Implication du remodelage de l’unité
neurovasculaire dans la maladie d’Alzheimer :
L’hypoperfusion cérébrale et le système de l’activateur
tissulaire du plasminogène
Mémoire
Maude Bordeleau
Maîtrise en Neurobiologie
Maître ès sciences (M.Sc.)
Québec, Canada
© Maude Bordeleau, 2016
Résumé
L’unité neurovasculaire (NVU) est centrale dans l’élimination de la β-amyloïde dont l’accumulation promeut le développement de la maladie d’Alzheimer (AD). Suivant une perturbation vasculaire, le bris ou l’altération de la barrière hématoencéphalique induit le remodelage de la NVU. Par exemple, les cellules endothéliales sécrètent l’activateur tissulaire du plasminogène (t-PA), ce qui module les cellules composant la NVU. C’est pourquoi, nous nous sommes intéressés à ce remodelage dans la AD en étudiant l’effet de l’hypoperfusion cérébrale chronique sévère (SCCH) et de l’administration du t-PA. Suite à la SCCH, les souris développant la AD, APPswe/PS1, démontrent un déclin cognitif plus important causé par un dysfonctionnement des microglies. En contre partie, nous avons observé une amélioration des fonctions cognitives des APPswe/PS1 suite à l’injection systémique du t-PA qui induit l’activation des microglies via la protéine apparentée au récepteur des protéines de faibles densité, LRP1, et promeut l’élimination de l’Aβ. Ainsi, nos résultats démontrent que le remodelage de la NVU peut aggraver la pathogenèse, mais également fournir des pistes de traitement.
Abstract
Brain remodeling by the neurovascular unit (NVU) has gain interest in disease such as Alzheimer’s disease (AD). Following vascular perturbation, NVU go through remodeling due to disruption or alteration of brain-blood barrier. One of the molecule inducing remodeling is the tissue-plasminogen activator (t-PA) released by endothelial cells. In fact, t-PA can act both as an enzyme and a cytokine. Thus, we studied the effect of vascular perturbation and t-PA system in AD. By developing a new model of a severe chronic cerebral hypoperfusion (SCCH), we demonstrate that SCCH aggravates memory loss in AD mice, APPswe/PS1, due to microglia dysfunction. Indeed, low glucose environment lowers microglia’s activity and phagocytosis capacity. On the other hand, systemic administration of t-PA improves cognition as well as decreases amyloid burden in APPswe/PS1. Acting as a cytokine, rt-PA binds LRP1 which induces microglia’s activation and promotes amyloid elimination. These data suggest that NVU remodeling occurring in AD may participate in the disease pathogenesis and provide new insight of treatment, such as rt-PA.
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste de tableaux ... xi
Liste de figures ... xiii
Abréviations ... xv Remerciements ... xix Avant-propos ... xxi 1. Introduction ... 1 1.1. Maladie d’Alzheimer ... 1 1.1.1. Tau ... 3 1.1.2. Amyloïde ... 4
1.1.2.1. Formation, élimination et dégradation de l’amyloïde ... 4
1.1.2.2. Agrégation de l’amyloïde ... 7
1.1.2.3. Hypothèse de la cascade amyloïde ... 7
1.1.3. Hypothèse vasculaire ... 9
1.2. Facteurs de risque de la maladie d’Alzheimer ... 9
1.2.1. Génétiques ... 9
1.2.2. Obésité ... 10
1.2.3. Diabète ... 10
1.2.4. Troubles vasculaires ... 11
1.3. Hypoperfusion ... 11
1.3.1. Régulation du flux sanguine et maladie d’Alzheimer ... 11
1.3.2. Oligémie versus ischémie ... 12
1.4. Système de l’activateur tissulaire du plasminogène ... 13
1.4.1. Fonction du système de l’activateur tissulaire du plasminogène ... 13
1.4.2. Système de l’activateur du plasminogène et maladie d’Alzheimer... 15
1.5. Unité neurovasculaire ... 16
1.5.1. Fonction de l’unité neurovasculaire ... 17
1.5.2. Remodelage de l’unité neurovasculaire et maladie d’Alzheimer ... 19
1.6. Hypothèses et objectifs ... 20
2. Severe chronic cerebral hypoperfusion induces microglial dysfunction leading to memory loss in APPswe/PS1 mice ... 23
2.1. Résumé ... 24
2.3. Introduction ... 25
2.4. Material and methods ... 26
2.4.1. Animals with severe chronic cerebral hypoperfusion ... 26
2.4.2. Behavior analysis ... 28
2.4.2.2. Two-object novel object recognition ... 28
2.4.2.3. Asymmetry cylinder test ... 29
2.4.2.4. Open field ... 29
2.4.3. Soluble Aβ1-40 and soluble Aβ1-42 ELISA ... 29
2.4.4. Immunofluorescence staining ... 30
2.4.5. Western blot analysis ... 30
2.4.6. Flow Cytometry ... 31
2.4.7. Immunohistochemistry ... 32
2.4.8. Nissl body staining ... 32
2.4.9. Fluoro-Jade B staining ... 33
2.4.10. In vitro experiments ... 33
2.4.10.1. Cell culture... 33
2.4.10.3. Griess assay ... 34
2.5. Results ... 35
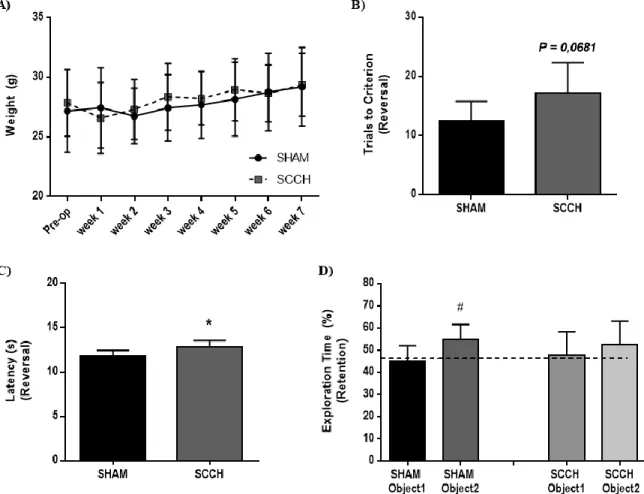
2.5.1. SCCH worsen memory impairment in APPswe/PS1 mice without affecting motor capacity ... 35
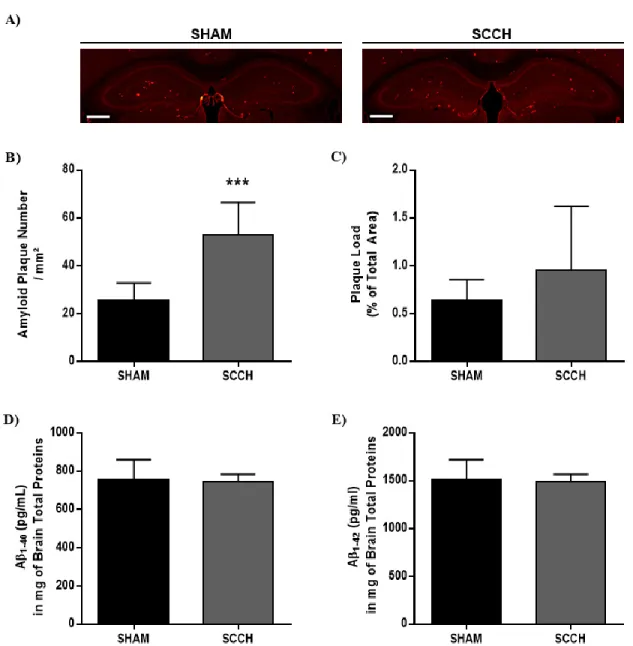
2.5.2. Memory loss in SCCH mice is associated with an increased number of parenchymal amyloid plaques ... 37
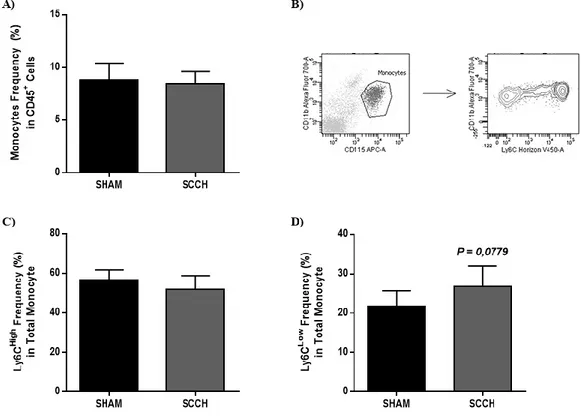
2.5.3. SCCH-linked trend towards an increased in the patrolling monocyte population ... 37
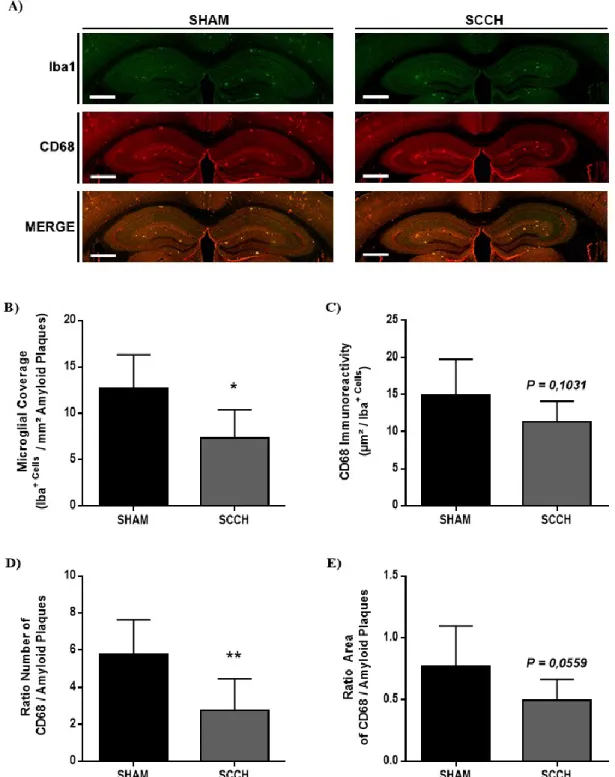
2.5.4. SCCH disrupts plaque coverage by microglia and alters microglial activation 39 2.5.5. Alteration of microglial function is caused by an impaired glucose metabolism ... 40
2.5.6. ERK pathway-dependent decrease in cell survival contributes to memory impairment in SCCH mice ... 42
2.6. Discussion ... 43
2.8. Acknowledgements ... 46
2.9. Grant support ... 46
3. Tissue-plasminogen activator attenuates Alzheimer’s disease-related pathology development in APPswe/PS1 mice ... 47
3.1. Résumé ... 48
3.2. Abstract ... 48
3.4. Materials and Methods ... 50
3.4.1. Animal experiments ... 50
3.4.2. Chimeric mice generation ... 51
3.4.3. Tissue collection ... 52
3.4.4. Immunofluorescence staining ... 52
3.4.5. IgG and albumin extravasation ... 53
3.4.6. Aβ plaques, microglia coverage and Aβ internalization by microglia quantification ... 53
3.4.7. In situ Hybridization ... 54
3.4.8. Soluble Aβ1–42 Enzyme-Linked Immunosorbent Assay (ELISA) ... 54
3.4.9. Brain microvessel isolation ... 54
3.4.10. Microglia’s isolation and analysis by flow cytometry ... 55
3.4.11. Protein extraction ... 56
3.4.12. Caseinase and gelatinase activity assays ... 56
3.4.13. Western blot analysis ... 56
3.4.14. Flow cytometry ... 57
3.4.15. Behavior analysis ... 57
3.4.16. In vitro experiments ... 58
3.4.16.1. Cells culture ... 58
3.4.16.2. Cell stimulation ... 58
3.4.16.3. Cell migration assay ... 58
3.4.16.4. Chemotaxis assay ... 59
3.4.16.5. Phagocytosis assay ... 59
3.4.16.6. Griess Assay ... 59
3.4.17. Statistics ... 60
3.5. Results ... 60
3.5.1. Activase® rt-PA regimen does not affect blood-brain barrier integrity and function ... 60
3.5.2. Activase® rt-PA slows the progression of AD-like pathology and behavioral deficits in APPswe/PS1 ... 62
3.5.3. The enzymatic activity is not responsible of rt-PA-induced clearance of Aβ ... 64
3.5.4. Activase® rt-PA modulates monocyte population phenotypes in a transient manner ... 66
3.5.5. The effects of Activase® rt-PA on resident microglia ... 66
3.5.6. Activase® rt-PA enhances BV2 microglial cell mobility and acts as chemoattractant molecule in a LRP1-dependent manner ... 69
3.5.7. The effects of Activase® rt-PA on the phagocytic capacity and oxidative stress
cascade in BV2 microglial cells ... 69
