HAL Id: tel-01647210
https://tel.archives-ouvertes.fr/tel-01647210
Submitted on 24 Nov 2017
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Estimation des taux de mutation : implications pour la
diversification et l’évolution du phytoplancton eucaryote
Marc Krasovec
To cite this version:
Marc Krasovec. Estimation des taux de mutation : implications pour la diversification et l’évolution du phytoplancton eucaryote. Génétique des plantes. Université Pierre et Marie Curie - Paris VI, 2016. Français. �NNT : 2016PA066371�. �tel-01647210�
1
Thèse de doctorat
Université Pierre et Marie Curie
Ecole doctorale « Complexité du vivant », ED 515
Estimation des taux de mutation :
implications pour la diversification et
l’évolution du phytoplancton eucaryote
Marc Krasovec
Le 19 Octobre 2016, à Banyuls sur mer
Gwenaël Piganeau Université Pierre et Marie Curie Directeur de thèse
Sophie Sanchez-Ferandin Université Pierre et Marie Curie Directeur de thèse
Vincent Laudet Université Pierre et Marie Curie Président de Jury
Laurent Duret CNRS, UMR 5558 Rapporteur
Olivier Tenaillon INSERM, UMR 1137 Rapporteur
3
Remerciements
Ma plus profonde gratitude va à mes deux directrices de thèse, Gwenaël Piganeau et Sophie Sanchez-Ferandin, pour m’avoir donné l’opportunité de réaliser cette thèse avec elles, et surtout pour le soutien indéfectible et permanent durant ces trois années. Au-delà de la grande qualité de l’encadrement qu’elles m’ont apporté, j’ai pris un grand plaisir à travailler avec elles pour leurs nombreuses qualités aussi bien professionnelles que relationnelles.
Ces trois années de thèse passées avec Gwenaël et Sophie ont été pour moi un grand épanouissement professionnel et personnel, et constituent une unique et excellente expérience pour ma vie future.
Je tiens également à adresser mes remerciements aux membres du jury pour avoir accepté d’évaluer mon travail, Laurent Duret, Olivier Tenaillon, Delphine Sicard et Vincent Laudet, ainsi que les membres de mes comités de thèse, Delphine Sicard, Jean-Paul Cadoret et Adam Erye-Walker.
Je tiens aussi à remercier le laboratoire de Biologie Intégrative des Organismes Marins et l’équipe de génomique environnemental du phytoplancton pour m’avoir accueilli et permis de réaliser cette thèse.
D’une manière plus générale, je remercie ma famille, en premier lieu ma mère Christine sans laquelle je ne serais jamais allé aussi loin aussi bien dans mes études que dans mes avancées personnelles, ainsi que mes frères et sœur Caroline, David, Frédéric (ou Lélic) et mon frère jumeau, Gabriel, comme moi grand admirateur des êtres vivants.
Aussi, nombreuses sont les personnes du laboratoire Arago qui m’ont aidé dans mes travaux, en sein de l’équipe, Nigel Grimsley, Hervé Moreau, Evelyne Derelle, Sheree Yau, et enfin une grande reconnaissance pour Elodie Desgranges et Claire Hemon, mes deux collègues de bureau et de laboratoire.
4 Je remercie également les membres de la plateforme cytométrie, David
Pecqueur et Christophe Salmeron, toujours disponibles pour venir à la rescousse d’un cytomètre en panne.
Pour finir, je remercie mes différents amis, Florian, Sylvain, Alex (vous vous reconnaitrez) et les doctorants du laboratoire pour les discussions, les soirées avec une mention spéciale pour les gaming-night, et les amitiés qui resteront bien après la fin de cette thèse.
Pour citer quelques noms, je remercie bien sûr mon cher collègue de thèse Hugo L et sa femme, Océane l’aristocrate, Sandrine et ses petits félins, Margot et son congénère larvaire qui ont toujours des bonbons à me donner, mon premier ministre imaginaire Mathieu que je remercie pour avoir effectué le déplacement, Hugo B pour les discussions philosophiques sur Homo sapiens, Marine, Tatiana et Remy, Mariana, Mathias qui va se faire séquencer, Nathalie, Brian et Elsa, Daniel.
A toutes les personnes évoquées ci-dessus, je vous suis reconnaissant d’avoir supporté mes discussions parfois inutiles et inintéressantes sur les chats, les chinchillas (dont Kalam et Glorfindel sont les plus beaux représentants), et mes idées sensiblement peu démocratiques.
5
SOMMAIRE
Liste des abréviations
7CHAPITRE 1: INTRODUCTION 9
1. Introduction générale 11
2. Les enjeux de la recherche sur les mutations 12
3. Les variations du taux de mutation 14
4. Les expériences d’accumulation de mutations 15
1. Les premières expériences de Terumi Mukai 15
2. L’effet des mutations sur la fitness 17
1. Les successeurs de Terumi Mukai 17
2. Paysage adaptatif 19
3. Interactions génotype-environnement 22
1. Les changements d’effet des mutations 22
2. Le stress et les hyper mutateurs 23
5. Les estimations directes du taux de mutation 24
1. Les variations inter génomiques du taux de mutation 24
1. La taille du génome 24
2. La taille efficace (Ne) 26
3. Le temps de génération 28
4. Le taux métabolique et la température
2. Les variations intra génomiques du taux de mutation 30
1. Le sens de la transcription et de la réplication 30
2. Le temps de réplication 31
3. Les régions codantes et le niveau d'expression 31
4. La composition en GC 31
6. Nouveaux modèles biologiques 35
1. L’importance écologique du phytoplancton 35
2. Présentation des espèces 36
1. Choix des modèles biologiques 36
2. Les Mamiellophyceae 40
3. Les Trebouxiophyceae 41
1. Présentation générale 41
2. Les transferts horizontaux de gènes 42
6
CHAPITRE 2: EFFETS DES MUTATIONS SUR LA FITNESS 47
CHAPITRE 3: LE TAUX DE MUTATION CHEZ LES MAMIELLOPHYCEAE 61
CHAPITRE 4: LES TRANSFERTS HORIZONTAUX DE GENES: LE CAS DE
PICOCHLORUM RCC4223 81
CHAPITRE 5: IMPACT DU TAUX DE MUTATION POUR
LES BIOTECHNOLOGIES 97
CHAPITRE 6: DISCUSSION ET CONCLUSION 113
1. Les variations de fitness indépendantes des mutations 115
1. La plasticité phénotypique 115
2. Les bactéries présentes dans les cultures d’O. tauri 117
3. Le rôle des variations structurelles sur le phénotype 118
2. Les limites à l’estimation du taux de mutation 120
3. Perspectives pour les EAMs 123
4. Conclusion générale 125
Annexes
127Listes des figures et des tableaux
175
Bibliographie
181Résumé
2147
Liste des abréviations
a : Effet de la mutation sur la fitnessADN : Acide désoxyribonucléique ARN : Acide ribonucléique
CV : Variation de l’effet des mutations
ΔV : Changement de variance de la donnée de fitness ΔM : Changement moyen de fitness par génération
EAM : Experience d’accumulation de mutations
G : Taille de génome
GCeq : Contenu en GC du génome à l’équilibre
Ge : Taille de génome codante
GxE : Interactions Genotype-Environement HGT : Horizontal gene transfer
Μb : Mega base
MMR : Mismatch repair
Ne : Taille efficace de population
OmV1 : Ostreococcus mediterraneus Virus 1 PFGE : Pulsed-field gel electrophoresis R1 : Taux de mutation de GC vers AT
R2 : Taux de mutation de AT vers GC
RCC : Roscoff culture collection ROS : Reactive oxygen species TCR : Transcription-coupled repair
U : Taux de mutation par génome
Uc : Taux de mutation caryotypique par génome
Ud : Taux de mutation délétères par génome
9
CHAPITRE 1:
INTRODUCTION
10
11
1.
