HAL Id: tel-01673785
https://tel.archives-ouvertes.fr/tel-01673785
Submitted on 1 Jan 2018
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Analyse du mécanisme et du rôle de l’inhibition de
l’autophagie par deux protéines complémentaires du
cytomégalovirus humain
Lina Mouna
To cite this version:
Lina Mouna. Analyse du mécanisme et du rôle de l’inhibition de l’autophagie par deux protéines complémentaires du cytomégalovirus humain. Virologie. Université Paris-Saclay, 2015. Français. �NNT : 2015SACLS153�. �tel-01673785�
NNT : 2015SACLS153
T
HESE DE DOCTORAT
DE
L’UNIVERSITE PARIS-SACLAY
Préparée à la faculté de pharmacie de l’université Paris-Sud
E
COLED
OCTORALEN°569
Innovation thérapeutique : du fondamental à l'appliqué
Spécialité de doctorat : Microbiologie
Par
Mme Lina MOUNA
Analyse du mécanisme et du rôle de l'inhibition de l'autophagie par deux
protéines complémentaires du cytomégalovirus humain
Thèse présentée et soutenue à Châtenay-Malabry, le 8 décembre 2015 :
Composition du Jury :
Mme IMBERT Berthe-Marie PU-PH / Université de Nantes - CHU de Nantes
Président/Rapporteur M MORIS Arnaud Directeur de recherche/CIMI
Paris
Rapporteur Mme BOTTI Joëlle MCF/Université Paris Diderot –
Institut Necker Enfants Malades
Examinatrice Mme BEAU Isabelle Chargée de Recherche /INSERM
UMR-S 1185
Directrice de thèse Mme ESCLATINE Audrey PU/Université Paris Sud - I2BC Co-directrice de thèse
1
Remerciements
Je tiens à remercier tout d’abord les membres du jury, particulièrement le professeur Berthe-Marie Imbert et le Dr Arnaud Moris qui ont accepté d’être rapporteurs de mon manuscrit et de participer au jury de ma thèse. Merci également au Dr Joëlle Botti d’avoir accepté d’assumer le rôle d’examinateur.
Je tiens à exprimer toute ma gratitude et mes remerciements les plus sincères à Audrey Esclatine qui m’a guidé dans mon travail et m’a aidé à trouver les solutions pour avancer dans la recherche, pendant le stage de Master 2 et tout au long de ma thèse. Egalement, je la remercie pour tes remarques, tes commentaires, tes critiques positives, ton suivi et pour ton enthousiasme pour la recherche. En fait, tes multiples explications ont apporté beaucoup à ma recherche.
J’aimerais profondément exprimer toute ma reconnaissance et mes sentiments les meilleurs à mon directeur de thèse Isabelle Beau. Je te remercie chaleureusement de m’avoir aidé et conseillé pendant ma thèse. Egalement, je te remercie pour ta disponibilité, tes connaissances et pour m’avoir fait connaître pas mal de techniques que je ne connaissais pas.
Merci beaucoup à vous les deux, vous étiez tout le temps à mon écoute pour mes problèmes professionnels et aussi personnels.
Je remercie tous les membres de l’ex UMR 984, en commençant par Marion, avec laquelle j’ai eu le plaisir d’essayer des méthodes innovantes pour que nos manipes marchent. Françoise pour son soutien et d’avoir consacré de temps tous les matins pour me demander si tout va bien. Marie-francoise pour les bons moments que nous avons passé surtout ce dernier temps. Merci David de m’avoir ‘protégé’ pendant l’écriture de ma thèse et de t’être gentil avec moi alors que ça te demandait beaucoup d’effort !. Matthieu, avec qui j’ai partagé le bureau et qui sait bien nous remettre à notre place. Idil, ma voisine de pays, merci pour les longues discussions et les bons moments que nous avons passé autour d'un café syrien-turque. Raymonde qui a amplifié une grande partie de mes constructions. Nadia, pour ces discussions pérennantes et sa volonté. Et récemment, une grande merci à Eva pour son aide pendant la révision de l’article surtout qu’elle a tous fait avec son gros ventre où le petit Adib se grandissait.
2
Un gros merci à Dorine. Grâce à toi Dodo j’ai pu obtenir le titre de Choupette 2013. Merci pour les nombreux bons moments passés ensemble autour des petits déjeuners le matin. Merci pour ton soutien, tes messages encouragants et ta présence dans ma vie comme une amie sur laquelle je peux compter toujours. Et bien sûr, Nicole, qui restait jusqu’au dernier jour de travail avant sa retraite un modèle pour tous pour son énergie et sa bonne humour. Nos réunions quotidiennes matinales dans le couloire avec Dodo et après David, étaient des moments uniques.
Merci aussi aux autres habitants de la tour E1, en commençant par les microbiologistes du 3ème. Une spéciale dédicace à Véronique qui était de mon coté surtout quand j’avais besoin de soutien. Nous avons partagé des moments difficiles sous la neige et la pluie et aussi des trajets plus tranquilles dans la voiture. Merci aussi à Jamil pour son aide moral et de ses discussions pérennantes à propos de notre cher pays la Syrie.
Un merci particulier avec une grande émotion à mon père et à ma famille en Syrie qui m’ont encouragés et soutenus tout au long de mes études. Merci, je vous aime gros comme la Terre. Une grande pensée à toi, maman, qui n’a pas pu voir l’aboutissement de mes études et qui aurait être fier de moi ce jour là. Que ton âme reste en paix………
Merci à la famille et aux cousins en France pour l‘amour que vous me portez.
Mes dernières pensées sont tournées vers ceux qui partagent ma vie. Mon cher Ahmad, pour ton soutien inconditionnel, ton encouragement pendant des longues soirées de travail et ton amour infini. Grâce à toi j’ai pu avancer. Mon trésor Taïm qui m’a accompagné dés ma première année de thèse.
Enfin, je voudrais remercier mon pays la Syrie qui’ m a financé tout au long de mes études supérieures en France afin d’obtenir le grade de docteur. Merci aussi à l’université Paris-Saclay, surtout la faculté de pharmacie à Châtenay-Malabry pour l’accueil.
3
Résumé
L’autophagie est un mécanisme constitutif et inductible de dégradation des composants cytoplasmiques afin de maintenir l’homéostasie cellulaire. Elle est souvent modulée par les virus car il s’agit également d’un mécanisme de défense antiviral. Elle peut avoir un rôle proviral quand elle est détournée et régulée par les virus. Nous avons précédemment observé au laboratoire que le cytomégalovirus humain (HCMV) stimule la formation des autophagosomes de manière précoce indépendamment de l’expression des protéines virales, puis qu’il entraine un blocage de l’autophagie aux temps tardifs. Dans ce travail, nous avons montré que ce virus a développé des stratégies impliquant la synthèse de deux protéines virales, IRS1 et TRS1, pour inhiber l’autophagie. De façon surprenante, nous avons également mis en évidence un rôle proviral de l’autophagie aux temps tardifs de l’infection par le HCMV. Nous avons pu montrer par des techniques de biochimie et d’imagerie cellulaire que l’expression aussi bien de TRS1 que d’IRS1 est capable de bloquer la formation des autophagosomes dans les cellules. Nous avons identifié le mécanisme d’action de ces protéines. Il est indépendant de la protéine kinase PKR mais nécessite une interaction avec Beclin 1, une protéine de la machinerie autophagique. Nous avons localisé le site d'interaction de Beclin 1 avec IRS1 et TRS1 (BBD pour Beclin 1 binding
domain) au niveau de leur région N-terminale. Ce domaine, conservé entre les deux protéines,
est nécessaire pour l’inhibition de l’autophagie. Le site d’interaction d’IRS1 a été identifié dans le domaine en superhélice (coiled-coil domain) CCD de Beclin 1.
