HAL Id: tel-01599337
https://tel.archives-ouvertes.fr/tel-01599337
Submitted on 2 Oct 2017HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are
pub-L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non,
en couches minces
Solange Temgoua
To cite this version:
Solange Temgoua. Optimisation et compréhension des dispositifs à base de Cu2ZnSn(Sx,Se1-x)4 pour applications photovoltaïques en couches minces. Matériaux. Université Pierre et Marie Curie - Paris VI, 2016. Français. �NNT : 2016PA066687�. �tel-01599337�
Thèse de Doctorat de l’Université Pierre et Marie Curie École Doctorale 397 - Physique - Chimie des Matériaux
Optimisation et compréhension des dispositifs à
base de Cu
2ZnSn(S
x,Se
1−x)
4pour applications
photovoltaïques en couches minces
Présentée par Solange TEMGOUA
Soutenue le 08 Novembre 2016 devant le jury composé de :
Alain LAFOND, Professeur Rapporteur
Abdelilah SLAOUI, Directeur de Recherche Rapporteur
Edgardo SAUCEDO, Docteur-Chercheur Examinateur
Charlotte PLATZER BJÖRKMAN, Professeur Examinateur
Sophie CASSAIGNON, Maitre de Conférences Examinateur
Negar NAGHAVI, Directeur de Recherche Directrice de Thèse
Table des matières
Introduction générale 1
1 Etat de l’art sur les kestérites Cu2ZnSn(S,Se)4 (CZTSSe) 5
1.1 Contexte général du photovoltaïque . . . 5
1.2 Les technologies photovoltaïques . . . 7
1.2.1 Disponibilité des ressources . . . 8
1.2.2 Question de la ressource en indium et en gallium : choix du Cu2ZnSn(S,Se)4 . . . 9
1.3 Les cellules solaires en couches minces à base de Cu2ZnSn(S,Se)4 . . . 10
1.3.1 La jonction P-N . . . 10
1.3.2 Structure des cellules solaires Cu2ZnSn(S,Se)4 . . . 13
1.3.3 Courbes courant-tension (I-V) . . . 13
1.4 Propriétés des matériaux de la famille des kestérites . . . 16
1.4.1 Propriétés cristallines . . . 16
1.4.2 Propriétés optoélectroniques . . . 17
1.4.3 Quels sont les verrous technologiques des absorbeurs kestérites ? 20 1.4.4 Techniques de synthèse des matériaux kestérites . . . 28
1.5 Conclusion . . . 30
2 Techniques expérimentales et méthodes d’analyses 31 2.1 Synthèse et caractérisation des absorbeurs . . . 31
2.1.1 Dépôt des précurseurs par pulvérisation cathodique . . . 31
2.1.2 Traitement thermique (recuit) des précurseurs . . . 36
2.2 Caractérisations des matériaux en couches minces . . . 38
2.2.1 Spectrométrie de fluorescence X . . . 38
2.2.2 Microscopie électronique à balayage . . . 39
2.2.3 Diffraction des Rayons X . . . 39
2.2.4 Spectroscopie Raman . . . 40
2.2.5 Spectroscopie d’émission optique par décharge luminescente (GD-OES) . . . 42
2.2.6 Quantification des absorbeurs CZTSSe . . . 42
2.3 Formulations de bain de traitement chimique . . . 43
2.4 Caractérisations des cellules solaires à base de CZTSSe . . . 44
2.4.1 Mesure des caractéristiques J-V des cellules solaires sous illu-mination . . . 44
2.5 Conclusion . . . 45
3 Mécanismes de formation de l’absorbeur CZTSSe 47 3.1 Formation de CZTSSe à partir des précurseurs métalliques . . . 48
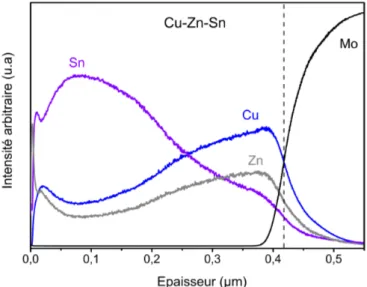
3.1.1 Propriétés du précurseur métallique Cu-Zn-Sn . . . 48
3.1.2 Recuit pur Se de précurseur Cu-Zn-Sn . . . 50
3.1.3 Recuit sulfosélénisant en une étape de précurseur métallique . 53 3.1.4 Recuit sulfosélénisant en deux étapes de précurseur métallique 55 3.1.5 Cellules solaires à base de CZTSSe obtenus par le recuit deux étapes . . . 58
3.2 Formation de CZTSSe à partir des précurseurs soufrés . . . 61
3.2.1 Effet de la température sur le mécanisme de formation du CZTSSe . . . 62
3.2.2 Récapitulatif du mécanisme de croissance . . . 77
3.2.3 Influence du palier de recuit sur la cristallisation du CZTSSe . 78 3.2.4 Conclusion partielle . . . 80
3.3 Impact du recuit sur les performances des cellules . . . 80
3.3.1 Compréhension de l’origine des pertes . . . 82
3.4 Conclusion . . . 84
4 Influence des propriétés de l’absorbeur 87 4.1 Effet de l’épaisseur du précurseur et de l’atmosphère de recuit sur les cellules CZTSSe . . . 87
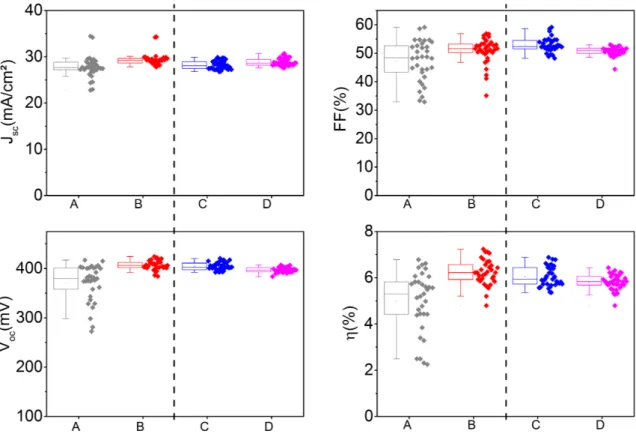
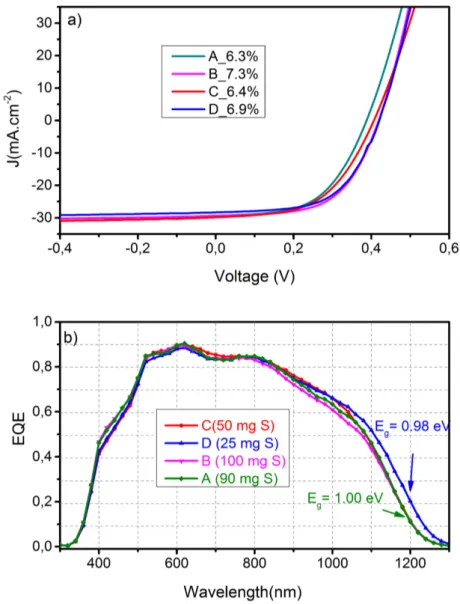
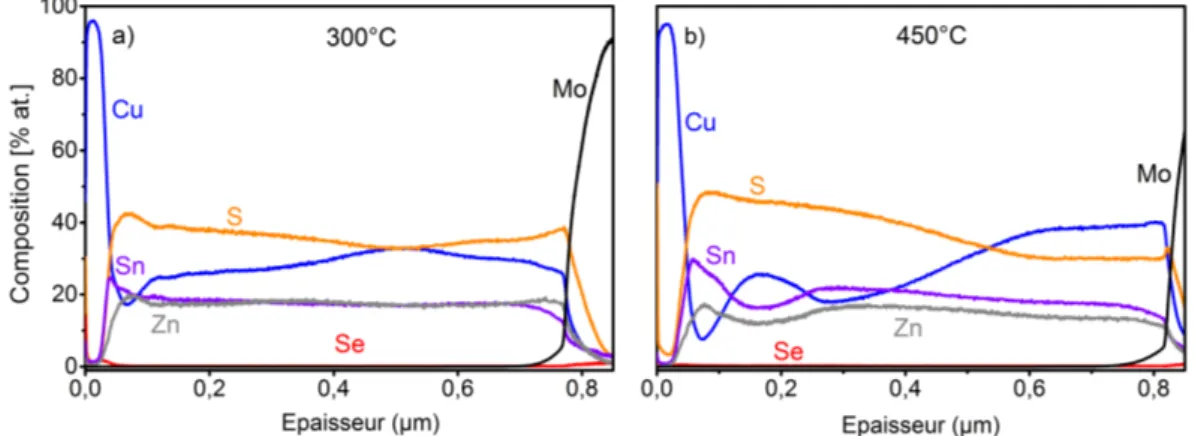
4.1.1 Analyse de composition des absorbeurs . . . 88
4.1.2 Analyse structurale des absorbeurs . . . 90
4.1.3 Impact de l’atmosphère de recuit et de l’épaisseur sur les pro-priétés des cellules solaires . . . 94
4.1.4 Conclusion partielle . . . 102
4.1.5 Traitements chimiques de surface . . . 102
4.2 Conclusion . . . 107
5 Contacts arrière à d’oxydes transparents conducteurs 109 5.1 Bref état de l’art et motivation . . . 109
5.1.1 Propriétés des TCO - quelques applications dans le photovol-taïque . . . 111
5.2 Synthèse du CZTSSe sur TCO : SnO2 :F(FTO) et ZnO :Al (AZO) . . 112
5.2.1 Impact des traitements thermiques sur les TCO . . . 113
5.2.2 Impact du TCO utilisé sur la morphologie des précurseurs . . 117
5.2.3 Etude du recuit des précurseurs Cu-Zn-Sn sur des substrats FTO et AZO . . . 121
5.2.4 Etude du recuit des précurseurs Cu-Zn-Sn-S sur TCO . . . 127
5.2.5 Caractérisations de cellules solaires sur TCO . . . 132
Table des matières
Conclusion et perspectives 137
6
Annexe A Spectrométrie de fluorescence
1417
Annexe B MEB/EDX
1437.1 Microscopie électronique à balayage (MEB) . . . 143 7.2 Analyse par Energie de dispersion de Rayons X . . . 143
8
Annexe C Diffraction de rayons X
1459
Annexe D La spectroscopie Raman
14710
Annexe E Spectroscopie d’émission optique
14911
Annexe F Analyse structurale par DRX
15112
Annexe G Le recuit mixte S et Se (une étape)
15313
Annexe H DRX en incidence rasante
15514
Annexe I Spectres Raman fittés
157Objectifs et challenges de cette thèse
Dans un contexte général d’augmentation de la demande énergétique et de préoccu-pation croissante face au réchauffement climatique et à la limitation des ressources naturelles, l’utilisation de l’énergie solaire est certaine d’augmenter. Selon l’Agence Internationale de l’Energie (AIE), la production d’énergie en 2050 engendrera une hausse de la température globale entre 2°C1 et 6°C2 [1]. D’après l’AIE, la puissance
photovoltaïque installée en 2050 pour la génération d’électricité sera comprise entre 200 GW et 7 TW.
