Construction d'un clone produisant des vecteurs
rétroviraux s'auto-inactivant pour le traitement de
l'épidermolyse bulleuse dystrophique récessive par
thérapie génique
Mémoire
Michael Boivin-Welch
Maîtrise en biologie cellulaire et moléculaire - avec mémoire
Maître ès sciences (M. Sc.)
Construction d'un clone produisant des vecteurs
rétroviraux s'auto-inactivant pour le traitement de
l'épidermolyse bulleuse dystrophique récessive par
thérapie génique
Mémoire
Michael Boivin-Welch
Sous la direction de :
Manuel Caruso, directeur de recherche
Lucie Germain, codirectrice de recherche
Résumé
L’épidermolyse bulleuse dystrophique récessive est une maladie génétique rare causée par des mutations dans COL7A1 codant pour le collagène de type VII. Cette protéine est produite par les kératinocytes de l’épiderme et les fibroblastes du derme et permet la formation des fibrilles d’ancrages dont le rôle est l’adhésion de l’épiderme au derme sous-jacent. Les patients atteints de l'EBDR souffrent de décollements de la peau et des muqueuses et aucun traitement ne permet de guérir cette maladie. Le but de ce projet est de construire un vecteur viral sécuritaire contenant l'ADNc de COL7A1 et de générer un clone de cellules productrices de virus à haut titre permettant de transduire assez de kératinocytes souches pour produire des peaux reconstruites et traiter les patients atteints de l'EBDR. Divers vecteurs rétroviraux "self-inactivated" exprimant l'ADNc de gfp ou de COL7A1 ont été générés. Les vecteurs GFP ont permis de déterminer que le remplacement de la région U3 du LTR 5' par l'enhancer et le promoteur du CMV, l'ajout de la séquence WPRE en 3' UTR du transgène, l'insertion de la séquence de polyadénylation du SV40 dans la région R du LTR 3' et l'ajout de la séquence de polyadénylation de bGH en aval du LTR 3', génèrent les titres viraux les plus élevés. L'ADNc de COL7A1 a été introduit dans un rétrovirus SIN optimisé puis transfecté dans une lignée d’encapsidation exprimant l’enveloppe Amphotropique 4070A. Un clone de cellules productrices produisant 9,8 x 105 particules virales par mL a été isolé. Le virus produit
par ce clone est capable de transduire les kératinocytes et fibroblastes de patients atteints de l'EBDR à un taux de transduction de 37 % et 24 % respectivement. En somme, un clone de cellules productrices d’un vecteur rétroviral sécuritaire portant COL7A1 a été généré et a le potentiel d’être utilisé pour la thérapie génique de l’EBDR.
Abstract
Recessive dystrophic epidermolysis bullosa is a rare genetic disorder caused by mutations in
COL7A1 encoding type VII collagen. This protein is produced by the epidermal keratinocytes and
dermal fibroblasts and allows the formation of anchor fibrils whose role is the adhesion of the epidermis to the underlying dermis. Patients with EBDR suffer from skin and mucosal detachments and no treatment can cure this disease. The goal of this project is to construct a safe viral vector containing COL7A1 cDNA and to generate a high-titer virus-producing cell clone in order to transduce enough keratinocytes stem cells to produce reconstructed skins to treat patients with EBDR. Various self-inactivated retroviral vectors expressing gfp or COL7A1 cDNA have been generated. The GFP vectors allowed us to determine that the replacement of the U3 region of the 5' LTR by the CMV enhancer and promoter, the addition of the WPRE sequence to the 3 'UTR of the transgene, the insertion of the SV40 polyadenylation sequence into the R region of 3' LTR and the addition of the polyadenylation sequence of the bGH downstream of the 3' LTR generate the highest viral titers.
COL7A1 cDNA was introduced into an optimized SIN retrovirus and transfected into a packaging cell
line expressing the Amphotropic envelope 4070A. A clone of packaging cells producing 9.8 x 105 viral
particles per mL was isolated. The virus produced by this clone is capable of transducing the keratinocytes and fibroblasts of patients with EBDR at a transduction rate of 37 % and 20 % respectively. In sum, a clone of cells producing a safe COL7A1 retroviral vector has been generated and has the potential of being used for gene therapy of EBDR.
TABLE DES MATIÈRES
Résumé ... III Abstract ... IV Liste des tableaux ... VII Liste des figures ... VIII Liste des abréviations ... IX Remerciements ... XII
Introduction ... 1
L'épidermolyse bulleuse ... 1
Les différents types de l’épidermolyse bulleuse ... 1
L’épidermolyse bulleuse simplexe ... 3
L’épidermolyse bulleuse jonctionnelle ... 4
L’épidermolyse bulleuse dystrophique ... 5
Le syndrome de Kindler ... 5
L’épidermolyse bulleuse dystrophique récessive ... 7
Structure et fonctions de l'hélice α1 du collagène de type VII ... 7
Maturation du collagène de type VII ... 9
Mutations dans le collagène de type VII ... 11
Cancer épidermoïde agressif ... 12