Nous avons caractérisé le rôle de TRS1 et IRS1 dans la modulation de l’autophagie dans le contexte de l’infection virale, en utilisant différents virus mutants : des virus dans lesquels on a supprimé soit le gène IRS1, soit le gène TRS1 et un virus dans lequel il manque les deux gènes IRS1 et TRS1. Les résultats obtenus suggèrent qu’IRS1 et TRS1 sont effectivement toutes les deux impliquées dans ce processus. Afin de mieux comprendre le rôle de l’interaction de ces protéines avec Beclin 1, nous avons étudié le phénotype d’un virus mutant qui n’exprime pas IRS1 et qui contient une délétion de la région BBD de TRS1. Nous avons montré que ce virus mutant ne se lie pas à Beclin 1 et qu’il ne bloque pas l’autophagie. De manière surprenante, il n’a pas de défaut de production virale, suggérant que l’inhibition de l'autophagie ne serait pas essentielle pour la réplication virale. Nous avons développé d’autres approches, comme l’utilisation de modulateurs pharmacologiques de l’autophagie ou de lentivirus hébergeant des shRNA, qui montrent que l’inhibition de l’autophagie est capable de diminuer la production virale et au contraire que sa stimulation l’augmente. Ces derniers résultats suggèrent que l’autophagie pourrait être bénéfique au HCMV dans certaines conditions.
4
Abstract
Autophagy is a constitutive and inducible mechanism of degradation of cytoplasmic components, in order to maintain the cellular homeostasis. Autophagy is often modulated by viruses, because it is also considered as an antiviral defense mechanism. It can have a beneficial role, when it is hijacked and regulated by viruses.
We have previously observed in our laboratory that the human cytomegalovirus (HCMV) stimulates autophagosome formation, at the early stage of infection, independently of viral protein expression then, later on, it blocks autophagy. In this work, we showed that this virus has developed strategies involving the synthesis of several viral proteins, such as IRS1 and TRS1, to inhibit autophagy. Surprisingly, we also demonstrated a proviral role of autophagy at late stages of infection with HCMV. We showed, through biochemical and cellular imaging technologies, that expression of both TRS1 and IRS1 is able to block the formation of autophagosomes. We identified the mechanism of action of these proteins. It is independent of the protein kinase PKR but requires interaction with Beclin 1, a protein of the autophagic machinery. We mapped the interaction site of Beclin 1 with IRS1 and TRS1 in their N-terminal region and called it BBD for Beclin 1-binding domain. This domain (BBD) is conserved between the two proteins and essential to inhibit autophagy. We also identified the site of interaction of IRS1 in the coiled-coil domain (CCD) of Beclin 1.
We characterized the role of IRS1 and TRS1 in the modulation of autophagy, in the context of viral infection, using different mutant viruses: viruses in which either the IRS1 or the TRS1 gene has been removed and a mutant virus lacking both IRS1 and TRS1 genes. Our results suggest that both IRS1 and TRS1 are involved in the regulation of this process. To better understand the role of the interaction of these proteins with Beclin 1, we studied the phenotype of a mutant virus that does not express IRS1 and which contains a deletion of the N-terminal region of TRS1. We showed that this mutant does not bind to Beclin 1 and is not able to block autophagy. Surprisingly, it has no defects in viral production, suggesting that inhibition of autophagy is not essential for viral replication. We developed other approaches, including the use of pharmacological modulators of autophagy or shRNA knockdown, which show that the inhibition of autophagy is able to reduce viral production and, on the contrary, that its stimulation increases it. These results suggest that autophagy may be beneficial to HCMV in certain conditions.
5
TABLE DES MATIERES
Remerciements………....1
Résumé……….3
Abstract……….4
Abréviations ……….8
Liste des figures……….11
Préambule………..13
DONNEES BIBLIOGRAPHIQUES……….17
1. LE CYTOMEGALOVIRUS HUMAIN (HCMV)……….19
A. Généralités ………..………19
B. Caractéristiques générales des -Herpesvirinae………...19
C. Epidémiologie………..20
i. Transmission horizontale………20
ii. Transmission verticale………21
D. Structure du virus………21 i. Génome viral………22 ii. Capside……….23 iii. Tégument………..23 iv. Enveloppe……….24 E. Cycle de réplication ………24 F. Manifestations cliniques……….30
i. Infection chez les sujets immunocompétents………..30
ii. Infection au cours de la grossesse et maladie congénitale………..30
iii. Infections opportunistes chez les sujets immunodéprimés………..31
G. Traitements antiviraux………32
H. Echappement aux mécanismes de défense antivirale………..35
i. Inhibition de l’apoptose………...35
ii. Modulation de l’immunité innée……….36
iii. Modulation de l’immunité adaptative………37
iv. Les cellules NK……….39
I. Latence……….41
2. LES PROTEINES IRS1 ET TRS1 DU HCMV………..43
6
B. Structures……….43
C. Fonctions………..44
3. L’AUTOPHAGIE………...47
A. Généralités sur l’autophagie : historique et définition………47
B. Mécanismes moléculaires de l’autophagie……….50
i. Initiation et formation du phagophore………...51
ii. Elongation et fermeture de l’autophagosome……….55
iii. Maturation de l’autophagosome………57
C. Régulation de l’autophagie :………..58
i. Régulation de l’autophagie par la voie de signalisation mTOR………58
ii. Régulation de l’autophagie par des voies de signalisation indépendantes de mTOR………63
iii. Régulation de l’autophagie par des voies de signalisation impliquant des facteurs de transcription……….64
D. Fonctions biologiques de l’autophagie……….………65
i. Rôles physiologiques………..65
ii. Rôles pathologiques………69
4. AUTOPHAGIE ET VIRUS………...73
A. Modulation de l’autophagie par les virus……….73
i. Rôle antiviral de l’autophagie………...73
ii. Rôle pro viral de l’autophagie………...75
iii. VIH (Virus de l'immunodéficience humaine)………..80
B. Interaction entre les infections herpétiques et l’autophagie…………...81
i. Cytomégalovirus humain………..………...81
ii. Herpès Simplex Virus type 1………...83
iii. Virus de la Varicelle et du Zona………..………87
iv. Epstein-Barr virus………...…………..87
v. Herpesvirus associé au Sarcome de Kaposi………..…….89
PRESENTATION DES TRAVAUX……….91
Introduction……….93
7 Discussion……….…...134 RESULTATS COMPLEMNTAIRES ………137 CONCLUSIONS ET PERSPECTIVES ………...145 TRAVAUX ANNEXE………..155 REFERENCES BIBLIOGRAPHIQUES………..175
8
Abréviations
AC : assembly Complex ARNdb ; ARN double brin AMPK : AMP-activated kinase ATG : AuTophaGy-related genes
ATU : autorisation temporaire d’utilisation
AMBRA1 : Activating Molecule in Beclin 1 Regulating Autophagy AWOL : Autophagic exit WithOut Lysis
BIF 1 : Bax-Interacting Factor 1
Barkor : Beclin 1 interacting autophagy related key regulator CMA : Chaperone-Mediated-Autophagy
CDV : cidofovir
CMH : complexe majeur d’histocompatibilité CVB3 : le virus Coxsackie B3
DAPK : death-associated protein kinase DENV : virus de la dengue
DFCP1 : double FYVE domain-containing protein 1 DRAM : Damage-Related 79Autophagy Modulator
ESCRT : Endosomal Sorting Complex Required for Transport EBV : le virus d'Epstein-Barr
EBNA1 : Epstein-Barr nuclear antigen 1
eIF2 : αsubunit of the eukaryotic translational initiation factor 2 ERGIC : Endoplasmic Reticulum-Golgi intermediate compartment GCV : ganciclovir
GCN2 : General Control Non-derepressible-2 HSV-1 et 2 : le virus herpès simplex 1 et 2 HCMV : le cytomégalovirus humain
HAART : Highly active antiretroviral therapy HRI : Heme-Regulated Inhibitor
HPIV3 : le virus parainfluenza de type 3 IAV : le virus de la grippe A
IRGM : immunity-related GTPase M IE : Immediate Early