L’avenir des différentes filières photovoltaïques PV dépend évidemment de leur ren-dement de conversion photovoltaique et de leur coût, mais plus important encore de la disponibilité des ressources. En effet, le prix et la rareté de certains éléments entrant dans la fabrication des dispositifs photovoltaiques sont de sérieux facteurs limitants au déploiement du photovoltaïque à grande échelle. A titre d’exemple, l’ar-gent (utilisé pour les contacts des cellules silicium), l’indium et le gallium sont les éléments limitants dans le cas du Cu (In,Ga)Se2 (CIGS), le tellurium dans le cas
du CdTe, le ruthenium pour les cellules à colorant pour ne citer que ceux-là. La question de la perennité de la production photovoltaïque devient donc pertinente et la nécessité de développer de nouvelles technologies à base d’éléments abondants et non-toxique s’impose ; ceci afin d’alléger la pression sur toutes les technologies photovoltaïques en terme de ressources.
La thèse de doctorat présentée ici s’inscrit dans cette stratégie de développement de matériaux abondants, non toxiques pour des applications photovoltaïques. Les composés de la famille des kestérites, Cu2ZnSnS4 (CZTS), Cu2ZnSnSe4 (CZTSe),
Cu2ZnSn(S,Se)4(CZTSSe) sont des candidats prometteurs. Nombreux groupes
tra-vaillent sur l’élaboration de ces matériaux via des techniques de synthèse sous vide [2, 3], des procédés atmosphériques [4, 5], les procédés une étape (croissance directe de l’absorbeur sur le substrat)[6] et deux étapes (dépôt du précurseur + recuit) [7]. Le procédé deux étapes semble être le plus adapté pour le développement de cellules solaires à base de CZTSSe. L’étape de recuit présente deux aspects importants : l’insertion du chalcogène (S ou Se) et la recristallisation des précurseurs. L’étude de cette étape de recuit et ses implications sur les propriétés finales de l’absorbeur est indispensable pour l’avenir de la filière kestérite.
Le chapitre 1 présente un bref état de l’art sur le positionnement du photovoltaïque sur le marché de la production énergétique mondiale, ainsi que les motivations de
1. En cas de déploiement accéléré des énergies renouvelables 2. Scénario conservateur
matériau seront détaillés.
Les différents outils analytiques de synthèse, et de caractérisation des absorbeurs et des dispositifs photovoltaïques finaux seront mentionnés dans le chapitre 2.
L’étude du chemin de réaction et de formation de la phase CZTSSe suivant la na-ture du précurseur, métallique ou soufrée sera détaillée dans le chapitre 3. En outre, l’influence de l’atmosphère de recuit sur l’incorporation des chalcogènes et la forma-tion des phases secondaires sera discutée. Des condiforma-tions de recuit optimales seront déterminées grâce à la compréhension de ces mécanismes de formation .
Le chapitre 4 présente l’impact des différents léviers sur lesquels nous avons travaillé afin d’améliorer à la fois les propriétés de l’absorbeur et des cellules solaires, notam-ment (i) l’impact de l’atmosphère de recuit, Se pur versus (S+Se), (ii) l’impact de l’épaisseur des précurseurs, (iii) les traitements chimiques de surface.
Le chapitre 5 porte sur le développement de nouvelles architectures de cellules so-laires à base des kestérites ; les cellules soso-laires bifaciales CZTSSe. Les résultats provenant d’une étude comparative préliminaire du recuit des matériaux kestérites déposés sur le molybdène et sur des oxides transparents conducteurs (TCO) comme contacts arrière seront abordés. Il s’agit notamment du SnO2 :F et ZnO :Al.
L’in-fluence du contact arrière sur les propriétés morphologiques, structurales ainsi que sur les propriétés électriques des cellules solaires sera présentée.
Enfin nous présenterons la conclusion de ce travail et les perspectives pour les travaux à venir.
1 Etat de l’art sur les kestérites
Cu
2
ZnSn(S,Se)
4
(CZTSSe)
1.1 Contexte général du photovoltaïque
La conjonction de l’instabilité des marchés des énergies fossiles et l’impératif de protection de l’environnement et de réduction des émissions de gaz à effet de serre imposent une révision des stratégies énergétiques. Ces problématiques ont constitué l’objectif de la COP 21 (30 novembre-12 décembre 2015 à Paris) qui consiste à ré-duire les émissions de gaz à effet de serre et contenir l’élévation de la température moyenne de la planète en dessous de 2°C d’ici 2100 [8]. Les énergies renouvelables constituent une réponse particulièrement adaptée aux besoins énergétiques consi-dérables des pays émergents qui assurent l’essentiel de la croissance mondiale. Les investissements dans les énergies renouvelables du secteur électrique ont dépassé les investissements nets dans les centrales électriques à combustibles fossiles. De nombreuses études montrent que le mix énergétique a évolué et une redistribution a été opérée. Le secteur électrique (dont l’hydroélectricité, l’éolien et le photovol-taïque) affiche la croissance la plus rapide ainsi que la plus forte augmentation de capacités d’énergies renouvelables [9]. Il est dominé par trois technologies : l’énergie éolienne, l’énergie solaire photovoltaïque et l’hydroélectricité. Dans les énergies re-nouvelables, l”hydroélectricité domine, suivie d’une évolution intéressante de l’éolien et du photovoltaique. La lumière produite par le soleil représente une source d’éner-gie inépuisable. En effet, la Terre reçoit en un an environ 7000 fois la consommation mondiale d’énergie de l’humanité.
Le photovoltaïque (PV) est l’une des sources d’énergie renouvelable qui sera la plus déployée à l’avenir avec des perspectives de croissance d’environ 40GW/an de 2015 à 2020. Cette forte progression de l’électricité d’origine solaire se traduit par une volonté manifeste d’intégrer cette ressource dans le mix énergétique. Selon l’Agence Internationale pour l’Energie concernant le photovoltaique, au moins 51 GW ont été installés dans le monde en 2015, soit 30 % de plus qu’en 2014 comme le montre la figure 1.1 . L’Europe a atteint 8.5 GW de capacité installée grâce au Royaume-Uni et à l’Allemagne qui totalisent à eux seuls plus de 5 GW. La demande sur le marché se déplace sur l’Asie qui représentera un marché 3 fois plus important que l’Europe sur la période 2015-2020. Les installations aux Etats-Unis ont crû de 56% en 2015, stimulées par le crédit d’impôt et les locations résidentielles. Plus intéressant car ils sont rarement détaillés, les pays émergents commencent à contribuer
significative-ment à la croissance mondiale : il y’a eu 1.5 GW dans les autres pays américains (amérique latine), 2.5 GW en Asie-Australie. L’Afrique et le Moyen-Orient compte pour 1 GW [10]. Cette augmentation est généralement la conséquence de politiques propres à chaque pays, rendant l’installation des panneaux solaires attractive. Selon l’entreprise américaine d’information économique IHS, les prédictions pour 2016 es-timent que les installations dans le monde atteindront les 70 GW de capacité. Les USA, la Chine et l’Inde totaliseront à eux seuls 9.3 GW sur les 10 GW attendus en 2016 [11].
En France, bien que le marché du photovoltaïque a connu un ralentissement des investissements depuis 2011, la capacité totale disponible était de 6.2 GW à la fin décembre 2015, soit une augmentation de 25 % par rapport à l’année précédente, avec une majorité (91%) de systèmes raccordés au réseau public de distribution [12].
Figure 1.1 – Evolution de la capacité annuelle des installations photovoltaiques dans le monde [10]. IEA PVPS : International Energy Agency Photovoltaic Power Systems Programme
1.2 Les technologies photovoltaïques
1.2 Les technologies photovoltaïques
Les cellules photovoltaïques sont classées en deux catégories : Les cellules à base de plaquette (“ wafer”) et les cellules en couches minces. Les cellules de type wafer sont fabriquées à base de plaquettes semiconductrices. Les cellules en couches minces consistent en des couches minces de semiconducteurs différents déposées sur un substrat de verre, plastique ou métallique. La figure 1.2 présente la structure typique des différentes types de cellules solaires.