Les traitements pour l’épidermolyse bulleuse dystrophique récessive ... 14
Thérapie pharmacologique ... 14
Thérapie cellulaire ... 16
L’édition du génome ... 17
Thérapie génique ... 18
Historique de la thérapie génique ... 18
Virologie ... 19
Différents vecteurs viraux disponibles pour la thérapie génique ... 19
Génome des rétrovirus ... 20
Maturation et infection des rétrovirus ... 22
Production de rétrovirus par une lignée d’encapsidation ... 25
Sécurité en thérapie génique ... 27
Apprentissages des essais cliniques antérieurs ... 27
Rétrovirus Self-Inactivating ... 28
Essai clinique avec sécurité améliorée ... 30
Thérapie génique de l’épidermolyse bulleuse dystrophique récessive ... 30
Cellules à transduire ... 30
Essais cliniques de thérapie génique de l’épidermolyse bulleuse ... 31
Hypothèse et objectifs ... 34
Chapitre 1. Matériels et méthodes ... 36
1.1 Construction des plasmides ... 36
1.2 Transformation ... 37
1.3 Culture cellulaire ... 37
1.4 Production de virus par transfection transitoire ... 38
1.5 Production de virus par transfection stable ... 38
1.7 Microscopie à fluorescence ... 40
1.8 Immunofluorescence et cytométrie en flux ... 40
1.9 Analyses statistiques ... 41
Chapitre 2. Résultats ... 42
2.1 Optimisation des rétrovirus SIN ... 42
2.1.1 Titres viraux des productions transitoires des vecteurs rétroviraux GFP ... 42
2.1.2 Titres viraux des productions stables des vecteurs rétroviraux GFP ... 47
2.1.3 Titres viraux des productions transitoires de vecteurs rétroviraux COL7A1 ... 49
2.2 Génération d'un clone producteur de virus COL7A1 à titre élevé ... 50
2.2.1 Génération des cellules productrices de virus ... 50
2.2.3 Isolement du meilleur clone ... 52
2.2.4 Sous-clonage et identification du moment optimal de récolte des surnageants viraux ... 53
2.3 Transduction de kératinocytes et fibroblastes de patients atteints de l'EBDR ... 54
Chapitre 3. Discussion ... 58
3.1 Optimisation des rétrovirus SIN ... 58
3.1.1 Titres viraux des productions transitoires des vecteurs rétroviraux GFP ... 58
3.1.2 Titres viraux des productions stables des vecteurs rétroviraux GFP ... 59
3.1.3 Titres viraux des productions transitoires de vecteurs rétroviraux COL7A1 ... 60
3.2 Génération d'un clone producteur de virus COL7A1 à titre élevé ... 61
3.2.1 Sélection de la lignée de titration ... 61
3.2.2 Isolement du meilleur clone ... 62
3.2.3 Sous-clonage et identification du moment optimal de récolte des surnageants viraux ... 63
3.3 Transduction de kératinocytes et fibroblastes de patients atteints de l'EBDR ... 63
3.3.1 Taux de transductions de kératinocytes et fibroblastes ... 63
3.3.2 Qualité des productions virales ... 64
3.3.3 Thérapie génique de l'épidermolyse bulleuse jonctionnelle et dystrophique ... 65
Conclusion ... 66
Perspectives ... 68
Liste des tableaux
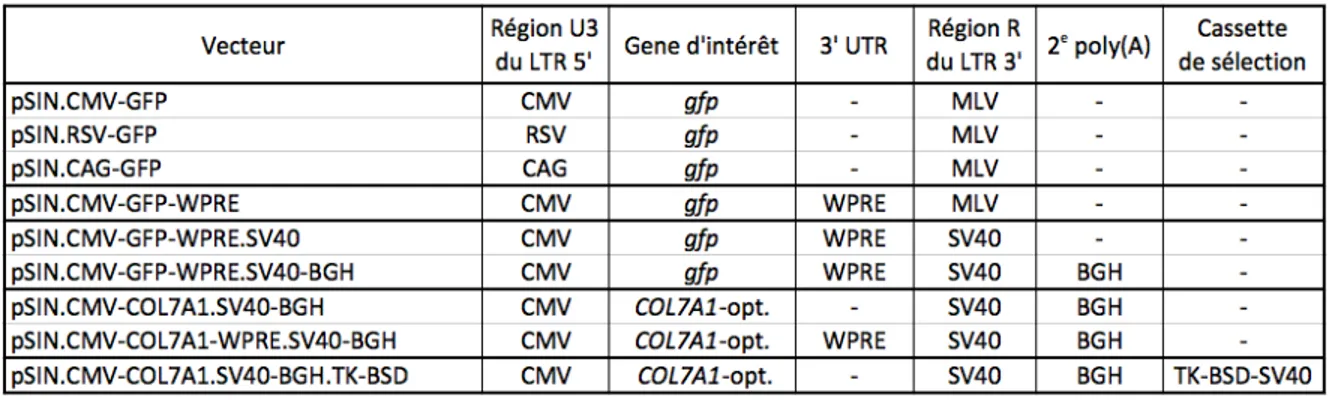
Tableau 1. Principaux virus utilisés en thérapie génique ... 20 Tableau 2. Plasmides codants pour les vecteurs rétroviraux SIN ... 43
Liste des figures
Figure 1. Couches de la peau et jonction dermo-épidermique par microscopie
électronique en transmission ... 3
Figure 2. Protéines impliquées dans les différents types d’épidermolyse bulleuse ... 6
Figure 3. Régions et domaines de l’hélice α1 du collagène de type VII ... 8
Figure 4. Maturation du collagène de type VII ... 11
Figure 5. Génome du Mo-MLV et ARNm de l'enveloppe ... 22
Figure 6. Structure et composants de la particule virale de rétrovirus ... 24
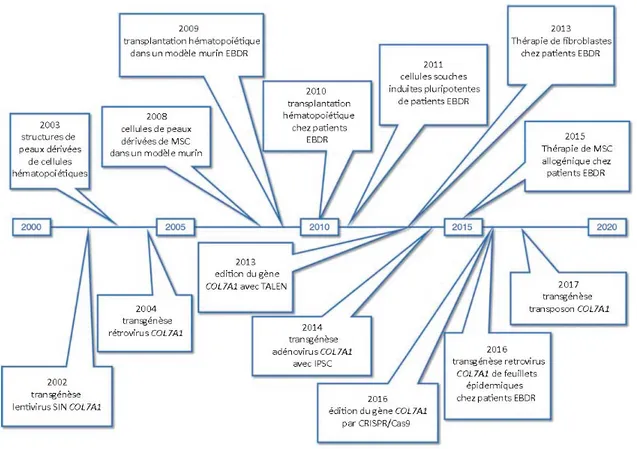
Figure 7. Les avancés de la thérapie de l’EBDR durant les 16 dernières années ... 34
Figure 8. Plan de conception des rétrovirus SIN ... 44
Figure 9. Analyse des titres viraux des vecteurs rétroviraux SIN exprimant GFP par cytométrie en flux ... 45
Figure 10. Analyse des titres viraux de vecteurs rétroviraux SIN exprimant GFP par cytométrie en flux ... 48
Figure 11. Analyse de l’impact de la séquence WPRE sur le titre viral de SIN.CMV-COL7A1.SV40-BGH par cytométrie en flux ... 50
Figure 12. Analyse de la fluorescence de COLVII dans différentes lignées cellulaires par cytométrie en flux ... 52
Figure 13. Criblage des clones 293Vec-Ampho exprimant SIN.CMV-COL7A1.SV40-BGH par cytométrie en flux et analyse de la fluorescence de COLVII de ces clones .... 53
Figure 14. Cinétique de la production virale et analyse de la fluorescence de COLVII des clones 293Vec-Ampho exprimant SIN.CMV-COL7A1.SV40-BGH par cytométrie en flux ... 54
Figure 15. Analyse de la fluorescence de COLVII dans des kératinocytes et fibroblastes par cytométrie en flux ... 56
Figure 16. Expression de GFP dans des monocultures de kératinocytes transduits par MFG-HYGRO.IRES-GFP par microscopie à fluorescence ... 57
Figure 17. Analyse de la fluorescence de GFP dans des kératinocytes et fibroblastes par cytométrie en flux ... 57
Liste des abréviations