3.5.8. The effects of Activase® rt-PA on the mobility and the phagocytic capacity of BV2 microglial cells is independent of its enzymatic activity. ... 71
3.6. Discussion ... 71 3.7. Conclusion ... 75 3.8. Acknowledgments ... 76 3.9. Funding ... 76 4. Discussion ... 77 5. Conclusions et perspectives ... 87 Références ... 89
Liste de tableaux
Liste de figures
Figure 1.1. Voies de transformation de l’APP………..5
Figure 1.2. Voie d’entrée et d’élimination de la β-amyloïde………..……..6
Figure 1.3. Schéma de l’hypothèse vasculaire de la maladie d’Alzheimer………..8
Figure 1.4. Structure et fonction de l’activateur tissulaire du plasminogène (t-PA)……….14
Figure 1.5. Représentation de l’unité neurovasculaire et de la microcirculation cérébrale…………17
Figure 2.1. Schema illustrating the SCCH surgery………....27
Figure 2.2. SCCH aggravates APPswe/PS1 memory loss………36
Figure 2.3. Number of amyloid plaques increase following SCCH without any change in amyloid burden………...38
Figure 2.4. A tendency of increased patrolling monocytes is observed following SCCH...39
Figure 2.5.SCCH alters microglial function in APPswe/PS1………41
Figure 2.6. Low glucose environment alter the activity and the phagocytosis capacity of microglia... ………42
Figure 2.7.SCCH lowers ERK1/2 activation………43
Figure. 3.1. Activase® rt-PA administration reduces Aβ aggregates and soluble Aβ1-42 levels in the brain………61
Figure. 3.2. Activase® rt-PA administration improves APPswe/PS1 mice cognitive functions…..62
Figure. 3.3. t-PA-associated perivascular proteases are not induced by Activase® rt-PA regimen..63
Figure. 3.4. Chronic Activase® rt-PA administration modulates monocyte subpopulation frequencies in the blood of APPswe/PS1 mice………64
Figure. 3.5. Acute Activase® rt-PA administration modulates monocyte subpopulation frequencies in the blood of wildtype mice………65
Figure. 3.6. Chronic Activase® rt-PA administration increases the number of resident microglia-associated to Aβ plaques and reduces the activation of stress-induced pathways……67
Figure. 3.7. Activase® rt-PA administration does not influence blood-derived monocyte infiltration into the brain parenchyma of APPswe/PS1 mice………68
Figure. 3.9. Activase® rt-PA decreases BV2 microglial cell intracellular stress and preserves their
phagocytic capacity………72
Figure 4.1. L’effet de l’hypoperfusion cérébrale chronique sévère (SCCH) sur le cerveau des APPswe/PS1………77
Figure 4.2. L’effet du traitement hebdomadaire de l’Activase ® rt-PA sur le cerveau des APPswe/PS1………82
Supplementary Figure 2.1. Motricity behavior in APPswe/PS1 after severe chronic cerebral hypoperfusion (SCCH)………109
Supplementary Figure 2.2. SCCH does not alter blood-brain barrier tightness………..110
Supplementary Figure 2.3. Absence of infiltred monocytes after SCCH………110
Supplementary Figure 2.4. SCCH seems to atrophy CA3 without neuronal death………111
Supplementary Figure. 3.1. BBB tightness is preserved after Activase® rt-PA administration…112 Supplementary Figure. 3.2. BBB integrity is preserved after Activase® rt-PA administration…..113
Supplementary Figure. 3.3. Endothelial transporters involved in Aβ transport across the BBB are not affected following Activase® rt-PA administration………114
Supplementary Figure. 3.4. Activase® rt-PA regimen does not modulate the brain levels of synaptophysin………115
Supplementary Figure. 3.5. Acute Activase® rt-PA administration modulates total monocyte frequency in the blood of APPswe/PS1 mice………116
Supplementary Figure. 3.6. Chronic Activase® rt-PA administration does not trigger a sustained inflammation in the brain of chimeric APPswe/PS1 mice…………116
Abréviations
1VO: 1-vessel occlusion, Ligature unilatérale de l’artère carotide commune
Aβ: Amyloid β-peptide, Protéine β-amyloïde
ABCB1: ATP-binding cassette sub-family B1, Transporteur à cassette liant l’ATP de la sous-famille B1
ACE: Angiotensin converting enzyme, Enzyme de conversion de l’angiotensine
AD: Alzheimer disease, Maladie d’Alzheimer
AMPA: α-amino-3-hydroxy-5-méthylisoazol-4-propionate
APH-1: Anterior pharynx 1, Protéine du pharynx antérieur défectueux 1
Apo: Apolipoprotéine
APP: Amyloid precursor protein, Protéine précurseure de l’amyloïde
APP-sα: APP-α soluble
APP-sβ: APP-β soluble
APPswe: Gène APP possédant la double mutation familiale Swedish (K670N, M671L)
APPswe/PS1: Souris transgénique double-mutante APP Swedish et PS1 A246E
ATP: Adénosine tri-phosphate
AVC: Accident vasculaire cérébral
BACE-1: β-site APP cleaving enzyme 1, Enzyme 1 de clivage du site β de l’APP
BBB: Blood-brain barrier, Barrière hémato-encéphalique
BCAO: Bilateral common carotid arthery occlusion, Ligature bilatérale des artères carotides communes
BCAS: Bilateral common carotid arthery stenosis, Sténose bilatérale des artères carotides communes
BDNF: Brain-derived neurotrophic factor
BSA: Bovin serum albumin, Albumine de serum bovin
CA: Cornu ammonis, Corne d’Ammon
CAA: Cerebral amyloid angiopathy, Angiopathie amyloïde cérébrale
CBF: Cerebral blood flux, Flux sanguin cérébral
CTFα: C-terminal fragment α, Fragment carboxy-terminal α
CTFβ: C-terminal fragment β, Fragment carboxy-terminal β
DAB: Diaminobenzidine
DMEM: Dulbeco’s modified eagle medium, Milieu Eagle modifié de Dulbeco
DMEM High: DMEM contenant 4500mg/L de glucose DMEM Low: DMEM contenant 1000mg/L de glucose
EDTA: Ethylenediaminetetraacetic acid, Acid éthylène diamine-tétraacétatique
ECL: Enhanced chimiluminescence solution
EGF: Epidermal growth factor, Facteur de croissance de l’épiderme
ERK: Extracellular signal-regulated kinase, Kinase régulée par les signaux extracellulaires
ELISA: Enzyme-linked immunosorbent assay, Essai d’immuno absorption enzymatique
FACS: Fluorescent-activated cells sorting, Tri de cellules activées par fluorescence
FBS: Fetal bovine serum, Sérum de veau fœtal
FDA: Food and Drug Association
FJB: Fluoro-jade B
FTD: Frontotemporal dementia, Démence fronto-temporale
GFP: Green fluorescent protein, Protéine fluorescente verte
GLUT1: Glucose transporter 1, Transporteur du glucose 1
HBSS: Hank’s balanced salt solution, Solution saline équilibrée de Hank
HIF-1α: Hypoxia-inducible factor 1α, Facteur de transcription 1α induit par l’hypoxie
HRP: Horseradish peroxidase, Peroxidase de raifort
IDE: Insulin degrading enzyme, Enzyme de dégradation de l’insuline
IgG: Immunoglobuline G
IL: Interleukine
KPBS: Potassium phosphate buffered saline, Saline tamponée au potassium et phosphate
LDL: Low-density lipoprotein, Lipoprotéine à faible densité
LPS: Lipopolysaccharide
LRP: LDL receptor-related protein, Protéine apparentée au récepteur des LDLs
LTD: Long-term depression, Dépression à long-terme
LTP: Long-term potentiation, Potentialisation à long-terme
MAPK: Mitogen-activated protein kinase, Protéine kinase activée par mitose
MCI: Mild cognitif impairment, Trouble cognitif léger
mGluR5: Metabotropic glutamate receptor 5 Récepteur métabotropique du glutamate 5 MMP: Matrix metalloproteinase, Métalloprotéase matricielle
ND: No cognitif deficit
NEP: Néprilysine
NGF: Nerve growth factor, Facteur de croissance des nerfs
NMDA: N-méthyl-D-aspartate
NVU: Neurovascular unit, Unité neurovasculaire
OMS: Organisation Mondiale de la Santé
PAI-1: Plasminogen activator inhibitor-1, Inhibiteur-1 des activateurs du plasminogène
PBS: Phosphate buffered saline, Saline tamponnée au phosphate
PDGF-CC: Platelet-derived growth factor-CC, Facteur de croissance CC dérivé des plaquettes.