IC : cytokines inflammatoires IFN : interféron
JNK1 (c-Jun N-terminal protein kinase 1)
LC3 : Microtubule-associated protein 1A/1B-light chain 3 LAMP-2A : Lysosome-Associated Membrane Protein type 2A LIR-1 : leucocytes Ig-like 1
LMP1 : Latent Membrane Protein 1 LUNA : latency unique nuclear antigen MICB : MHC class I-related chain B MCMV : CMV murin
9 MAPK : Mitogen-activated protein kinase
MBV : Maribavir
mTOR : mammalian Target Of Rapamycin MCP : protéine majeure de capside
NEDA : nuclear envelope-derived autophagy NF-κB : nuclear factor-kappa B
NKG2D : Natural-killer group 2, member D NIEPs : particules enveloppés non-infectieuses
NOD2 : nucleotide-binding oligomerization domain-containing-2 NRBF2 : nuclear receptor binding factor 2
NS1 : Non-structural protein 1
OIS : oncogene-induced senescence
PAMPs : motifs moléculaires associés aux pathogènes PKR : protéine kinase dépendante de l’ARN double brin PI3P : phosphatidylinositol triphosphate
PI3K : phosphatidylinositol 3-kinase PP1 : phosphatase 1 alpha
PGHS : protéoglycanes héparanes sulfates p.i : post infection
PFA : foscarnet
PE : phosphatidyl-éthanolamine
PTEN : phosphatase and TENsin homolog deleted on chromosome ten PINK 1 : PTEN-induced putative kinase 1
PV : le poliovirus
Rag : Ras-related GTPases RE : réticulum endoplasmique
RTA : replication and transcription activator
RUBICON : Run domain cystein rich domain containing Beclin 1 Interacting protein SCP : petite protéine de capside
SNC : système nerveux central SNC : système nerveux central
STAT3 : signal tranducer and activator of transcription 3 STING : stimulator of interferon genes
SNAREs : Soluble N ethylmaleimide sensitive factor Attachment protein Receptor TAP : transporter associated with antigen processing
TLR2 : Toll like receptor 2 TGN : Trans-Golgi Network
TNFR1 : Tumor Necrosis Factor Receptor
TAP : Transporter associated with antigen processing TNF : tumor necrosis factor
TLR : Toll-like receptors
UPR : stress du réticulum endoplasmique Unfolded Protein Response UL : unique long
10 UVRAG : UV irradiation resistance associated gene Val-GCV : Valganciclovir
vFLIP : FLICE-like inhibitor protein VHB : virus de l’hépatite B
vICA : viral inhibitor of caspase-8-induced apoptosis vMIA : mitochondria-localized inhibitor of apoptosis Vps34,15 : Vacuolar protein sorting 15 et 34 Vps35 : Vacuolar protein sorting-35
11
Liste des figures
Figure 1 : Structure du cytomégalovirus humain………..22
Figure 2 : Représentation schématique du génome du HCMV………...22
Figure 3 : Cycle de réplication du HCMV………..25
Figure 4 : Mécanisme d’entrée du HCMV au niveau de la membrane cellulaire………27
Figure 5 : Physiopathologie de l’infection par le HCMV………...30
Figure 6 : Mode d’action des inhibiteurs de l’ADN polymérase virale UL54………33
Figure 7 : Antiviraux en développement contre le HCMV………...34
Figure 8 : Modulation de la présentation des antigènes via le CMH de classe I et II par le HCMV………...38
Figure 9 : Inhibition de l’expression du CMH de classe II par le HCMV………...39
Figure 10 : Modulation de la réponse des cellules NK par le HCMV………41
Figure 11 : Organisation et structure d’IRS1/TRS1……….43
Figure 12 : Voie de signalisation PKR et sa régulation par HCMV et HSV-1………..44
Figure 13 : Les différentes formes d’autophagie………..48
Figure 14 : Les différentes étapes de l’autophagie………..50
Figure 15 : Origine du phagophore ou la membrane d’isolation………51
Figure 16 : Formation de l’omégasome et de l’autophagosome………53
Figure 17 : Régulation du complexe ULK par mTOR………..54
Figure 18 : La structure de la protéine Beclin 1………....55
Figure 19 : Le système de conjugaison de l’ubiquitine et les deux systèmes de conjugaison autophagique………..56
Figure 20 : La protéine SNARE STX17 permet la fusion autophagosome-lysosome………57
Figure 21 : Régulation de l’autophagie par la voie mTOR………..59
Figure 22 : Différents complexes Beclin 1-Vps34 et leurs fonctions……….60
Figure 23 : Différents partenaires de Beclin 1………..62
Figure 24 : Rôles physiologiques et pathologiques de l'autophagie……….65
Figure 25 : Régulation de l’autophagie par les virus………77
Figure 26 : Modulation de l’autophagie par le HCMV au cours du cycle viral……….82
Figure 27 : Manipulation de l’autophagie par les herpesvirus………86
Figure 28 : La fixation du virus aux PGHS est nécessaire à la stimulation de l’autophagie…...139
Figure 29 : Impact des anticorps neutralisants anti-TLR2 sur l’autophagie induite par UV-HCMV………141
Figure 30 : Etude de l’autophagie dans les celles HEK293 infectées………142
Figure 31 : Implication du stress oxydatif dans l’induction précoce de l’autophagie………143
Figure 32 : Stimulation de l’autophagie aux temps précoces de l’infection………...149
Figure 33 : Expression des protéines IRS1/TRS1 au cours de l’infection……….151
13
15
L’autophagie est un processus catabolique, hautement conservée, servant à la dégradation des composants cytosoliques, des organites endommagés, des agrégats protéiques et des bactéries intracellulaires, à travers une voie dépendante du lysosome. Elle peut être induite en réponse à des conditions de stress. En outre, l'autophagie a été décrite comme impliquée dans les réponses immunitaires innées et adaptatives. Plusieurs études ont montré que certains micro-organismes peuvent être éliminés par la voie autophagique dans un processus connu sous le nom de xénophagie. L’autophagie participe également à la production de peptides antigéniques microbiens ou endogènes via les lysosomes et favorise la présentation des antigènes par les CMH de classe I et II. Toutefois, plusieurs agents pathogènes, notamment les virus, ont développé différentes stratégies pour éviter ou exploiter l’autophagie afin d’assurer leur survie et l’autophagie joue dans ce dernier cas un rôle proviral.
Le cytomégalovirus humain (HCMV) est un virus pathogène opportuniste qui, comme les autres membres de la famille des Herpesviridae, a la capacité de persister chez l'hôte dans un état latent, après la primo-infection. L’infection par le HCMV des patients immunodéprimés entraîne une morbidité et une mortalité importantes sans traitement, en particulier chez les receveurs de greffe, alors que l'infection chez les sujets immunocompétents est généralement asymptomatique ou paucisymptomatique.
Il avait été montré au laboratoire que le HCMV module l’autophagie dans les fibroblastes humains de deux manières opposées. Quelques heures après l’infection, le HCMV stimule l’autophagie indépendamment de la synthèse des protéines virales. Dans un second temps l’autophagie est inhibée sous la dépendance de protéines virales néosynthétisées. Les objectifs de cette thèse ont été de mieux caractériser les mécanismes moléculaires développés par le virus pour inhiber l’autophagie et d’étudier l’impact de l’autophagie sur le HCMV.
Lors de l’infection par le HCMV, plusieurs réponses cellulaires ont été mises en place pour contrôler l’infection. Cependant, le HCMV a développé des mécanismes pour contrecarrer ces défenses cellulaires en inhibant l’apoptose, en limitant la présentation aux lymphocytes T des antigènes via le CMH de classe I et II ou encore en supprimant la reconnaissance des cellules infectées par les cellules NK. Parmi les mécanismes antiviraux mis en place par la cellule, on trouve également l’arrêt de la synthèse protéique par activation de la voie PKR (protéine kinase dépendante de l’ARN double brin) suite à la production d’ARN double brin (ARNdb) et d’interféron (IFN), qui aboutit à l’inhibition de la réplication virale. Cependant, ce virus code plusieurs protéines virales qui lui permettent d’échapper à tous ces mécanismes.