AAV ADN ADNc Ampho ARN ARNg ARNm ARNt bGH BSD CA CAG CMV COLVII CRISPR CSP DMEM EB EBD EBDR EBJ EBS EF-C EF1-α ENV GAG GAG-POL GALV GFP IFM IN JDE kpb LB LTR MA MFG Mo-MLV MOI MSC NC NC1Adeno-Associated Virus (virus associés-aux-adénovirus)
Acide désoxyribonucléique
Acide désoxyribonucléique complémentaire Amphotropique
Acide ribonucléique
Acide ribonucléique génomique Acide ribonucléique messager Acide ribonucléique de transfert
Bovine Growth Hormone (hormone de croissance bovine)
Blasticidine S Déaminase Capside
Promoteur synthétique: CMV, β-Actine, β-Globine Cytomégalovirus
Protéine collagène de type VII
Clustered Regularly Interspaced Short Palindromic Repeats (courtes répétitions
palindromiques groupées et régulièrement espacées) Codons-stops prématurés
Dulbecco’s Modified Eagle’s Medium (milieu minimum essentiel de Eagle)
Épidermolyse bulleuse
Épidermolyse bulleuse dystrophique
Épidermolyse bulleuse dystrophique récessive Épidermolyse bulleuse jonctionnelle
Épidermolyse bulleuse simplexe Peptide polycationique
Elongation Factor 1-alpha (Facteur d'élongation 1-alpha)
Glycoprotéine d'enveloppe
Group-Specific Antigen (polyprotéine GAG)
Group-Specific Antigen-Polymerase (polyprotéine GAG-POL) Gibbon Ape Leukemia Virus (virus de la leucémie du singe gibbon) Green Fluorescent Protein (protéine fluorescente verte)
Intensité de fluorescence moyenne Intégrase
Jonction dermo-épidermique Kilo paire de base
Lame basale
Long Terminal Repeat (séquence terminale longue répétée)
Matrice
Vecteur rétroviral
Moloney Murine Leukemia Virus (virus de la leucémie murine de Moloney) Multiplicity of Infection (multiplicité d'infection)
Mesenchymal Stem Cell (cellules souches mésenchymateuses)
Nucléocapside
NC2 nt Opt PB pb PBS PEI POL PPT PR R RBD RCR RD114 RER RH RSV RT SA SD SIN SK SNV SU SV40 TALEN TK TLR TM U3 U5 UI UTR VSV-G VWFA1 VWFA2 WPRE X-SCID ZFN
Noncollagenous 2 (non collagénique 2)
Nucléotides Optimisé Polybrène Paire de base
Primer Binding Site (site de liaison de l’amorce)
Polyethylenimine
Polymerase (Polyprotéine POL)
Polypurine Tract (séquence riche en purine)
Protéase virale
Repeat Sequence (séquence répétée)
Receptor-Binding Domain (domaine de liaison au récepteur)
Rétrovirus compétent pour la recombinaison Enveloppe de virus endogène de chat Réticulum endoplasmique rugueux Recombinaison homologue
Rous Sarcoma Virus (virus du sarcome de Rous) Reverse Transcriptase (transcriptase inverse) Splice-Acceptor (site accepteur d’épissage) Splice-Donor (site donneur d’épissage) Self-Inactivating (s’auto-inactivant)
Syndrome de Kindler
Spleen Necrosis Virus (virus de la nécrose de la rate)
Sous-unité de surface
Simian Virus 40 (virus simien 40)
Transcription Activator-Like Effector Nuclease (nucléases effectrices ressemblant aux
activateurs de transcription)
Promoteur thymidine kinase du virus herpes simplex de type 1
Toll-Like Receptor (récepteur Toll-like)
Sous-unité transmembranaire
Unique Sequence at 3’ End of Genome (séquence unique à l'extrémité 3' du génome) Unique Sequence at 5’ End of Genome (séquence unique à l'extrémité 5' du génome)
Unités Infectieuses
Untranslated region (région non-transcrite)
Vesicular Somatitis Virus G protein (protéine G du virus de la stomatite vésiculaire) von Willebrand factor A1 (facteur A1 de von Willebrand)
von Willebrand factor A2 (facteur A2 de von Willebrand)
Woodchuck Hepatitis Virus Post-transcriptional Regulatory Element (élément
régulateur post-transcriptionnel du virus de l’hépatite de la marmotte)
X-Linked Severe Combined Immunodeficiency (déficit immunitaire combiné sévère lié
à l’X)
Dédicace
Je dédie ce mémoire à tous les enfants atteints d'Épidermolyse bulleuse et À la mémoire de Jonathan Pitre. Jonathan était atteint d'épidermolyse bulleuse dystrophique récessive. Il était ambassadeur de cette maladie et reconnu pour sa vision très positive de la vie. Jonathan est décédé le 4 avril 2018 des complications de cette maladie avant qu'un traitement n'ait vu le jour.
Remerciements
Je tiens à remercier mon directeur de recherche le Dr. Manuel Caruso pour la formation qu’il m’a donnée et les échanges que nous avons eus qui m’ont permis de grandir en tant que chercheur. Son côté humain et sa patience auront teinté mon image du domaine de la recherche. Je le remercie sincèrement pour son soutien au cours ma maîtrise. Je remercie également ma codirectrice de recherche la Dr. Lucie Germain pour les commentaires constructifs qu'elle m’a partagés et qui ont aidé à améliorer ma formation ainsi que pour son soutien.
Je remercie les organismes qui ont subventionné ce projet, dont la fondation GO et les Fonds de Recherche du Québec en Santé ainsi que le Dr. Didier Trono pour le plasmide pMD2.G et la Dre. Elena Pope pour les cellules de patients atteints de l'EBDR. Je remercie les évaluateurs de ce mémoire: Manuel Caruso, Lucie Germain, Samer Hussein et Jacques Tremblay pour leurs
corrections et suggestions.