PEN-2: Preseniline enhancer 2, Protéine activatrice de la préséniline 2
PET: Positron emission tomography, Tomographie à émission de positrons
PFA: Paraformaldéhyde
PgP: P-glycoprotéine
PrP: Prion protein, Protéine prionique
PS: Préséniline
RAGE: Receptor for advanced glycation endproducts, Récepteur des produits finaux de glycosylation avancée
RT: Room temperature
rt-PA: Recombinant-tisssue-type plasminogen activator, Activateur tissulaire du plasminogène recombinant
SAPK/JNK: Stress-activated protein kinases /Jun amino-terminal kinases, Protéine kinase active
par le stress / Kinase c-Jun N-terminal
SCCH: Severe chronic cerebral hypoperfusion, Hypoperfusion cérébrale chronique sévère
S.D.: Standard deviation, Écart-type
SD: Severe cognitif deficit
SDS-PAGE: SDS-polyacrylamide gel electrophoresis, Gel d’électrophorèse SDS-polyacrylamide S.E.M.: Standard error of the mean, Écart-type de la moyenne
sLRP: LRP soluble
SNC: Système nerveux central
TLR: Toll-like receptor, Récepteur de type Toll
Remerciements
Au cours de ces deux dernières années, j’ai côtoyé un ensemble de gens qui m’ont offert le soutien nécessaire pour réussir et aboutir dans mes projets entrepris. Je tiens donc à souligner leur contribution qui a été très marquante.
Tout d’abord, je voudrais remercier Serge Rivest, Ph.D., qui m’a offert la chance de travailler au sein de son équipe, ainsi que Frédérique Calon, Ph.D., et Denis Soulet, Ph.D., qui ont accepté de faire partie de mon comité d’encadrement. Tous m’ont offert de précieux conseils en tant que scientifique. Ils m’ont guidé pour l’avancement de mes projets vers la bonne direction. Surtout au cours des derniers mois, Dr. Rivest a toujours été prêt pour discuter des derniers détails et ce, malgré son horaire chargé. Je voudrais également souligner l’énorme contribution d’Ayman ElAli, Ph.D., en tant superviseur de projet. Servant de guide et de collègue, Dr. ElAli m’a offert un soutien continu pour que je retire la meilleure expérience qui soit de ma maîtrise. Il a été sans conteste un mentor important qui a permis l’avancement de mes projets et stimulé ma curiosité scientifique. Au cours de ma maîtrise, j’ai également eu la chance de profiter de l’expertise de plusieurs personnes qui m’ont conseillé lorsque j’avais des interrogations. Nataly Laflamme, M.Sc., Marie-Michèle Plante, M.Sc., Paul Préfontaine, M.Sc., Antoine Lampron, Ph.D., Jean-Philippe Michaud, Ph.D., Peter Thériault, M.Sc., et Audrey LeBehot, Ph.D., m’ont tous offert leur avis lorsque je leur posais une panoplie de questions. J’ai également eu l’opportunité de connaitre brièvement Marc-André Bellavance, Ph.D., et Antoine Larochelle, M.Sc.
Sur un plan plus personnel, je voudrais remercier Yannick Tremblay, B.Sc., Édith Godbout-Miville, B.Sc., Catherine Fontaine-Lavallée, B.Sc., Catherine Gilbert, B.Sc., André-Pascal Roy, B.Sc., Prenitha Innatious, M.Sc., avec qui j’ai discuté de science comme de tout et n’importe quoi. Comparse dans l’acheminement de nos travails respectifs, discuter avec eux a toujours allégé la morosité d’une série de résultats négatifs. Je dois également souligner le support de ma famille et amis qui m’ont aidé à décrocher lorsque j’en avais besoin ce qui m’a permis d’être toujours positive dans mon activité de recherche.
Finalement, un dernier merci pour toutes ces personnes qui, sans le savoir, m’ont permis d’avoir une expérience dont je me souviendrai et de m’apprendre tant de choses autant sur le plan scientifique que personnel. Merci.
Avant-propos
L’article intitulé Severe chronic cerebral hypoperfusion induces microglial dysfunction leading to memory loss in APPswe/PS1 mice est en revision pour le journal Oncotarget. Cet article représente le second chapitre du mémoire. Lors de ce projet, j’ai effectué l’intégralité des expériences mise à part le comportement qui a été réalisé par Mohammed Filali. J’ai également accompli l’analyse des résultats, puis interprété ceux-ci en discutant avec Ayman ElAli. Suite à la discussion scientifique, j’ai rédigé intégralement l’article scientifique qui a ensuite été corrigé par Ayman ElAli et Serge Rivest. Le projet lui-même a été conçu et encadré par Ayman ElAli et Serge Rivest.
L’article intitulé Tissue-plasminogen activator attenuates Alzheimer’s disease-related pathology development in APPswe/PS1 mice a été publié en ligne dans Neurospychopharmacology le 9 septembre 2015 (ElAli, A., Bordeleau, M., Thériault, P., Filali, M., Lampron, A. et Rivest, S. Neuropsychopharmacology. doi:10.1038/npp.2015.279.). Celui-ci constitue le troisième chapitre de ce mémoire. Dans ce projet, j’ai effectué l’ensemble de l’étude in vitro décrivant le mécanisme d’action du t-PA, l’analyse et l’interprétation des résultats, en plus de la rédaction du matériel et méthode correspondant. J’ai également coupé les tissus et effectué les marquages sur les tissus (FJB, hybridation in situ, immunofluorescence). Peter Thériault a effectué l’expérience de FACS, aider à l’analyse et rédiger la partie matériel et méthode correspondante. Mohammed Filali ont effectué les tests de comportement, de même que la rédaction du matériel et méthode associée. Ayman ElAli a effectué le reste des mannipulation in vivo, ainsi de l’analyse des résultats et l’interprétation de l’ensemble des résultats in vivo. L’article a été principalement écrit par Ayman ElAli, puis corrigé par l’ensemble des co-auteurs. Le projet lui-même a été conçu et encadré par Ayman ElAli et Serge Rivest
Chapitre 1
1. Introduction
1.1. Maladie d’Alzheimer
La maladie d’Alzheimer (AD) est un désordre neurodégénératif se développant au cours d’une vie et représentant la forme de démence la plus répandue (1). Au cours des dernières années, L’Organisation Mondiale de la Santé (OMS) a estimé que le nombre de gens atteints par la AD s’élève à 30 millions ; nombre qui devrait tripler au cours des prochaines décennies (2,3). Chez la population âgée de plus de 85 ans, 1 personne sur 3 développe des symptômes de la AD (3).