Figure 1.2 – Structures typiques des cellules photovoltaïques classées en deux ca-tégories :les cellules à base de wafer et les cellules en couches minces. Les filières sont généralement classées d’après le nom de la couche absorbante de la cellule. La figure a été adaptée d’après [13]
Les filières telles que le silicium cristallin (c-Si), l’arséniure de gallium (GaAs), les multijonctions III-V sont regroupées dans la catégorie dite wafer, tandis que les fi-lières telles que le silicium amorphe (a-Si :H), le telluriure de cadmium (CdTe), le Cu2(In,Ga)Se2 (CIGS), le Cu2ZnSn(S,Se)4(CZTSSe), les cellules à colorants
orga-niques (DSCC), ou les cellules à base de pérovskites constituent la catégorie couches minces. La réduction du coût de fabrication des cellules photovoltaïques est la prin-cipale raison pour laquelle les cellules en couches minces ont vu le jour. En effet, les cellules à base de c-Si requièrent des épaisseurs de l’ordre de 100 µm car le Si est un semiconducteur à bande interdite indirecte et possède en outre un faible coefficient d’absorption. Les cellules en couches minces permettent d’absorber 10 à 100 fois plus de lumière avec des couches d’à peine quelques µm d’épaisseurs. La réduction de matière utilisée est donc un avantage clé pour les filières couches minces.
Une comparaison des performances photovoltaïques des filières développées est résu-mée sur la figure 1.3 Le Si cristallin reste en tête avec les meilleures performances de
cellules et de modules.Il représente environ 90% de la part du marché des modules commercialisés [13]. Cependant les cellules en couches minces évoluent rapidement, notamment pour le CdTe et le CIGSe. Elles représentent à l’heure actuelle environ 10% de la part du marché.
Figure 1.3 – Comparaison des performances des filières photovoltaïques commer-cialisées [14]. Les meilleurs rendements de cellules et modules sont illustrés. Les données sont extraites dans [15]
Pour qu’un module soit compétitif, le prix du module doit passer sous la barre de 0.7 $/W. C’est le cas pour les technologies Si et CdTe dont les prix moyens en 2016 varient entre 0.45-0.69 $/W [16] pour le Si, et 0.58 $/W pour le CdTe [17]. L’aug-mentation du volume de production, l’amélioration du rendement de conversion photovoltaïque et des coûts de production ont permi de réduire les prix des modules CIGS à environ 0.4 $/W [18]. Ce qui place la filière CIGS comme l’alternative la plus compétitive au Si dans la course à la réduction des prix
1.2.1 Disponibilité des ressources
Chaque technologie photovoltaïque possède un ou plusieurs éléments critiques, tels que l’argent utilisé pour les contacts des cellules à base de Si cristallin, l’indium et la gallium pour le CIGS, le tellurium pour le CdTe, le ruthénium pour les cellules à colorants, le germanium, l’or pour les III-V [19, 20]. Les métaux tels que le cadmium, le gallium, l’indium, le tellurium sont des produits dérivés mineurs de la production d’autres métaux tels que le cuivre, le zinc, l’aluminium. Leur disponibilité est donc fortement dépendante de la production des métaux. Il se pose donc la question
1.2 Les technologies photovoltaïques
pertinente de la pérennité de l’industrie photovoltaïque, et la nécessité de développer des nouveaux matériaux performants à base d’éléments abondants et non toxiques s’impose.
1.2.2 Question de la ressource en indium et en gallium : choix
du Cu
2ZnSn(S,Se)
4L’indium est présent en faible quantité dans les gisements de zinc [21]. Il faut envi-ron une tonne de zinc pour extraire une dizaine de gramme d’indium. La production mondiale actuelle d’indium est de l’ordre de 600 t par an [22, 23], et une frac-tion significative de cette producfrac-tion est utilisée dans l’industrie microélectronique. Les écrans à cristaux liquides (en anglais LCD), GPS (Global Positioning System), smartphones, tablettes, ordinateurs...contiennent en moyenne un gramme d’indium, sans oublier les panneaux solaires à base de CIGS. Les écrans LCD utilisent 65% de la production mondiale en In, alors que le photovoltaïque en consomme environ 5% [21]. L’industrie microélectronique a explosé ces 15 dernières années, multipliant le cours de l’indium par 10 entre 2000 et 2005. Son prix est passé de 100$/kg en 2000 à 1000$/kg en 2005 [24]. Bien que la forte demande en indium a poussé les industries de recyclage à développer une activité de récupération de ce métal, les quantités demeurent encore relativement faibles. Pour une tonne de déchet d’écran LCD, 174g d’indium peuvent être recupérés [25].
Les wafers de GaAs et GaN sont très utilisés dans les circuits intégrés, et des dispo-sitifs optoélectroniques qui incluent les diodes laser. Environ 57% de la production de gallium est utilisé dans les circuits intégrés [23]. Les circuits intégrés à base de GaAs sont très utilisés dans les applications de défense à cause de leurs propriétés uniques pour ces applications.
Table 1.1 – Production mondiale des principaux métaux sources et des produits dérivés en 2015. Les données proviennent de U.S. Geological Survey 2016 [23]
Métaux primaires Produits dérivés Production du produit dérivé
(en tonnes métriques)
Prix du métal raffiné ($/Kg)
Aluminium, Zinc Gallium (Ga) 160 295
Zinc Indium (In) 755 460
Zinc Cadmium (Cd) 24200 1.8
Cuivre Tellurium (Te) > 120 89
Cuivre Sélénium (Se) >2340 17
Cuivre (Cu) 18700 5.8
Zinc (Zn) 13400 2.3
Etain (Sn) 294000 1.7
Le tableau 1.1 présente la production annuelle des métaux primaires, ainsi que les produis dérivés raffinés à la fin 2015 d’après U.S. Geological Survey. Le prix de certains éléments augmente jusqu’à 80 fois, le prix du métal primaire, comme dans le cas de l’indium.
Des prévisions concernant l’avenir du CIGS ont été réalisées par plusieurs auteurs [21, 26] . Fthenakis et al [21] ont évalué pour une production annuelle d’une puis-sance de 20 GW, il n’y aurait pas de blocage jusqu’en 2020. Au-delà, il devient plus qu’urgent d’économiser de l’indium. Une solution intéressante considérée par l’auteur est de réduire l’épaisseur de la couche de CIGS tout en augmentant les ren-dements. Si l’on imagine un scénario où l’on passe d’une cellule standard de CIGS d’épaisseur 1.6 µm d’efficacité 11.2% à une cellule d’épaisseur 1 µm d’éfficacité à 16%, la quantité d’indium passerait de 83 tonnes/GW à 11-20 tonnes/ GW en 2020, ce qui permet d’envisager la poursuite du développement à grande échelle de la fi-lière CIGS [21]. Cette stratégie est étudiée au laboratoire IRDEP dans le cade du projet ANR UltraCISM, où des épaisseurs en deça de 300 nm sont ciblées avec des
configurations adaptées pour augmenter les rendements.
Une autre alternative également étudiée à l’IRDEP permettant de réduire la consom-mation d’indium est de travailler avec des microcellules sous concentration lumi-neuse. En intercalant un dispositif concentrateur (à base de lentilles ou mirroirs) entre le soleil et la cellule, une surface beaucoup plus petite peut être utilisée et ainsi augmenter le rendement des cellules entre 30 et 40% au minimum.
Le développement de nouveaux matériaux à base d’éléments plus abondants et non toxiques est également une voie très prometteuse pour perenniser les filières photo-voltaïques à base de chalcogénures. Le CZTSSe est le matériau le plus engageant pour le développement d’une nouvelle filière photovoltaïque. Sa production ne se-rait pas dépendente de la disponiblité des éléments, car le cuivre, le zinc, l’étain, le soufre/sélénium ne présente pas de restrictions en termes de disponibillité.
Les matériaux de la famille des kestérites, Cu2ZnSnS4(CZTS), Cu2ZnSnSe4(CZTSe)
et Cu2ZnSn(Sx,Se1-x)4 (CZTSSe) sont particulièrement attractifs pour un
dévelop-pement à grande échelle du photovoltaïque, en plus de leur avantages économiques intéressants. L’intérêt pour ces matériaux est d’autant plus grandissant que des ré-sultats encourageants ont été récemment obtenus par Kim et al. dont les meilleurs rendements sont à 12.7% [27].
1.3 Les cellules solaires en couches minces à base de
Cu
2ZnSn(S,Se)
41.3.1 La jonction P-N
Les dispositifs PV ou cellules solaires sont constitués d’une supperposition de ma-tériaux semiconducteurs. Le principe de fonctionnement d’une cellule solaire est
1.3 Les cellules solaires en couches minces à base de Cu2ZnSn(S,Se)4
basé sur les propriétés électriques des semiconducteurs. Les semiconducteurs sont constitués d’atomes liés ente eux dans une structure périodique et ordonnée (réseau cristallin) et d’électrons faiblement liés. Les semiconducteurs sont caractérisés par une conductivité électrique intermédiaire entre celle d’un métal (conducteur) et d’un isolant. Lorsque tous les électrons sont dans des états d’énergie liés, ie dans la bande de valence, la conduction est nulle et le semiconducteur se comporte comme un iso-lant. Si un électron est excité avec une énergie supérieure ou égale égale à celle de la bande interdite, par excitation thermique ou absorption d’une source lumineuse comme c’est le cas pour une cellule photovoltaïque, l’électron est libéré de son état lié dans la bande de valence (BV) pour un état libre dans la bande de conduction (BC). L’absence d’électron dans la bande de valence est vue comme une quasi parti-cule de charge positive appelée trou. Ce sont ces partiparti-cules chargées libres (électrons et trous) qui sont responsables de la conduction électrique dans les semiconducteurs. Dans un semiconducteur à la température ambiante, il y’a suffisamment d’électrons excités dans la bande de conduction, et le semiconducteur est conducteur.