16
Parmi ces protéines, il a été mis en évidence que les protéines IRS1 et TRS1 bloquent directement PKR et empêchent l'arrêt de la synthèse protéique. Un virus mutant HCMV n’exprimant ni TRS1 ni IRS1 ne se réplique pas car la voie PKR est toujours activée. Il est intéressant de noter que l’activation de PKR a également été décrite comme impliquée dans la stimulation de l’autophagie. Nous avons donc exploré les fonctions d’IRS1 et de TRS1 comme régulateurs de l’autophagie
Dans ce manuscrit, je présenterai tout d’abord des données bibliographiques sur le HCMV et ses protéines virales IRS1 et TRS1. Je décrirai ensuite le processus autophagique et sa régulation. Enfin, l’interaction entre l’autophagie et les virus, notamment les herpesvirus, sera présentée dans la quatrième partie du manuscrit. Après ces parties bibliographiques, mes travaux de thèse seront exposés sous forme d’un article actuellement en révision favorable. Deux autres articles dans lesquels j’ai participé pendant ma thèse, ont été ajoutés en annexe. Pour conclure, j’exposerai dans la dernière partie les conclusions de ce travail et les perspectives qu’il ouvre.
17
Données
19
1. LE CYTOMEGALOVIRUS HUMAIN (HCMV)
A. Généralités
Le HCMV est un virus ubiquitaire qui possède une distribution mondiale. Il infecte une grande majorité de la population et est responsable de maladies opportunistes. Il appartient à la famille des Herpesviridae.
Les Herpesvirus qui infectent l’homme sont divisés en trois groupes :
Alphaherpesvirinae : trois virus humains appartiennent à cette sous-famille. Lesvirus herpès simplex HSV-1 et 2 (HHV-1 et HHV-2), le virus de la varicelle et du zona (VZV ou HHV-3).
Betaherpesvirinae : cette sous-famille contient trois virus infectant l’homme : Lecytomégalovirus humain (HCMV ou HHV-5), et les herpesvirus humains 6 et 7 (HHV-6 et HHV-7). Ils ont un long cycle réplicatif et une gamme d'hôtes restreinte.
Gammaherpesvirinae : les deux virus appartenant à cette sous-famille sontcapables d’établir une latence dans les lymphocytes et sont responsables de cancers comme le virus d'Epstein-Barr (EBV ou HHV-4) et l'herpesvirus humain 8 associé au Sarcome de Kaposi (KSHV ou HHV-8).
Alors que l’infection par le HCMV chez les hôtes immunocompétents est le plus souvent cliniquement silencieuse et sans gravité, il peut provoquer de graves infections chez certains individus, comme les personnes immunodéprimées. Le HCMV est aussi la première cause d’infections congénitales. Des infections primaires (primo-infections) et secondaires (réactivations et récurrences) sont possibles durant la grossesse et peuvent entrainer des complications chez les fœtus et les nouveau-nés.
Ce virus, comme tous les herpesvirus, a la possibilité de persister dans l’organisme de l’hôte à vie. L’immunité est acquise mais elle n’est pas complètement protectrice. Après la primo-infection, le HCMV n’est jamais éliminé et reste en latence à vie dans son hôte. De ce fait, le virus est sporadiquement excrété, ce qui constitue une source récurrente importante de transmission (Mocarski, Shenk et al. 2013). Le HCMV a une haute spécificité d’espèce et l’homme est son unique réservoir.
B. Caractéristiques générales
Il existe plusieurs cytomégalovirus qui infectent différents mammifères. Les cytomégalovirus isolés chez une espèce de mammifères sont plus facilement cultivés dans des fibroblastes de la même espèce. Lors de l'infection naturelle, le HCMV infecte les cellules épithéliales, les cellules
20
myéloïdes (monocytes, macrophages et cellules dendritiques), les fibroblastes et les cellules endothéliales. Les cellules infectées par le HCMV présentent un effet cytopathique caractéristique avec des inclusions nucléaires et cytoplasmiques. Les inclusions cytoplasmiques sont associées à un compartiment distinct du complexe d'assemblage viral (Assembly Complex ou AC) (Das, Vasanji et al. 2007). Les fibroblastes humains (par exemple, les cellules MRC5) sont couramment utilisés pour l'isolement et la propagation du HCMV. L’utilisation de ces cellules permissives a été essentielle pour comprendre les fonctions des gènes viraux et comment ils contrôlent les différentes étapes de la réplication. La latence du HCMV s’établit naturellement dans les cellules hématopoïétiques dérivées de la moelle osseuse, où l'ADN viral est présent à très faible nombre de copies (Bego and St Jeor 2006).
C. Epidémiologie : prévalence et transmission
L’infection par le HCMV est ubiquitaire, endémique et survient tout au long de l’année sans variations saisonnières. Des épidémies n’ont pas été décrites. Les taux de séropositivité dans la population varient beaucoup selon des facteurs géographiques, ethniques et les conditions socioéconomiques. La plupart des études épidémiologiques ont été faites sur les femmes en âge de procréer, à cause de l’importance en Santé Publique des infections congénitales au cours de la grossesse. Au niveau mondial, le taux de prévalence est plus élevé chez les femmes que chez les hommes et il augmente avec l’âge. La prévalence globale de l'infection est aux alentours de 50% dans les pays développés alors qu’elle arrive à 99% dans les pays en voie de développement. Aux Etats-Unis et en Europe, la prévalence est plus grande dans la population des couches socio-économiques basses (Mocarski, Shenk et al. 2013).
i. Transmission horizontale
Le HCMV est un virus enveloppé et de ce fait, fragile. La transmission du virus ne se produit pas par voie respiratoire et elle demande un contact direct avec les sécrétions corporelles infectées comme l’urine, le sperme, le lait, la salive, les secrétions cervicales ou les larmes. La transmission est plus élevée à la suite de contacts étroits comme lors de rapport sexuels, entre enfants ou au contact d’enfants. L’excrétion virale persiste des années. Suite à l'acquisition initiale du HCMV, le virus se réplique et provoque une infection systémique ou virémie et diffuse pour infecter les cellules épithéliales tubulaires des organes de sécrétion telle que les glandes salivaires et les reins, qui secrètent les virus produits (Mocarski, Shenk et al. 2013). La transmission horizontale se produit également par transfusion ou par greffe de moelle et surtout par la transplantation d'organes de donneurs séropositifs asymptomatiques. Le risque de transmission par transfusion sanguine est quasiment nul maintenant grâce à l’utilisation de
21
produits sanguins testés séronégatifs ou de sang filtré (déleucocyté) (Bowden, Slichter et al. 1995; Allain, Stramer et al. 2009).
ii. Transmission verticale
Le HCMV est la première cause d’infection congénitale avec un mode de transmission in utero à travers le placenta qui entraine l'infection du fœtus pendant la grossesse. Le HCMV se propage de trois manières : par voie transplacentaire, au cours de l’accouchement (intrapartum) et par le lait maternel. L’infection par voie transplacentaire peut provoquer une maladie congénitale. La transmission in utero peut se produire soit au cours d’une primo-infection soit au cours d’une infection récurrente (Stagno and Britt 2006). Les infections récurrentes sont dues, dans la plupart des cas, à la réinfection par une nouvelle souche ou rarement à la réactivation du virus endogène (Boppana, Rivera et al. 2001; Kenneson and Cannon 2007).
Le taux de transmission est constant tout au long de la grossesse, il ne dépend pas de l'âge gestationnel et il est accompagné d’un grand risque de développer des séquelles si la primo-infection survient pendant le premier trimestre de la grossesse (Stagno, Pass et al. 1986; Adler and Marshall 2007; De Paschale, Agrappi et al. 2009). En cas de réactivation ou de réinfection par le HCMV, les anticorps maternels ne préviennent pas complètement la transmission virale au fœtus, ce qui entraine une infection congénitale mais en général moins grave.
La transmission au moment de l’accouchement et par le lait maternel ne provoque pas de malformations, mais l’infection peut être symptomatique, notamment chez les nouveau-nés prématurés de très faible poids à la naissance (Mocarski, Shenk et al. 2013). Les conséquences de l'infection fœtale par le HCMV et la fréquence de cette infection en font l’infection congénitale la plus grave en France.