Celle-ci a été diagnostiquée pour la première fois, en 1907, par Aloïs Alzheimer. Il avait alors observé la présence de plaques denses dans la parenchyme, appelé plaques séniles, et de neurofibrilles (4). Il est maintenant connu que ces plaques et neurofibrilles sont respectivement entraînées par la déposition du peptide β-amyloïde (Aβ) (5,6), dérivé de la protéine précurseure de l’amyloïde (APP), et l’hyperphosphorylation de tau, une protéine associée aux microtubules, qui forme des filaments (7,8). Ces phénomènes moléculaires entraînent principalement un dysfonctionnement de la capacité mnésique qui s’instaure graduellement. Braak et Braak ont distingué six paliers (I à VI) à cette progression selon les régions du cerveau touchées. Les premiers changements neuropathologiques débutent dans le cortex entorhinal (I-II), puis s’étendent aux régions limbiques (III-IV) et, aux aires associatives temporales, pariétales et frontales du néocortex (V-VI) (9,10). Récemment, un stade préclinique de la AD a été identifié. Celui-ci se décrit par un trouble cognitif léger (MCI) (11) se manifestant quelques années avant le diagnostic de démence et étant notamment associés avec plusieurs problèmes causé par le remodelage du cerveau (Section
1.5.2 – Remodelage de l’unité neurovasculaire et maladie d’Alzheimer) (1). Durant cette
période, le patient présente des troubles cognitifs, mais trop légers pour interférer dans sa vie de tous les jours (1). Les premières manifestations cliniques de la AD, soit post-diagnostic, démontrent généralement une dysfonction de la mémoire de travail et de la mémoire sémantique, en plus de la sensibilité aux distractions. Il a été également reporté que, lors de la phase initiale, les malades d’Alzheimer peuvent être dépressifs ou apathiques. Dans un stade plus avancé, ceux-ci peuvent devenir confus, désorientés et avoir des comportements anormaux (1) associés à une perturbation du cycle circadien (12). Ils peuvent également présenter des symptômes atypiques tels des troubles moteurs (1), du langage (1,13) et visuels (13).
Il existe deux formes de la AD, soit la familiale (précoce) et la sporadique (tardive). La AD familiale est observée lors de l’expression génomique de polymorphismes génétiques qui favorisent l’expression de l’amyloïde ou altèrent le ratio entre les différentes formes d’Aβ, soit l’Aβ1-42, qui
tend à s’agréger, et l’Aβ1-40, qui est la forme plus commune (14,15). Ces variations génétiques ont
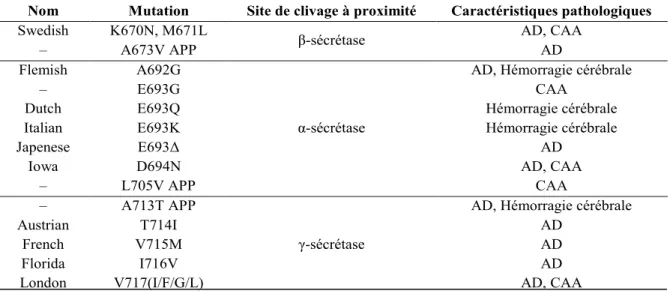
été observées au niveau des gènes codant l’APP, la préséniline (PS)-1 et la PS-2. Situés sur le chromosome 21, 20 polymorphismes du gène APP (Tableau I) ont été identifiés, ce qui correspond seulement à 5% des cas familiaux. Les cas précoces sont donc principalement causés par des mutations au chromosome 14 au niveau des gènes PS-1 et PS-2. Quant à ceux-ci, 120 et 8 variations génomiques ont respectivement été répertoriées (16). Les patients qui les possèdent présentent des symptômes de la AD pour la première fois vers 40 ans (17). Toutefois, près de 95% des cas de la AD sont observés dans la population âgée d’au moins 65 ans qui possède la forme tardive de cette pathologie (18).
À partir des mutations identifiées chez les patients atteints de la forme familiale, des modèles animaux ont été développés chez la drosophile, le poisson P. marinus, le ver C. elegans et la souris. Ces modèles permettent de reproduire les aspects généraux de la pathogenèse tels la cascade de progression, ainsi que les modulateurs et les gènes influencant la AD (16). Les premiers modèles murins furent développés par l’insertion des gènes humains APP Dutch et APP Flemish. Ces modèles présentèrent une amyloïdose cérébrale à un âge très avancé, vers 18 mois (14). Par la suite,
Nom Mutation Site de clivage à proximité Caractéristiques pathologiques
Swedish K670N, M671L
β-sécrétase AD, CAA
– A673V APP AD
Flemish A692G
α-sécrétase
AD, Hémorragie cérébrale
– E693G CAA
Dutch E693Q Hémorragie cérébrale
Italian E693K Hémorragie cérébrale
Japenese E693Δ AD
Iowa D694N AD, CAA
– L705V APP CAA
– A713T APP
γ-sécrétase
AD, Hémorragie cérébrale
Austrian T714I AD
French V715M AD
Florida I716V AD
London V717(I/F/G/L) AD, CAA
Tableau I. Formes mutantes du gène APP découvertes dans les cas familiaux. Les mutations du gène
APP sont réparties selon leur position de l’acide aminé muté. Pour chaque mutation, le site de clivage à proximité de la mutation est indiqué de même que certaines caractéristiques pathologiques générales. AD: Maladie d’Alzheimer, CAA: Angiopathie amyloïde cérébrale (16).
Hsiao et ses collègues développèrent une souris «knock-in» où le gène APP Swedish (APPswe) est inséré dans le génome par le vecteur de protéine prionique (PrP) du hamster. Cette lignée transgénique murine fut appelée Tg2576. Contrairement aux modèles précédents, les souris Tg2576 possèdent de nombreuses plaques en plus d’un trouble cognitif dès 9 mois (19). La Tg2576, une autre lignée transgénique, APP23, exprime le gène APPswe humain, mais sous le promoteur mThy1.2 (16). Ces derniers modèles développent également des dépôts vasculaires d’amyloïde caractéristiques de l’angiopathie amyloïde cérébrale (CAA) (14,16) qui est très commune chez les malades d’Alzheimer (20).
Un autre modèle transgénique intéressant est la lignée double mutante APPswe/PS1 A246E (APPswe/PS1). Plus précisément, les souris APPswe/PS1 expriment davantage d’Aβ1-42 que les
souris mutantes APPswe et PS1 A246E (21). De plus, dès 3-4 mois, les femelles APPswe/PS1 démontrent des dépôts d’amyloïde (22) alors que, chez les mâles, l’accumulation robuste est évidente seulement à partir de 6 mois (23). Comparativement aux autres modèles de la AD, la pathologie se développe plus rapidement chez les APPswe/PS1. Suivant l’accumulation de plaques séniles, il a été observé que, dans ce modèle, il y a une diminution de l’expression de plusieurs protéines synaptiques (e.g. AMPA, Arc, Erg1, NR2A/B, PSD95) et de facteurs neurotrophiques (e.g. BDNF) (23). À un stade plus avancé, le nombre de plaques devient plus important et couvre près de 80% de l’hippocampe chez certains animaux (21).
1.1.1. Tau
La AD est caractérisée par la formation de neurofibrilles (7,8) localisés dans le corps cellulaire, les dentrites apicales et distales des neurones, ou encore associés à des plaques amyloïdes dans les neurites anormaux (24,25). Ce type de lésions neurofibrillaires est également observé dans d’autres troubles neurodégénératifs tels la maladie de Pick, la dégénération cortico basale, la paralysie supranucléaire progressive et la démence fronto-temporale (FTD) (24,25). Parmi ces troubles neurodégénératif, la maladie de Pick et la FTD décrivent la neurodégénération des lobes frontal et temporal respectivement associé avec une démence progressive présentant des lésions neurodégénérative riches en tau et des déficits moteurs pouvant contribuer au développement de la maladie de Parkinson (16). Tout comme la FTD, la dégénération cortico-basale et la paralysie supranucléaire sont également des troubles neurodégénératifs moteurs entrainant respectivement un déficit moteur induit par la perte de neurones corticales et extrapyramidales, et une paralysie occulaire (16)(24,25). Les premières mutations de tau ont été identifiés chez les patients ayant la FTD (16).
La protéine tau semble ainsi participer à la neurodégénération. Plusieurs modèles ont donc été développés pour étudier son rôle lors de maladies. L’expression de l’isoforme de tau humain la plus courte de 352 acides aminés (26) ou la plus longue de 441 acides aminés (27), chez des souris Alzheimer, induit respectivement la formation tardive, vers 18-20 mois (26), ou précoce (27) de neurofibrilles. Chez les Tg2576, l’intégration de tau muté, tau P301L, augmente massivement le nombre de neurofibrilles notamment au niveau du cortex entorhinal et l’amygdale, les régions vulnérables à la déposition d’amyloïde, et ce, sans modifier l’expression de tau (28). En ce sens, Geula et al ont démontré la capacité de l’Aβ1-42 synthétique à induire la formation de neurofibrilles
chez le singe rhésus âgé (29). En effet, les oligomères d’Aβ sont capables d’indure la phosphorylation de tau par l’activation de protéines kinases, soit GSK3β, CDK5, MARK et MAPK (30). Lorsque hyperphosphorylé, tau est clivé à son domaine de liaison aux microtubules ce qui le dissocie des microtubules. Il devient soluble et est sécrété par les cellules (31). L’hyperphosphorylation de tau permet également son entrée dans les épines dendritiques où il se localise anormalement (32). Une fois entré dans les neurones, la conformative native de tau soluble favorise la formation de filament hélicoïdaux neurotoxiques, puis l’agrégation de ces filaments en neurofibrilles ce qui contribue au dysfonctionnement synaptique (33,34). Outre l’amyloïde, la phosphorylation de tau est également induite après un choc thermique, l’hypoxie ou la privation de glucose (30). Bien que la phosphorylation de tau à des sites supplémentaires soit associée à un déficit synaptique, le processus d’hyperphosphorylation reste à approfondir (35).