Figure 1.4 – Représentation schématique de la structure électronique de bande d’un semiconducteur. 1) Un photon incident hν> Eg. 2) Promotion d’un électron
de la BV vers la BC, créant un trou dans la BV. 3) Porteurs libres (électrons de la BC et trous de la BV) de se déplacer et participant à la conduction du matériau. Un semiconducteur sera dit de type P (N) lorsque les porteurs libres majoritaires
sont des trous (électrons). Le niveau de Fermi pour un semiconducteur P (N) est proche de la bande de valence (bande de conduction). L’énergie de ces électrons est décrite par une structure électronique dite en bande, comme visible sur la figure 1.4. L’énergie de bande interdite (bandgap en anglais) est l’énergie qui sépare les bande d’énergie inférieure ou bande de valence (BV) et la bande d’énergie supérieur ou bande de conduction (BC).
La meilleure solution pour séparer les charges électriques consiste à utiliser un champ électrique. C’est pourquoi le fonctionnement même des cellules photovoltaïques re-pose sur l’utilisation de la jonction P-N. Dans une jonction P-N, les propriétés électriques dépendent fortement de l’alignement de bandes à l’interface des deux matériaux semiconducteurs constituants la jonction. Il a été démontré que l’aligne-ment de bande1 favorable pour l’obtention de meilleures performances est de type
« spike » [28, 29]. Des études expérimentales réalisées par Haight et al ont conclu que l’alignement de bandes entre CZTSSe et CdS est de type spike, et était faiblement dépendant de [S]/([S]+[Se]) [30]. La figure 1.5 représente un diagramme de bande entre une couche de CZTSe et de CdS.
Figure 1.5 – Diagramme de bande d’une cellule type CZTSe/CdS. Les différents mécanismes de transport sont illustrés. [Adaptée de [31]
1. Il existe deux types d’alignement de bande ; le « spike » où le minimum de la BC de la couche n est supérieur au minimum de la BC de la couche p. Le cas contraire, le « cliff » correspond au cas où le minimum de la BC de la couche n est inférieur au minimum de la BC de la couche p.
1.3 Les cellules solaires en couches minces à base de Cu2ZnSn(S,Se)4
1.3.2 Structure des cellules solaires Cu
2ZnSn(S,Se)
4La structure des cellules CZTSSe est basée sur un empilement verre/Mo/CZTSSe/CdS/i-ZnO/ZnO :Al. Celle-ci est présentée dans la figure 1.6.
Figure 1.6 – Structure en coupe d’une cellule CZTSSe : image en coupe transverse MEB (a), schéma explicatif (b)
Dans ce type de structure, les couches constitutant l’empilement sont : - un substrat de verre sodocalcique ~ 2-3 mm d’épaisseur
- une couche de molydène (Mo) (0.5-1 µm) utilisée comme contact arrière - l’absorbeur CZTSSe ( 1.5- 2.5 µm) qui est un semiconducteur de type p
- une fine couche (50 nm) à base de sulfure de cadmium CdS, In2S3, Zn (O,S) de
type n appelée couramment la couche tampon
- la couche fenêtre comme contact avant (400 nm) constituée de ZnO intrinsèque et ZnO :Al
- la couche antireflet pour réduire les pertes optiques par reflexion.
1.3.3 Courbes courant-tension (I-V)
Les caractéristiques courant-tension ou courbes I(V) illustrent le comportement élec-trique d’une jonction p-n idéale à la lumière et à l’obscurité (figure 1.7a) . Afin d’étudier les propriétés d’une cellule solaire, un circuit équivalent est utilisé comme modèle d’étude de la cellule ( figure 1.7b).
Figure 1.7 – Caractéristiques J(V) d’une jonction P-N idéale sous illumination (a). Modèle de circuit équivalent (b).
Le courant qui traverse une cellule sous illumination peut être exprimé sous la forme de l’équation ci-dessous :
J = Jph− J0(e(qV/nkBT)−1) (1.1)
où Jphest le photocourant (A/m2) et
J = J0(e(qV/nkBT)−1) (1.2)
est la densité de courant de diode à l’obscurité. Le terme en exponentiel caractérise le courant à travers une diode. J0 est la densité de courant de saturation inverse
ou courant de fuite. Il est dû au transport des porteurs minoritaires par le champ électrique crée dans la zone de charge d’espace. Le courant de saturation permet de mesurer les recombinaisons dans la cellule. V est la tension appliquée en Volts, q la valeur absolue de la charge de l’électron (1.6x10−19C), k
B la constante de
Boltz-mannn et T la température en Kelvin. n est le facteur d’idéalité de la cellule, qui renseigne sur la qualité de la jonction et le type de recombinaison dans la cellule. Il varie entre 1 et 2.
Par ailleurs, en fonctionnement réel, une cellule est le siège de phénomènes résistifs dûs aux matériaux utilisés et à leur fabrication :
1.3 Les cellules solaires en couches minces à base de Cu2ZnSn(S,Se)4
— Les résistances de contact et de connexion associées à une résistance série Rs
— Les résistances dûes aux courants de fuite dans la diode et aux effets de bord dans la jonction associés à une résistance parallèle Rsh
Physiquement, ces effets résistifs sont représentés en ajoutant une seconde diode en parallèle et deux résistances sur le schéma électrique équivalent de la cellule photovoltaïque (figure 1.5c). L’expression de la caractéristique I-V devient alors :
J = Jph− J01(e(V +JRs)/nkbT −1) + J02(e(V +JRs)/nkBT −1) −
V + JRs
Rsh
(1.3) Dans les cellules de bonne qualité, la résistance shunt est supérieure à 10 kΩ et la résistance série est inférieure à 1Ω.
Les courbes J(V) donnent accès à quatre paramètres qui renseignent sur les per-formances de la cellule solaire. Les caractéristiques courant-tension à l’obscurité donnent également des informations sur les propriétés des cellules solaires, via la détermination des paramètres de perte (Rs, Rsh, n, J0) qui dictent les performances
électriques des cellules. Le rendement de conversion photovoltaïque d’une cellule so-laire est donné par le ratio entre la puissance maximale que peut délivrée le dispositif sur la puissance du rayonnement incident. Il est donné par la relation
η
=
P maxP in (1.4)La puissance maximale générée par une cellule solaire est donnée par le produit
P max = Imax × V max (1.5)
D’autres paramètres significatifs permettant de définir les performances sont le Voc,
le Jsc, le FF. Le Voc représente la tension maximale que peut délivrer la cellule en
circuit ouvert. Il représente aussi la différence des quasi-niveaux de Fermi. Plus le Vocd’une cellule est élévé, plus la qualité de la cellule est meilleure, car plus faible est
le taux de recombinaison des porteurs photogénérés. Le Jsc est le courant maximal
dans la cellule lorsqu’elle est en court-circuit. Ce courant résulte de la collecte des porteurs photogénérés dans la cellule. Le facteur de forme FF est le rapport entre la puissance maximale fournie par la cellule sur le produit Jsc x Voc, soit :
F F = J max × V max J sc × V oc (1.6)
En remplaçant le produit Jmaxx Vmaxdans l’équation (1.4), le rendement de
conver-sion photovoltaïque en pourcentage d’une cellule solaire s’exprime en fonction des paramètres définis plus haut :
η(%) = F F × J sc × V oc
1.4 Propriétés des matériaux de la famille des
kestérites
1.4.1 Propriétés cristallines
Les matériaux CZTSSe cristallisent dans une structure tétragonale qui peut être de type kestérite (groupe d’espace I4) [32, 33, 34] ou stannite (groupe d’espace I42−m−)
[33]. Cette structure cristalline dérive de la structure cubique Zn blende. La figure 1.7 illuste les différents types de structures cristallines. Partant de la structure Zn blende (groupe II-VI), la structure tétragonale chalcopyrite est obtenue par une substitution croisée des éléments, tout en maintenant la règle de l’octet (figure 1.b) : Zn (II) est substitué par Cu (I) et Ga (III) et en doublant l’unité de maille . En substitutant le Ga (III) par le Zn (II) et Sn (IV), les structures kestérite et stannite sont obtenues (figure 1.7 c-d) [35]. La principale différence entre ces deux structures réside dans la distribution des cations : dans la structure kestérite, les couches de cations s’alternent suivant l’ordre CuSn, CuZn, CuSn et CuZn à z =0,1/4,1/2, 3/4 respectivement. Par contre, la structure stannite présente un ordre d’empilement des couches cationiques ZnSn, Cu2, ZnSn, Cu2. Une autre différence structurale est la
position des anions. Dans la structure stannite, les anions sont situés sur des mirroirs de plan (110), alors que dans la structure kestérite, cette symmétrie est perdue.
1.4 Propriétés des matériaux de la famille des kestérites
Figure 1.8 – Représentations des structures critsallines de la Zn blende (a), la chalcopyrite (b), la kestérite (c) et la stannite (d). Adapté de [35]
A cause de leurs rayons atomiques extrêmement voisins, le Cu (128 pm) et le Zn (134 pm) peuvent occuper des positions aléatoires dans les plans z=1/4 et 3/4. Cette variante est appelée kestérite désordonnée. La structure kestérite est considérée comme étant plus stable que la stannite. C’est cette structure qui sera considérée dans la suite de cette thèse.
