Exciton States of Dynamic DNA Double Helices: Alternating dCdG Sequences
Texte intégral
Figure
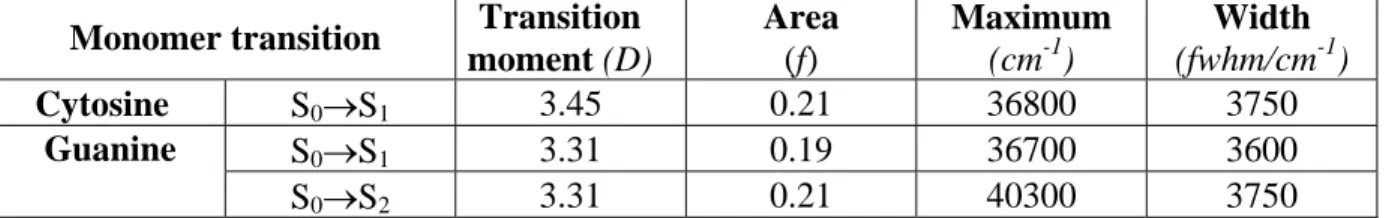

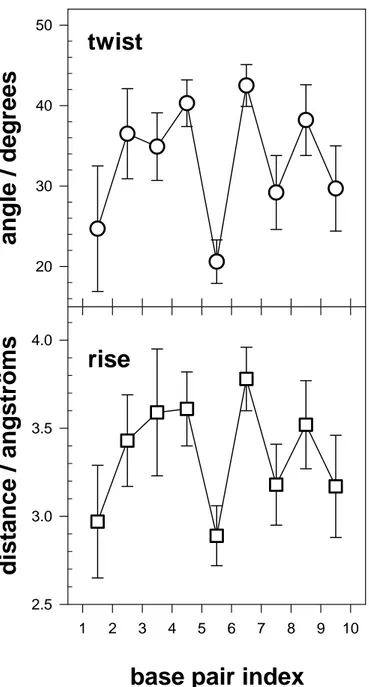
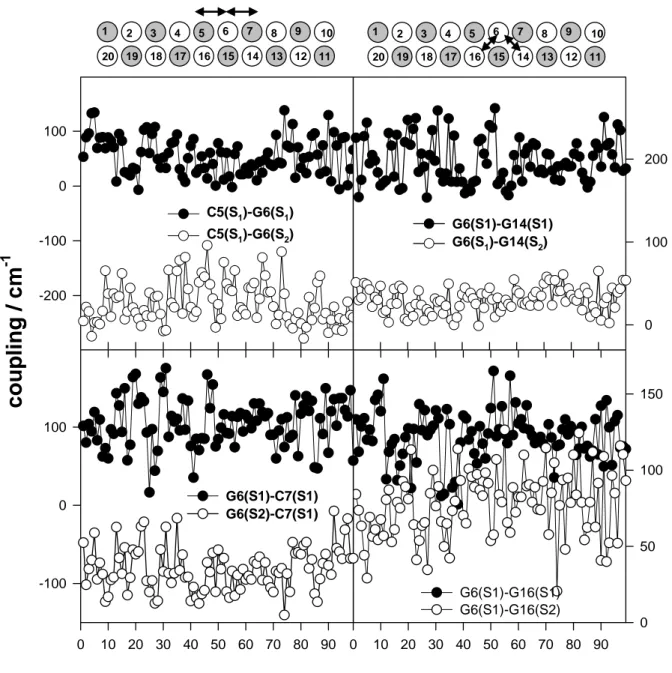