Introduction générale
Depuis la publication de l’origine des espèces et du principe de la sélection naturelle par Charles Darwin en 1859, des générations de biologistes ont étudié les questions fondamentales qui entourent l’évolution et la diversité du vivant. A cette époque, la génétique n’est pas connue et Darwin ignore les mécanismes qui génèrent la variabilité et la diversité soumises à la sélection naturelle. Les lois de Mendel sont redécouvertes en 1900, et en 1902 Walter Sutton propose la théorie chromosomique de l’hérédité. L’existence des mutations est démontrée en 1911 par Thomas Morgan en réalisant des expériences sur des drosophiles. Les mutations sont le moteur de l’évolution et constituent la base du potentiel adaptatif des espèces car elles constituent la principale source de diversité sur laquelle peut agir la sélection. Les biologistes s’intéressent donc depuis longtemps aux rôles des mutations, et les découvertes du début du 20ème siècle vont aboutir à la théorie synthétique de l’évolution, en particulier avec les travaux de Sewall Wright, John B. S. Haldane, Hermann J. Muller ou Julian Huxley (Haldane, 1949, 1937; Muller, 1928; Wright, 1932). La découverte de l’ADN et de sa structure (Watson and Crick, 1953) ouvrira la voie aux technologies de séquençage qui permettent d’observer directement l’apparition des mutations sur un génome, leurs fréquences et leurs distributions. Leurs effets sur la capacité de survie sont également explorés (Eyre-Walker and Keightley, 2007; Haldane, 1937; Muller, 1950) pour comprendre les différents processus évolutifs et adaptatifs des êtres vivants. La théorie neutraliste de l’évolution de Kimura dans les années 1960 apporte une nouvelle vision de l’évolution avec la mise en avant du hasard comme force aussi importante que la sélection, la dérive génétique (Kimura, 1991, 1987, 1968). Il s’agit de la variation aléatoire des fréquences alléliques dans une population (Charlesworth, 2009), indépendamment de la sélection ou des migrations. La dérive est plus forte dans des populations de petite taille, et donc de faible taille efficace (Wright, 1931), et peut aller à l’encontre de la sélection naturelle (Charlesworth, 2009; Willi et al., 2006). Les mutations sont soumises à ces forces évolutives et le taux de mutation subit lui même la sélection naturelle ou le hasard de la dérive génétique.
12
2.
Les enjeux de la recherche sur les mutations
La diversité que nous pouvons observer sur Terre au sein des trois empires du vivant que sont les bactéries, les archées et les eucaryotes est issue des processus de sélection et de mutations. Les mutations sont une altération de la molécule d’ADN, à un niveau ponctuel ou chromosomique. Cette altération peut être le remplacement d’un nucléotide par un autre, une insertion ou une délétion de séquence, une cassure, une duplication, un réarrangement chromosomique ou autres modifications de l’ADN. Nous pouvons distinguer deux origines aux mutations: les mutations issues des erreurs de réplication d’une part, et issues de facteurs mutagènes d’autres part (rayonnement ultra violet, stress oxydatifs ou radioactivité par exemple); voir la revue de Maki, 2002 (Maki, 2002) et la Figure 1.
Les mutations constituent un large enjeu pour la recherche en biologie et en médecine. En recherche fondamentale, elles sont étudiées pour répondre à des questions centrales sur l’évolution et les capacités d’adaptation des espèces. La diversité génétique, en partie issues des mutations, est étudiée en écologie pour la conservation des espèces menacées (Ellegren and Galtier, 2016). En médecine, elles sont étudiées en raison de leurs implications dans différentes maladies génétiques et cancers (Ding et al., 2015; Salk et al., 2010). Deux points essentiels intéressent particulièrement les évolutionnistes et la communauté scientifique en général:
Le premier est de savoir comment les mutations impactent la fitness des organismes, c’est à dire leurs capacités de survie et de reproduction. L’effet des mutations se définit alors comme avantageux (la fitness augmente), neutre (la fitness ne change pas) ou délétère (la fitness diminue).
Le second point est de comprendre à quelles fréquences les mutations apparaissent, et quels facteurs influencent le taux de mutation et ces éventuelles variations aux différentes échelles.
"$! Nous verrons donc dans un premier temps l’état de l’art sur notre compréhension des effets des mutations sur la fitness et leurs rôles dans l’adaptation, suivis d’une liste non exhaustive des facteurs qui expliquent en partie les variations inter et intra génomiques du taux de mutation.
Ce travail de thèse s’inscrit pleinement dans ces deux problématiques, par l’étude du taux de mutation et de l’effet des mutations sur la fitness en considérant cinq espèces d’algues vertes (chlorophytes, plantae, eucaryotes) comme modèles biologiques.
Figure 1. Processus de mutations, modifié de Gao et al., (Gao et al., 2016). Les mutations sont
issues des erreurs de réplication ou des facteurs mutationnels indépendants de la réplication. Dans les deux cas, des mécanismes de réparations existent pour corriger une partie de ces mutations. Si la mutation n’est pas réparée, elle peut être transmise ou non à la descendance en fonction du mode de reproduction de l’organisme. ADN intact Lésion ADN intact Pas de dommages Mutation Réparation correcte Réparation non correcte ou partielle Absence de réparation Lésion Mutagènes endogènes ou exogènes Dommage ADN pré-réplication Réplication Résultat post-réplication Pas de mutation Mutation Erreur de réplication Mutation létale Arrêt de la réplication
14
3.
Les variations du taux de mutation
Au début des années 1960 apparaît la notion d’horloge moléculaire (Bromham and Penny, 2003). L’horloge moléculaire avance l’hypothèse d’une apparition constante et continue des mutations dans un génome. Cette horloge moléculaire sera utilisée pour dater les phylogénies, mais des études vont invalider cette hypothèse, avec des variations inter taxons (Britten, 1986; Bromham, 2009) et intra taxon (Bousquet et al., 1992; Bromham et al., 1996) du taux de mutation. De plus, les données actuelles montrent des variations importantes au sein d’une même espèce, par exemple en fonction du mode de reproduction, où le taux de mutation est plus fort dans une population asexuée (Henry et al., 2012; Neiman et al., 2010). C’est aussi le cas pour différentes souches de Chlamydomonas reinhardtii (Ness et al., 2015b) avec une variation d’un facteur 10 entre les taux de mutations les plus bas et les plus hauts. En plus de ces variations inter espèces, il existent des variations intra génomiques du taux de mutation, comme dans le génome mitochondrial des angiospermes (Laroche et al., 1997), ou entre les organelles et l’ADN nucléaire comme chez la drosophile ou Caenorhabditis elegans (Denver et al., 2000; Haag-Liautard et al., 2008; Smith, 2015; Xu et al., 2012). Une revue chez les mammifères expose les nombreuses variations du taux de mutation dans un génome (Hodgkinson and Eyre-Walker, 2011), que ce soit aux échelles de sites adjacents, ou de chromosomes entiers. Nous savons par exemple que le taux de mutation est plus élevé au niveau du chromosome sexuel Y les chimpanzés par rapport aux autres chromosomes (Consortium, 2005; Ebersberger et al., 2002). Au niveau intra chromosomique, il a été montré que certains trinucléotides mutent préférentiellement par rapport à d’autres (Ness et al., 2015b; Sung et al., 2015), ou que les régions avec de petites séquences répétées mutent plus rapidement que le reste du génome (Ma et al., 2012; Tesson et al., 2013). Ces variations du taux de mutation mettent en avant l’importance de comprendre quelles forces évolutives l’impactent et le font varier aux échelles inter et intra génomiques. Ces types de résultats sont en partie obtenus par une étude directe du taux de mutation, via les expériences d’accumulation de mutations (EAM). C’est cette approche qui est utilisée dans ce travail de thèse sur les cinq espèces modèles.
15
4.
Les expériences d’accumulation de mutations
1.
