (nF) 2.3 1.5 τ (µs) 5 0.5
TABLE I. Best fit values for the components of the equivalent circuit of Figure 7, for suffixes refer to text.
model, this results in two different values for RSP.
With impedance matching taken into account, the model circuit fits well the experimental values of the non-switching pulse. The steady state low R is consis-tent with the high-current measured by static
electri-cal experiment17. For the switching pulse P− to P+,
the Pt/BTO/NSTO stack resistance R, is higher, con-sistent with the lower current through the capacitor measured in Ref. 17. The high to low current
behav-ior when switching from P− to P+ is due to the
crease of the electron barrier height at the bottom in-terface (BTO/NSTO) of the capacitor (see Figure 7 of Ref. 17 for the polarization-dependent band lineups of the Pt/BTO/NSTO heterostructure).
Turning now to the transient behaviour, for the
non-switching pulse P− to P−, both rise (charge) and fall
(discharge) sequence fits well the experimental binding
energy shifts. However, for the P− to P+pulse, although
the P+ simulation curve fits well the rising edge of the
core level binding energies, it does not reproduce at all the falling edge because of the time-resolution of our ex-periment compared with the electron dynamics of po-larization switching. At the end of the downward pulse,
when the voltage goes from Vtop= 0.35 V back to 0.85 V,
the BTO film switches back to the P− state. The
dy-namic of polarization switching in thin films can be in the nanosecond range depending on the pulse
character-istics8, the ferroelectric layer properties24 and the
elec-trodes12. This is faster than our time resolution so that,
with respect to the spectral acquisition time, the stack
instantaneously switches to the P− state at t = 7.8 µs.
The simulation of the P+ to P- switch in Fig. 7 is
cal-culated from the parameters obtained for the P+ state
which predict a time constant ten times larger than for
the P−state. Figure 8 shows a close-up of the switch back
to P− following the P+ pulse. When the parameters of
the P− steady state are used the fit is much better,
re-producing quantitatively the experimental data (red line
for Pt 4f7/2, black line for Pd 3d5/2on Fig. 8). The time
constant of the RC component (τ = RC) estimated from
the steady state fit parameters is 0.5 µs (5 µs) for the P−
(P+) state.
The switching time is therefore sub-µs, in agreement
with literature9,19,25. The much higher value of τ
de-duced from the steady P+ state may well reflect the RC
circuit load rather than a material property.
-0.4
0.0
0.4 BE shift (eV)
9
6
3
0
Time (µs)
1.35
0.85
0.35
V
top(V)
FIG. 5. Time-dependent evolution of the binding energy (BE) shifts for the Pd 3d5/2 (blue circles), Pt 4f7/2 (green squares)
and Ba 3d5/2(red diamonds). The blue dotted curve shows the pulse train applied on the top electrode.
+
-Pd
Pt
R
SPR
PPR
C
FIG. 6. Equivalent circuit used in the PSpice simulation soft-ware. The components values are summarized in Table I.
-0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 BE shift (eV) 9 6 3 0 Time (µs) Pd 3d5/2 (exp.) Pd 3d5/2 (sim.) Pt 4f7/2 (exp.) Pt 4f7/2 (sim.)
FIG. 7. Results of the electrical simulations (plain lines) of the experimental Pd 3d5/2(blue circles) and Pt 4f7/2(green
squares) evolution. The simulated curves are generated from
the parameters of P− (Table I, right column) from t = 0
to t = 5.8 µs and from the parameters of P+ (Table I, left
column) from t = 5.8 to t = 9.2 µs.
barium core-level is more complex because it cannot eas-ily be related to a well-defined probing point in the model
0.6 0.4 0.2 0.0 BE shift (eV) 9 8 7 Time (µs)
FIG. 8. Results of the electrical simulations for the fall se-quence of the P−to P+ pulse. Pd 3d5/2and Pt 4f7/2
exper-imental binding energy are shown in blue circles and green squares respectively. The simulated plain curves of the fall
sequence are generated from the parameters of the P− state
(black line for Pd 3d5/2, red line for Pt 4f7/2). The simulated
dotted curves are generated from the parameters of the P+
state.
+
-Pd
Pt
Ba
R
SPR
PPR
intR
bu lkC
C
intFIG. 9. Capacitance model for modeling the barium core-level behavior.
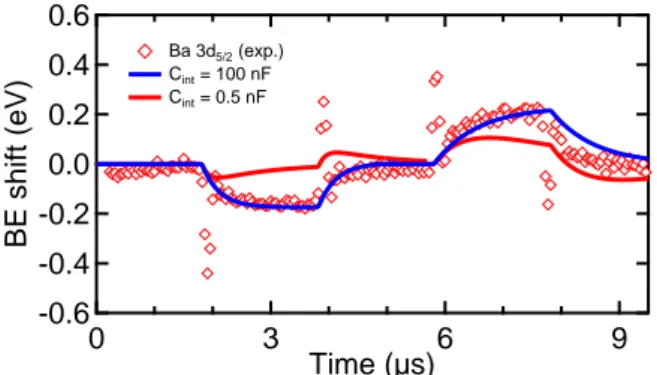
polarization-6 -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 BE shift (eV) 9 6 3 0 Time (µs) Ba 3d5/2 (exp.) Cint = 100 nF Cint = 0.5 nF
FIG. 10. Results of the electrical simulations (plain lines)
of the experimental Ba 3d5/2 (red diamonds) evolution. The
simulated curves are generated with Cint = 0.5 nF (red line)
and Cint= 100 nF (blue line).