1.1.2. Amyloïde
1.1.2.1. Formation, élimination et dégradation de l’amyloïde
L’Aβ est dérivée de l’APP. Cette glycoprotéine associée à la membrane est transformée par deux voies dont l’une génère l’amyloïde et l’autre non (Fig. 1.1). Lors de cette dernière, l’APP est clivée par l’α-sécrétase au niveau de son domaine Aβ sécrétant la portion amino-terminale, le fragment soluble APP-α (APP-sα), et prévenant la formation d’amyloïde. L’Aβ, quant à elle, provient du clivage séquentiel de l’APP par la β-sécrétase et le complexe γ-sécrétase (36). La β-sécrétase, BACE-1, clive l’APP libérant le fragment soluble APP-β (APP-sβ) du fragment carboxy-terminal β (CTFβ) (36,37). Le CTFβ est alors clivé par le complexe γ-sécrétase qui est composé de la PS, la nicastrine, protéine du pharynx antérieur défectueux 1 (APH-1) et la protéine activatrice de la préséniline (PEN-2) (36,38). La longueur de fragment d’Aβ, variant entre 39 et 43 acides aminés (6), est définie par le clivage via la γ-sécrétase (37). La forme la plus commune obtenue par ce clivage est du peptide Aβ1-40 (36). L’Aβ est produite directement au cerveau, mais également à la périphérie. L’Aβ circulante entre au cerveau grâce à l’action du récepteur des produits finaux de
glycosylation avancée (RAGE) (Fig. 1.2) (39,40) et ce, uniquement au niveau du système nerveux central (SNC) (41). En effet, Roberts et ses collègues (2014) détectèrent aucune fluctuation des concentrations d’amyloïde au niveau des veines périphériques signifiant que l’entrée et la sortie de l’Aβ s’effectue uniquement au niveau du système nerveux central (SNC) (41).
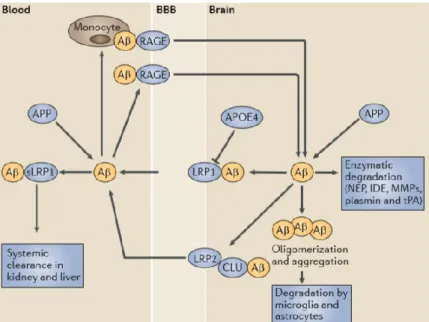
L’élimination de l’Aβ devient alors un phénomène intrinsèque à l’homéostasie. L’amyloïde est éliminée du cerveau par divers mécanismes (Fig. 1.2) (42). Notamment, les cellules de l’unité neurovasculaire (NVU) (Section 1.5 – Unité neurovasculaire) sécrètent des protéases contribuant à sa dégradation (43), telles que la plasmine, la néprilysine (NEP), la NEP2, l’enzyme de dégradation de l’insuline (IDE), l’enzyme de conversion de l’angiotensine (ACE), les métalloprotéases matricielles (MMPs), etc. (44). De plus, certaines cellules de la NVU, astrocytes (43,45), péricytes (46), et microglies (43,47), peuvent internaliser l’Aβ. Cette internalisation est généralement dépendante de la protéine 1 apparentée au récepteur des lipoprotéines à faible densité (LDLs), LRP1. Une fois lié, LRP1 promeut l’internalisation de son ligand et le dirige vers les lysosomes où il sera dégradé (48,49). De plus, LRP1 et la p-glycoprotéine (PgP) contribuent à la transcytose rapide de l’amyloïde soluble, de l’Aβ associée à l’α-macroglobuline (42,50) et de l’Aβ associée à l’apolipoprotéine (Apo) E2 ou 3 (51) à travers la barrière hémato-encéphalique (BBB) au
Figure 1.1. Voies de transformation de l’APP. L’APP via deux cascades, l’une non-amyloïdogénique et
l’autre amyloïdogénique. L’APP est clivée par l’α-sécrétase formant deux fragments, APP-sα et CTFα. l’APP peut également être endocyter, puis clivé par la β-sécrétase produisant le fragment APP-sβ et le CTFβ. Celui-ci forme alors l’Aβ lorsque clivé par la γ-sécrétase. APP-sα: L’amyloïde est ensuite sécrétée dans le milieu extracellulaire où son accumulation promeut la formation de plaques (44) [tiré intégralement].
sang veineux (48,49) et à travers le plexus choroïde au fluide cérébrospinal qui est ensuite réabsorbée par les veines cérébrales (52). Une équipe en particulier , permirent entre-autre d’évaluer l’importance de l’élimination de l’Aβ du CNS à travers la BBB à 25% et celle via les plexus choroïdes également à 25% (41). En plus d’exprimer LRP1 (53,54), les cellules de la BBB expriment ABCB1 (50,55) et LRP2 (56) modulant le transport de l’Aβ. Plus précisément, LRP2 orchestre l’influx et l’efflux du complexe formé par l’Aβ et la clusterine, également appelée ApoJ (56). Tout comme LRP1, LRP2 est impliqué au niveau de la dégradation de l’Aβ par endocytose, de même que son élimination par la BBB lorsque celle-ci est associée à l’ApoJ (57). L’Aβ peut former un complexe avec l’ApoE. Celle-ci possède trois isoformes: ApoE2, ApoE3 et ApoE4 (58), dont chacun module différemment l’élimination de l’Aβ. Comparativement à l’ApoE2 et l’ApoE3, le complexe de l’Aβ avec l’ApoE4 est internalisé via le récepteur LRP1 et éliminé du cerveau beaucoup plus lentement (51,59,60). Ainsi, l’ApoE4 a un effet de rétention de l’Aβ au cerveau. Suite au passage à travers la BBB, l’Aβ se lie à LRP soluble (sLRP) qui est produit par le clivage de LRP par la β-sécrétase (61). LRPs agit alors comme un «siphon périphérique» captant l’Aβ du cerveau dans le sang et la transportant jusqu’au foie (62) où elle sera dégradée (62,63).
Figure 1.2. Voie d’entrée et d’élmination de la β-amyloïde. Les voies d’entrées et d’élimination de l’Aβ
démontre l’importance de l’unité neurovasculaire à son homéostasie. L’Aβ est produite à partir de l’APP au cerveau et à la périphérie. L’Aβ est dégradée enzymatiquement par la néprylisine, l’IDE, les métalloprotéinase et la plasmine. L’Aβ non-dégradée peut s’oligomériser et être dégradée par les microglies et les astrocytes. L’efflux de l’Aβ est médiée par LRP1/2 alors que l’influx est modulé par RAGE. L’Aβ s’associe à LRP1 soluble dans la circulation sanguine. L’élimination systémique de l’Aβ se produit au niveau du foie. CLU: Clusterine. (42) [tiré intégralement].
1.1.2.2. Agrégation de l’amyloïde
Toutefois, avec l’âge ou en condition pathologique, ces voies d’élimination de l’Aβ sont altérées (64). Par exemple, l’activité enzymatique de l’IDE et de la NEP diminuent avec l’âge (65). De plus, l’activité de la NEP2 (66) et l’expression de la NEP (67) diminuent chez les patients déments. Chez les malades d’Alzheimer, il a également été observé que LRPs est oxydé (62) en plus d’une expression accrue de RAGE à la surface des cellules endothéliales de la BBB (53). Ces deux phénomènes contribuent respectivement à la réduction de la liaison de l’Aβ à LRPs oxydé (62) et l’augmentation de l’entrée d’Aβ au cerveau (53). Avec l’âge et lors de la AD, la capacité des microglies à dégrader l’Aβ décroît (68). L’ensemble de ces altérations contribue à l’accumulation de l’Aβ et la formation d’agrégats (53). Grâce à une étude de spectrométrie de masse, la dynamique de nucléation des plaques amyloïdes a pu être décrite. Bernstein et ses collègues ont déterminés que l’Aβ1-40 formait un dimère puis un tétramère, alors que l’Aβ1-42 formait des dimères, tétramères,
hexamères, puis dodécamères. Cette structure de dodécamère constitue le noyau des agrégats pouvant former, par un processus lent, des protofibrilles (69). Les plaques amyloïdes sont formées principalement de ce nucléus, en plus d’autres protéines dont l’α1-antichymotrypsine (70), l’ApoE (71), le protéoglycane à héparane sulfate (71) et la thrombospondine (72). Avec le temps, ces plaques amyloïdes grossissent et s’organisent en feuillet-β jusqu’à se stabiliser tel que confirmé par leur suivi longitudinal in vivo (73).
1.1.2.3. Hypothèse de la cascade amyloïde
Outre la AD, la déposition d’amyloïde est commune à plusieurs troubles neurodégénératifs tels le syndrome de Down (74), la maladie de Parkinson, la maladie d’Huntington (75), la démence vasculaire, la CAA et l’atrophie corticale postérieure (76), ce qui suggère une étroite relation entre la neurodégénérescence et l’amyloïde. Cette théorie est d’ailleurs renforcie par plusieurs évidences génétiques sur le gène APP identifiées chez des malades Alzheimer et dont, lorsqu’exprimé chez des modèles animales, suffisent au développement de la AD (16). De ce fait, les scientifiques ont émis l’hypothèse de la cascade amyloïde. Elle postule que l’Aβ initie une cascade cellulaire provoquant la perte neuronale et des dommages neuronaux qui ont initialement été attribués au nombre de plaques séniles (36). Toutefois, le déclin cognitif ne corrèle pas avec les dépôts d’amyloïde et la perte synaptique (77). Dès lors, l’hypothèse de la cascade amyloïde a été révisée (36,78). Il a alors été proposé que l’Aβ soluble, oligomère et dodécamère (69), serait à l’origine de la neurotoxicité (79,80). Les oligomères d’Aβ induisent l’hyperphosphorylation de tau (Section
1.1.1 – Tau) (29,30). Certes, ils peuvent également interagir avec plusieurs protéines neuronales
calciques (81), les récepteurs ionotropiques AMPA (82), les récepteurs ionotropiques NMDA (83) et les récepteurs métabotropiques du glutamate 5 (mGluR5) (84). En se liant aux neurones, l’amyloïde induit une signalisation aberrante (e.g .voie Wtn) un influx calcique anormal (81), en plus d’une activité et d’une plasticité synaptique altérées (81,85). Dans leur ensemble, les effets induits par les oligomères d’Aβ contribuent au déclin cognitif.