1.4.2 Propriétés optoélectroniques
Les propriétés optoélectroniques d’un absorbeur doivent satisfaire un certain nombre de critères nécessaires pour une meilleure éfficacité outre l’abondance des éléments rentrants dans la composition du matériau déjà présentée dans la section précédente. Parmi ces critères on peut citer :
— L’absorption du matériau : le coefficient d’absorption détermine la pro-fondeur dans le matériau à laquelle les photons peuvent pénétrer avant d’être absorbés. Plus celui-ci est élevé, meilleure sera l’absorption de la lumière et moindre sera l’épaisseur de l’absorbeur utilisée. Les matériaux CZTSSe ont
un cefficient d’absorption > 104cm−1 qui est convenable pour développer des
cellules solaires en couches minces [36, 37, 38] .
— Le dopage : le but du dopage est de pouvoir manipuler le type de la conduc-tivité électrique, la mobilité, la durée de vie des porteurs dans un semicon-ducteur par addition contrôlée d’impuretés dans le matériau. Dans les cellules solaires à base de chalcogénures comme le CIGSSe ou le CZTSSe, le dopage et dans une plus large mesure la densité des défauts dépendent fortement de la stoéchiométrie. A cause du nombre assez important des éléments (multi-valents) constituants ces matériaux, en l’occurence 5, le nombre de défauts possibles pouvant être créer à cause d’un écart à la stoéchiométrie est élevé [34, 39, 40]. Les matériaux kestérites de composition stoéchiométrique ont une conductivité intrinsèque de type p principalement due à l’antisite accep-teur CuZn[41, 42] et aux lacunes de VCud’après les calculs ab-initio. La figure
1.9 représente la densité de dopage mesurée expérimentalement en fonction de la composition de l’absorbeur CZTS en termes des ratios [Cu]/([Zn]+[Sn]) et [Zn]/[Sn].
Figure 1.9 – Valeurs expérimentales du taux de dopage en fonction des ratios [Cu]/([Zn]+[Sn]) et[Zn]/[Sn] mesurées par effet Hall et C(V) dans les absorbeurs CZTS.[43]
D’après cette figure, le taux de dopage augmente avec la concentration en Cu. En effet, travailler sous condition Cu-riche favorise la formation de phases telles que Cux(S,Se)y qui sont très conductrices [44]. Les compositions correspondantes aux
meilleures celllules reportées sont [Cu]/([Zn]+[Sn]) =0.8-0.9 et [Zn]/[Sn]=1.1-1.2, soit Cu-pauvre, Zn-riche [45, 42, 46] [46]. Sur la figure 1.9, ces compositions corres-pondent à des taux de dopage compris entre 1015et 1016cm−3 qui sont des valeurs
1.4 Propriétés des matériaux de la famille des kestérites des dopages des meilleures cellules.
— L’énergie de bande interdite : pour produire des cellules performantes d’après la limite théorique de Schokley et Queisser, l’énergie de la bande interdite doit être située autour de 1.34 eV pour une jonction P-N, ce qui correspond à un maximum d’éfficacité théorique de 33.7%. La famille des semiconducteurs CZTSSe ont l’avantage d’avoir une bande interdite directe : le CZTSe a une bande interdite de 1.0 eV et celui du CZTS est de 1.5 eV [46]. La structure électronique de bandes des kestérites montrent que les états de la bande de conduction (BC) résultent du couplage des états s de Sn avec les états p des chalcogènes S ou Se. ceux de la bande de valence (BV) résultent du couplage des états 3d du Cu avec les états p des chalcogènes [47]. De ce fait, trois stratégies de modulation du gap découlent de ces conclusions : 1. Il est possible de fixer un seul type de chalcogènes et faire des substitutions
cationiques en remplaçant soit le Cu (pour jouer au niveau de la BV) ou le Sn (pour jouer au niveau de la BC). Des travaux récents encourageants sur la substitution partielle du Cu par de l’argent montrent qu’il est pos-sible de controler le gap et d’améliorer le Voc[48, 49]et d’atteindre 7,2%. La
substitution Cu-Li se revèle également prometteuse pour augmenter la bande interdite et donc le Voc avec des rendements atteignants 7% [50]. Par ailleurs,
la substitution partielle de Sn par Ge est d’autant plus prometteuse qu’elle permet d’atteindre des rendements supérieurs à 10% [51].
2. D’autre part il est possible de faire varier uniquement les proportions des chalcogènes dans le matériau en laissant fixe les cations. Le rôle des chalco-gènes dans les bandes de conduction et de valence montre que modifier le ratio S/Se conduit à un décalage de ces deux bandes et donc sur la largeur de la bande interdite. Sur la figure 1.9, il peut être constaté que l’alignement de bandes entre CZTSe et CZTS est de type I2 [34], en particulier, le décalage
des bandes de conduction est d’environ ~ 0.35 eV, et le décalage des bandes de valance est d’environ ~ 0.15 eV [34]. Ceci confirme bien que la variation du taux de [S]/([S]+[Se]) affecte majoritairement la bande de conduction par rapport à la bande de valence. En outre, la figure 1.10 illustre la variation quasi-linéaire de l’énergie de bande interdite avec le ratio[S]/([S]+[Se]). C’est l’approche la plus répandue dans la communauté scientifique pour moduler la largeur de la bande interdite : à savoir le contrôle du ratio [S]/([S]+[Se]). C’est également celle qui donne les meilleures performances actuelles 12.7% [27].
3. La dernière stratégie est de combiner les deux à savoir substituer simultané-ment des cations et des anions. Par exemple, le cadmium (Cd) pour substituer le Zn [52], le germanium (Ge) pour substituer Sn [53, 54, 51, 55].
2. la bande valence est plus élevée et la bande de conduction est plus faible du coté du CZTSe par rapport au CZTS
Figure 1.10 – Variation de l’énergie de bande interdite du CZTSxSe1-x en fonction du taux x=[S]/([S]+[Se]) [56]
1.4.3 Quels sont les verrous technologiques des absorbeurs
kestérites ?
Les cellules solaires à base de kestérites sont encore à leurs stades de développement : l’augmentation des rendements de cellules solaires aussi bien que le développement d’un procédé de synthèse économiquement viable sont les clés du succès futur d’une technologie photovoltaïque. Bien que des progrès sur la compréhension de ce ma-tériau et les performances des cellules aient beaucoup évolué ces dernières années, un certain nombre de problèmes fondamentaux sur ce matériau doivent encore être résolus avant que la filière des kestérites ne deviennent compétitives par rapport aux technologies déjà présentes sur le marché.
1.4.3.1 Mécanismes de perte dans les cellules solaires à base des kestérites Il était attendu que la structure kestérite ait les mêmes propriétés optoélectroniques que la structure chalcopyrite (CIGS) à cause de leurs structures cristallines similaires et des substitutions isovalentes qui différencient les deux matériaux. Dans le but de jeter les fondations sur une discussion sur les mécanismes de pertes dans les cellules solaires à base des kestérites, nous avons illustré sur la figure 1.11 les caractéristiques des meilleures cellules reportées dans la littérature (rendements > 5%) afin de les comparer à des cellules idéales sans pertes d’après la limite de Shockley et Queisser (abbrégée LSQ sur la figure).
1.4 Propriétés des matériaux de la famille des kestérites
Sur la figure 1.11a où sont reportés les rendements des cellules solaires actuelles, les performances sont encore très loin de celles prévues par la théorie bien que l’intervalle d’énergie de bande interdite soit favorable. Les densités de courant de court-circuit (Jsc) de ces cellules sont comparées à des cellules idéales ( dans la LSQ) sur la figure
1.11b). En tenant compte des pertes sur le courant d’une cellule idéale, on considère que l’effet d’une couche antireflet (CAR) provoque des pertes optiques estimées à 15% (valeur basée d’après les cellules record CIGSe) contre une valeur de 24% en l’absence de cette couche (courbe en vert). Le déficit du Voc des cellules CZTSSe
et CIGS est comparé à celui qu’on obtiendrait dans la LSQ sur la figure 1.11c. Le déficit du Voc peut être défini comme étant l’écart entre le Voc et le rapport Eg/q
d’une cellule.
Figure 1.11 – (a) Rendements des cellules solaires idéales dans la limite de Scho-ckley et Queisser (LSQ). (b) Comparaison entre les efficacités des cellules CZTSSe publiées en fonction de leur gaps. (c) Densités de courants (Jsc) des cellules
CZTSSe en fonction de la bande interdite, comparées à des cellules dans la LSQ avec ou sans pertes optiques. (d) Comparaison entre les pertes de Voc pour les
meilleures cellules CZTSSe et CIGSe en fonction de la bande interdite. Figure adaptée de [57].
est le déficit de Voc. Les valeurs de Voc pour le CZTSSe sont très inférieures (~
400 mV) à celles correspondantes pour le CIGSe. Ce déficit est d’environ 500 mV pour les cellules CIGS record [58], alors qu’il chute jusqu’à plus de 650 mV pour les cellules CZTSSe [59].
Dans la littérature, plusieurs hypothèses ont été émises pour essayer de comprendre cette perte de Voc. Elles sont regroupées en trois grandes catégories : (1) l’activité des
défauts, (2) la complexité du diagramme de phase des kestérites et, (3) les interfaces avant et arrière [58].