Je remercie Karim Ghani qui a généré plusieurs constructions plasmidiques, produit des vecteurs rétroviraux de manière transitoire et stable et effectué certaines titrations qui auront permis de faire avancer le projet et pour son partage au quotidien de connaissances avec les vecteurs rétroviraux. Je remercie Angela Dakiw-Piaceski qui a participé à ma formation, et pour son aide à générer les images de microscopie à fluorescence. Je remercie Amélie Morissette pour les échanges stimulants que nous avons eus et ses précieux conseils. Je remercie Rina Guinard pour la
transmission de son savoir en culture cellulaire. Je remercie Francis Bisson pour les commentaires constructifs sur mon mémoire. Je tiens à remercier les membres du Laboratoire de la Dr. Germain: Caroline Simard-Bisson, Danielle Larouche, Amélie Lavoie, Sergio Cortez-Ghio, Gaëtan Le-Bel, Benjamin Goyer et Israel Martel qui auront contribué chacune et chacun à ma formation avec leurs questionnement critique et commentaires constructifs à de multiples reprises. Je remercie ma famille pour leur soutien et leur compréhension au travers mes études.
À vous tous, je dis: « Merci ».
Introduction
L'épidermolyse bulleuse
Les différents types de l’épidermolyse bulleuse
L’épidermolyse bulleuse (EB) est une maladie génétique rare de la peau décrite pour la première fois en 1886 (1). Sa prévalence est de 11,07 cas par millions et près d’un demi-million de personnes sont atteintes dans le monde, bien que ce nombre soit sous-estimé, car certains cas plus bénins sont non diagnostiqués (2, 3). Le diagnostic est établi à la naissance dans la majorité des cas (2, 4, 5). Cette maladie est caractérisée par une fragilité extrême de la peau et des muqueuses menant à la formation de cloques et à son érosion (2, 6, 7). Dépendamment du type de l'EB et du type de mutations associés, la sévérité de la maladie va varier. Dans les cas les plus graves de l'EB, certains nouveau-nés mourront dans les premiers jours de leur vie suite aux complications d’une peau dénudée de son épithélium menant à des déséquilibres métaboliques et à des infections (8, 9). En général, l'EB peut entraîner le développement de cloques au niveau des mains et des pieds, alors que dans certains cas, des complications peuvent perdurer tout au long de la vie du patient au niveau des yeux, du système respiratoire et des muscles en plus de la peau (2, 6, 10). Pour les gens atteints de l’EB, ce bris facile de la peau et des muqueuses entraîne une difficulté à mastiquer et à avaler menant à la malnutrition, de l’anémie due aux pertes de sang, un retard de croissance et un ralentissement dans la guérison des plaies, auxquels viennent s’ajouter des problèmes sociaux et psychologiques (6, 11). De plus, un taux élevé de patients souffrant de l’EB généralisé (sur tout le corps) seront atteints d’un carcinome épithélial métastatique avant l’âge de 40 ans (12, 13).
La peau est composée de trois couches : l’épiderme, le derme et l’hypoderme (Figure 1A). Dans le cas de l’EB, seuls l’épiderme et le derme sont en cause. L’épiderme est principalement composé de kératinocytes maintenus entre eux par des desmosomes et ancrés à la lame basale (LB) cutanée via des hémidesmosomes. Ces desmosomes et hémidesmosomes sont rattachés entre eux par un réseau de kératines intracellulaires. La couche immédiate de kératinocytes ancrés à la LB cutanée se nomme la couche basale. Suite à la division des kératinocytes de la couche basale, les cellules se différencieront et formeront la couche épineuse, la couche granuleuse, la couche claire
dans certaines régions de la peau, et la couche cornée, où ils seront alors différenciés en cornéocytes (Figure 1B) (14, 15). Sous l'épiderme se trouve le derme qui est composé de deux couches: soit le derme papillaire, qui est la couche en contact avec la LB cutanée, sous lequel se trouve le derme réticulaire (Figure 1A). Entre la couche basale et le derme papillaire se trouve la LB cutanée qui est une structure complexe composée de protéines, telles que celles de la matrice extracellulaire, dont le collagène et la laminine, ainsi que de glycoprotéines et de sucres (Figure 1C). La fonction principale de la LB cutanée est l’adhésion de ces deux couches de la peau via des hémidesmosomes et des intégrines (2, 6). La LB cutanée est divisée en deux régions : la lamina
lucida et la lamina densa. La lamina lucida est la région de la LB qui est en contact avec l’épiderme
et porte son nom de par sa structure plutôt transparente, lorsqu’observée au microscope électronique à transmission, comparativement à la lamina densa qui est en contact avec le derme et qui a une structure de forte densité. Sous la lamina densa se trouvent les fibrilles d'ancrages qui interagissent avec le collagène de type I et participent au réseau de protéines reliant l'épiderme au derme (Figure 2).
Trois consensus internationaux ont été édités suite à des rencontres tenues depuis 1988 afin d’améliorer le diagnostic et la classification des différents types de l’EB (4, 16). Selon le niveau ultrastructurel où les cloques se développent dans la peau, l’EB a ainsi été divisée en quatre grandes classes : EB simplexe (EBS), EB jonctionnelle (EBJ), EB dystrophique (EBD) et syndrome de Kindler (SK) (16). L’EB simplexe (EBS) est caractérisée par une rupture de la peau au niveau de l’épiderme (4). La peau de l’EBJ se rompt dans la lamina lucida ou à sa jonction, alors que pour l’EBD, le bris s’effectue sous la lamina densa au niveau du derme papillaire (2, 4). Le syndrome de Kindler est caractérisé par un détachement de la peau à l’une de ces trois régions ultrastructurelles (épiderme,
lamina lucida ou sous la lamina densa) et d’un redoublement de la LB (4, 17). Les types d'EB
peuvent ensuite être divisés en plusieurs sous-catégories, d’abord selon les symptômes cliniques observés, puis selon l’endroit où les mutations sont retrouvées dans les gènes spécifiques à l’EB (4, 16). Dix-huit gènes ont jusqu’à présent été identifiés comme étant causaux dans la maladie de l’EB (2). Une étude parue en 2017 a rapporté la découverte d’un 19e gène susceptible de causer l’EB :
KLHL24 (18). Les gènes en cause dans la maladie d’EB codent pour des protéines majoritairement
structurelles de localisation extra et intracellulaire. Ces protéines sont retrouvées dans la LB cutanée, à la surface des cellules du derme et de l’épiderme ou en continuité avec ce réseau protéique au niveau intracellulaire dans le cas des kératinocytes (Figure 2) (2, 6, 7).