L’Aβ module également l’expression de gènes critiques à l’apprentissage, la mémoire et la neuroprotection (86). Par exemple, les fibrilles amyloïdes inhibent l’expression de la neuroligine, ce qui promeut la neurodégénération (87). Mis à part les oligomères et les fibrilles, l’amyloïde intracellulaire contribue également à la toxicité au sein de la mitochondrie résultant en un dysfonctionnement mitochondrial reconnu chez les malades d’Alzheimer (88). L’implication des fibrilles, des oligomères insolubles et de l’amyloïde intracellulaire reste à explorer afin de préciser cette hypothèse.
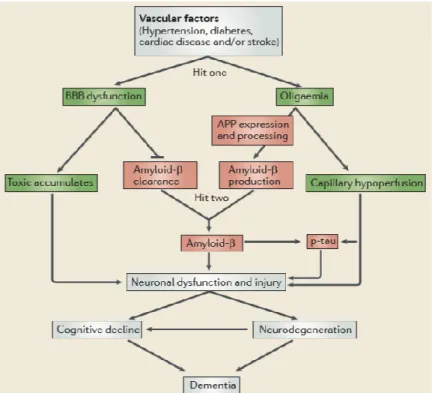
Figure 1.3. Schéma de l’hypothèse vasculaire de la maladie d’Alzheimer. Les facteurs vasculaires
provoquent le premier dommage: la dysfonction de la BBB et l’oligémie. La dysfonction de la BBB provoque une accumulation de substances toxiques et une diminution de l’élimination de l’Aβ. L’oligémie entraîne une hypoperfusion capillaire en plus d’une augmentation de l’expression et de la transformation de l’APP. Cela provoque le deuxième dommage : l’augmentation du niveau d’Aβ. L’augmentation de l’Aβ entraîne une hyperphosphorylation de tau et, favorise le dysfonctionnement neuronal conduisant à long terme à la démence (42) [tiré intégralement].
1.1.3. Hypothèse vasculaire
Récemment, une autre hypothèse, considérant les facteurs de risque de la AD, a vu le jour, soit l’hypothèse vasculaire (Fig. 1.3). Elle suppose qu’un dommage initial de la microcirculation cérébrale initie le dysfonctionnement neuronal amyloidogénique. Cette voie non-amyloidogénique comporte une rupture de la BBB, induisant une perte d’étanchéité et la sécrétion de molécules neurotoxiques. De plus, le dommage vasculaire occasionne une oligémie pouvant provoquer des ischémies focales. Ces deux phénomènes provoquent une augmentation de l’Aβ respectivement par la perte de l’élimination et par une élévation de la production de l’Aβ (42). En effet, des études ont démontré qu’un contexte hypoxique ou ischémique favorisait la conversion de l’APP en Aβ en induisant l’augmentation de l’activité de la β-sécrétase et de la γ-sécrétase, en plus de l’expression de la β-sécrétase (89). Loin de contredire l’hypothèse de la cascade amyloïde, l’hypothèse vasculaire considère la dynamique de la pathogenèse de la AD. Cependant, le processus pathologique lui-même est encore peu connu. L’étude des comorbidités telles que les troubles vasculaires permettent de dévoiler la cascade pathologique de la AD.
1.2. Facteurs de risque de la maladie d’Alzheimer
La AD possède une étiologie complexe qui progresse sur une vie. L’âge est le facteur ayant le plus d’incidence sur la progression de la forme sporadique (90). De plus, la génétique (91,92), l’obésité, le diabète (93,94), l’accident vasculaire cérébral (AVC) (93–96), l’hypertension (93–95), l’hypotension (95), les maladies coronariennes (95,96), la fibrillation auriculaire (97), l’athérosclérose (93,98) et l’hypercholestérolémie (93) représentent des facteurs de risque de la AD tardive. Nous résumerons brièvement l’implication de la génétique, soit l’incidence des différents isoformes d’ApoE, de l’obésité, du diabète et des troubles vasculaires dans la pathogenèse de la AD.
1.2.1. Génétiques
ApoE4 constitue le facteur génétique principal de la AD tardive. Les personnes porteuses de l’allèle ApoE4 présentent 4 à 10 fois plus de risque de développer la AD (91,92). Chez l’homme, le gène ApoE se localise sur le chromosome 19 (92) pour lequel 3 allèles du même locus génétique ont été identifiées, ApoE2, ApoE3 et ApoE4 (58,92). L’isoforme ApoE4 est la plus primitive (99) et la moins prévalente (100). Au sein de la population, 30%, 60% et 10% de la population possède respectivement l’isoforme ApoE2, ApoE3 ou ApoE4 (100). Ces isoformes varient d’une substitution d’un seul acide aminé au niveau de deux résidus: ApoE2 (Cys112, Cys158), ApoE3
(Cys112, Arg158) et ApoE4 (Arg112, Arg158) (101). Ces mutations ont des effets majeurs sur sa structure et sa fonction (101).
L’ApoE est produite principalement par les astrocytes au cerveau (102) et par le foie à la périphérie. Circulant dans le sang, elle est intégrée aux lipoprotéines de très faible densité et aux chylomicrons. L’ApoE régule le transport du cholestérol et des lipides. Elle module également l’élimination des lipoprotéines plasmiques par les récepteurs LDL (103). Tel qu’évoqué plus tôt, l’ApoE module le transport de l’Aβ selon l’isoforme exprimée. L’ApoE2 et l’ApoE3 facilitent l’élimination de l’Aβ, alors que l’ApoE4 favorise sa rétention (51). L’ApoE2 possède une structure plus stable due à la substitution d’acides aminés, ce qui lui confère un effet protecteur contre la AD (101). L’ApoE2 et l’ApoE3 contribuent également à la plasticité synaptique et la réparation neuronale (100). À l’opposé, l’ApoE4 est étroitement associée au dysfonction de la BBB (104), à l’augmentation de l’incidence des maladies vasculaires (105), de la CAA (106) et celle de la AD (91,92).
1.2.2. Obésité
Chez la personne obèse, les tissus adipeux, plus importants, produisent des cytokines qui seront sécrétées dans la circulation sanguine (e.g. TNFα, IL-6). Ces cytokines circulantes peuvent altérer la fonction endothéliale et contribuer à la résistance à l’insuline (107). En cours de vie, ce phénomène résulte en une augmentation du risque de diabète de type 2 (108) et des maladies cardiovasculaires dont l’hypertension artérielle (109). Ces maladies secondaires à l’obésité sont reconnues comme ayant des effets nocifs sur le cerveau (94). De ce fait, il a été reporté que l’obésité à mi-vie est associée à un déclin cognitif en fin de vie (93,94,110).
1.2.3. Diabète
Le diabète favorise le déclin cognitif, ce qui promeut la AD (93,94). Sonnen et ses collaborateurs ont reporté une aggravation de la démence chez les malades d’Alzheimer diabétiques non contrôlés par rapport à ceux qui sont traités (111). De ce fait, lors d’épisodes d’hyperinsulinémie, le niveau d’insuline cérébrale augmente. L’insuline cérébrale compétionne alors avec l’Aβ extracellulaire en tant que ligand de l’IDE, ainsi la dégradation de l’amyloïde par l’IDE est réduite (112). L’hyperinsulinémie entraîne également une altération de la signalisation de l’insuline, du stress oxydatif et des mécanismes inflammatoires pouvant notamment contribuer au déclin cognitif (113). De plus, le diabète peut entraîner des complications dont les maladies vasculaires, les néphropathies, les neuropathies et les rétinopathies (114). L’augmentation de l’incidence de démence chez les diabétiques a été d’ailleurs attribuée aux maladies cardiovasculaires (115).
1.2.4. Troubles vasculaires
En plus d’être des facteurs de risque de la AD, l’expression d’ApoE4, l’obésité et le diabète accroient les risques de développer des troubles vasculaires (105,109,115). Plusieurs études épidémiologiques, cliniques et animales ont démontré que les troubles vasculaires systémiques tout comme les maladies cardiovasculaires en mi-vie contribuent à la défaillance cognitive (93,116). Nous comptons parmi ces pathologies: l’AVC (93–96), l’athérosclérose (93,98), la fibrillation artriale (97), l’hypercholestérolémie (93), l’hypertension (93–95,117,118), l’hypotension (95,117,118) et les maladies coronariennes (95,96).
