Caractérisation structurale et dynamique d’UbKEKS, une ubiquitine nouvellement identifiée et encodée dans un pseudogène
85
0
0
Texte intégral
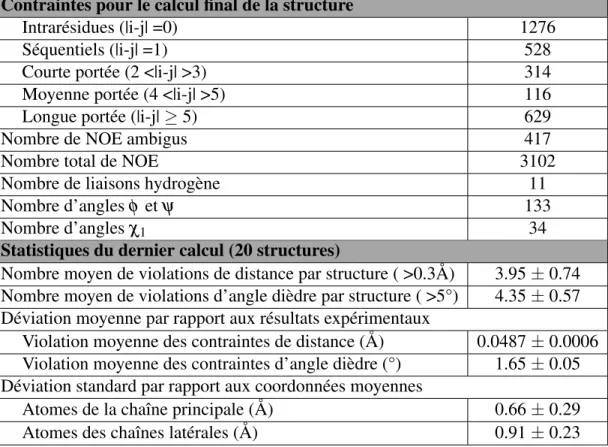
Figure
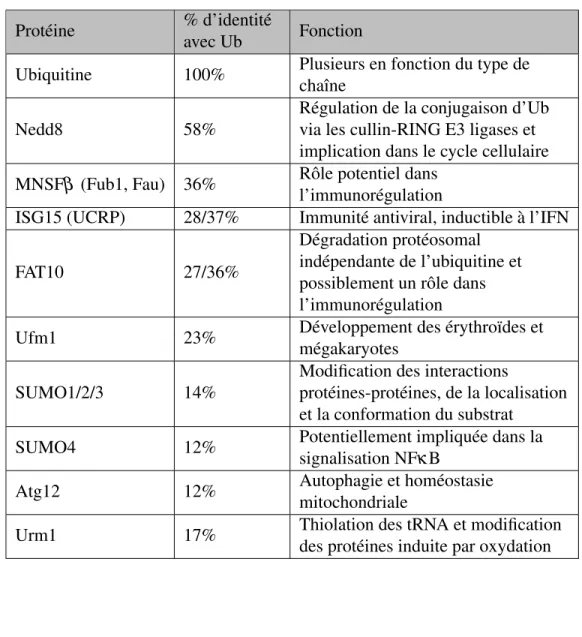
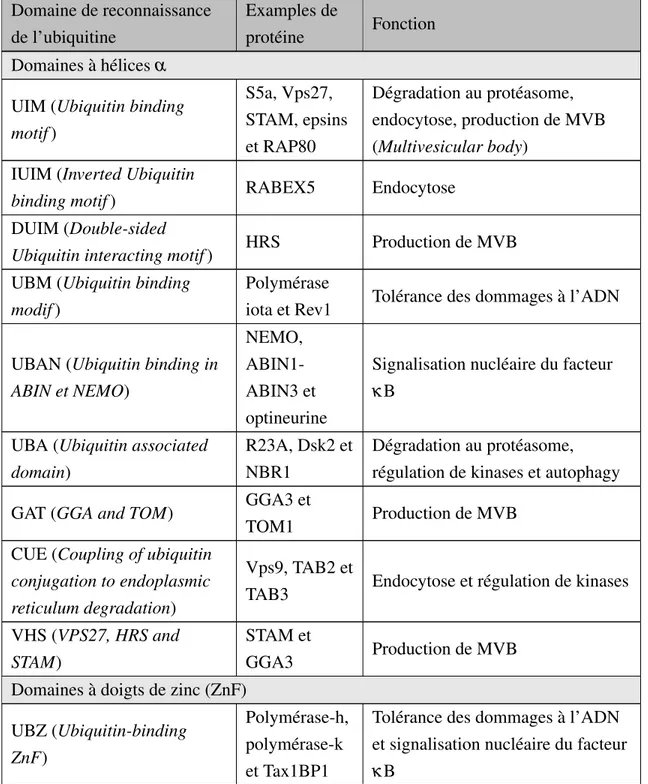
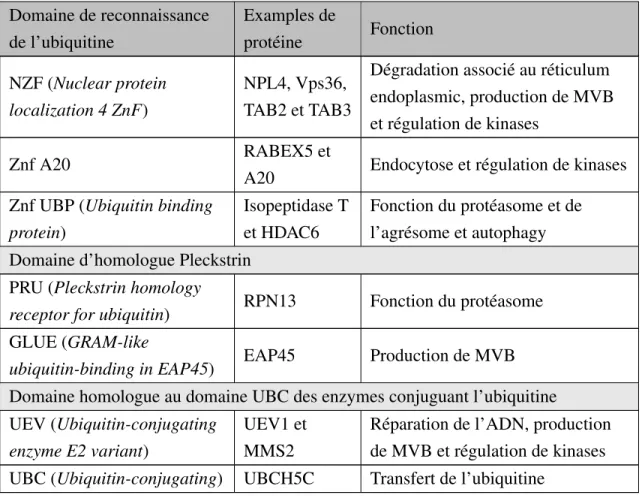
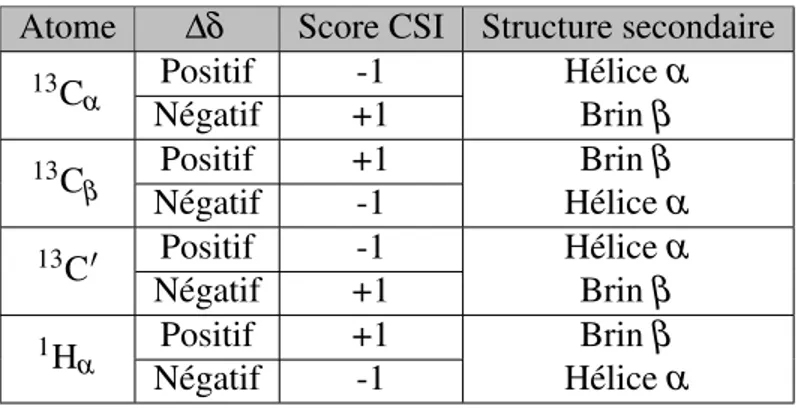
+2
Documents relatifs