Figure 1. Couches de la peau et jonction dermo-épidermique par microscopie électronique en transmission. (A) La peau est composée de l'épiderme, du derme, divisé en derme papillaire et derme
réticulaire, et de l'hypoderme. Figure adaptée de (19). (B) L'épiderme est composé d'une couche basale ancrée à la lamina lucida et d'une couche épineuse, granuleuse et cornée selon le niveau de différentiation des kératinocytes. La couche claire est retrouvée, au niveau de la paume des mains et des pieds. Figure adaptée de (20). (C) Les principaux éléments de la jonction dermo-épidermique (JDE) sont visibles par microscopie électronique en transmission. Les filaments intermédiaires sont visibles dans les kératinocytes de l’épiderme. Les hémidesmosomes permettent de relier les filaments intermédiaires et d’attacher les kératinocytes de l’épiderme à la matrice extracellulaire qui compose la lamina lucida et la lamina densa. Sous la lamina densa se trouvent les fibrilles d’ancrages essentielles à l’adhésion du derme à l'épiderme. Sous les fibrilles d’ancrages se trouve la matrice du derme enrichie de fibrilles de collagène. Figure adaptée de (6).
L’épidermolyse bulleuse simplexe
Dans le cas de l’EBS, la kératine 5 ou 14 est souvent mutée ce qui affecte l’intégrité de la structure des kératinocytes de la couche basale en fragilisant le réseau de filaments intermédiaires (21, 22). La plectine et la dystonine, des protéines adaptatrices des hémidesmosomes reliant les filaments intermédiaires de kératines 5 et 14 aux hémidesmosomes (Figure 2), sont également dysfonctionnelles dans cette classe de l’EB diminuant la force d’adhésion entre l’épiderme et la LB
desmosomes qui relient les filaments de kératines aux desmosomes (Figure 2), sont mutées dans l’EBS ce qui diminue l’adhésion entre les kératinocytes adjacents (25-27). L’exophiline 5, une protéine effectrice de la GTPase Rab27 (Figure 2), se retrouve également mutée dans l'EBS ce qui affecte le transport intracellulaire de vésicules vers la membrane plasmique (28). La transglutaminase 5, une enzyme extracellulaire transférant des groupements polyamines aux protéines permettant la réticulation des filaments de kératine et des protéines spécifiques à la différenciation et menant à la formation de l’enveloppe cornée des kératinocytes est, elle aussi, non-fonctionnelle dans certains cas d’EBS (Figure 2) (29). La protéine Kelch-like 24, nouvellement associée à l’EBS, fait partie du complexe E3 ubiquitine ligase et médie l’ubiquitination de la kératine 14 contrôlant ses niveaux d'expression durant la différenciation de kératinocytes (Figure 2) (18, 30). Elle peut, lorsque mutée, stimuler la dégradation excessive de cette kératine et affaiblir le cytosquelette résultant en l’érosion facile de l’épiderme (18, 30).
L’épidermolyse bulleuse jonctionnelle
Dans l’EBJ, les protéines mutées sont des récepteurs membranaires et des composants de la matrice extracellulaire. Par exemple, une des deux sous-unités de l’intégrine α6β4, retrouvée sur
les hémidesmosomes des cellules épithéliales et responsable de la liaison à la laminine (Figure 2), peut parfois être mutée et mener au détachement de l'épiderme au derme sous-jacent (31, 32). La sous-unité α3 des intégrines permet, avec la sous-unité β1, de former le récepteur aux intégrines
α3β1, qui lie la laminine-332 et 511 permettant de créer des points focaux d'adhésion (Figure 2) (33).
Lorsque la sous-unité α3 des intégrines est absente, l'adhésion de l'épiderme est réduite, des
ruptures de l'épiderme peuvent se produire au niveau de la lamina lucida et l'assemblage et le remodelage de la LB sont alors diminués (34). Des mutations dans la chaîne α3, β3 ou γ2 de la
laminine-332, protéine formant les filaments d’ancrage dans la lamina lucida avec son association au collagène de type IV (Figure 2), peuvent nuire à son interaction avec différentes protéines de la matrice extracellulaire lors de son organisation et diminuer l’adhésion de l'épiderme au derme sous-jacent (35-37). L’hélice α1 du collagène de type XVII qui est localisée dans la LB peut, lorsque mutée,
mener à une réduction du nombre d'hémidesmosomes et qui seront alors malformés résultant en la séparation du tissu au-dessus de la lamina densa (Figure 2) (38).
L’épidermolyse bulleuse dystrophique
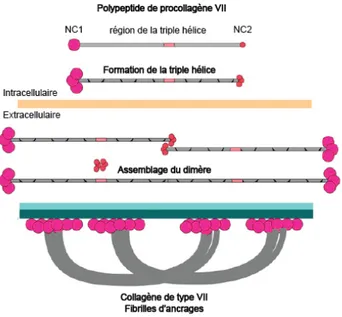
L’EBD est causée par la mutation d’une protéine retrouvée immédiatement sous la LB cutanée. Dans le cas de l’EBD, il s'agit de l’hélice α1 du collagène de type VII (39-41). Cette dernière
s'assemble en homotrimère pour former la triple hélice de collagène VII qui est nécessaire à la formation des fibrilles d’ancrage retrouvées sous la lamina densa (2, 39, 42, 43). Les fibrilles d'ancrage sont la forme mature du collagène de type VII et permettent l'adhésion de l'épiderme au derme via un réseau protéique avec entre autres, mais non-exclusivement la laminine-332 et l'intégrine α6β4 (Figure 2). Selon l’endroit où se trouve la mutation dans la séquence du gène du
collagène de type VII, COL7A1, la protéine peut devenir non-fonctionnelle et les fibrilles d’ancrages peuvent ne pas être formées correctement, ou voir pas du tout, affectant l'intégrité de la JDE à des niveaux d'intensité variés (44). Cela se traduit par une fragilité extrême de la peau, entraînant l'érosion de l'épiderme originant de sous la LB cutanée, ainsi que la formation de cloques, la "pseudosyndactylie" et la formation de cancer épithéliaux (12, 41, 45).
Le syndrome de Kindler
Le syndrome de Kindler est causé par la mutation d'une protéine de localisation intracellulaire : la kindline-1 (46). Cette protéine sert d’adaptateur entre le cytosquelette d’actine et l’intégrine α3β1 des points focaux d’adhésion attachés à la laminine-511 et est responsable de
l’établissement de la polarité des cellules souches épithéliales (47, 48). La kindline-1 régule également la prolifération et la différenciation des cellules souches épithéliales par son activation de l’intégrine αVβ6 qui active le TGF-β et maintient la voie de signalisation WNT inactive (2, 49, 50).
Lorsque la kindline-1 n’est pas présente, suite à un stress au niveau de la peau, il a été constaté que les kératinocytes augmentent leur expression de cytokines ce qui engendre une réponse inflammatoire menant à la différenciation de fibroblastes en myofibroblastes qui sécrètent alors des protéines de la matrice extracellulaire (51). La répétition de tels cycles de stress sur la peau de patients atteints du SK mène à : une fibrose de la peau, la formation de cloques, une photosensibilité de la peau, de la poïkilodermie et un dédoublement de la LB (47, 51).