1.3. Hypoperfusion
Afin de bien fonctionner, le cerveau nécessite un approvisionnement constant et régulé de nutriments et d’oxygène lequel est orchestré par le CBF. En fait, le cerveau est un des organes les plus actif et consomme jusqu’à ~20% de l’oxygène et ~25% du glucose consommés par le corps ce qui correspond à 20% du débit cardiaque (119). Qui plus ait, depuis la dernière décennie, l’importance de la perfusion sanguine cérébrale a été à de nombreuses reprises démontrée et ce, par les effets délétères qu’à une réduction du CBF. La cas échéant, nous observons une perturbation des effecteurs de la mémoire (120) conduisant à une altération de l’apprentissage et de la mémoire (121).
1.3.1. Régulation du flux sanguine et maladie d’Alzheimer
En condition physiologique, la NVU effectue des ajustements vasculaires afin de maintenir le CBF stable. Elle régule égalment la distribution du sang selon les demandes énergétiques (121). Cette régulation du CBF est appelée couplage neurovasculaire (119,121). En cas de troubles vasculaires, des événements cellulaires et moléculaires sont déclenchés qui entraînent un dysfonctionnement de la NVU (95) suivi d’une réduction du flux cérébral sanguin (CBF) (95,96,98,117,122). La NVU est alors incapable de combler les demandes énergétiques des régions actives du cerveau (121) et de contrôler le CBF (95). Cette perte de l’autorégulation s’observe par des fluctuations du flux saguin chez les souris transgéniques Alzheimer (123), les malades d’Alzheimer (118,124) et ceux MCI (124,125). Suite à ces évidences, plusieurs ont tenté de démontrer la relation entre l’hypoperfusion et la démence. Une étude intéressante à grande échelle a permis de confirmer cette hypothèse (126). D’autres études épidémiologiques suggèrent une contribution de l’hypoperfusion dans la pathogenèse de la AD (127,128). Par la technique de tomographie à émission de positrons (PET), Hunt et ses collègues ont également démontré une réduction du métabolisme du glucose cérébral chez des individus MCI situant l’altération vasculaire antérieure à la démence (129). Selon
l’hypoperfusion cérébrale, il a été observé une crise énergétique (95,130) où l’apport en glucose et en oxygène est réduit aux régions vulnérables (131). La déficience en glucose, causée par l’hypoperfusion, induit un stress oxydatif et réticulaire aux neurones hippocampaux et corticaux restreignant leur production d’ATP (132). Ainsi, l’hypoperfusion induit un dysfonctionnement neuronal (133) ce qui altère l’intégrité et la structure du cerveau (93,134) et, contribue à la neurodégénérescence et au déclin cognitif (95,130) ; phénomènes qui aggravent la AD (42). L’hypoperfusion induit également l’altération de l’élimination et/ou du transport de l’Aβ (93,121). Par conséquent, l’hypoperfusion induite par les troubles vasculaires initierait ou accélèrerait la cascade pathologique de la AD (42,135).
1.3.2. Oligémie versus ischémie
Il existe deux formes d’hypoperfusion cérébrale: l’oligémie et l’ischémie. L’oligémie décrit un processus lent pouvant prendre des mois ou des années à s’intaller, alors que l’ischémie réfère à une réduction assez rapide et soudaine du flux sanguin entraînant la mort de cellules au niveau d’une lésion dite ischémique (136). Suite à l’oligémie induite par la ligature unilatérale de l’artère carotide commune (1VO), le déclin cognitif est exacerbé chez les Tg2576 (137) et les APPswe/PS1 (138). Le même effet synergique a été observé chez les J20/APP (139) soumises à une ischémie induite par la sténose bilatérale des artères carotides communes (BCAS).
L’oligémie induit une réduction modérée du CBF associée à une diminution de la synthèse protéique (121,140). Certaines anomalies de la NVU ont également été observées suite à l’oligémie, dont l’altération de l’interaction intermodale axone-glie (141), l’épaississement de la membrane basale et la déposition de collagène sur la BBB (142). L’altération de la capacité mnésique décrite est causée par un dysfonctionnement neuronal (138,140), puisqu’aucune mort cellulaire n’est observée. Quant à l’ischémie, la réduction du CBF est plus importante provoquant généralement une hypoxie (42). L’hypoxie entraîne l’expression du facteur de transcription, HIF-1α, qui augmente l’expression de BACE-1 (89) et diminue l’expression de la neprilysine (143). De ce fait, les souris Tg swe/dutch/iowa soumises à la BCAS développent plus rapidement des dépôts amyloïdes associés à un stress ischémique (144). Lorsque la réduction du CBF est supérieure à 50%, celle-ci promeut un dysfonctionnement de la synthèse de l’ATP et une altération de l’activité neuronale (119). De surcroît, l’ischémie altère l’expression des protéines neurotrophiques et neuronales (145–147). Suite à l’ischémie, l’expression de MMP-2 augmente (148) alors que l’expression de claudine V et occludine diminuent (149), occasionnant une altération de la BBB. La rupture de la BBB interrompt le transport membranaire normal des nutriments, des vitamines et des
électrolytes, nuisant encore une fois au fonctionnement neuronal (150). Contrairement à l’oligémie, plusieurs études, basées sur le modèle de l’occlusion bilatérale des artères carotides communes (BCAO) ou de la BCAS, ont démontré que les dommages de la matière blanche et la perte neuronale sont à l’origine du déclin cognitif (139,149,151–157). Ces évidences indiquent un rôle central de l’hypoperfusion cérébrale, l’oligémie et l’ischémie, dans la pathogenèse de la AD. Bien que davantage d’études portent sur l’ischémie, la relation entre la AD et l’oligémie représente un sujet à approfondir, puisque les changements du CBF observés avec le vieillissement ressemble davantage à l’oligémie (130,142).
1.4. Système de l’activateur tissulaire du plasminogène
Suite à un stress vasculaire, tel que la formation d’un caillot sanguin ou thrombus occludant un vaisseau sanguin, l’activateur tissulaire du plasminogène (t-PA), une sérine protéase, est produit et libéré par les cellules endothéliales afin de lyser le caillot (158). Il s’ensuit la conversion par clivage protéolytique du plasminogène en plasmine par le t-PA (159–161). La plasmine contribue alors à dégrader le thrombus en dégradant la fibrine (162) et la laminine (163). Ce système du plasminogène est régulé à plusieurs niveaux. L’inhibiteur 1 des activateurs du plasminogène (PAI-1) inhibe le t-PA (159,160,164), alors que la neuroserpine interagit uniquement avec le t-PA (159,160,165,166). Quant à la plasmine, elle est inhibée par anti-plasmine et l’α2-macroglobuline (159,160). Le complexe formé avec les protéines inhibitrices est généralement internalisé par les cellules via LRP1, puis dégradé (167). Dans la circulation, le t-PA possède une demi-vie très courte, d’environ 5 minutes (168,169), suivant laquelle il est éliminé de la circulation par LRP1 au niveau du foie pour y être dégradé (170). Jusqu’à ce jour, une forme recombinante du t-PA (rt-PA) représente le seul traitement pour l’AVC ischémique (171), l’un des facteurs de risque de la AD (93–96).
1.4.1. Fonction du système de l’activateur tissulaire du plasminogène
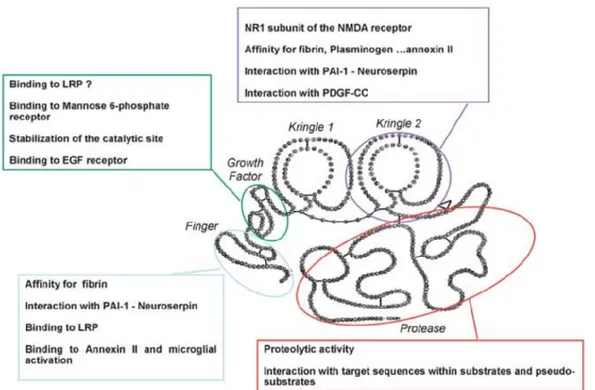
Présent endogéniquement dans la circulation comme enzyme thrombolytique (172), le t-PA est également exprimé au SNC par les astrocytes, les microglies et les neurones, soit à l’amygdale (173), au cervelet (174), au corps calleux (175), à l’hippocampe (165,173,174,176,177), à l’hypothalamus (173,176), etc. Au niveau du SNC, la majorité du t-PA qui agit sur celui-ci est produit endogéniquement et ce, par les cellules endothéliales de la BBB (158). Or, le t-PA circulant et exogène au cerveau peut également agir au niveau de celui-ci directement en traversant la BBB par transcytose médié par LRP1 (178). Plus précisément, le t-PA est une glycoprotéine de 69 kDa formée d’une seule chaîne de polypeptide organisé en 5 domaines: Kringle 1, Kringle 2, epidermal
growth factor(EGF)-like, en doigt et protéase (Figure 1.5) (179,180). Ce sont d’ailleurs ces différents domaines qui lui confèrent son effet pléiotropique au sein du SNC. Le t-PA possède une activité thrombolytique (162,163). Son activité enzymatique lui confère également des propriétés neuroprotectrices. En effet, au SNC, cette sérine protéase contribue notamment à moduler la plasticité synaptique (181–186), la potentialisation à long-terme (LTP) (170,173,187,188) et la dépression à long-terme (LTD), mais également la perméabilité de la BBB (170,179,189,190) et la réponse inflammatoire du cerveau (191–197).