Les premières expériences de Terumi Mukai
Les premières estimations du taux de mutation datent des années 1960 avec les expériences d’accumulation de mutations (EAM) de Terumi Mukai (Keightley and Eyre-Walker, 1999; Mukai, 1964) sur la drosophile, bien que les premières expériences portant sur les mutations ont été développées une cinquantaine d’années plus tôt par Muller (Crow and Abrahamson, 1997; Muller, 1927). A cette époque, l’estimation du taux de mutation ne se fait pas par séquençage, en raison de l’absence des technologies modernes, mais par l’estimation du taux de mutation délétères (Ud) à partir de données de fitness. Le principe des expériences
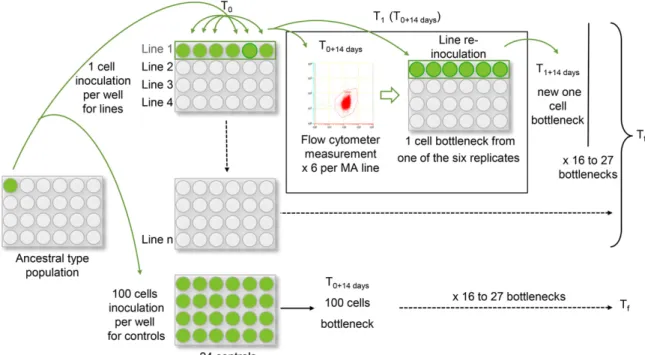
d’accumulation de mutations est de maintenir des lignées filles issues d’une lignée mère pendant un certain nombre de générations et de comparer les lignées filles en fin d’expérience avec le type ancestral (Figure 2). Durant les expériences d’accumulation de mutations, une série de goulots d’étranglements est nécessaire pour maintenir une taille efficace (Ne) la plus faible possible dans les lignées. La
taille efficace d’une population, notion introduite par Sewall Wright en 1931 (Wright, 1931), est la part de la population qui participe à la reproduction, ou la taille théorique qu’aurait la population dans un cas idéal (c’est à dire une population avec reproduction aléatoire, la panmixie) qui aurait la même diversité que la population réelle. Plus la taille efficace de la population est grande, plus la sélection est efficace. Inversement, plus la taille efficace est faible plus la dérive génétique sera forte. Réduire la taille efficace dans les lignées permet donc d’éliminer au maximum la sélection naturelle et d’estimer le taux de mutation avant sélection. Nous avons donc accès à la totalité des mutations (exception faite des mutations létales), définissant le taux de mutations spontanées (Drake et al., 1998). C’est pour cette raison qu’une étude de la diversité existante dans une population ou une espèce est insuffisante pour estimer le taux de mutations spontanées car seule la diversité après sélection est mesurée. Dans le cas de la drosophile, en raison de la diploïdie et de la reproduction sexuée, la lignée mère est généralement consanguine homozygote avant de commencer l’expérience (Keightley et al., 2014a, 2014b, 2009).
"'!
Figure 2. Schéma d’une expérience d’accumulation de mutations. Les lignées sont maintenues avec
une succession de goulots d’étranglements. En fin d’expérience, une comparaison de fitness ou une comparaison génomique permet d’étudier l’effet des mutations et leurs distributions dans le génome.
De cette façon, des variations de fitness dues à la recombinaison de plusieurs allèles lors de la méiose sont évitées. Les locus portant tous le même allèle, l’hypothèse est faite que seules les mutations créent une variation de fitness.
Avec les données de fitness, estimées par le succès de reproduction (nombre d’œufs et nombre d’éclosions), Mukai développe une méthode statistique et calcule les paramètres de mutation:
!! ! !" ! ! !! ! !!!!!!! ! !"#!!!!! !" ! !!!! !!! !!!! ! !! ! !!!
Où a est l’effet de la mutation, !V le changement de variance de la donnée de fitness par génération et !M le changement moyen de fitness par génération.
!V et !M peuvent être estimés directement par régression sur les données de
fitness mesurées pendant l’expérience. L’augmentation de la variance de la valeur de fitness résulte de l’impact des mutations qui vont faire changer la fitness dans le cas de mutations avantageuses ou délétères. Mukai estime Ud=0.34 mutations
délétères par génome par génération comme première estimation d’un taux de mutation délétère chez un organisme et E(a)=0.027 comme baisse moyenne de fitness par génération. Ce taux de mutation est le taux de mutation minimal, car il ne prend en compte que les mutations délétères (seule la baisse de fitness est prise en
N Lignées Type ancestral Contrôle Type ancestral Mutation
17 compte pour les calculs des paramètres de mutations). De plus, il estime le taux de mutation létal à 0.006 mutations par génération.
Suite à la méthode de Mukai, une autre méthode, par maximum likelihood (Fry et al., 1999; Keightley, 1994; Keightley and Bataillon, 2000; Keightley and Caballero, 1997) a été développée. Elle permet notamment d’utiliser des données de fitness issues d’une EAM comme la méthode de Mukai, mais avec une variance plus faible.
4.
2. L’effet des mutations sur la fitness
1. Les successeurs de Terumi MukaiLa méthode statistique de Mukai est utilisée par différents biologistes pour estimer Ud chez différents organismes modèles. Une revue a été publiée en 2009
(Halligan and Keightley, 2009). D’une manière générale, il est constaté une baisse de la fitness chez les lignées mutantes au cours des générations pour toutes les espèces qui ont été testées, dont quelques exemples sont cités ci-dessous:
- Drosophila melanogaster (Fernández and López-Fanjul, 1996; Fry, 2004,
2001, Fry et al., 1999, 1996; Fry and Heinsohn, 2002; Houle et al., 1992; Huey et al., 2003; Keightley, 1994; Schrider et al., 2013; Wang et al., 2014);
- Caenorhabditis elegans (Ajie et al., 2005; Baer et al., 2006, 2006; Davies et
al., 1999; Estes et al., 2004; Katju et al., 2014; Vassilieva et al., 2000; Vassilieva and Lynch, 1999);
- Saccharomyces cerevisiae (Korona, 1999; Wloch et al., 2001; Zeyl and
DeVisser, 2001);
- Daphnia pulex (Deng and Lynch, 1997; Korona, 1999, 1999; Latta et al.,
2013; Schaack et al., 2013);
- Arabidopsis thaliana (Deng and Lynch, 1997; Rutter et al., 2012; Schultz et
18 Il existe aussi des organismes un peu moins étudiés par EAMs, mais de plus en plus de données sont disponibles sur tout l’arbre du vivant; Chlamydomonas
reinhardtii (Morgan et al., 2014), Tetrahymena thermophila (Brito et al., 2010), Dictyostelium discoideum (Hall et al., 2013), Escherichia coli (Cao et al., 2014;
Kibota and Lynch, 1996).
De ce fait, il est avancé que la majorité des mutations sont délétères, c’est à dire qu’elles diminuent la capacité de survie. L’impact des mutations délétères dans une population ou chez un individu est défini comme le poids des mutations délétères, ou fardeau génétique (Agrawal and Whitlock, 2012; Charlesworth et al., 1990): c’est la différence de fitness qu’il existe entre la fitness optimale et la fitness réelle. Les mutations délétères sont normalement supprimées par la sélection naturelle, mais la dérive peut les maintenir ou les fixer dans une population. L’effet des mutations délétères sur les populations a largement été exploré (Agrawal and Whitlock, 2012; Charlesworth and Charlesworth, 1998; Kondrashov, 1995, 1988, Lande, 1994, 1988; Lynch et al., 1999), de même que l’estimation par des méthodes statistiques des paramètres mutationnels dans les populations naturelles (Deng et al., 2002; Deng and Lynch, 1996; Li and Deng, 2005). Une population de petite taille efficace est plus sensible aux mutations délétères en raison de la faible efficacité de la sélection naturelle (Eyre-Walker et al., 2002; Higgins and Lynch, 2001; Houle, 1992; Lande, 1998; Lynch et al., 1995; Lynch and Gabriel, 1990; Willi et al., 2006). Si la sélection est trop faible, elle ne permet pas une purge efficace des mutations délétères. Cela peut avoir un impact sur les espèces menacées avec de faibles tailles de population: la dérive et les mutations délétères peuvent accentuer la perte de diversité et de viabilité d’une population.
Cependant, le taux de mutation « optimal » résulte d’un compromis entre le coût des mutations délétères et le bénéfice de mutations avantageuses (Wielgoss et al., 2013). La taille efficace d’une population joue donc un rôle essentiel dans la force de sélection et la capacité adaptative de cette population (Gossmann et al., 2012). Ainsi, la probabilité de fixation d’une mutation dans une population va dépendre de l’intensité de la dérive et de la sélection, et de l’effet de cette mutation sur la survie (neutre, avantageux ou délétère).
19
4.
2.
2. Paysage adaptatif et distribution de l’effet des mutationsComme nous venons de voir, la majorité des mutations semble être délétère, mais une partie est neutre ou avantageuse (Hall et al., 2008; Joseph and Hall, 2004). La distribution de fitness des mutations vient en partie du niveau de fitness d’un génome dans un environnement donné. En faisant l’hypothèse qu’il existe un niveau de fitness maximal possible dans un environnement, la proportion de mutations délétères augmente si la fitness du génome se rapproche du maximum. L’ensemble des fitness possibles se définit comme le paysage adaptatif (Orr, 2005; Petren, 2013), une notion introduite par Sewall Wright et Fisher (Mousseau and Roff, 1987; Edwards, 2000; Zhang, 2012). Il existe de nombreuses théories sur les modèles de paysages adaptatifs possibles, en particulier le « single-peak » (Wright, 1932), le « rugged » ou vallée (Martin and Wainwright, 2013; Steinberg and Ostermeier, 2016) ou le « holey » (Gavrilets, 1997).