Figure 2. Protéines impliquées dans les différents types d’épidermolyse bulleuse. L’épidermolyse
bulleuse simplexe est due à des mutations des protéines retrouvées au niveau de l’épiderme dont la kératine 5, la kératine 14, la plectine, la dystonine, la desmoplakine, la plakophiline 1, la plakoglobine, l’exophiline 5, la transglutaminase 5 ou la protéine Kelch-like 24. L’épidermolyse bulleuse jonctionnelle est causée par des mutations dans des protéines retrouvées dans la lamina lucida ou à sa jonction dont les chaînes α6 ouβ4 de
l’intégrine α6β4, la chaîne α3 des intégrines, la chaîne α3, β3 ou γ2 de la laminine-332 ou l’hélice α1 du
collagène de type XVII. L’épidermolyse bulleuse dystrophique est due à des mutations dans une protéine retrouvée sous la lamina densa soit l’hélice α1 du collagène de type VII. Le syndrome de Kindler est dû à des
mutations dans une protéine retrouvée dans les kératinocytes et les fibroblastes soit la Kindline-1.
Ainsi, 19 gènes sont en cause dans la maladie d’EB, mais la présence de mutation(s) dans ceux-ci ne signifie pas de manière absolue que la maladie se développera. En effet, certaines
protéines sauvages, identifiées comme pouvant causer l’EB lorsque mutée, parviendront à compenser la perte de fonction de protéines produites par un allèle différent et l’individu ne sera alors pas affecté par l’EB. Toutefois la mutation dans un seul allèle de certaines protéines pouvant causer l’EB peut aussi se traduire par la manifestation de la maladie (6). C'est le cas de KRT5, impliqué dans l’EBS, dont une seule copie mutée de l'allèle est capable de provoquer la maladie (2). Certaines mutations dans la kératine 14, la plectine, l’intégrine β4 et le collagène de type VII sont également reconnues pour être autosomale dominante et peuvent causer l’EB (2, 52). Également, des mutations dans la kératine 14 ainsi que dans la plectine ont été identifiées comme semi-dominantes, alors que tous les gènes impliqués dans l'EB, à l’exception de KRT5, peuvent être transmis de manière autosomale récessive (2).
L’épidermolyse bulleuse dystrophique récessive
L’EBD est causée par des mutations dans un allèle du gène codant pour le collagène de type VII: COL7A1, alors que l'EBDR est causée par des mutations dans les deux allèles de COL7A1. Comme les symptômes de l'EBDR sont souvent généralisés cela augmente la nécessité de traiter cette maladie avant les autres types de l'EB. Le COL7A1 est localisé sur le bras court du chromosome 3 au locus 3p21.31 et contient 118 exons totalisant 31 195 paires de bases (pb), d’après la version du génome humain GRCh38/hg38 (53, 54). L’acide ribonucléique messager (ARNm) encodé par COL7A1 fait 9287 nucléotides (nt) et la protéine traduite, sous forme de propeptide, a une longueur de 2944 acides aminés (50, 55). En 2018, un total de 659 mutations différentes avaient été identifiées dans ce gène chez 1039 patients à travers le monde (56).
Structure et fonctions de l'hélice α1 du collagène de type VII
Le COL7A1 est exprimé par les kératinocytes et les fibroblastes dermiques, mais son niveau d’expression est plus élevé dans les kératinocytes (57, 58). Au total, 2 isoformes de la protéine sont produites dont une possède une délétion de 31 acides aminés à partir de la position 1869 (50, 55). L’hélice α1 du collagène de type VII est composée de quatre grandes régions. De l’acide aminé 17 à 1253 se trouve la région NC1 (acronyme de la terminologie anglaise pour Noncollagenous 1; non
région interrompue entre l’acide aminé 1254 et 1477. De l’acide aminé 2785 à 2944 se trouve la région NC2 (acronyme de la terminologie anglaise pour Noncollagenous 2; non collagénique 2) (Figure 3) (50).
Figure 3. Régions et domaines de l’hélice α1 du collagène de type VII. Dans la région non collagénique 1
(NC1) (acides aminés 17 à 1253) se trouve le domaine von Willebrand factor A1 (VWFA1) suivi de neuf domaines de fibronectine de type III (Fib) et d’un domaine von Willebrand factor A2 (VWFA2). La région interrompue (acides aminés 1254 à 1477) est présente dans la région de la triple hélice (acides aminés 1254 à 2784) dans laquelle sont retrouvées les répétitions de la triple hélice représentées par des cercles roses. La région non collagénique 2 (NC2) (acides aminés 2785 à 2944) contient le domaine Kunitz inhibiteur de la trypsine pancréatique représenté par un carré rose.
La région NC1 est elle même sous-divisée en plusieurs domaines. D’abord, les seize premiers acides aminés codent pour un peptide signal permettant l’exportation du polypeptide fraîchement synthétisé de l’hélice α1 du collagène de type VII dans le réticulum endoplasmique rugueux (RER) (50). De l’acide aminé 38 à 211 se trouve le domaine VWFA1 (acronyme de la terminologie anglaise pour von Willebrand factor A1; facteur A1 de von Willebrand) (50). Cette région adopte une structure en feuillet-β ouvert-tordu flanquée d’hélices alpha et possède un domaine MIDAS (Metal-Ion Dependent Adhesion Site) pour la liaison de protéines (59, 60). Un second domaine, VWFA2 (acronyme de la terminologie anglaise pour von Willebrand factor A2; facteur A2 de von Willebrand) est retrouvé entre les acides aminés 1054 et 1229 (50). Neuf domaines FN3 (Fibronectin Type III Domain) sont retrouvés entre les domaines VWFA1 et VWFA2 (50). Ces derniers contiennent une séquence de 3 acides aminés : Arg-Gly-Asp (RGD) prédite pour permettre une liaison avec les intégrines et permettant une liaison avec la laminine-332 (61-63). La région NC1 sert ainsi de zone d’adhésion avec une variété de protéines, dont le collagène de type I et de type IV ainsi que la laminine-332 et 511 (64-66).