? L’activité des défauts
La complexité des défauts intrinsèques aux composés kestérites se reflète sur leurs faibles Voc. Les courants de saturation3 J0 sont de l’ordre de 10−6 à 10−3 mA.cm−2
[60], soit 3 ordres de grandeur au moins plus élevés que leurs correspondants CIGS [61]. Dans le cas du CZTSSe, les défauts responsables des recombinaisons incluent des défauts profonds (situés en milieu de la bande interdite) et des centres recom-binants aux interfaces. Cependant suivant que l’absorbeur est du CZTS, du CZTSe ou du CZTSSe, les mécanismes de recombination dominants ne sont pas les mêmes [57]. Pour les cellules à base de CZTSe, le mécanisme de recombinaison dominant a lieu dans le bulk de l’absorbeur, tandis que pour les cellules à base de CZTSSe, les recombinaisons sont dominantes aux interfaces [57]. D’autres travaux mettent en exergue d’autres facteurs limitants le Voc comme les fluctuations de potentiel
induites par la forte densité de défauts compensés dans les kestérites [62, 63], un contact CZTSSe/Mo non ohmique pour des CZTSSe faiblement dopés (~1015cm−3)
et des “tail states” qui causent un piègeage du niveau de Fermi aux interfaces [64]. La question de savoir finalement jusqu’à quel point les causes énumérées ci-dessus (défauts profonds ou compensés, centres recombinants aux interfaces) contribuent au déficit de Voc reste encore irrésolue.
? La complexité du diagramme de phase des kestérites
L’élaboration d’une phase kestérite pure n’est pas une tâche aisée à cause du faible écart à la stoéchiométrie tolérée par ces matériaux, comme cela est observé sur la figure 1.12. Les calculs ab-initio revèlent que la zone de stabilité d’une phase pure de CZTSSe stoéchiométrique pendant sa synthèse est très étroite [32] ; ce qui signi-fie que le nombre de phases secondaires qui peuvent potentiellement être formées en compétition avec la phase pure de CZTSSe est élevé [65]. L’obtention de cel-lules solaires performantes à base de CZTSSe requière une phase pure de CZTSSe de stoéchiométrie Cu-pauvre, Zn-riche. Or il est davantage plus probable de for-mer des phases secondaires que la composition idéale pour les cellules solaires est sous-stoéchiométrique en Cu et sur-stoéchiométrique en Zn. En outre, suivant qu’il s’agisse de CZTS, CZTSe ou CZTSSe, les conséquences des écarts à la stoechio-métrie sont différents. Les travaux de Choubrac et al. sur l’impact sur la structure
3. Or il est connu que les courants de saturation sont des indicateurs de taux de recombinaison dans la cellule. Plus ils sont élevés, plus la cellule souffre de recombinaisons.
1.4 Propriétés des matériaux de la famille des kestérites
des écarts à la stoechiométrie ont révélé que pour les composés CZTS, de faibles inhomogénéités de dépôt pouvaient conduire à des grains de composition très dif-férentes et donc plus facilement mener à des phases secondaires. Ce qui n’est pas le cas des composés à base de Se (CZTSe) dont la région d’existence est beaucoup plus grande : en d’autres termes des fluctuations de compositions du même ordre peuvent conduire à une phase de CZTSe avec une probabilité plus faible de former des phases secondaires [66].
Figure 1.12 – Coupe en section du diagramme de phase du CZTSSe avec la zone de formation de la phase pure (ellipse en bleu), la zone des meilleurs rendements (ellipse en rouge), et les différentes phases secondaires détectées dans la littérature. Adaptée de [67]
En outre, parmi les possibles phases secondaires qui peuvent être formées, certaines sont conductrices, d’autres induisent des shunts dans la cellule :
— les phases de Cux-S(e) sont très conductrices et provoquent des court-circuits
dans la cellule. Elles peuvent aussi être responsables de l’augmentation de la concentration des trous dans le CZTSSe [68].
— Les phases SnSe/SnSe2 peuvent causer des pertes de résistances shunt et de
Voc [69].
— Si elles sont présentes à la surface, les phases de ZnSe (ZnS)peuvent bloquer le transport du courant [70].
— Les phases de Cu2SnSe3 ont un effet néfaste sur le FF, le Voc [71].
? La problématique des substances volatiles
Un facteur important est que certains phases volatiles se forment pendant la crois-sance du CZTSSe, rendant encore plus difficile le contrôle de la composition de l’absorbeur. Les chalcogènes S et Se ne sont pas les seuls éléments volatiles, mais le SnS, SnSe, Zn sont aussi très volatiles comme le montre la figure 1.13. De ce fait,
à faible pression et haute température, le contrôle de la la quantité de Sn et de Zn devient problématique.
Figure 1.13 – Pressions de vapeur saturante du Cu, Zn,Sn, S, Se et des binaires SnS, SnSe, ZnS, ZnSe. Adapté de [67]
? L’optimisation des interfaces
La qualité des interfaces avant et arrière dans l’empilement d’une cellule solaire est déterminante pour les performances des cellules, car les interfaces sont le siège de recombinaisons qui lorsqu’elles sont très élevées affectent les rendements. Le déve-loppement de cellules solaires à base des kestérites a été une réplication de l’archi-tecture des cellules solaires à base de CIGS, en d’autres termes toutes les autres couches impliquées dans une cellule CIGS sont les mêmes utilisées dans une cellule CZTSSe. Cette replication directe qui ne tient pas en réalité compte des différences entre les deux matériaux pose cependant quelques problèmes aux interfaces qui sont expliquées dans les paragraphes suivants.
Le Mo est couramment utilisé pour les contacts arrières des cellules solaires à base de CIGS. Sa grande stabilité thermique pour les procédés à haute température, son excellente adhésion au substrat de verre et sa non-miscibilité avec le Cu en font le métal le plus adapté pour ce type de cellules [72]. De même la couche de MoSe2
formée à l’interface CIGS/Mo améliore la qualité ohmique de l’interface [73]. Dans le cas des kestérites, le Mo se revèle avoir une importance encore plus significa-tive lors de la croissance du CZTSSe ; l’apparition des cavités à l’interface avec le Mo [74], la formation de phases secondaires résistives (ZnS, SnS) [75] en plus du MoSe2
1.4 Propriétés des matériaux de la famille des kestérites
ont été menés pour améliorer la qualité de cette interface en insérant des couches sacrificielles pour améliorer la qualité de l’interface Mo/CZTSSe. Par exemple, il a été reporté qu’une fine couche de TiN (< 20 nm) améliore le Jsc, le Voc et le FF [76].
De même une fine couche de ZnO (10 nm) permet d’améliorer la morphologie de l’interface et réduit les cavités qui sont généralement observées entre le Mo et l’ab-sorbeur [77]. Les résistances séries sont réduites de 3.7 Ω.cm2 à 0.1Ω.cm2. Un autre
candidat intéressant est une couche fine de carbone évaporée ; le carbone adhère aux parois internes des vides et joue le rôle de connexion entre le CZTSSe et le Mo, ce qui réduit les résistances séries et augmente le photocourrant [78].
Le jonction P-N est faite entre l’absorbeur CZTSSe et la couche tampon CdS. Tout comme dans le cas du CIGS, l’alignement de bandes est de type spike, avec une hauteur de barrière ≤ 0, 4eV . Au delà de 0,5 eV, l’interface absorbeur/CdS se comporte comme une barrière bloquant le transport des charges minoritaires[28]. Des études ab-initio sur la nature de cette interface démontrent qu’elle est de type spike pour les absorbeurs mixtes S/Se[79] , mais les valeurs expérimentales reportées dans la littérature divergent ; certainement parce que les surfaces des absorbeurs sont très dépendantes des méthodes de synthèse et des traitements de surface [30, 80]. 1.4.3.2 La chimie de réaction des kestérites.
La réaction de formation d’une phase CZTSe (CZTS) est une réaction d’équilibre chimique solide-gaz à haute température (>500°C) proposée par Scragg [81] :
2 Cu2Se(s) + 2 ZnSe(s) + 2 SnSe(g) + Se2(g) ↔ 2 Cu2ZnSnS(e)4(s) (1.1)
Il a été observé expérimentalement que la composition des absorbeurs après recuit était plus faible en Sn que les précurseurs initiaux [82]. Cela a permi de comprendre qu’en réalité la réaction d’équilibre du CZTSe est un équilibre non pas avec un seul gaz (le chalcogène), mais avec deux espèces gazeuses, le Se et le SnSe. En effet la seule hypothèse de perte de Sn pendant le recuit est qu’il forme avec le Se du SnSe qui est une phase volatile se formant à faible pression partielle de chalcogène aux températures typiques de recuit (500°C-600°C). D’après cette équation, il est possible de déplacer le sens de la réaction en contrôlant la pression partielle du SnSe et du chalcogène (Principe de Le Chatelier). Afin de maximiser la quantité de kestérite produit, il faut avoir une atmopshère de recuit riche en SnSe et en Se. Des calculs théoriques montrent qu’il est plus facile de créer des lacunes de Se dans le CZTSe que dans le CIGSe pour les mêmes conditions de recuit [83, 84, 85] [85] : Cela justifie pourquoi la stabilité de la phase CZTSe est plus faible que celle du CIGSe [81]. Ces conclusions sont aussi valables pour le CZTS.