Grâce à son activité enzymatique, le t-PA convertit des pro-neurotrophines en leur forme active (e.g. BDNF, NGF) (198,199) et participe au remodelage neuronal par la production de vésicules contenant la synaptophysine (183). Par la digestion des protéines de la matrice extracellulaire, le t-PA module la pousse axonale et, donc, la plasticité synaptique (185). Certes, certains des effets neurotrophiques du t-PA sont plutôt orchestrés par son action de cytokine qui est elle-même médiée
Figure 1.4. Structure et fonction de l’activateur tissulaire du plasminogène (t-PA). Le t-PA est composé
de 5 domaines: Kringle 1, Kringle 2, facteur de croissance, doigt de zinc et protéase. Le domaine Kringle 2 interagit avec l’unité NR1 du récepteur NMDA. Le domaine de facteur de croissance peut se lier au récepteur d’EGF et le récepteur du mannose-6-phosphate. Le domaine Kringle 2 et le domaine en doigt possèdent plusieurs ligands, soit la fibrine, le plasminogène, l’annexine II et les antagonistes du t-PA (PAI-1 et neuroserpine). Le domaine en doigt serait également le domaine liant LRP et potentiellement le domaine du facteur de croissance. PDGF-CC: Facteur de croissance CC dérivé des plaquettes. (174) [tiré intégralement].
par LRP1 (200). Le t-PA promeut également l’élongation des neurites par l’induction de kinases trophiques tel que ERK, la protéine kinase C et PI3K/Akt (201). La modulation de voie de signalisation par le t-PA lui confère entre-autre un effet anti-apoptotique sur les oligodendrocytes (175) et module l’apoptose neuronale (202,203). Toutefois, une activité excessive du t-PA a été associée à l’induction de la mort neuronale (204). Kim et ses collègues ont démontré que le t-PA empêche la mort par stress oxydatif des neurones par son action de cytokine, mais n’interrompt pas l’apoptose et l’excitotoxicité de ceux-ci (205). D’autres travaux proposent plutôt que le t-PA même induit l’excitotoxicité (163,175,206). Celle-ci peut être induite par le clivage du récepteur NMDA (175,206) au niveau de sa sous-unité NR1 à laquelle le t-PA se lie (207). En plus, le t-PA se lie à LRP (208) ce qui induit des signaux intracellulaires et promeut le flux calcique altèrant transmission (209,210). Ces dernières actions par le t-PA contribue alors à la LTD (211).
De nombreuses études mettent en évidence la modulation de la perméabilité de la BBB par le t-PA circulant (170,179,189,190). En se liant à LRP1 (170,212), le t-PA déclenche des signaux intracellulaires menant également à l’activation de MMP2/9 (212–215). Le t-PA module la perméabilité de la BBB suite à un stimulus (e.g. ischémie) qui, lorsque soutenu, s’avère nocif et peut provoquer la rupture de la BBB (216,217) et l’extravasation de fluide dans l’espace périvasculaire causant un œdème (217). En fait, lors de l’administration à l’extérieur de la fenêtre d’intervention thérapeutique, 4,5 heures suivant l’AVC (218), ou la sur-administration (219) du rt-PA suite à un AVC se produit, davantage d’effets néfastes que bénéfiques sont observés. En effet, l’administration tardive et le sur-dosage du t-PA augmentent les risques d’hémorrhagies cérébrales (219–221).
Par sa liaison avec LRP1 (222) ou l’annexine II (195), le t-PA est également apte à moduler l’inflammation en induisant le recrutement des macrophages (196), l’activité microgliale (192–195), en plus de la production de cytokines pro-inflammatoire (191–194). Une étude avait également reporté un rôle de cytokine anti-inflammatoire au t-PA (197). Cette activation des microglies s’avère nécessaire à la neurodégénération excitotoxique des neurones hippocampaux quoiqu’insuffisante à l’initier (177). Par conséquent, le t-PA possède plusieurs rôles et dépendemment du contexte son activité devient bénéfique ou néfaste pour le cerveau.
1.4.2. Système de l’activateur du plasminogène et maladie d’Alzheimer
Dans les modèles transgéniques murins, le t-PA est fortement exprimé autour des plaques denses amyloïdes résultant en leur arrêt de croissance ou leur dégradation (162,223,224). Les agrégats d’Aβ de type fibrilles stimulent l’expression du t-PA (225) ce qui entraîne la dégradation de
l’amyloïde via la plasmine (225,226). Le t-PA contribue également à la dégradation de l’Aβ indirectement par l’induction de l’activation de MMP2/9 (213–215). De plus, l’Aβ régule positivement la neuroserpine (227) qui intéragit directement avec l’Aβ et limite la formation de fibrilles (228). Des études avec des souris «knock-out» du PAI-1 (229) et de la neuroserpine (230) révèlent une réduction de la quantité d’amyloïde soulignant l’importance du t-PA dans l’élimination de l’Aβ. Cet effet du t-PA a également été observé suite à l’inhibition pharmacologique de régulateur de la plasmine (231). Dans cet ordre d’idée, la déplétion du t-PA augmente le nombre de plaques en plus d’aggraver les déficits cognitifs chez les Tg2576/t-PA+/– (232). Malheureusement,
avec le temps, le système du plasminogène devient inefficace à dégrader des dépôts d’amyloïde (162). En effet, le t-PA réduit dramatiquement avec l’âge et la AD (162,175,198,233,234). Quoiqu’une certaine étude n’a pas observé d’altération du système du t-PA (235), la majorité des travaux effectués chez les patients et les souris transgéniques démontrent une diminution de l’expression et de l’activité du t-PA (162,227,233,234) et de la plasmine (226,227), alors que celle du PAI-1 (162,236) et de la neuroserpine (227) augmentent. Dans ces conditions, la déposition de fibrine augmente (237,238). Il s’en suit alors des dommages neurovasculaires, en plus d’une réaction inflammatoire (239). La dysfonction du t-PA entraîne également des déficits sévères de la plasticité synaptique (211) ce qui contribue à la progression de la AD. Ainsi, le t-PA et les protéines du système du plasminogène représentent des cibles thérapeutiques potentielles.
1.5. Unité neurovasculaire
Tel que brièvement mentionné plutôt, la NVU (Fig. 1.5) représente l’unité intrinsèque au sein de laquelle les cellules de la BBB (cellules endothéliales et péricytes), les cellules gliales (astrocytes, oligodendrocytes et microglies) et les neurones interagissent et communiquent étroitement entre eux (119,121). Les cellules endothéliales composent l’endothélium des microvaisseaux où elles forment des jonctions serrées et adhérentes qui limitent les échanges passifs (119,240). Les péricytes entourent près de 80% de la surface de ces microvaisseaux (241) via un contact peg-socket essentiel à leur maintien, qui constitue des projections cytoplasmiques entrelancées ancrant les péricytes sur les cellules endothéliales (242). Les pieds astrocytaires entourent les cellules endothéliales complétant la BBB et permettant la communication entre les neurones et les cellules de la BBB (243), en plus de maintenir la fonction de la celle-ci (244). L’intégrité de cette unité fonctionnelle est essentielle au bon fonctionnement du cerveau (121). La NVU contribue notamment à la maintenance de l’homéostasie cérébrale, au couplage neurovasculaire (119,121,243,245,246), à la perméabilité de la BBB (119,240,247), etc.
1.5.1. Fonction de l’unité neurovasculaire
Par exemple, la BBB forme une barrière physique et sélective (247) régulant les échanges de métabolites et de nutriments nécessaires au bon fonctionnement neuronal (119). La BBB est donc cruciale à la maintenance de la composition du fluide interstitiel du cerveau, mais également à l’élimination de macromolécules potentiellement toxiques (119) telles l’Aβ (68).
Les péricytes, quant à eux, sont des cellules contractiles modulant le CBF via la constriction de la paroi des vaisseaux sanguins (246). Ils expriment d’ailleurs plusieurs protéines associées à la contraction dont l’α-actine spécifique aux muscles lisses, la tropomyosine et la desmine (248). Ils stabilisent également les capillaires nouvellement formés (249). En effet, ils jouent un rôle important dans la régulation de la prolifération, la survie et la migration des cellules endothéliales, en plus de moduler les connections des vaisseaux cérébraux (250). Ils prennent part à l’inflammation du fait de l’expression de récepteur de l’immunité innée (251) et leur capacité de recrutement de leucocytes au site d’inflammation (252,253). Qui plus est, les péricytes possèdent une activité semblable aux macrophages induite par la signalisation de récepteurs tels le récepteur de type Toll(TLR)-4 (254,255) et LRP1 (46). Cela leur permet d’ailleurs d’internaliser l’Aβ pour le dégrader (46).
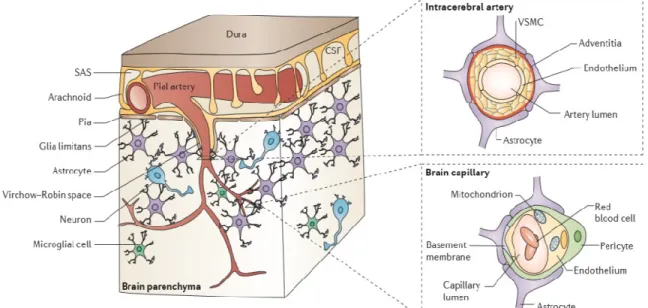
Figure 1.5. Représentation de l’unité neurovasculaire et de la microcirculation cérébrale. Les artères
piales, situées dans l’espace sous-arachnoïdie, se divisent en capillaires innervant le parenchyme. Les cellules endothéliales entourées par les péricytes et en contact avec les pieds astrocytaires forment une barrière étanche, la BBB. La BBB interagie avec les neurones et les microglies ce qui forme une unité fonctionnelle, la NVU. SAS: Espace subarachnoïde, VSMC: Cellules vasculaires des muscles lisses. (42) [tiré intégralement].