Le « single-peak », le plus simple, est un pic de fitness avec un maximum possible (Figure 3). Dans ce cas, la fitness du génome, en fonction des mutations et de l’épigénétique (Kaity et al., 2008), va se déplacer sur le pic de fitness entre le maximum et le minimum. Plus la fitness du génome est proche du maximum, plus les mutations auront de fortes probabilités d’être délétères et, inversement, un génome avec un niveau de fitness bas va compter plus de mutations avantageuses (Tenaillon et al., 2016). Enfin, si la fitness du génome est trop basse, il peut simplement être éliminé par la sélection.
Le second modèle est le modèle « holey » (Gavrilets, 1997), où la fitness maximale est définie comme le plancher du paysage adaptatif. Les mutations avantageuses ne font que maintenir le génome à ce niveau. Ce plancher est marqué par des puits de fitness, dans lesquelles le génome « tombe » en cas de mutations délétères.
20 Enfin, le modèle le plus souvent accepté, et qui a déjà été mis en évidence chez les bactéries (Nahum et al., 2015) ou des espèces comme un téléostéen du genre Cyprinodon (Martin and Wainwright, 2013), est le « rugged ». Dans ce cas, il existe de nombreux pics de fitness avec des vallées ou des plateaux sur lesquels le génome va se déplacer. De plus, une vallée entre des pics de fitness peut entrainer une différenciation de deux populations, d’où l’importance de cette hypothèse en évolution. Avec ce modèle, une population avec une faible taille efficace peut atteindre un pic de fitness plus élevé qu’une population avec une taille efficace plus grande (Rozen et al., 2008). A cause de l’efficacité de la sélection, une grande population atteindra rapidement le sommet de fitness le plus proche. En revanche, une population avec une petite taille efficace pourrait atteindre un pic de fitness plus élevé, car la dérive génétique déplace la population dans le paysage de fitness.
Dans tous les cas, quel que soit le model admis, ce sont les mutations qui vont principalement augmenter ou diminuer la fitness du génome sur le paysage adaptatif et permettre l’accession à une fitness supérieure dans le cas de mutations avantageuses. Par ailleurs, le paysage adaptatif est spécifiquement défini pour un génotype et un environnement. La position d’un génome est donc le résultat de l’interaction génotype-environnement et du contexte génétique. On a donc une variation du paysage adaptatif suite à une variation environnementale (Matuszewski et al., 2014) ou le long d’un gradient environnemental (Laughlin and Messier, 2015), avec un compromis d’adaptation (Elena and Lenski, 2003) entre les environnements (Figure 4).
Au delà des mutations avantageuses ou délétères, les mutations neutres sont tout aussi importantes en raison de la variation de la distribution de la fitness des mutations. Les mutations avantageuses peuvent voir leurs impacts augmentés ou diminués, et les mutations neutres dans un cas peuvent avoir un effet dans d’autres conditions. Le changement de distribution de fitness des mutations neutres met en avant l’importance de la variation existante comme base d’adaptation immédiate à un changement environnemental (Barrett and Schluter, 2008; Hermisson and Pennings, 2005). On parle de la « standing genetic variability ».
#"!
Figure 3. Représentation du « fitness landscape ». La fitness du génotype (en bleu) va bouger et
changer en fonction de l’effet des mutations qui vont apparaître. Plus la fitness du génotype est proche du maximum, plus les mutations auront de probabilité d’être délétères. De même une variation environnementale peut entrainer un déplacement du génotype sur le paysage de fitness. La difference entre la position du génotype et le maximum de fitness est definie comme le poids des mutations délétères.
Figure 4. Changement de fitness d’un genotype entre environnements. La variation de l’effet des
mutations et de la variabilité entre envrironnements. La figure est reprise de Santiago et Richards, 2003 (Elena and Lenski, 2003). Le genotype 1 est specialisé dans l’environnement A mais peu adapté au B, inversement pour le génotype 2, alors que le génotype 3 est généraliste.
Mutation
- neutre
- avantageuse
- délétère
Fitness du
génome
Position du génome
Fitness
!"#$%&%
!"#$%'%
()"$%*%
()"$%+%
()"$%,%
22
4.
2.
3. Interaction génotype-environnement1. Les changements d’effet des mutations
Différentes études sur des lignées mutantes issues d’expériences d’accumulation de mutations ont tenté d’explorer les effets d’un changement environnemental sur la distribution de fitness des mutations en comparant les fitness d’une même lignée, notamment chez la drosophile (Fry et al., 1996, p. 19996; Fry and Heinsohn, 2002; Kondrashov and Houle, 1994; Korona, 1999), le nématode
Caenorhabditis elegans (Baer et al., 2006), ou la plante Arabidopsis thaliana (Rutter
et al., 2012). Comme nous l’avons vu dans le paragraphe précédent sur le paysage adaptatif, nous nous attendons à des changements de fitness selon les conditions. En laboratoire, différentes variables facilement contrôlables peuvent être testées; on peut citer le cas de la disponibilité en ressource (Chang and Shaw, 2003) et de la luminosité (Kavanaugh and Shaw, 2005) chez Arabidopsis thaliana. De ces études, nous pouvons estimer les paramètres de mutations comme pour les EMAs et les comparer pour émettre des hypothèses sur les implications biologiques des mutations, définies au nombre de trois (Martin and Lenormand, 2006).
Premièrement, un changement de U (nombre de mutations par génome par génération) traduit une différence d’effet des mutations, avec des mutations qui ont un effet détectable dans une condition mais neutre (ou non détectable suivant le caractère de fitness considéré) dans une autre. On peut par exemple penser à des changements d’expression de gènes entre deux conditions.
Deuxièmement, une variation de a (effet moyen d’une mutation par génération) indique un changement de l’intensité de la sélection car les effets d’une mutation varient. Or, plus l’effet de la mutation est fort, plus la sélection pourra influer sur la fréquence de cette mutation dans une population : on attend une plus forte contre sélection d’une mutation délétère qui a un plus fort impact sur la fitness.
Enfin, si CV varie (c’est à dire la variation de l’effet des mutations), on s’attend à avoir un effet du stress sur les mutations. Ainsi, si une population est adaptée à une condition et est proche du maximum de fitness, l’effet des mutations sera le plus souvent délétère. Mais en cas de stress, la population n’est plus à son optimum de fitness, ce qui fera varier l’effet des nouvelles mutations.
23
4.
2.
3. Interaction génotype-environnement2. Le stress et les hyper mutateurs
En cas de stress, il est traditionnellement admis que les mutations délétères ont un impact plus fort sur la fitness (Elena and de Visser, 2003). C’est notamment le cas dans une étude pourtant sur l’impact du stress chez des lignées issues d’expériences d’accumulation de mutations chez Chlamydomonas reinhardtii (Kraemer et al., 2015). Cependant, il est à noter que cette observation n’est pas systématique et différents articles tendent à montrer que le stress n’a pas d’effet sur l’ampleur de l’impact des mutations délétères (Andrew et al., 2015; Kishony and Leibler, 2003). L'impact du stress sur les effets des mutations peut être caractérisé de trois façons (Elena et de Visser, 2003): tout d'abord, la mutation peut être délétère sans conditions, avec une augmentation de l'effet délétère avec le stress; Ensuite, la mutation peut être conditionnellement neutre, c’est à dire neutre dans certaines conditions et délétère dans d'autres; Enfin, la mutation peut être conditionnellement bénéfique: avantageuse dans certaines conditions, mais délétère ou neutre dans d'autres.
En cas de stress, chez les bactéries, il a été mis en évidence la présence d’allèles mutateurs qui vont avoir un impact sur le taux de mutation en l’augmentant significativement (Couce et al., 2013; Taddei et al., 1997). Ce type de mécanismes est avantageux dans un environnement défavorable. Les nouvelles mutations vont apparaître plus fréquemment, ce qui augmente la probabilité des mutations avantageuses et donc l’adaptation (Giraud et al., 2001; Sniegowski et al., 1997; Tenaillon et al., 1999). Il n’y a pas d’allèles mutateurs connus chez les eucaryotes, mais il semble que chez la drosophile, les individus moins adaptés à un nouvel environnement ont un taux de mutation plus élevé que les autres (Sharp and Agrawal, 2012). Cette observation est également faite chez la plante A. thaliana (Jiang et al., 2014) ou chez la levure (Shor et al., 2013). Cette augmentation est toutefois moins significative que dans le cas des hyper-mutateurs bactériens.