induced interface dipole but also for the non trivial in-terface chemistry. As a first approximation, we model the top interface as an additional RC circuit as shown in Fig-ure 9. The total capacitor resistance R must be equal to
the sum of interface resistance Rint and remaining bulk
resistance Rbulk. The photoemission probing depth at
1100 eV photon energy is of 3-4 nm so that only electrons emitted from the top interface and the first barium lay-ers are collected. Therefore, a model adding a separate contribution due to the bottom interface is not neces-sary here. Figure 10 shows the simulation of the barium binding energy shifts for two extreme values of the
in-terface capacitance Cint. The model correctly fits the
spectroscopic data when the capacitance value is 100 nF, i.e. when the interface RC circuit has a time constant well above the pulse train period of 10 µs. For the
low-est value (Cint = 0.5 nF), the simulation is qualitatively
incorrect since the simulated signal reaches a maximum before decreasing, which is clearly not the case of the experimental signal. Therefore, to fit the data, the in-terface capacitance of the model has to be much higher than the bulk capacitance. This is an extremely impor-tant result because it places a lower limit on the interface
capacitance, Cint. The Ba 3d transient core level response
to the polarization switch therefore confirms the poten-tially important role of the interface capacitance in thin
film ferroelectric based devices.26We note that this does
not necessarily mean that polarization switching time in the whole capacitor stack is 10 µs but that close to the electrode, within the framework of an RC model, there is a significant interface capacitance.
A more sophisticated quantitative analysis is not pos-sible since the RC circuit we use to simulate the stack
actually reduces to a simple resistance Rint at the
fre-quencies used (Rint= 50 Ω (1000 Ω) and Rbulk= 300 Ω
(1200 Ω) for P− (P+) states).
The result is in qualitative agreement with the first-principles-based theoretical results of Stengel et al. in Ref. 26. Using a series capacitance model, they found a high value for the interface capacitance relatively to
the bulk capacitance (Cint≈ 200×Cbulk) of the Pt/BTO
interface. Though the theoretical system
(symmet-ric Pt/BTO/Pt structure) is different from our exper-imental stack (asymmetric Pt/BTO/NSTO), the semi-quantitative agreement is encouraging and might pro-mote similar experiments with different pulse
frequen-cies or other metal/FE interfaces. The SrRuO3/BaTiO3
interface should show a very different behavior for in-stance26.
Further information on the relationship between the interface electronic structure and capacitance could be obtained by improved signal to noise ratio which would allow a quantitative analysis of the two components of the Ba 3d core level spectrum. It is known that structural changes such as rumpling and interplanar relaxation at the surface can be correlated with the core level shift of
the Ba 3d high binding energy component27. The use of
hard X-ray photoemission would also improve the count-ing statistics and allow to better identify interface and bulk film contributions to the electronic structure and hence the interface capacitance.
IV. CONCLUSION
We have provided a proof of concept for the investiga-tion of realistic devices in operating condiinvestiga-tions with time-resolved photoemission spectroscopy. The chemical, elec-tronic and depth sensitivity of core level photoemission is used to probe the transient response of different parts of the upper electrode/ferroelectric interface to voltage pulse induced polarization switching. With the spatial and time resolution available, valuable information on the physics of the interface is accessible.
The linear dielectric response of the electronic struc-ture is measured using time-resolved photoelectron spec-troscopy and agrees quantitatively with a simple RC
cir-cuit model. The non-linear response of the electronic
structure due to the polarization switch is demonstrated by the time-resolved response of the characteristic core levels of the electrode and the ferroelectric. Adjustment of the RC circuit model allows a first estimation of the Pt/BTO interface capacitance. The experiment shows the interface capacitance is at least 100 times higher than the bulk capacitance of the BTO film, in quali-tative agreement with theoretical predictions from the literature.
Using the temporal structure of x-ray pulses, time-resolved photoemission experiments can reach the
pi-cosecond resolution18 and thus access valuable
informa-tion on the electron dynamics of polarizainforma-tion
switch-ing itself. Moreover, in our case the spatial
resolu-tion is limited by the beam spot size, approximately
100 × 100 µm2 and prevent from studying realistic
mi-crochips. However, the development of photoelectron
emission microscopy with in-situ bias application, time-resolved detectors and pump/probe setups using high-harmonic generation lasers might lead the way for
in-operando chemical/electronic analysis of realistic devices at typical operating frequencies.