Les recuits des absorbeurs kestérites ne se font pas en réalité dans des enceintes totalement fermées, mais plutôt dans des boites graphites qui sont une sorte de puit
de Se. Le graphite est suffisamment poreux pour piéger ou faire diffuser du Se vers l’extérieur qui se retrouve généralement condensé vers les parois froides du four. Dans ces conditions de recuit hors équilibre, deux approches permettent de limiter la perte de SnSe :
? L’approche thermodynamique qui consiste simplement à compenser la perte
des espèces volatiles. Les travaux de Scragg et al. ont démontré que le Se et le SnSe doivent être suffisament présents dans l’atmosphère de recuit et que le produit de leur concentration est un facteur crucial [86] . Ainsi pendant le recuit, l’évaporation simultanée de SnSe et de Se augmente rapidement leur pression partielle au dessus de la couche et empêche leur évaporation du film. Des travaux de Redinger et al. ont confirmé ces résultats : en ajoutant du S et du SnS pendant le recuit dans une boite graphite, il a été obtenu des cellules avec plus de 5% de rendement, comparé à 0% de rendement en l’absence de SnS [87].
? L’approche cinétique qui consiste à limiter la perte de SnSe et de Se via une
étape limitante de diffusion comme par exemple travailler à haute pression de fond dans le four. En effet une forte pression de fond d’un gaz inerte ralentit la diffusion des espèces volatiles (et donc réduit leur vitesse d’évaporation) et augmente leur pression partielle au dessus de la surface de l’absorbeur. Diverses auteurs dans la littérature ont démontré que la perte de Sn était très infime lorsque le recuit du CZTS à haute température était effectué sous atmosphère N2+H2S [88, 89, 82].
1.4.3.3 Impact des phases secondaires et techniques de décapage
La présence de phases secondaires locales est néfaste pour les performances des cellules et est partiellement responsable de la perte de Voc. De ce fait, la question
de décapage des phases secondaires à la surface des absorbeurs est fondamentale pour améliorer les performances des cellules. Plusieurs formulations de traitement de surface ont été développées par différents groupes pour enlever les phases secondaires à la surface des kestérites, comme illustré sur le tableau 1.2. Le décapage par voie humide présente plusieurs avantages : 1) enlever les phases d’oxides à la surface, 2) enlever de manière sélective les phases secondaires à la surface, 3) passiver la surface.
1.4 Propriétés des matériaux de la famille des kestérites
Table 1.2 – Recettes de décapages des phases secondaires reportées dans la littérature.
Solution de décapage (concentration)
T (°C) Temps (min)
Phases secondaires ciblées
KCN (5% wt) 1 min Oxides natifs [90]
KCN (10 % wt) T.amb. 3 Cu2-xS [91, 92]
KCN (10% wt) T.amb. 30 Cu2Se [93]
KCN (5% wt), KOH (0.5% wt)
20 2 CuxSe, Se0,CuxSnSey,
SnSe2,SnO2[94, 95] (NH4)2S (20% wt) T.amb 15 CuxSe [96] (NH4)2S (4-22 % wt) T.amb 1 Sn(S,Se) [97]
Br2-MeOH (0.02M) 1 Phases à base de Cu, Sn, [71]
HCl (5% v/v) 75 5 ZnS [98]
KMnO4 (0.01M)/ H2SO4
(1M)+Na2S (1M)
T.amb 1 ZnSe [99]
1.4.3.4 Décapage des phases secondaires — Phases secondaires à base de Cu
De manière générale, les solutions acqueuses de cyanure de sodium ou potassium (NaCN ou KCN) permettent d’enlever les phases de CuxS(e) à la surface des
ab-sorbeurs CIGS [100, 101, 102]. A cause de leur nature semi-metallique ( [p]>1020
cm−3), les phases de Cu
xS(e) sont connues pour causer des chemins de court-circuits
au déplacement des charges. Ces phases peuvent également augmenter la concentra-tion des porteurs majoritaires dans le CZTSSe. Le traitement de KCN (ou NaCN) permet aussi de réduire les recombinaisons à l’interface absorber/CdS. Bär et al. ont montré expérimentalement que le KCN permettait d’augementer légèrement la lar-geur de la bande interdite à l’interface CZTS/CdS et donc d’augmenter la hauteur de barrière d’énergie aux recombinaisons [80].
— Phases secondaires à base de Sn
Xie et al. ont proposé un traitement très efficace à base de sulfure d’ammonium (NH4)2S permettant d’attaquer de manière sélective les phases de Sn(S,Se) à la
surface du CZTSS [97]. En plus d’éliminer les phases secondaires, le S contenu dans le (NH4)2S forme des liaisons avec les atomes de l’extrême surface de l’absorbeur, et
de ce fait passive la surface de l’absorbeur. Ce traitement a également été utilisé par Buffière et al. comme alternative au KCN4 pour le traitement des phases de Cu
xSe
dans la CIGSe [96].
— Phases secondaires à base de Zn
Pour décaper les phases de ZnS, Fairbrother et al. ont utilisé un traitement à l’acide chlorhydrique à chaud [98]. En plus de d’éliminer le ZnS, le HCl élimine également
les oxides à la surface. Cependant, ce traitement ne s’est pas avéré concluant pour les phases de ZnSe. Une autre solution de décapage des phases de ZnSe a été trouvée par Lopez-Marino et al. [99]. Elle consiste en un mélange de (KMnO4+ H2SO4)
+ Na2S. Les performances des cellules après ce traitement sont considérablement
améliorées.
1.4.4 Techniques de synthèse des matériaux kestérites
Le CZTSSe en couche mince peut être déposé par des techniques dites sous-vides et celles dites humides. Des revues détaillées sur les voies de synthèse des kestérites ont été présentées dans la littérature [46, 103]. Le tableau 1.3 (adapté de [67] ) regroupe non de manière exhaustive les techniques de synthèse par différents groupes (laboratoires et industriels) dont les cellules solaires ont des rendements supérieurs à 7%. Comme le montre ce tableau, la majorité de ces absorbeurs sont obtenus à partir des procédés à deux étapes, excepté la coévaporation par IBM qui est une voie une seule étape.
Une première observation du tableau 1.3 montre que tous les paramètres électriques sont dépendants des procédés de synthèse. Par exemple, le Voc et le FF présentent
des variations pouvant dépasser 17%.
En général, le chemin de réaction est dépendant de la technique de synthèse. Par exemple, dans le cas de la sélénisation des empilements métalliques Cu/Zn/Sn, l’ap-parition immédiate des phases de CuxSe suivie des autres binaires conduisent à
l’apparition du CZTSe au-delà de 370°C [117]. Dans le cas de précurseurs de nano-particules à base de nanocristaux de ZnS(e)(N2H4) et de bimétalliques Cu2Sn(Se,S)x,
aucun des binaires de CuxSe ou SnSex n’ont été détectés pendant la formation des
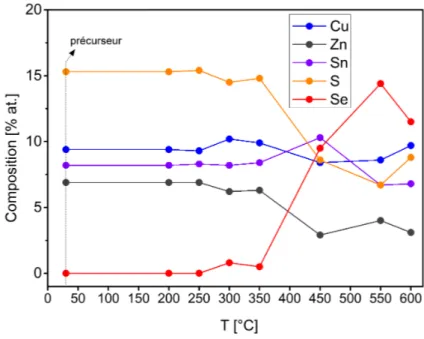
absorbeurs CZTSSe [118]. Pour des précurseurs métalliques co-électrodéposés Cu-Zn-Sn, des binaires soufrés se forment au préalable, suivi des ternaires de Cu2SnS3
qui réagissent avec le ZnS pour former le CZTS [119]. Ces études amènent à conclure qu’autant les mécanismes des réaction des kestérites sont variés, autant les phases secondaires susceptibles de rester stables après recuit sont nombreuses, et par consé-quent leurs impacts sur les performances des cellules seront diversifiés. Les meilleures performances des cellules solaires à base de kestérites sont obtenues via un procédé de synthèse en deux étapes, dont les précurseurs contiennent déjà un chalcogène [27]. Ceci est probablement dû au fait qu’il est plus simple de contrôler la compo-sition des espèces métalliques, à savoir Cu pauvre, Zn riche lors de l’élaboration du précurseur. En effet, lors des procédés deux étapes, les précurseurs sont déposés à température ambiante, ou à une température n’excèdant pas 200°C pour homo-généiser l’alliage constituant le précurseur. Dès 350°C, il a été observé que l’étain s’évapore sous forme de SnS pour la phase CZTS [120, 121]. De même, les travaux de Redinger et al, montrent que la perte de Zn devient très importante dès 500 °C lors de la coévaporation de CZTSe [122]. Ils montrent aussi que les quantités de Zn et de Sn lors de la coévaporation dépendent fortement de la pression partielle de Se.
1.4 Propriétés des matériaux de la famille des kestérites T able 1.3 – Tableau récapitulat if des rendemen ts de cellule s à base de kestérites sup érieurs à 7% T ec hniques de syn thèse Instituts ou industriels Absorb eurs couc he s tamp ons Eff (%) Vo c (m V) Jsc (mA/cm 2 ) FF (%) Surface (cm 2 ) Eg (eV) Références Dép ôts sous vide Co-év ap oration IBM CZTSe CdS 11.6 423 40.6 67.3 0.43 [104] Sputtering Solar Fron tier CZTSSe CdS 11.0 516 34.1 63 14 [105] Sputtering IMEC CZTSe 10.4 394 39.7 66.4 0.52 [106] Co-év ap oration NREL CZTSe CdS 9.8 380 37.6 68.9 0.41 [6, 107] Sputtering Solar Fron tier CZTS CdS 9.2 708 21.6 60 14 1.5 [108] Sputtering IREC CZTSe CdS 8.2 392 32.4 64.4 0.25 [109] Sputtering IRDEP CZTSe CdS 8.0 460 28.6 60.8 0.1 Sputtering Uppsala CZTS CdS 7.9 667 19.6 60 0.5 [110] Sputtering IRDEP CZTSSe CdS 7.1 404 31.6 56.5 0.1 [69] Sputtering CEA CZTSSe CdS 7.0 376 34 55 0.5 [111] Dép ôts par v oie h umide Hydrazine IBM CZTSSe In2 S3 /CdS 12.7 466 38.9 69.8 0.42 1.06 [27] Nano-particules EMP A CZTSSe CdS 10.8 0.25 [112] Nano-particules IMRA CZTSSe CdS 11.2 479 36.5 63.8 0.3 [113] Monograins Crystalsol CZTSSe CdS 8.4 703 17.8 61.3 [114] Nano-particules A ca de m y of Sciences China CZTSe CdS 8.0 408 33.4 58.8 0.35 [115] Electro dép ôt East China Norm. Univ CZTS CdS 7.1 369 32.4 58.5 0.45 [116]
Ces deux facteurs pourraient justifier pourquoi le procédé en deux étapes est le plus répandu pour la réalisation des absorbeurs à base de kestérites.