24
5. Les estimations directes du taux de mutation
1.
Les variations inter génomiques du taux de mutation
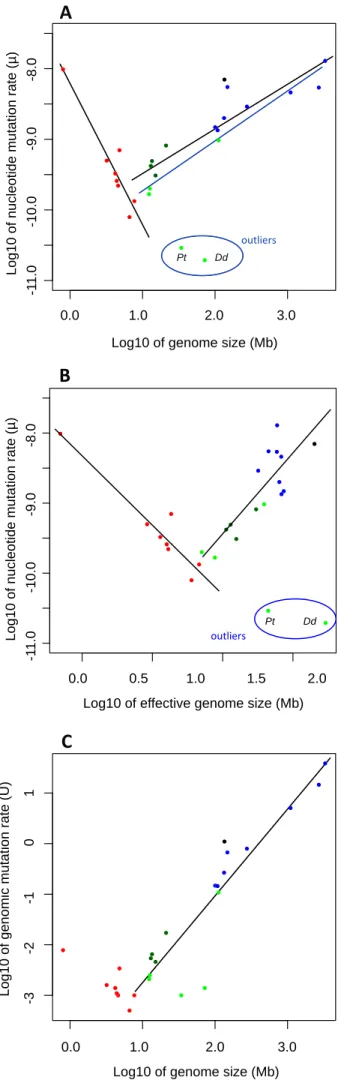
1. La taille du génomeDe nos jours, les nouvelles générations de séquenceurs permettent d’estimer directement le taux de mutation en comparant les génomes de début et de fin d’EAM (voir le Tableau 1 pour les estimations actuelles). Une base de données en ligne est également disponible (Wei et al., 2014). Ces estimations ont permis de formuler différentes hypothèses sur les facteurs biologiques et écologiques qui agissent sur l’évolution du taux de mutation.
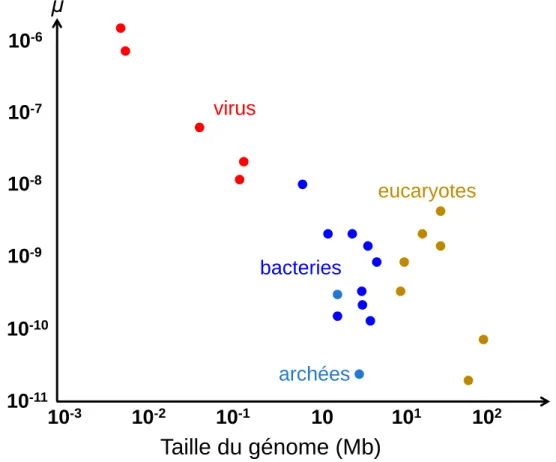
Parmi les premiers articles, Drake propose en 1991, à partir de données sur des organismes unicellulaires, un nombre de mutations constant par génome (Drake, 1991; Drake et al., 1998). Cette constante serait de U=0.0033 mutations par génome par réplication chez les microorganismes. Il s’agit plutôt de formuler que le taux de mutation U varie moins par rapport au niveau de variation des taux de mutation µ et des tailles des génomes. De ce fait, le taux de mutation diminue avec l’augmentation de la taille du génome pour garder le nombre de mutations U par génome constant à chaque réplication (Figure 5). Dans le cas d’un taux de mutation qui ne diminue pas avec l’augmentation de la taille du génome, nous obtenons un nombre de mutations par génome croissant. Cela augmente la probabilité d’apparition de mutations délétères à chaque réplication, ce qui peut compromettre la survie. Cette relation ne semble toutefois pas concerner les eucaryotes, où le taux de mutation augmente avec la taille du génome (Smeds et al., 2016; Sung et al., 2012a).
25
Tableau 1. Les taux de mutations spontanées estimés par des expériences d'accumulation de
mutations. Dans ce tableau, seules les estimations de taux de mutation par séquençage du génome entier sont spécifiées. G est la taille du génome en Mb, µ est le taux de mutation par nucléotide par génome par génération et U est le nombre de mutations par génome par génération. Dans ce tableau n’apparaissent pas les mesures de taux de mutation obtenues avec des lignées artificiellement mutantes (suppression d’un mécanisme de réparation de l’ADN) ou issues d’un pédigrées, comme chez l’homme ou la souris (ces données sont disponibles dans le chapitre 3, tableau S10).
Espèces G µ U Références
Arabidopsis thaliana Col-0 157.0 7.00E-09 1.0990 (Ossowski et al., 2010)
Caenorhabditis elegans N2 100.3 2.50E-09 0.2508 (Denver et al., 2009)
Caenorhabditis elegans N2 100.3 3.10E-09 0.3109 (Denver et al., 2009)
Caenorhabditis elegans N2 100.3 1.33E-09 0.1334 (Denver et al., 2012)
Caenorhabditis elegans PB306 100.3 1.62E-09 0.1625 (Denver et al., 2012)
Caenorhabditis briggsae PB800 108.4 1.44E-09 0.1561 (Denver et al., 2012)
Caenorhabditis briggsae HK104 108.4 1.23E-09 0.1333 (Denver et al., 2012)
Pristionchus pacificus PS312 133.1 2.0E-09 0.2663 (Weller et al., 2014)
Drosophila melanogaster Madrid 122.0 3.50E-09 0.4270 (Keightley et al., 2009)
Drosophila melanogaster Florida 122.0 5.49E-09 0.6698 (Schrider et al., 2013)
Drosophila melanogaster Florida 122.0 2.80E-09 0.3416 (Keightley et al., 2014a)
Heliconius melpomene 273.8 2.90E-09 0.7940 (Keightley et al., 2014b)
Chlamydomonas reinhardtii CC-2937 112 2.08E-10 0.0233 (Ness et al., 2012)
Chlamydomonas reinhardtii CC-124 112 6.76E-11 0.0076 (Sung et al., 2012a)
Chlamydomonas reinhardtii 112 9.63E-10 0.1079 (Ness et al., 2015b)
Paramecium tetraurelia d4-2 72.1 1.94E-11 0.0014 (Sung et al., 2012b)
Saccharomyces cerevisiae FY10 12.3 3.30E-10 0.0041 (Lynch et al., 2008)
Saccharomyces cerevisiae 12.3 1.67E-10 0.0021 (Zhu et al., 2014)
Schizoaccharomyces pombe ED668 12.6 2.00E-10 0.0025 (Farlow et al., 2015)
Schizoaccharomyces pombe 12.6 1.70E-10 0.0021 (Behringer and Hall, 2015)
Dictyostelium discoideum AX4 34.2 2.90E-11 0.0010 (Saxer et al., 2012)
Burkholderia cenocepacia HI2424 7.7 1.33E-10 0.0010 (Dillon et al., 2015)
Escherichia coli 3k 4.6 1.88E-10 0.0009 (Lee et al., 2012)
Escherichia coli 6k 4.6 2.45E-10 0.0011 (Lee et al., 2012)
Mesoplasma florum L1 0.8 9.78E-09 0.0078 (Sung et al., 2012a)
Mycobacterium tuberculosis H37Rv 4.4 2.58E-10 0.0011 (Ford et al., 2011)
Salmonella typhimurium LT2 4.8 7.00E-10 0.0034 (Lind and Andersson, 2008)
Bacillus subtilis 4.2 3.28E-10 0.0014 (Sung et al., 2015)
Pseudomonas aeruginosa 6.6 7.92E-11 0.0005 (Dettman et al., 2016)
Deinococcus radiodurans BBA816 3.2 4.99E-10 0.0016 (Long et al., 2015a)
#'!
Figure 5. Relation entre le taux de mutation et la taille du génome. Figure reprise de Sung, 2012
(Sung et al., 2012a). On observe une diminution du taux de mutation avec une augmentation de la taille du génome chez les microorganismes. Cela se traduit par des apparitions peu fréquentes des mutations délétères dans les plus grands génomes.
5.
1.
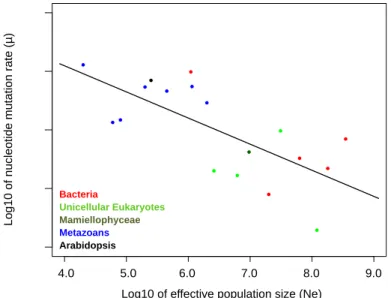
2. La taille efficace (Ne)Un autre facteur essentiel est la taille efficace qui va conditionner l’intensité de sélection à laquelle sera soumis le taux de mutation (Charlesworth, 2009; Lanfear et al., 2014), avec la notion de barrière de dérive (Martincorena and Luscombe, 2013; Sung et al., 2012a). Selon Lynch, le taux de mutation est plus faible chez les microorganismes en raison de leur grande taille efficace de population qui permet une sélection efficace (Lynch, 2010a). Le taux de mutation pourrait cependant être attendu plus petit chez les organismes multicellulaires en raison des dommages somatiques liés aux mutations délétères (Lynch, 2008), en particulier les cancers (Cowin et al., 2010; Knudson, 2000). Chez les organismes pluricellulaires, le taux de
Taille du génome (Mb)
!