ACKNOWLEDGMENTS
J.R. is funded by a CEA Ph.D. Grant CFR. This work, partly realized on Nanolyon platform, was supported by
ANR project Surf-FER, ANR-10-BLAN-1012 and Minos, ANR-07-BLAN-0312. We acknowledge SOLEIL for pro-vision of synchrotron radiation facilities. We thank J. Leroy, S. Foucquart and C. Chauvet for technical assis-tance.
∗
Now at: Synchrotron-SOLEIL, BP 48, Saint-Aubin,
F91192 Gif sur Yvette CEDEX, France
†
Now at: IHP, Im Technologiepark 25, D-15236 Frankfurt (Oder), Germany
‡
Correspondence should be addressed to [email protected]
1
R. Ramesh and N. A. Spaldin, Nat Mater 6, 21 (2007).
2 D. G. Schlom, L. Chen, X. Pan, A. Schmehl, and M. A.
Zurbuchen, Journal of the American Ceramic Society 91, 2429 (2008).
3 N. Balke, M. Gajek, A. K. Tagantsev, L. W. Martin, Y.-H.
Chu, R. Ramesh, and S. V. Kalinin, Advanced Functional Materials 20, 3466 (2010).
4 W. J. Merz, Physical Review 95, 690 (1954).
5 Y. Ishibashi and Y. Takagi, Journal of the Physical Society
of Japan 31, 506 (1971).
6 M. Avrami, The Journal of Chemical Physics 7, 1103
(1939).
7
A. Kolmogoroff, Izv. Akad. Nauk SSSR Ser. Mat. 1, 355 (1937).
8 A. K. Tagantsev, I. Stolichnov, N. Setter, J. S. Cross, and
M. Tsukada, Physical Review B 66, 214109 (2002).
9
A. Gruverman, D. Wu, and J. F. Scott, Physical Review Letters 100, 097601 (2008).
10
S. Zhukov, Y. A. Genenko, O. Hirsch, J. Glaum,
T. Granzow, and H. von Seggern, Physical Review B 82, 014109 (2010).
11
T. K. Song, S. Aggarwal, Y. Gallais, B. Nagaraj,
R. Ramesh, and J. T. Evans, Applied Physics Letters 73, 3366 (1998).
12 T. H. Kim, S. H. Baek, S. M. Yang, Y. S. Kim, B. C. Jeon,
D. Lee, J.-S. Chung, C. B. Eom, J.-G. Yoon, and T. W. Noh, Applied Physics Letters 99, 012905 (2011).
13 S. Gorfman, O. Schmidt, M. Ziolkowski, M. von
Kozierowski, and U. Pietsch, Journal of Applied Physics 108, 064911 (2010).
14 T. Nagata, M. Haemori, Y. Yamashita, H. Yoshikawa,
Y. Iwashita, K. Kobayashi, and T. Chikyow, Applied
Physics Letters 97, 082902 (2010).
15
S. Suzer, E. Abelev, and S. L. Bernasek, Applied Surface Science 256, 1296 (2009).
16 F. Chen and A. Klein, Physical Review B 86, 094105
(2012).
17
J. E. Rault, G. Agnus, T. Maroutian, V. Pillard,
P. Lecoeur, G. Niu, B. Vilquin, M. G. Silly, A. Bendounan, F. Sirotti, and N. Barrett, Physical Review B 87, 155146 (2013).
18 N. Bergeard, M. G. Silly, D. Krizmancic, C. Chauvet,
M. Guzzo, J. P. Ricaud, M. Izquierdo, L. Stebel, P. Pit-tana, R. Sergo, G. Cautero, G. Dufour, F. Rochet, and F. Sirotti, Journal of Synchrotron Radiation 18, 245 (2011).
19
P. K. Larsen, G. L. M. Kampsch¨oer, M. J. E. Ulenaers,
G. A. C. M. Spierings, and R. Cuppens, Applied Physics Letters 59, 611 (1991).
20
F. Polack, M. Silly, C. Chauvet, B. Lagarde, N. Bergeard, M. Izquierdo, O. Chubar, D. Krizmancic, M. Ribbens, J.-P. Duval, C. Basset, S. Kubsky, and F. Sirotti, AIP Confer-ence Proceedings 1234, 185 (2010).
21
S. D. Traynor, T. D. Hadnagy, and L. Kammerdiner, In-tegrated Ferroelectrics 16, 63 (1997).
22
S. Doniach and M. Sunjic, Journal of Physics C: Solid State Physics 3, 285 (1970).
23 Orcad package from Cadence Design Systems:
http://www.cadence.com/products/orcad/pages/default.aspx.
24
A. Musleh Alrub and L.-H. Ong, Journal of Applied Physics 109, 084109 (2011).
25 J. Y. Jo, H. S. Han, J.-G. Yoon, T. K. Song, S.-H.
Kim, and T. W. Noh, Physical Review Letters 99, 267602 (2007).
26 M. Stengel, D. Vanderbilt, and N. A. Spaldin, Nat Mater
8, 392 (2009).
27
A. Pancotti, J. Wang, P. Chen, L. Tortech, C.-M. Teodor-escu, E. Frantzeskakis, and N. Barrett, Physical Review B 87, 184116 (2013).