La région de la triple hélice est majoritairement une répétition des acides aminés G-X-Y (où X et Y représentent n’importe quel acide aminé) (42). Cette répétition de séquence permet la formation d’une chaîne polypeptidique sous forme d’hélice de pas gauche capable de former une triple hélice superenroulée de pas droit (42). Des répétitions de sept triplets par deux tours d’hélice, nommées une symétrie 7/2, ainsi que des répétitions de dix triplets en trois tours, nommées symétrie 10/3, ont été observées dans différents fragments de collagène (67, 68). Ainsi, dépendamment du nombre de répétitions de ce triplet qui est variable dans une même chaîne de collagène, la torsion de l’hélice sera plus ou moins serrée, ce qui peut affecter l’assemblage du collagène en fibrilles ou sa liaison à d’autres molécules (69).
Dans la région NC2 se trouvent trois domaines procurant au procollagène VII différentes fonctions. Au début de la région NC2 se trouve un domaine d’hélice-α coiled-coil qui permet aux chaînes de procollagène de s’associer en structure en forme de corde (70). Basée sur des expériences faites sur le collagène de types I et III, une séquence discontinue de 15 acides aminés serait aussi présente près du centre de la région NC2 (71). Elle permettrait la reconnaissance spécifique d’autres chaînes de procollagène assurant le bon pairage des différentes chaînes et est essentielle à la formation de la triple hélice de collagène (71). Entre l’acide aminé 2872 et 2944 se trouve le domaine Kunitz inhibiteur de la trypsine pancréatique (50). La fonction principale de ce domaine est l’inhibition des protéases de la famille S1 ; des sérines protéases possédant une sérine nucléophile au site actif leur permettant d’attaquer le fragment carbonyle de résidus d’arginine ou de lysine (chargés positivement) et de phénylalanine, tryptophane et tyrosine (hydrophobiques), présents dans des liaisons peptidiques de substrats, pour former un intermédiaire acyle-enzyme (72, 73). Ainsi la région NC2 du procollagène VII sert de point d’ancrage pour la liaison de chaînes de procollagène et a la capacité d'inhiber les protéases retrouvées à proximité de la LB.
Maturation du collagène de type VII
Diverses étapes sont nécessaires afin que la protéine collagène de type VII (COLVII) parvienne à sa forme mature hors de la cellule. La synthèse du polypeptide de l'hélice α1 du collagène de type VII débute au RER et grâce à une séquence signal, le polypeptide pourra y pénétrer (74). La séquence signal est clivée par une peptidase lorsque le polypeptide émerge dans la
prolines et lysines de la région hélicale des molécules de procollagène VII sont ensuite hydroxylées (50, 74-76). Les prolines et hydroxyprolines forment fréquemment des liaisons peptidiques en cis dues à la présence de groupements propyle et hydroxypropyle et seront inversées en conformation
trans par des propyles cis-trans isomérases afin que la triple hélice de procollagène puisse être
formée (77). Des monomères de galactose, puis de glucose sont ensuite ajoutés aux hydroxylysines (o-glycosylation) par une galactosyl transférase et une glucosyl transférase respectivement (74, 75). La région NC2 servira de point d’attachement pour trois chaînes de procollagène VII grâce à la présence de ponts disulfure intra et inter-chaînes entre les cystéines, produits par l'enzyme disulfure isomérase (74, 78). Cette étape est nécessaire à l'enroulement des chaînes de procollagène de l’extrémité C-terminale vers l’extrémité N-terminale, aidé des chaperones BiP et GRP94, et permet d'éviter de mauvais alignements entre les répétitions de séquence G-X-Y, permettant de générer la triple hélice de procollagène VII dont les extrémités ne sont pas enroulées (79, 80). L’adhésion entre les trois chaînes de procollagène VII est possible grâce à la présence de ponts hydrogènes entre les glycines et les hydroxyprolines présentes dans la région de la triple hélice alors que l’agrégation prématurée de molécules de procollagène dans le RER est prévenue par la chaperone HSP47 (81, 82). Certaines asparagines dans la région NC1 du procollagène VII subiront l’ajout d’oligosaccharides par une oligosaccharyl transférase (N-glycosylation) permettant d’assurer la sécrétion du procollagène de type VII (50, 66, 74). Suite à ces modifications post-traductionnelles, la protéine transmembranaire du RE, TANGO1, via son domaine SH3 extrudant dans la lumière du RE, va s’associer au procollagène de type VII et le charger dans les vésicules de transfert recouvertes des protéines adaptatrices SEC23A/24C et des protéines de manteau SEC13/31 (83-88). L’ubiquitine ligase CUL3 et sa protéine adaptatrice KLHL12 vont ubiquitiner SEC31 ce qui pourrait aider à la génération des vésicules de COPII de l’ordre de 200 à 500 nm, qui sont de taille supérieure aux vésicules standard de COPII (60-80 nm), permettant au procollagène VII d’une longueur de 424 nm d’y être transporté (39, 89). SLY1, une protéine interagissant avec TANGO1 et les t-SNAREs spécifiques au RE (syntaxine 17 et syntaxine 18), permettrait ensuite la fusion des vésicules de procollagène VII avec les membranes du compartiment intermédiaire du réticulum endoplasmique-golgi et serait aussi impliquée dans la croissance de larges vésicules transportant le procollagène VII (86). Dans l’appareil de Golgi, le procollagène VII serait empaqueté dans des vésicules de sécrétion par des mécanismes peu connus, mais qui pourraient impliquer la GTPase Rab10 et la protéine Arf-GAP Crag (90). Le procollagène VII est ensuite sécrété hors des fibroblastes dermiques et des
kératinocytes épidermiques sous forme de faisceaux de procollagène (90). Via leur extrémité NC2, deux molécules de procollagène VII vont s’associer dans l’espace extracellulaire et former un dimère anti-parallèle maintenu par des ponts di-sulfures sur une région de 60 nm puis la région NC2 sera clivée et plusieurs dimères de COLVII pourront ensuite s’assembler latéralement et former des fibres d’ancrages (Figure 4) (2, 43). Les régions NC1 permettront l’interaction avec la laminine-332 ou 511 et le collagène de type IV, retrouvé dans la lamina densa, générant ainsi des fibrilles d’ancrages au niveau du derme (2, 65). Les fibrilles d’ancrage piégeront des fibres de collagène de type I grâce au domaine VWFA2 de la région NC1 assurant l’intégrité de la membrane basale et donc l'adhésion de l’épiderme au derme sous-jacent (2, 65, 91).
Figure 4. Maturation du collagène de type VII. À l’intérieur de la cellule, l’hélice de procollagène VII va
s’enrouler à partir de l’extrémité C-terminale et former une triple hélice de procollagène VII. Une fois sécrété, le procollagène VII va dimériser au niveau des régions NC2 qui seront ensuite clivées. Plusieurs dimères vont s’assembler pour former les fibrilles d’ancrages qui sont la forme mature du collagène de type VII. Figure adaptée de (2).