10
-1110
-1010
-910
-810
-710
-610
-310
-210
-110
10
110
2virus
archées
eucaryotes
bacteries
#(! mutation varie en fonction des tissus (Lynch, 2010a), et le taux de mutation dans la lignée germinale est inférieur aux taux de mutation des cellules somatiques (Lynch and Hagner, 2015), limitant la transmission de mutations délétères au générations suivantes. Le coût des mutations délétères va pousser vers la sélection d’un taux de mutation faible, avec un taux de mutation théorique défini comme optimal (Figure 6). Mais ce taux de mutation optimal n’est jamais atteint en raison de la dérive génétique. Il existe donc une limite, dite la « barrière de dérive », qui empêche d’atteindre un taux de mutation optimal par la sélection, qui est un compromis entre l’adaptation, le coût des mutations délétères sur la fitness et le coût de réplication (Martincorena and Luscombe, 2013). Pour conclure, le taux de mutation réel est le plus proche du taux de mutation optimal chez les organismes à grande taille efficace (comme les microorganismes), que celui des organismes à taille efficace de population plus faible, comme les métazoaires (Figure 7).
Figure 6. La barrière de dérive et le coût de la réplication. Figure reprise de Martincorena et
Luscombe, 2013 (Martincorena and Luscombe, 2013). La dérive impose une limite à la sélection du taux de mutation, qui ne peut atteindre le taux de mutation optimal, défini comme le compromis entre le coût des mutations délétères et le coût de la fidélité de réplication. Les espèces avec une grande taille efficace sont plus susceptibles de se rapprocher du taux de mutation optimal.
Coût de fitness
Taux de
mutation optimal
Coût des mutations
délétères
Taux de mutation
!
Coût de
réplication
Limite
de
dérive
Taux de mutation
observé
#)!
Figure 7. Relation entre taille efficace et taux de mutations. Figure reprise de Ness, 2012 (Ness et al.,
2012). La taille efficace de la population définit l’efficacité de la sélection, et donc la capacité à atteindre le taux de mutation le plus bas possible pour limiter l’apparition des mutations délétères.
5.
1.
3. Le temps de générationLe temps de génération étant variable en fonction des caractéristiques biologiques ou écologiques des espèces, le nombre de mutations qui apparaissent par unité de temps varie également. Des études ont tenté de comprendre l’influence du temps de génération sur le taux de mutation (Laird et al., 1969), notamment chez les vertébrés (Martin and Palumbi, 1993; Mooers and Harvey, 1994). D’une manière générale, il est observé une diminution du taux de mutation avec l’augmentation du temps de génération (Tableau 2). Des études plus récentes sur les mollusques (Thomas et al., 2010) ou les bactéries (Weller and Wu, 2015) tendent à confirmer cette hypothèse. Cela signifie une plus forte capacité à créer de nouvelles mutations et donc à s’adapter pour les espèces à temps de génération court.
Taille efficace
104 105 106 107 108 10-10 10-9 10-8 10-7 M. domesticus H. sapiens C. elegans A. thaliana P. falciparum N. crassa C. reinhardtii S. serevisiae D. melanogaster!
29
Tableau 2. Corrélation entre le temps de génération et le taux de mutation. Données de Martin et
Palumbi (Martin and Palumbi, 1993). On observe une diminution du taux de mutation avec une augmentation du temps de génération et une baisse du taux métabolique.
Espèces Substitutions par site par
milliard d'années Taux métabolique (O2/kg/h) Temps de génération (jours) Douroucouli 2.1 450 880 Singe araignée 1.9 415 1 700 Macaque 1.8 430 1 095 Gibbon 1.7 370 3 410 Orang-outang 1.2 230 4 290 Gorille 1.2 200 3 438 Chimpanzé 1.2 220 3 190 Humain 1.1 210 6 200
5.
1.
4. Le taux métabolique et la températureEn plus du temps de génération vu précédemment, Martin et ses collaborateurs montrent une augmentation du taux de mutation avec une augmentation du taux métabolique mesurée par la respiration (Martin and Palumbi, 1993). Cette augmentation est en général expliquée par la plus importante production d’espèces réactives d’oxygène (ROS) qui induisent un stress oxydatif (Baer et al., 2007). Les ROS, s’ils sont produits en trop grand nombre par l’organisme, peuvent provoquer des mutations, en particulier par l’oxydation de la guanine (Foster et al., 2015) ou la déamination de la cytosine (Cooke et al., 2003; Dizdaroglu, 1992; Hurst and Williams, 2000).
Au delà du taux métabolique, la température pourrait également influer sur le taux de mutation (Lewis et al., 2016; Wolfenden, 2014). Selon Wolfenden, les sources hydrothermales auraient pu être un accélérateur pour l’évolution en raison de la forte température qui augmente la vitesse des réactions enzymatiques, comme l’hydrolyse des peptides, et l’instabilité de l’ADN. Ainsi, les réactions chimiques provoquant des changements irréversibles auraient été plus fréquentes, notamment
30 les déaminations hydrolytiques des cytosines et adénines qui deviennent des uraciles et xanthines (Wolfenden, 2014).
En lien avec les deux points précédents, il existe une théorie dite « metabolic theory of ecology », qui propose une accélération de l’évolution moléculaire avec la température. Cela se traduit par plus de spéciations et divergences en régions tropicales, chez plusieurs taxons, dont les plantes, les amphibiens ou les mammifères (Gillman et al., 2010; Mittelbach et al., 2007; Rolland et al., 2014; Wright et al., 2010).
5.
2. Les variations intra génomiques du taux de mutation
1. Le sens de la transcription et de la réplicationUne étude sur Bacillus subtilis (Paul et al., 2013) suggère une hétérogénéité du taux de mutation en fonction du sens de la réplication et de la transcription sur le brin d’ADN. Une augmentation du taux de mutation est observée dans les zones dites de «conflit réplication-transcription». Le taux de mutation est plus élevé dans les gènes orientés inversement au sens de réplication, ce qui signifie une variation du taux d’évolution entre gènes. Ce «conflit réplication-transcription» a également été mis en évidence par d’autres études portant sur des lignées issues d’expériences d’accumulations de mutations (Schroeder et al., 2016). Le taux de mutation est également plus fort dans certaines zones, appelés « points chauds de mutations ». Chez les bactéries, notamment Escherichia coli, ces points chauds ont été localisés au niveau des points de blocage ou collision entre les fourches de réplication des deux brins matrices et non matrices (Foster et al., 2013). De même, la réplication est plus ou moins fidèle selon l’orientation des brins sens et anti-sens, ce qui a également une influence sur le taux de mutation (Fijalkowska et al., 1998).
31
5.
2.
2. Le temps de réplicationAu-delà du sens de la réplication, le temps de réplication induit un taux de mutation plus fort en fin de réplication, phénomène bien connu chez les mammifères (Chen et al., 2010; Stamatoyannopoulos et al., 2009). C’est à dire que plus le temps de réplication est long, plus le taux de mutation peut être élevé en fin de réplication. Les deux hypothèses avancées pour expliquer ce phénomène sont la diminution du stock de nucléotides disponibles et la perte d’efficacité des mécanismes de réparation de l’ADN (MisMatch Repair ou MMR). Les MMR permettent de réduire l’apparition des mutations au cours de la réplication (Fukui and Fukui, 2010; Jiricny, 2006; Kunkel and Erie, 2015; Li, 2008). Nous savons, par des expériences d’accumulation de mutations avec des lignées artificiellement déficientes en MMR (Denver et al., 2005; Jiricny, 2006; Lang et al., 2013; Lee et al., 2012; Long et al., 2015b; Sung et al., 2015), que ces mécanismes de réparation réduisent d’environ un facteur 100 le taux de mutation et peuvent changer le sens et la distribution des mutations en fonction de leur activation. Ce biais mutationnel en fin de réplication existe aussi chez les bactéries (Hudson et al., 2002) et chez la levure (Lujan et al., 2014). Le type et le taux d’erreur lors de la réplication peuvent également dépendre du type d’ADN polymérase. En effet, les différentes ADN polymérases n’ont pas les mêmes niveaux de fidélité, induisant plus ou moins d’erreurs (Hestand et al., 2016; Kunkel and Bebenek, 2000). Chez les eucaryotes, il existe par exemple de nombreuses polymérases avec des fonctions et capacités enzymatiques différentes (Hubscher et al., 2002).