D. Structure du virus
Le HCMV a la structure caractéristique de tous les Herpesvirus : un génome sous forme d’ADN bicaténaire linéaire se trouve à l'intérieur d’une capside icosaédrique hautement stable, composée de 162 capsomères. La nucléocapside est entourée par une enveloppe dérivée des membranes de la cellule hôte et contenant des glycoprotéines virales qui permettent l’attachement et l’entrée du virus dans les cellules. Les Herpesvirus ont également un tégument. C’est une couche bien épaisse entre l'enveloppe et la capside, constituée de phosphoprotéines (Figure 1). Le virion du HCMV est le plus grand et le plus complexe structurellement de tous les herpesvirus, avec un diamètre de 200 à 300 nm (Mocarski and Courcelle 2001)
.
22 Figure 1 : Structure du cytomégalovirus humain.
Le HCMV est un virus à ADN double brin linéaire avec une capside icosaédrique. La capside est entourée d'un tégument, lui-même entourée de l'enveloppe virale.
i. Génome viral
Le HCMV a un génome linéaire de 236 kb qui contiendrait 751 cadres de lecture (Open Reading
Frames ORF) (Stern-Ginossar, Weisburd et al. 2012). Il est constitué de deux segments : L
(long) et S (short) (Figure 2). Chacun de ces segments comporte une partie unique long UL et
une partie unique short Us. Ces dernières sont encadrées par deux régions répétées terminales
(TRL et TRS) et internes (IRL et IRS). Au cours de la réplication, quatre formes isomères de cette
molécule d’ADN apparaissent, par changement d’orientation relative des deux segments UL et
US (Davison, Dolan et al. 2003) (Figure 2). Ce processus d’isomérisation n’est pas indispensable
pour la réplication (Sauer, Wang et al. 2010). Le génome viral contient une origine de réplication d'ADN (ori Lyt) située entre les gènes UL57 et UL69, au milieu de la région UL. Cette position est
conservée chez tous les - herpesvirinae. L’Ori Lyt est nécessaire pour la réplication de l'ADN
viral (Anders, Kacica et al. 1992).
Figure 2 : Représentation schématique du génome du HCMV.
UL (Unique Long), US (Unique Short), TRL (Terminal Repeat Long), TRS (Terminal Repeat Short), IRL (Internal Repeat Long), IRS (Internal Repeat Short). A, B, C et D correspondent aux quatre isomères.
23 ii. Capside
La capside d’HCMV est une capside icosaédrique composée de 162 capsomères (150 hexons et 12 pentons). Elle est formée de quatre protéines : la protéine majeure de capside (MCP ou UL86) composant les hexons et la plupart des pentons, la protéine mineure de capside (mCP ou UL46), la protéine mineure de fixation de la capside (mC-BP ou UL85) et la petite protéine de capside (SCP ou UL48A) (Mocarski, Shenk et al. 2013). Les protéines MCP et mCP sont les éléments les plus abondants de la capside. Contrairement à HSV-1, où SCP n’est pas indispensable, toutes les protéines de capside du HCMV sont essentielles pour la réplication virale (Britt 2007). De nombreuses protéines interviennent dans l‘assemblage de la capside et l‘encapsidation de l’ADN viral néo synthétisé. La protéine portale PORT ou UL104 constitue un pore au niveau de la capside permettant l’encapsidation du génome et la libération de l'ADN viral en se liant avec les protéines d'encapsidation ou terminases codées par UL89 et UL56 (Dittmer and Bogner 2005).
iii. Tégument
Le tégument est une couche épaisse de protéines virales qui se trouve entre la capside et l’enveloppe. Il est composé d'au moins 32 protéines virales, dont beaucoup sont phosphorylées. Ces protéines ont diverses activités, du conditionnement de la cellule hôte au début de l'infection à l'orchestration de la phase finale de l'assemblage des virions (Kalejta 2008).
Au début de l'infection, des protéines comme UL47 et UL48 ont probablement un rôle dans le transport des particules virales par les microtubules, l‘ancrage au niveau du pore nucléaire de la nucléocapside, ainsi que dans la décapsidation et l‘entrée de l’ADN viral dans le noyau (Bechtel and Shenk 2002) (Yu, Silva et al. 2003).
La protéine UL83 ou pp65 est la protéine de tégument la plus abondante et le constituant principal des corps denses. Les corps denses sont des particules non infectieuses qui sont formés d’une enveloppe virale et de protéines de tégument alors qu’ils ne contiennent ni capside, ni génome. UL83 est hautement immunogène et sa détection dans les polynucléaires circulants est utilisée dans un test de diagnostic (antigénémie pp65). Après l’entrée du virus, pp65 se localise dans le noyau des cellules infectées, où elle joue un rôle d’immunomodulateur, qui freine la réponse cellulaire à l’interféron (IFN) (Kalejta 2008). Bien qu’elle soit abondante et ait un rôle potentiellement important lors de l'infection naturelle, pp65 n’est pas indispensable pour la réplication dans les cellules en culture (Mocarski, Shenk et al. 2013). La protéine pp71 (UL82) est un homologue d’UL83 qui se localise aussi dans le noyau après l'entrée du virus. Elle recrute la machinerie cellulaire de transcription pour activer la transcription des gènes très précoces (Petrik, Schmitt et al. 2007) via la dégradation ou la relocalisation des répresseurs de
24
la transcription, tels que la protéine cellulaire Daxx (Penkert and Kalejta 2012). La protéine du tégument pp150 reste liée à la capside (Yu, Shah et al.) et joue un rôle essentiel dans la stabilité des nucléocapsides matures pendant la translocation du noyau vers le cytoplasme puis leur transport dans le compartiment d’assemblage (Tandon and Mocarski 2008).
La protéine UL48, également liée à la capside (Yu, Shah et al.), semble stabiliser les nucléocapsides. Quant à la protéine kinase ppUL97 (ou VPK ou UL97), elle semble jouer un rôle central dans l’infection en agissant à différents niveaux, dans les étapes précoces de l'infection et également dans la maturation et la sortie de la capside du noyau ou «nuclear egress» (Prichard 2009). Certaines protéines du tégument, comme ppUL99 ou pp28, facilitent l’assemblage et la sortie des virus de la cellule.
La famille US22 code pour les protéines de tégument UL23, UL24, UL25, UL26, UL29-28, UL36, UL43, IRS1, US22, US23, US24, US26 et TRS1. Ces protéines seraient impliquées dans les premières étapes qui suivent la pénétration du virus (Adair, Douglas et al. 2002).
iv. Enveloppe
Les virions, les corps denses et les NIEPs (particules enveloppés non-infectieuses) sont entourés d’une bicouche lipidique dérivée du compartiment ERGIC (Endoplasmic
Reticulum-Golgi intermediate compartment) (Britt 2007; Tandon and Mocarski 2012). Plus de 20 différentes
glycoprotéines virales sont insérées dans cette enveloppe. Les plus connues sont les glycoprotéines B, H, M/N, L, O et 48 (respectivement notées gB, gH, gM/N, gL, gO et gp48). Elles sont relativement bien conservées chez les herpesvirus. Seulement 5 de cette vingtaine de glycoprotéines sont essentielles pour la réplication virale in vitro. Il s’agit de gB (UL55), de gM/gN (UL100/UL73) et de gH/gL (UL75/UL115). L’enveloppe confère au virion une sensibilité particulière aux solvants des lipides, au pH bas et à la chaleur.
E. Cycle de réplication
La pathogenèse du HCMV implique une réplication productive dans une grande variété de types cellulaires (Revello and Gerna 2010), tels que les cellules épithéliales, endothéliales, neuronales, myéloïdes et fibroblastiques (Sinzger, Digel et al. 2008). Cependant, la plupart des études in vitro effectuées pour comprendre les fonctions des gènes lors de la réplication virale ont été faites dans des fibroblastes. La réplication virale dans des cellules autres que les fibroblastes nécessite l'utilisation de souches virales spécifiques qui conservent les caractéristiques des isolats cliniques. Généralement, les isolats cliniques sont plus difficiles à cultiver que les souches de laboratoire, qui ont été adaptées dans les fibroblastes. Le cycle de réplication du HCMV est long dans les fibroblastes et demande de 48h à 72h pour arriver à
25
l’étape finale de la maturation et la libération du virus. Après l’entrée du virus, trois classes de gènes, très précoces (IE ou ), précoces (DE ou ) et tardives (L ou ) sont exprimées séquentiellement de manière coordonnée au cours du cycle viral.