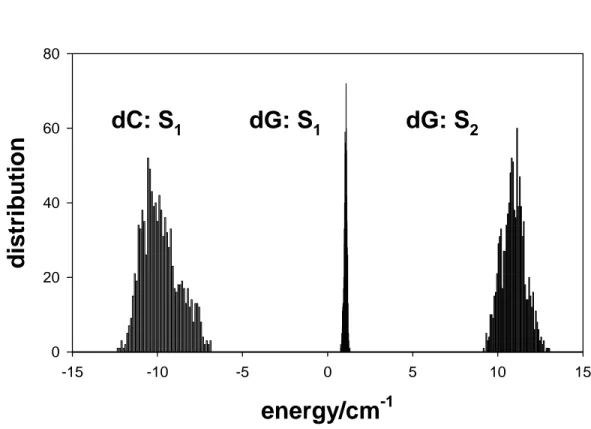
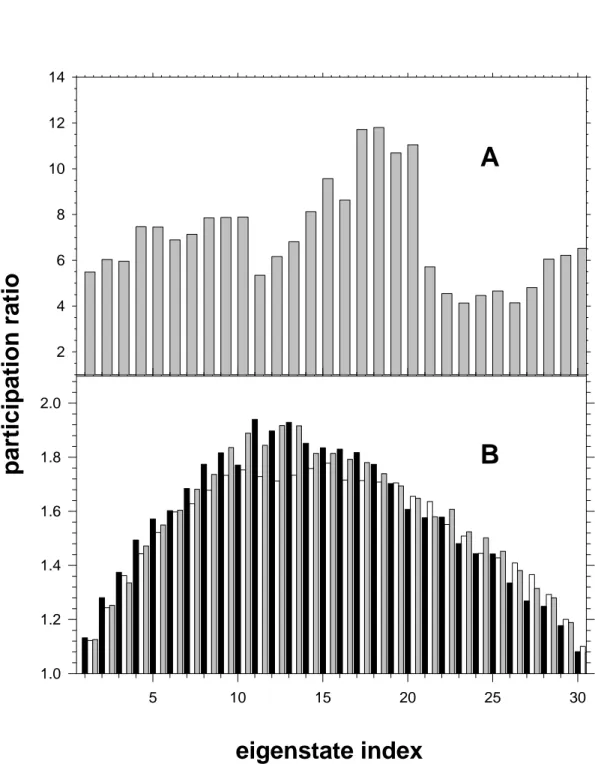
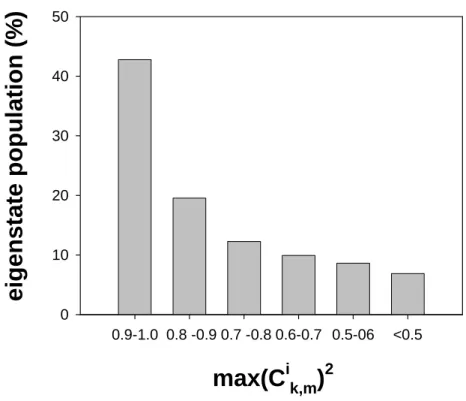
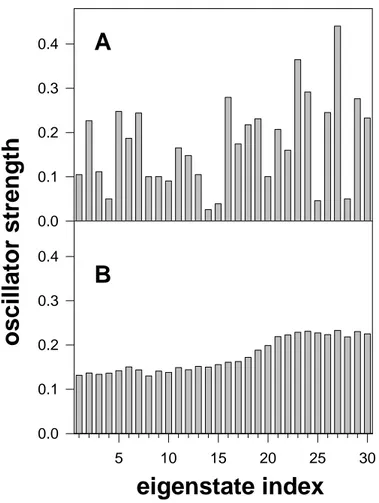

Documents relatifs
The decoding of the data revealed that the activity of an ensemble of simultaneously recorded neurons presents a robust sequence of states related to the
ﺎﺴﯿﺋر دﻮﻟﻮﻣ شﻮﺑﺎط /د
L’acceptation et la défusion l’amène à ne plus réagir comme d’habitude mais à observer l’émotion, les pensées, l’éventuelle tendance à l’action (y compris la
III.4 Résultats de la simulation Dans cette partie les résultats de simulation et les résultats expérimentaux de la commande sans capteur mécanique de la machine asynchrone
Information on the bending rigid- ity (persistence length) of the nucleoprotein complex and Hfq mediated bridging between different segments of the DNA molecule is then derived from
Abstract .- Proton spin-lattice relaxation times (T1) in CU(NO~)~ .e20 have been measured as a func- tion of the applied magnetic field at different temperatures. The
Cette étude nous montre que la Dimedone et l’ortho-phénylènediamine deux produits commerciaux et bon marché, réagissent pour donner l’intermédiaire
Port cities and global legacies – Alice MAH (2014) Book review – César Ducruet..