32
5.
2.
3. Les régions codantes et le niveau d'expressionUne troisième raison aux variations intra génomiques vient de la différence du taux de mutation entre les régions codantes et non codantes du génome, et le niveau d’expression. Le taux de mutation semble en effet plus faible dans les gènes fortement exprimés (Eyre-Walker and Bulmer, 1995; Martincorena et al., 2012). Les expériences d’accumulation de mutations ont permis de le confirmer, notamment chez la levure (Zhu et al., 2014). Deux explications peuvent être avancées pour l’expliquer. Les MMR, qui peuvent être plus efficaces en région codante (Foster et al., 2015), et les transcription-coupled repairs (TCR), capables de réparation dans les régions fortement exprimées (Hanawalt and Spivak, 2008).
Cependant différentes étude faites chez Escherichia coli (Beletskii and Bhagwat, 1996; Chen and Zhang, 2013; Klapacz and Bhagwat, 2002) contredisent ces résultats et montrent que les gènes fortement exprimés mutent plus rapidement que les autres. L’une des hypothèses avancées est le lien entre le taux de transcription et la mutabilité de la région transcrite: le processus de transcription peut perturber la réplication (Kim and Jinks-Robertson, 2012), comme vu dans le paragraphe traitant du sens de la réplication et de la transcription.
5.
2.
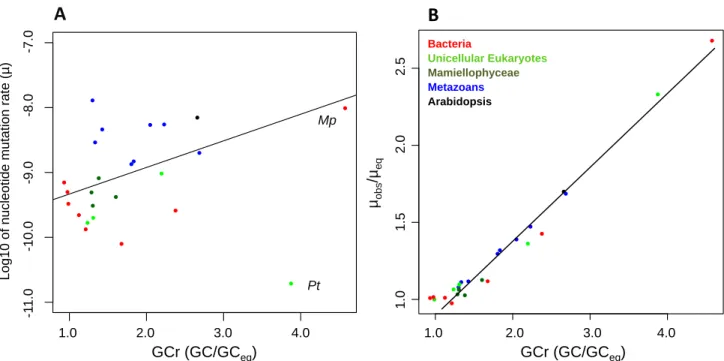
4. La composition en GCEnfin, la composition en GC a une influence sur le taux de mutation, par le biais de la proportion transversions/transitions et la proportion des mutations A-T vers G-C ou inversement. Hershberg et Petrov ont montré un biais mutationnel chez les bactéries (Hershberg and Petrov, 2010), avec une proportion plus importante de mutations depuis les nucléotides G-C vers A-T par rapport aux autres types de substitutions. Les expériences d’accumulation de mutations montrent généralement aussi un biais de mutation de G-C vers A-T, chez Caenorhabditis elegans (Denver et al., 2009), Arabidopsis thaliana (Ossowski et al., 2010), Escherichia coli (Lee et al., 2012), Salmonella typhimurium (Lind and Andersson, 2008), par exemple. Cette observation n’est cependant pas systématique chez les bactéries, avec deux contre-exemples (Dillon et al., 2015; Long et al., 2015a), voir le Tableau 3. Certains types
33 de mutations fréquentes comme la déamination de la cytosine (Coulondre et al., 1978; Fryxell and Zuckerkandl, 2000) et l’oxydation de la guanine sont connus pour induire des mutations de G-C vers A-T (Cooke et al., 2003; Dizdaroglu, 1992). Ce biais mutationnel est bien connu chez les mammifères, où les sites CpG, c’est à dire les dinucléotides CG, mutent plus rapidement que le reste du génome (Hodgkinson and Eyre-Walker, 2011). Le taux de mutation des sites CpG est ~10 fois plus important que pour les autres sites. De ce fait, les dinucléotides CpG ne sont présents qu’a 20% de leur fréquence attendue dans le génome humain (Lander et al., 2001). Cependant, cette relation n’est pas aussi simple, car il existe aussi chez les mammifères des régions dite « CpG islands », très riches en GC (Bird, 1986). Or, dans ces régions d’environ 1kb, le taux de mutation est inferieurs à celui des CpGs situés ailleurs dans le génome (Cohen et al., 2011). Cela s’expliquerait par la stabilité de la méthylation des cytosines, influencer par la richesse des nucléotides adjacents en GC (Elango et al., 2008).
Face à cela, nous pouvons nous attendre à observer une baisse de la teneur en GC dans certains génomes au cours des générations. Or, chez les bactéries, certains génomes sont très riches en GC (proche de 70%). Cela s’explique en partie par la sélection pour des codons optimaux plus riches en GC (Hildebrand et al., 2010). En effet, même des mutations synonymes peuvent avoir un impact sur la fitness, comme démontré précédemment (Glémin, 2010) en raison du biais d’usage du code (Ikemura, 1981). Le biais d’usage du code se traduit par l’utilisation préférentielle de certains codons par rapport à d’autres, notamment en fonction de la quantité de séquences qui codent pour l’ARN de transfert associé à ces codons.
La conversion génique biaisée peut aussi expliquer une augmentation de la teneur en GC d’un génome (Chen et al., 2007; Duret and Galtier, 2009; Galtier et al., 2001; Glémin et al., 2015; Mugal et al., 2013). Il s’agit d’un biais de réparation des mésappariements, généralement lors de la recombinaison, qui conduit à enrichissement en GC du génome. Ce phénomène semble aussi présent chez les bactéries (Lassalle et al., 2015), qui recombinent moins que les eucaryotes étudiés généralement pour la conversion génique biaisée.
34 De plus, des variations dans l’orientation des mutations ont également été observées entre le génome nucléaire et le génome des organelles chez
Chlamydomonas reinhardtii (Ness et al., 2015a). Chez Chlamydomonas reinhardtii,
la composition en GC des organelles (47%) est plus faible que celle du génome nucléaire (63%), ce qui peut expliquer en partie cette différence.
Pour prédire le nombre de mutations en fonction de la composition du génome, il est utile de calculer le GC% à l’équilibre (Sueoka, 1962), c’est à dire la teneur en GC du génome pour laquelle il y a autant de mutations de type G-C vers A-T que A-T vers G-C. Sachant que les mutations sont biaisées de G-C vers A-T, le taux de mutation est en général plus fort pour les nucléotides G et C que pour A et T. Le GCeq se calcule avec les équations suivantes :
𝑅!=(GC→AT)𝐺𝐶 ! , 𝑅!= (AT→GC) 𝐴𝑇! , GCeq = 𝑅! 𝑅!+ 𝑅!
avec GC→AT le nombre de mutations de type G-C vers A-T et 𝐺𝐶! le nombre de G et C dans le génome.
Tableau 3. Le biais de GC vers AT, dans la dernière colonne du tableau, est le rapport du taux de
mutation de GC vers AT sur celui de AT vers GC. Les données sont reprises de Dillon et collaborateurs en 2015 (Dillon et al., 2015).
Espèce (%GC) A/T->T/A G/C->C/G A/T->G/C G/C->A/T Biais vers AT
B. cenocepacia (67) 2.67 2.38 12.23 9.95 0.81 E. coli (51) 2.8 2.88 15.38 18.79 1.22 M. florum (27) 15.67 185.36 62.68 1000.97 15.97 H. sapiens (45) 129 295 581 1219 2.1 D. melanogaster (42) 98.06 74.52 149.14 643.95 4.32 S. cerevisiae (38) 3.03 7.82 12.43 27.55 2.22 A. thaliana (36) 43.56 123.63 165.52 1035.38 6.26 C. elegans (35) 17.5 16.89 24.19 101.32 4.19
35
6.
Nouveaux modèles biologiques
1.