Le cycle de réplication comprend plusieurs étapes (Figure 3) : i. L’attachement et l’entrée du virus dans la cellule ii. La réplication du génome
iii. L’assemblage des particules virales et la sortie des virions
Figure 3 : Cycle de réplication du HCMV.
1-Après la fixation sur les héparanes sulfates à la membrane plasmique, le virus interagit avec des récepteurs spécifiques, ce qui entraine la fusion de l’enveloppe virale avec la membrane et la libération de la nucléocapside et les protéines de tégument dans le cytoplasme. 2- La nucléocapside migre le long des microtubules vers le noyau où l’ADN virale est libéré. 3- La synthèse séquentielle des protéines très précoces et précoces. 4- La réplication virale puis synthèse des protéines tardives et formation de nouvelles nucléocapsides. 5- Les nucléocapsides acquerront leur enveloppe au niveau de la membrane nucléaire ou du TGN (Trans-Golgi Network). 6-Les virions sont libérés par exocytose au niveau de la membrane plasmique.
i. L’attachement et l’entrée du virus sur la cellule En général, l’entrée du virus se produit en plusieurs étapes : 1) La liaison à des récepteurs spécifiques à la surface cellulaire.
26
2) La fusion de l'enveloppe virale avec les membranes cellulaires pour libérer la nucléocapside dans le cytoplasme, soit directement au niveau des membranes plasmiques (dans les fibroblastes) ou après endocytose (dans les cellules endothéliales et épithéliales).
3) La translocation de la nucléocapside vers le noyau grâce au cytosquelette. La nucléocapside va ensuite interagir avec les pores nucléaires, ce qui permet la libération du génome dans le noyau. Simultanément mais indépendamment de la translocation de la nucléocapside vers le noyau, les protéines du tégument sont libérées dans le cytosol et elles assurent différentes fonctions, comme la modulation de la réponse innée de l'hôte et l'orchestration de la transcription des gènes très précoces IE (Immediate Early). La plupart des cellules semblerait être permissives à l‘infection par le HCMV, étant donné que l‘on peut détecter le virus dans la quasi-totalité des organes d‘un individu infecté.
Le HCMV interagit avec plusieurs récepteurs de manière successive avant de pénétrer dans la cellule (Compton 1995). Le contact initial du virion avec les cellules fait intervenir des protéoglycanes héparanes sulfates (PGHS) (Figure 4) (Compton, Nowlin et al. 1993), un phénomène relativement conservé dans la voie d'entrée des herpesvirus. Le HCMV complète l’attachement ainsi que l'étape de la fusion à l’aide des glycoprotéines gB et gH/gL (Lopper and Compton 2004). D'autre complexes glycoprotéiques sont impliqués dans l’entrée du virus, comme le trimère gB/gH/gO et le pentamère composé de gH/gL/UL128/UL130/UL131A, qui facilite l'entrée dans les cellules épithéliales et les cellules endothéliales (Ryckman, Chase et al. 2008). Dans les fibroblastes, le HCMV pénètre par fusion directe avec la membrane plasmique, de manière indépendante du pH (Compton, Nepomuceno et al. 1992). Cependant, il pénètre dans les cellules endothéliales et les cellules épithéliales par un mécanisme d’endocytose dépendant du pH (Bodaghi, Slobbe-van Drunen et al. 1999; Ryckman, Rainish et al. 2008). L'entrée du HCMV ne se produit pas au hasard sur la membrane plasmiques mais spécifiquement au niveau des radeaux lipidiques (Wang, Huang et al. 2005).
Les récepteurs cellulaires impliqués dans l’entrée du HCMV ne sont pas encore parfaitement connus. A cet égard, la 2-microglobuline, l’annexine II et l’aminopeptidase neutre (CD13), initialement identifiés comme étant des récepteurs du HCMV, ne sont en fait pas ou peu impliqués dans l'entrée virale (Pietropaolo and Compton 1999). Il a été rapporté que le HCMV est capable d’infecter les fibroblastes en se liant à l’EGFR (epidermal growth factor receptor) et que cette interaction se fait par l’intermédiaire de gB (Wang, Huong et al. 2003). Plus récemment, il a été montré que le HCMV se lie à l’EGFR exprimé à la surface des cellules endothéliales, induisant ainsi une cascade de signalisation impliquant la phosphatidylinositol
3-27
kinase (PI3K) et des MAPK (Bentz and Yurochko 2008). En 2009, son rôle en tant que récepteur du HCMV a été également étendu aux monocytes (Chan, Nogalski et al. 2009). Cependant, il reste controversé car le groupe de T. Compton a montré que l’EGFR n’est pas impliqué dans l’entrée du HCMV dans les fibroblastes et dans une lignée de cellules endothéliales (Isaacson, Feire et al. 2007). Son rôle pourrait ainsi dépendre du type cellulaire.
Les intégrines cellulaires 2β1, 61 et αVβ3 fonctionneraient aussi comme récepteurs du HCMV, via un domaine hautement conservé de type « désintégrin-like » (Feire, Koss et al. 2004). L’intégrine αVβ3 pourrait également être un co-récepteur du HCMV et se lier à l’EGFR de façon synergique (Wang, Huang et al. 2005). Les intégrines pourraient donc soit interagir seules avec le HCMV comme un récepteur primaire, soit jouer un rôle de co-récepteur et interagir avec un autre récepteur (Isaacson, Feire et al. 2007). Il a été montré que l’interaction entre gB et la β3 intégrine après l’attachement du virus à la surface de la cellule est un processus déterminant de la réponse interféron (IFN) et que le blocage de cette interaction en utilisant une gB incapable d’interagir avec la β3 intégrine, diminue le signal IFN, mais pas l’activation des cytokines inflammatoires (Juckem, Boehme et al. 2008). Il a été rapporté plus récemment que le PDGFR (platelet-derived growth factor-α receptor) pourrait jouer le rôle de récepteur pour le HCMV (Soroceanu, Akhavan et al. 2008).
En revanche, il semble que le TLR2 (Toll like receptor 2) ne soit pas impliqué dans l’entrée du HCMV (Juckem, Boehme et al. 2008) mais son interaction avec gB induit la sécrétion de cytokines inflammatoires (Compton, Kurt-Jones et al. 2003).
Figure 4 : Mécanisme d’entrée du HCMV au niveau de la membrane cellulaire
Le virus se fixe initialement sur la cellule grâce aux héparanes sulfates via la glycoprotéine d’enveloppe gB, ce qui facilite la fixation à des récepteurs plus spécifiques. Le PDGFR semblerait être le récepteur du HCMV le plus probable, permettant l’entrée du virus dans la cellule. L’intégrine αvβ3 joue le rôle de corécepteur. Les TLR ne sont pas impliqués dans l’entrée mais leur activation entraine la production des
28
cytokines inflammatoires. D’autres récepteurs cellulaires avaient été proposés comme le CMH de classe I, l’annexine II, mais leur rôle en tant que récepteurs est remis en cause. Le rôle d’EGFR en tant que récepteur est controversé et dépend du type cellulaire.
ii. La réplication du génome
La transcription et la traduction du génome viral du CMV se déroulent en trois étapes séquentielles et coordonnées. La première ou la phase très précoce (Immediate Early, IE) permet de détourner le métabolisme cellulaire au profit de la réplication virale, d’inhiber la réplication de l’ADN cellulaire et de déclencher la phase précoce. Les gènes très précoces sont soit exprimés pour initier une cascade d’expression des gènes pour une infection lytique, soit réprimés pour établir la latence. L'initiation de la transcription des gènes IE débute en l’absence de l’expression de novo de protéines virales. La transcription des gènes IE1 et IE2 (immediate
early protein 1 et 2), qui sont les gènes les plus abondants parmi les gènes très précoces, est
sous le contrôle du promoteur MIEP (Major Immediate Early Promoter). Grâce à des sites d’interaction avec le NF-κB (nuclear factor-kappa B) dans le MIEP, une signalisation pro-inflammatoire induite par l'entrée du virus est effectivement exploitée pour activer l’expression des gènes IE1/IE2 (Caposio, Luganini et al. 2007; Isaacson, Juckem et al. 2008; Isern, Gustems et al. 2011). Le MIEP est activé par des protéines cellulaires et virales, comme la protéine du tégument pp71 (pUL82) qui entre avec la particule virale. En l’absence de pp71, un blocage général de la transcription des gènes IE peut se produire (Bresnahan and Shenk 2000) et contribue à la mise en place de la latence virale (Saffert and Kalejta 2007).