1.5 Conclusion
Au cours de ce chapitre, nous avons présenté l’ensemble du contexte de la conver-sion photovoltaïque et le positionnement de la filière kestérite à laquelle est consacrée ce travail. Outre les aspects généraux concernant les propriétés structurales, opto-électroniques des matériaux kestérites et des cellules solaires correspondantes, nous avons également abordé les aspects pratiques de synthèse de ces matériaux. La syn-thèse des kestérites n’est pas une tâche aisée, principalement dûe au matériau en soi qui présente un domaine de stabilité faible et une grande probabilité de formation de phases secondaires.
2 Techniques expérimentales et
méthodes d’analyses
2.1 Synthèse et caractérisation des absorbeurs
Comme nous l’avons mentionné au chapitre précédent, les absorbeurs sont synthé-tisés par un procédé dit deux étapes, où les précurseurs à base de Cu, Zn, Sn et/ou S sont déposés par pulvérisation cathodique (sputtering en anglais) et par la suite subissent un traitement thermique sous atmosphère de chalcogènes (S et Se) pour obtenir la phase désirée CZTSSe. Dans ce chapitre, les techniques de synthèse des précurseurs, les différents procédés de recuit, ainsi que les moyens de caractérisation des absorbeurs et des cellules solaires fabriquées seront présentés.
2.1.1 Dépôt des précurseurs par pulvérisation cathodique
2.1.1.1 Généralités sur le sputteringLa pulvérisation cathodique fait partie des nombreux procédés affiliés à la famille des dépôts sous vide dits PVD (physical vapor deposition). Très utilisée dans l’industrie, elle permet de déposer une large variété de matériaux. Le principe de fonctionnement de la pulvérisation cathodique repose sur la création d’un plasma localisé autour de la cible du matériau :
(1) Un gaz inerte est injecté à faible débit au sein de l’enceinte.
(2) La cible (cathode) est polarisée permettant la circulation d’un champ électrique entre cette dernière et l’anode reliée à la masse. Un plasma froid est crée suite à l’ionisation du gaz inerte.
(3) Les ions de ce plasma sont accélérés vers la cible, acquérant de l’énergie cinétique qu’ils libèrent en impactant la surface de la cathode qui est alors pulvérisée.
(4) La matière éjectée se dépose alors sur le substrat, en vis-à-vis selon une trajectoire diffuse liée au libre parcours moyen des espèces.
2.1.1.2 Sputtering de Cu-Zn-Sn-(S) à l’IRDEP
L’ensemble des précurseurs réalisés au cours de cette thèse ont été déposés dans une machine de pulvérisation cathodique du fabricant PLASSYS. Cette machine de
référence MP500S, est de type radiofréquence (RF) Magnetron. Le générateur RF (f=13.56 MHz) est adapté pour pulvériser des cibles isolantes. Dans le sputtering magnétron, les lignes de champ sont confinées au niveau de la cible, ce qui donne lieu à une abrasion circulaire. La machine MP500S est constituée de 2 chambres indépendantes : un sas de chargement qui contient un porte substrat pouvant ac-cueillir des échantillons 5x5 cm2; une enceinte de dépôt où sont placées les trois
cibles et où sont éffectués les dépôts sous un flux d’argon. Cette configuration de chambre est optimale pour, faciliter l’introduction des échantillons dans le réacteur, réduire les temps de pompage et la contamination de la chambre de dépôt. Sur la figure 2.1 est représentée un schéma de l’enceinte de dépôt (a) et du bâti tout entier (b). Les trois cathodes sont disposées suivant une configuration confocale. Dans ce type de configuration, les cibles sont placées de manière à former un cercle avec un point focal commun. Le substrat est alors placé au voisinage de ce point focal et en rotation autour de son propre axe pour garantir un dépôt uniforme des différentes cibles.
Figure 2.1 – Représentation schématique de l’enceinte de dépôt (a) et du bâti entier de pulvérisation cathodique (b).
La co-pulvérisation cathodique (co-sputtering) est la pulvérisation simultanée de plusieurs cibles. Les cibles utilisées de Cu, ZnS et SnS sont fournies par Neyco, de pureté 99.99%, et d’épaisseur 3 pouces. Il est important de préciser les raisons du choix d’une cible de Cu, plutôt qu’une cible de CuS ou Cu2S. En effet, nous avons
2.1 Synthèse et caractérisation des absorbeurs
rencontré de sérieux problèmes avec les cibles de cuivre soufrées, car celles-ci étaient instables et se brisaient systématiquement au bout de quelques dépôts seulement. Le co-sputtering est une des méthodes propices au dépôt des précurseurs uniformes et homogènes, car elle favorise l’interdiffusion des espèces présentes dans le précur-seur. De plus, nous avons choisi cette méthode contrairement à des dépôts successifs des différentes couches « stack » pour deux raisons principales : la première raison est un gain considérable de temps de synthèse. En effet à épaisseurs finales égales ~ 700 nm, un précurseur Cu-Zn-Sn-S déposé en stack nécessite entre 2h et 2h30 min de durée contre 1h pour un dépôt par co-sputtering. Et dans le cas des précurseurs Cu-Zn-Sn, le constat est le même. Environ 1h30 min de temps de dépôt pour les précurseurs en stack contre 30 min pour ceux en co-sputtering. La seconde raison est que nous avons voulu éviter des effects d’influence d’ordre des couches sur l’incor-poration des chalcogènes pendant le recuit. Il a été démontré que la position du Cu et de Sn affectent différemment l’incorporation des chalcogènes [123, 124]. En plus, elle permet de nous affranchir des étapes de pré-recuit « pre-alloying », autour de 200°C, généralement employées lors des dépôts de précurseurs en forme de couches successives « stack » [67]. En effet, ces traitements de pré-recuits peuvent s’avérer néfastes pour le précurseur, notamment lorsque la durée est longue (> 10min). Dans ce cas, on observe une ségrégation de phases dans le précurseur [125] ou la formation de bulles notamment dûe à la basse température de fusion de l’étain (~230°C) [67]. En effectuant un co-dépôt de Cu, Zn et Sn, cela favorise l’obtention d’un précur-seur bien homogène tout en limitant la perte de Zn, et par conséquent à une phase CZTSSe bien homogène en composition.
2.1.1.3 Conditions de dépôt des précurseurs
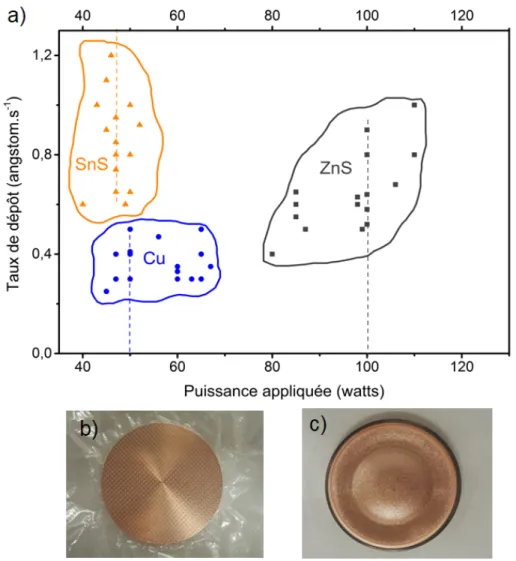
Avant de commencer le procédé, les taux de dépôt (en angstrom/s) de chaque cible sont mesurés grâce à la microbalance. Ainsi, il est non seulement possible de connaitre en fonction de la puissance appliquée le taux de dépôt de chaque cible, mais aussi d’estimer l’épaisseur de la couche déposée après une certaine durée. Par ailleurs, les taux de dépôts peuvent varier en fonction de plusieurs paramètres. Par exemple, la pression d’argon, la puissance appliquée, la distance de la cible au sub-strat, l’ancienneté de la cible. Sur la figure 2.2 ont été reportées les variations des taux de dépôt en fonction de la puissance appliquée pour ce qui est des cibles de Cu, ZnS, SnS (a) et la comparison entre une cible de Cu neuve (b) et une cible de Cu en cours d’utilisation (c).
![Figure 1.10 – Variation de l’énergie de bande interdite du CZTS x Se 1-x en fonction du taux x=[S]/([S]+[Se]) [56]](https://thumb-eu.123doks.com/thumbv2/123doknet/14718304.569381/29.892.241.704.122.462/figure-variation-énergie-bande-interdite-czts-fonction-taux.webp)
![Figure 2.5 – Spectres Raman de quelques alliages CZTSSe avec différents taux de S/(S+Se) [129]](https://thumb-eu.123doks.com/thumbv2/123doknet/14718304.569381/50.892.232.631.228.583/figure-spectres-raman-alliages-cztsse-taux.webp)