L’importance écologique du phytoplancton
Le phytoplancton est composé de la partie photosynthétique du plancton, présente à l’échelle mondiale dans tous les écosystèmes aquatiques (de Vargas et al., 2015). Ce n’est cependant pas un terme qui désigne un groupe monophylétique, et ne constitue donc pas un groupe naturel d’organismes au sens évolutif. Il inclut des organismes issus de différents règnes, parmi les eucaryotes et les bactéries, notamment les cyanobactéries. Le phytoplancton eucaryote est très diversifié et se retrouve dans tous les règnes excepté les unicontes (qui comprennent entre autres les fungis et les métazoaires). La production primaire du phytoplancton constitue environ la moitié de la production terrestre (Field et al., 1998) et la base de la plupart des écosystèmes océaniques (Li, 1994; Worden et al., 2004; Jardillier et al., 2010). Le phytoplancton est donc essentiel pour les transferts trophiques, et joue également un rôle fondamental dans les cycles biogéochimiques de la planète (Worden et al., 2015). Par exemple, les diatomées sont responsables d’environ 40% de la production primaire océanique (Boyd and Newton, 1995) et jouent un rôle clé dans les cycles biogéochimiques, comme l’export de carbone (Boyd and Newton, 1999; Buesseler, 1998).
Dans le cadre de ce travail de thèse, nous nous intéressons aux Chlorophytae (Friedl and Rybalka, 2012; Leliaert et al., 2012; Lewis and McCourt, 2004), ou «algues vertes», qui regroupent 4 300 espèces dans le règne eucaryote de la lignée verte (archaeplastidae ou plantae). La photosynthèse est apparue dans la lignée verte avec la première endosymbiose par transfert du chloroplaste d’une cyanobactérie il y a environ 1.6 milliard d’années dans une cellule eucaryote (Yoon et al., 2004). Parmi les chlorophytes, il existe une importante diversité de forme de vie (De Clerck et al., 2012): des espèces unicellulaires, pluricellulaires, d’eaux douces, marines ou saumâtres, des espèces coloniales ou non, et des espèces symbiotiques.
36
6.
2. Présentation des espèces
1. Choix des modèles biologiques
L’objectif du travail de doctorat est d’acquérir une meilleure compréhension des processus évolutifs et adaptatifs du pico-phytoplancton eucaryote. Il faut souligner l’importance des progrès que de telles expérimentations permettent aujourd’hui dans les recherches menées par la communauté scientifique sur l’évolution. Cette thèse apporte une importante contribution à la littérature existante sur les expériences d’accumulation de mutations et permet d’évaluer le potentiel adaptatif d’un groupe écologique majeur.
Pour cela, nous avons choisi cinq espèces d’algues vertes (Figure 9):
Ostreococcus tauri RCC4221 (Blanc-Mathieu et al., 2014; Derelle et al., 2006), Ostreococcus mediterraneus RCC2590 (Subirana et al., 2013), Bathycoccus prasinos RCC1105 (Moreau et al., 2012), Micromonas pusilla RCC299 (Worden et
al., 2009) et Picochlorum sp. RCC4223. Toutes appartiennent à la classe des Mamiellophyceae (Marin and Melkonian, 2010), sauf le genre Picochlorum qui appartient à la classe des Trebouxiophyceae (Henley et al., 2004); voir l’arbre phylogénétique, Figure 8. Les fiches détaillées des souches sont disponibles en Annexes. Cinq raisons nous ont orienté vers ces choix:
Premièrement, la culture de toutes ces espèces est bien connue en laboratoire, dans du milieu L1 (voir la composition du L1 en annexe) à 20 °C, avec un cycle jour-nuit de 8h-16h. Les cultures sont clonales, mais pas axéniques, c’est-à-dire qu’elles contiennent des bactéries. La maîtrise de la culture est une étape essentielle pour la mise en place d’expériences et de protocoles avec ces espèces. Pour les expériences, toutes les souches proviennent de la Roscoff Culture Collection (http://roscoff-culture-collection.org/), une banque de microorganismes basée en France et disponible pour la recherche.
37 Deuxièmement, ces espèces sont devenues des modèles d’étude avec une importante bibliographie qui nous donne accès à différentes informations biologiques ou écologiques. C’est surtout le cas des Mamiellophyceae, avec quelques exemples relatés dans la littérature (Abby et al., 2014; Blanc-Mathieu et al., 2014, 2013; Demir-Hilton et al., 2011; Grimsley et al., 2010; Jancek et al., 2008; Palenik et al., 2007; Piganeau et al., 2011b; Rodríguez et al., 2005; Šlapeta et al., 2006; Sullivan et al., 2015).
Troisièmement, en lien avec l’argument précédent, le génome de ces espèces a été entièrement séquencé, ce qui est essentiel pour une étude du taux de mutation. Les génomes et données associées sont disponibles sur deux sites, ORCAE (Sterck et al., 2012) pour l’annotation et Picoplaza (Vandepoele et al., 2013) pour l’analyse comparative des génomes. Ce n’est cependant pas le cas pour
Picochlorum RCC4223, dont l’assemblage et l’annotation du génome font partie du
travail de thèse.
Quatrièmement, elles possèdent une large diversité génétique et génomique: un petit génome haploïde de 13 à 21 Mb (Tableau 4), avec des variations en GC qui vont de 46 à 63%. Ces différences génomiques nous intéressent précisément dans le cadre des EAMs pour tester les différentes hypothèses exposées en seconde partie de cette introduction.
Tableau 4. Diversité génomique des espèces utilisées pour les expériences d’accumulation de
mutations. La diversité génétique de nos modèles, notamment la composition en GC et la taille du génome, nous intéressent pour tester leurs rôles dans la variation du taux de mutation.
Espèces RCC Génome (Mb) %GC Gènes Génome codant (%)
Ostreococcus tauri 4221 12.5 56 8 116 81.21
Ostreococcus mediterraneus 2590 13.5 69 7 492 84.25
Bathycoccus prasinos 1105 15.1 48 7 847 83.09
Micromonas pusilla 299 21.0 63 10 286 81.85
38 Enfin, les algues vertes d’une manière générale font l’objet de recherches pour leur potentiel biotechnologique (Becker, 2007; Brennan and Owende, 2010; Chisti, 2007; Mata et al., 2010). La possibilité d’exploiter les lipides des algues, notamment pour la recherche de biocarburant (Brennan and Owende, 2010; Hannon et al., 2010), a poussé de nombreux chercheurs à optimiser les protocoles de production ou d’extraction des lipides chez certaines espèces, notamment chez les Trebouxiophyceae (Dassey and Theegala, 2013; Garzon-Sanabria et al., 2012; Gerken et al., 2013; S.-J. Park et al., 2012; Tran et al., 2014; Yang et al., 2014; Zhu and Dunford, 2013). Une étude récente a également mis en évidence une potentielle application médicale (Black et al., 2014) du genre Nannochloris. Brièvement, les
Nannochloris eukaryotum (ou Picochlorum eukaryotum) pénètrent spontanément
dans des cellules humaines de l’épithélium pigmentaire de la rétine. Les algues qui entrent sont viables et la photosynthèse est active, avec division cellulaire. Ces cellules de la rétine jouent un rôle crucial dans la formation du réseau vasculaire de la rétine en régulant l’expression de la production de facteurs de croissance vasculaire, qui est fonction de la concentration en dioxygène. Plusieurs pathologies oculaires sont liées à des problèmes de régulation de ces facteurs de croissance. La production de dioxygène via la photosynthèse par les Nannochloris entrées dans les cellules de la rétine semble donc, pour les auteurs, une piste à explorer.
La connaissance du taux de mutation est particulièrement importante ici, en raison de son utilité pour les recherches d’évolution expérimentale qui peuvent être utilisé pour sélectionner des lignées d’intérêts. C’est la raison pour laquelle un chapitre sera consacré à cette question, en se focalisant sur l’espèce Picochlorum RCC4223.
$*!
Figure 8.Arbre phylogénétique des Chlorophyta, repris de Marin et Melkonian (Marin and Melkonian,
2010), réalisé à partir des séquences qui codent l’ARNr 18S. Les Mamiellophyceae constituent un groupe basal ayant divergé de façon précoce alors que les Trebouxiophyceae sont plus dérivés.
Picochlorum RCC4223 Picochlorum oklahomensis Chlorella vulgaris Dunaliella salina Chlamydomonas reinhardtii Oltmannsiellopsis viridis Acrosiphonia sp. Tetraselmis striata Nephroselmis astigmatica Nephroselmis rotunda Micromonas pusilla Ostreococcus tauri Pyramimonas disomata Pyramimonas olivacea
Coleochaete nitellarum (groupe externe) Prasinoderma coloniale Picocystis salinarum Monomastix minuta Bathycoccus prasinos Crustomastix stigmata Ulvophycea Chlorophycea Trebouxiophycea Nephroselmidophycea Mamiellophycea Pyramimonadales Acrosiphonia