A la suite du pic d’expression des protéines très précoces régulatrices et avant la réplication du génome, la phase précoce débute. Les protéines précoces comprennent les enzymes et protéines nécessaires à la synthèse de l’ADN viral. Ces gènes ont plusieurs fonctions dans la maturation et la sortie des virions. (Browne and Shenk 2003; Abate, Watanabe et al. 2004). La réplication de l‘ADN viral dépend de plusieurs protéines, notamment de l‘ADN polymérase virale UL54 et de sa protéine accessoire UL44. L'expression des gènes tardifs est maximale après la réplication de l'ADN viral. Les gènes tardifs codent principalement pour des protéines structurales et contrôlent la maturation de la capside, l'assemblage de l’ADN, la maturation du virion et des corps denses et finalement la sortie du virus de la cellule. La synthèse de l'ADN a lieu dans le noyau des fibroblastes infectés, en commençant entre 14 et 16h pi, et peut atteindre plus de 10000 copies par cellule au moment où les virions commencent à se former (Mocarski, Shenk et al. 2013).
29
iii. L’assemblage des particules virales et la sortie des virions
Les caractéristiques de base de la formation de la capside et la maturation sont communes chez les herpesvirus. Le processus commence lorsque cinq composants de la capside conservés chez les herpesvirus, (MCP, TRI1, TRI2, SCP- UL48A -, et PORT UL104) forment une coquille de procapside autour d'un précurseur de la protéine d'assemblage (AP ou UL80.5) qui est responsable à la fois de la translocation de MCP vers le noyau et de l’initiation de l’assemblage de la capside (Gibson 2008). Les protéines du tégument sont ajoutées aux nucléocapsides séquentiellement dans le noyau et cet assemblage se poursuit dans le cytoplasme. Ceci assure la stabilité de la nucléocapside au cours de sa translocation vers le cytoplasme et contrôle son trafic vers des sites d'enveloppement dans le cytoplasme. Sept structures virales différentes sont libérées par les cellules infectées. Parmi elles, on trouve des virions complets, des NIEPs et des corps denses (Topilko and Michelson 1994).
L’ADN viral produit a une forme de concatémères qui sont de longues molécules d'ADN constituées d'un même monomère répété séparé par des séquences pac (Cis-acting packaging
element). Ensuite, une étape d’encapsidation se déroule où les concatémères seront clivés et
empaquetés dans les capsides. Ce processus fait intervenir des protéines d'encapsidation ou terminases (UL51, UL52, UL56, UL77, UL89 et UL104 ou PORT).
Un complexe terminase, comprenant les protéines UL89 et UL56, s‘associe à un pentamère de la protéine portale UL104, qui forme un pore au niveau de la capside permettant l‘introduction de l‘extrémité libre du concatémère d‘ADN viral néoformé. La protéine UL56 se fixe sur les séquences pac, ce qui active le premier clivage de l‘ADN et la fixation du complexe UL56/ADN à UL89. Puis le complexe UL56/UL89/ADN se fixe à UL104, permettant le transfert de l‘ADN dans la capside (Mocarski, Shenk et al. 2013).
Pour expliquer la sortie dans le milieu extracellulaire du virion, deux hypothèses co-existent. L’hypothèse la plus ancienne est qu’une fois dans le réticulum endoplasmique (RE), le virion conserverait son enveloppe acquise après bourgeonnement au niveau de la membrane interne nucléaire et qu’il se dirigerait vers l’extérieur de la cellule, via l’appareil de Golgi et le TGN. La deuxième hypothèse, la plus acceptée aujourd’hui, est que les capsides acquièrent une première enveloppe transitoire en traversant la membrane nucléaire interne. La fusion de cette enveloppe virale avec la membrane nucléaire externe a pour résultat le désenveloppement de la nucléocapside et sa translocation dans le cytoplasme. La majorité des protéines du tégument sont alors ajoutées aux nucléocapsides, qui obtiennent leur enveloppe finale par bourgeonnement dans des vésicules dérivées de l’appareil de Golgi
(Mettenleiter, Klupp et al.
30
2006)
. Quand les virions sont formés, ils sont ensuite transportés vers la surface cellulaire dans des petites vésicules à l'aide de la machinerie cellulaire d'exocytose.F. Manifestations cliniques
Au cours de l’infection par le HCMV, la physiopathologie dépend du stade de l’infection ; la primo-infection, la réinfection et la réactivation (Figure 5).
Figure 5 : Physiopathologie de l’infection par le HCMV (Alain, Hantz et al. 2008).
i. Infection chez les adultes immunocompétents
En règle générale, la primo infection par le HCMV chez les sujets immunocompétents est cliniquement asymptomatique. L’infection symptomatique donne parfois de la fièvre ou un syndrome mononucléosique, qui est caractérisé par une fièvre élevée (entre 39 et 40°C) pendant plus de 10 jours, des malaises, des myalgies, des céphalées et de la fatigue. Le syndrome mononucléosique peut se produire également chez les enfants. Les enfants sont moins susceptibles d'être fébriles mais plus susceptibles d'avoir une hépatomégalie ou une splénomégalie (Pannuti, Vilas Boas et al. 1985). La primo-infection peut cependant aboutir à des manifestations cliniques diverses mais peu fréquentes telles que des arthralgies et arthrites, colites ulcératives, pneumopathies, méningites aseptiques et myocardites.
ii. Infection au cours de la grossesse et maladie congénitale
L’infection congénitale par le HCMV est la plus fréquente des infections congénitales en France et toucherait environ 0,5% des naissances. Cependant l’atteinte sévère n’est symptomatique à
31
la naissance que chez 10 % à 15 % des fœtus infectés. Dans ces cas, il existe une atteinte de plusieurs organes, et en particulier des atteintes neurologiques et sensorielles. L’infection chez les enfants symptomatiques est associée de façon variable à une hypotrophie, une hépatite, une hépatosplénomégalie, et des signes d’atteinte du système nerveux central (SNC) qui peuvent aboutir, dans les cas les plus graves, à des séquelles neuro-sensorielles et à un retard psychomoteur sévère. Cette forme grave est létale dans 30 % des cas (Malm and Engman 2007). Que l’enfant soit symptomatique initialement ou pas, il peut apparaître ultérieurement des anomalies notamment cérébrales et sensorielles, comme la surdité (Fowler, McCollister et al. 1997; Ivarsson, Lernmark et al. 1997).
L’incidence de la primo-infection maternelle pendant la grossesse est de 1% à 4% et le taux de transmission mère-fœtus est alors de 30 % à 40% contre 1% au cours d’une infection récurrente (Boppana, Rivera et al. 2001).
Le HCMV a un tropisme particulier pour le SNC, entraînant des atteintes plus ou moins sévères selon la date de l’infection en anténatal. Si l’infection du fœtus survient durant le premier trimestre de la grossesse, les lésions cérébrales retrouvées seront plus fréquemment des
anomalies de structuration (Fowler, Stagno et al. 1992). Les nouveau-nés infectés
congénitalement excrètent du HCMV dans l'urine et d'autres fluides corporels à des niveaux élevés. Cependant, des niveaux élevés de virus ne sont pas toujours corrélés à une maladie congénitale grave, même s’il existe un lien entre la charge virale sanguine élevée à la naissance et la surdité (Boppana, Fowler et al. 2005; Lanari, Lazzarotto et al. 2006).
iii. Infections opportunistes chez les sujets immunodéprimés
Le HCMV est un important agent pathogène opportuniste affectant les patients immunodéprimés. L'infection est fréquente suite à la réactivation du virus latent, ou lors d’une réinfection des patients qui ont eu une infection passée ou encore à l'initiation d'une primo-infection.
Au cours du SIDA
Avant l'utilisation de la thérapie antirétrovirale hautement active, environ 20 à 40% des adultes atteints du SIDA développait une maladie due au HCMV. Chez les personnes infectées par le VIH (virus de l’immunodéficience humaine), le risque de maladie à HCMV est lié à un faible taux de lymphocytes T CD4+. L'utilisation des HAART (Highly active antiretroviral therapy), qui entrainent une meilleure reconstitution ou préservation des cellules T CD4 immunitaires, a considérablement réduit l'occurrence de la virémie et de la maladie à HCMV (Deayton, Mocroft et al. 1999; O'Sullivan, Drew et al. 1999). Cependant, dans les pays en voie de développement,
32
cette infection continue d’être problématique, du fait d’un accès limité aux thérapies efficaces. Les manifestations cliniques les plus importantes sont les rétinites suivies par des atteintes du tube digestif, comme l’œsophagite et la colite. On peut retrouver des présentations cliniques moins courantes comme des encéphalites, des neuropathies périphériques, des pneumopathies, des gastrites et des hépatites.
Après transplantation d’organe solide ou de moelle osseuse
Les patients transplantés reçoivent un traitement immunosuppresseur qui limite le risque de rejet du greffon. Ce traitement favorise la réplication du HCMV à partir d’une contamination par le greffon ou d’une réactivation d’une souche latente. L’infection à cytomégalovirus constitue ainsi une cause majeure de morbidité et de mortalité chez les patients transplantés d’organes solides, de moelle osseuse ou de cellules souches hématopoïétiques. La gestion de l'infection par le HCMV reste un problème majeur et constitue un composant important du coût global de la transplantation. Le plus haut risque d’infection s’observe lorsqu'un receveur séronégatif est transplanté avec un organe séropositif. L’infection est associée à une diminution de la survie du patient et au rejet du greffon.
Les manifestations cliniques sont les résultats de la lyse cellulaire suite à la réplication virale ou sont dues aux effets indirects de cytokines et de chimiokines produites au cours de la réponse immunitaire déclenchée par l’infection (Nordoy, Muller et al. 2000). L’infection sévère chez un patient transplanté se manifeste notamment par de la fièvre ≥ 38°C et l’un des signes suivants : malaise, leucopénie, une arthralgie, lymphocytose atypique, thrombocytopénie, élévation des transaminases, occasionnellement par une éruption cutanée. La maladie est dite invasive lorsqu’il y a une dysfonction d’organe avec preuve de la présence virale dans cet organe. Bien que les traitements antiviraux arrivent à prévenir les formes sévères, des effets indirects de l’infection peuvent se produire chez ces patients, y compris un risque accru d’infections opportunistes par des champignons, des bactéries et des virus. D’autres conséquences peuvent survenir comme le rejet d’organe solide ou un retard à la prise de greffe en cas d’allogreffe de moelle (Fishman, Emery et al. 2007).
G. Traitements antiviraux
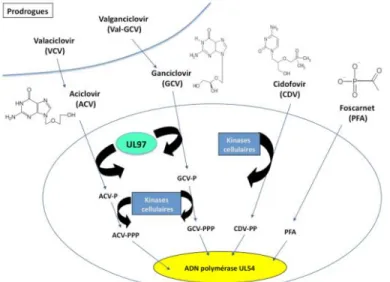
Plusieurs molécules sont disponibles pour les traitements curatifs et préventifs des infections : le ganciclovir ou GCV (Cymevan®), le Valganciclovir Val-GCV (Rovalcyte®), le cidofovir ou CDV (Vistide®, non commercialisé actuellement), le foscarnet ou PFA (Foscavir®). Toutes ces molécules inhibent l’activité de l’ADN polymérase virale UL54 (Figure 6) et sont donc sans action sur le virus latent. Ces molécules possèdent une toxicité non négligeable qui limite leur utilisation.
33
Les personnes transplantées et immunodéprimées obtiennent de bons résultats après l’utilisation de GCV. Cependant l’apparition de résistance est assez fréquente chez ces patients. Le GCV subit une première phosphorylation par la phosphotransférase UL97 puis deux autres phosphorylations par des kinases cellulaires pour arriver à son état actif. Le Val-GCV, une pro-drogue du GCV, est aussi couramment utilisée et a une biodisponibilité 10 fois supérieure à celle du GCV. Ces deux molécules ont une toxicité hématologique, essentiellement neutropénique. L’Aciclovir (Zovirax®) et son ester le Valaciclovir (Zelitrex®), bien que moins actifs sur le HCMV que le GCV, sont utilisés en prophylaxie après greffe d'organe, chez des patients séropositifs pour le HCMV.
Figure 6 : Mode d’action des inhibiteurs de l’ADN polymérase virale UL54 (Hantz, Mazeron et al. 2009).
Le PFA est capable, sans aucune modification supplémentaire, d’inhiber sélectivement le site de liaison au pyrophosphate sur l'ADN polymérase virale UL54. Son utilisation est associée avec une toxicité rénale et des troubles digestifs.
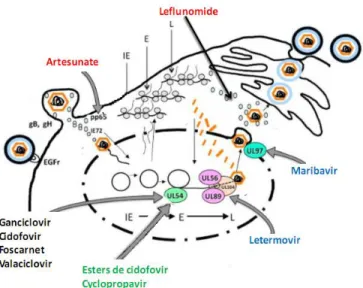
De nouvelles molécules antivirales sont actuellement en développement. Il y avait une nécessité pour deux raisons : 1), il est nécessaire de trouver des alternatives moins toxiques, notamment en prophylaxie et en traitement d’entretien, après la baisse de la charge virale. 2) en raison de l’apparition de résistance du HCMV aux molécules actuelles, résistances qui peuvent être croisées car ces antiviraux ciblent tous l’ADN polymérase, il est nécessaire de trouver des antiviraux qui ont des cibles différentes (Figure 7).
34
Figure 7 : Antiviraux en développement contre le HCMV d’après (Hantz, Mazeron et al. 2009). L’Artésunate, un traitement antipaludéen à l’origine, est efficace contre les souches sauvages d’HCMV et celles résistantes au GCV (Kaptein, Efferth et al. 2006). Il semblerait qu’il inhibe les protéines très précoces du HCMV. Il a été rapporté qu’après un traitement de 7 jours d’artésunate, la charge virale est significativement diminuée chez un receveur de moelle présentant une infection résistante au GCV et au PFA (Shapira, Resnick et al. 2008; Wolf, Shimoni et al. 2011). Une étude récente rapporte que l’artésunate a également donné de bons résultats chez trois patients transplantés en échec thérapeutique sur cinq (Germi, Mariette et al. 2014). Actuellement, l’artésunate est en autorisation temporaire d’utilisation (ATU) et, comme la forme orale n’est plus disponible en ATU, il s’administre par voie intraveineuse. Il est bien toleré, avec une toxicité modérée in vivo. Il serait plutôt à utiliser en prophylaxie, mais comme il est efficace sur les souches résistantes, il pourrait être utile en cas d’impasse thérapeutique.
Des recherches sont également en cours sur le leflunomide, un inhibiteur de la synthèse des pyrimidines utilisé dans le traitement de la polyarthrite rhumatoïde. Il est efficace sur le HCMV in
vitro et sur modèle animal. Il inhibe les étapes tardives du cycle viral et notamment l’acquisition
du tégument du virion. Il pourrait être utile en seconde intention (« rescue therapy »), chez des patients infectés par des souches résistantes au GCV (Ciszek, Mucha et al. 2014).
Le Maribavir (MBV) est une molécule inhibitrice spécifique de la kinase virale UL97. Il inhibe la sortie des capsides virales du noyau. Le MBV a très peu d’effets secondaires et il n’y a pas de résistance croisée avec les autres inhibiteurs. Son efficacité clinique en prophylaxie n’est pas totalement convaincante et nécessite des doses élevées. Il reste actif contre les souches résistantes au GCV, laissant penser que cette molécule pourrait être utile dans le traitement des