Mutations dans le collagène de type VII
Différentes mutations ont été identifiés chez le COL7A1 (4, 44). Ces mutations sont transmises de manière autosomale dominante ou récessive et la forme récessive de la maladie (EBDR) est reconnue pour manifester des symptômes plus graves que la forme dominante puisque les deux allèles sont touchés (4, 92). Toutefois, les mutations dans le COL7A1 sont souvent
dominantes, car pour une expression équivalente de l'allèle sauvage et muté, 7/8 des molécules trimériques contiendront au moins une hélice α1 du collagène de type VII muté et seulement 1/8 seront composés de peptides normaux (42, 93).
Des codons stop prématurés (CSP) dans la région NC1 de COL7A1 sont parmi les mutations les plus sévères, car la protéine ne sera pratiquement pas exprimée diminuant directement le nombre de fibrilles d'ancrage formées et résultant en l'expression la plus prononcée du phénotype lorsque le patient est homozygote pour de telles mutations (41, 94). Des mutations faux sens dans la région NC1 de COLVII peuvent diminuer l'adhésion des fibrilles d'ancrages avec la laminine-332, 511 ou le collagène de type IV (2, 64, 65). Lorsque les mutations changent le nombre de triplet G-X-Y dans la région de la triple hélice, la torsion de l’hélice alpha peut être modifiée ayant pour conséquence de diminuer la stabilité de la protéine et d'altérer la formation des fibrilles d'ancrages (42, 69). La substitution de glycines dans les séquences G-X-Y de la région de la triple hélice du COLVII est associée à la forme dominante de la maladie et peut résulter en la formation d’un coude dans cette région et défavoriser l’assemblage des fibrilles d’ancrages (42, 44, 95). De plus, des mutations dans les lysines et les prolines peuvent diminuer l’adhésion entre les chaînes α de la triple hélice de procollagène VII ou entre les dimères et nuire à l’assemblage des fibrilles d’ancrages ainsi que diminuer leur temps de demi-vie (76, 96). D'autre part, des mutations dans les résidus cystéines situés dans la région NC2 peuvent nuire à la dimérisation des homotrimères de procollagène VII ou au clivage de cette région et faire diminuer le nombre de fibrilles d’ancrages formées (44, 71, 96). En somme, diverses régions de l'hélice α1 du collagène de type VII peuvent être mutées affectant la formation, la stabilité et la fonction des fibrilles d’ancrages, ce qui peut en partie expliquer la sévérité de la maladie.
Cancer épidermoïde agressif
Des mutations dans COL7A1 vont occasionner des érosions de l’épiderme déclenchant une réponse inflammatoire pouvant causer des carcinomes épidermoïdes agressifs (97, 98). Suivant un bris de l’épiderme, un caillot sera formé et les plaquettes vont attirer des neutrophiles et des monocytes. Le recrutement de ces cellules générera un milieu riche en cytokines et en facteurs de croissance dont : les récepteurs à l’interleukine 4, 13RA1, IL-6, CXCL13, CCL2, CCL5 et les gènes induits par INF-α et TGF-β permettant la prolifération et la migration de cellules au niveau de la plaie
(45, 99). Toutefois, lorsque le COLVII est muté, la laminine-332 ne se dépose plus aussi bien au niveau de la lamina densa (100). L’activation de la voie de signalisation de la laminine-332/intégrine α6β4 (JNK2 et AKT) s’en trouve alors compromise ce qui nuit à la migration des kératinocytes vers la plaie prolongeant ainsi la durée de la réponse inflammatoire favorisant un microenvironnement tumoral (100).
Lorsque la barrière épithéliale est détruite, des infections par Staphylococcus aureus et
Pseudomonas aeruginosa sont fréquentes et font augmenter les niveaux d’inflammation en plus de
pouvoir causer des septicémies ou mener à des des infections chroniques (9). Or, il a été montré que les flagelles de certaines bactéries peuvent activer le récepteur TLR5 (acronyme de la terminologie anglaise pour Toll-Like Receptor; récepteur Toll-like) et réguler positivement le facteur de transcription HMGB-1 permettant l’activation de fibroblastes et de kératinocytes, l’angiogénèse et la formation de cicatrices (101). Lors du processus de guérison des plaies, HMGB-1 va transloquer du noyau vers le cytoplasme et sera sécrété par des fibroblastes et kératinocytes destinés à mourir ou qui nécrosent et par des cellules activées du système immunitaire inné (101, 102). HMGB-1 agira alors comme une "alarmine" en se liant aux récepteurs TLR2 et TLR4 et à RAGE et stimulera la sécrétion de cytokines pro-inflammatoires ainsi que le recrutement de cellules inflammatoires respectivement contribuant davantage au développement d'un microenvironnement tumoral (101, 102).
La voie de signalisation TGF-β pourrait jouer un rôle important dans le développement tumoral chez les patients atteints de l'EBDR. Chez les mammifères, trois isoformes de TGF-β existent : TGF-β1, TGF-β2 et TGF-β3. Les deux premières isoformes ont une activité pro-fibrotique alors que TGF-β3 possède une activité anti-fibrotique (103). La sécrétion de TGF-β par différentes cellules au niveau de la plaie va stimuler la différenciation de fibroblastes en myofibroblastes alors que TGF-β1 va entraîner la contraction des myofibroblastes permettant la fermeture de la plaie, ce qui libérera davantage de TGF-β emmagasiné dans la MEC et augmentera la différenciation de fibroblastes en myofibroblastes (104-106). Lors de la réparation des tissus, les myofibroblastes produisent différentes protéines et glycoprotéines (107). En condition pathologique comme avec l'EBDR, des cycles répétés de bris de la peau entraîneront une sécrétion de collagène de type I, dû à l'activité de TGF-β1, menant à une fibrose qui stimulerait la tumorigénèse chez ces patients (99, 108). De plus, TGF-β active la voie canonique impliquant les protéines SMAD, dont SMAD3 qui peut
se lier aux promoteurs des micro-ARN miR-29 (miR-29a, miR-29b et miR-29c) et faire diminuer leur expression (109-111). Une diminution de miR-29c active la synthèse de composants de la MEC contribuant à la fibrose et via une boucle de rétroaction positive, la MEC fibrotique réprime l'expression de miR-29c (112, 113). De plus, la ténascine-C, une glycoprotéine extracellulaire exprimée de manière transitoire lors de la guérison des plaies, a été retrouvée surexprimée dans les tissus fibrotiques de souris et peut activer de façon persistante les récepteurs TLR4 des fibroblastes stimulant également leur activation et la sécrétion de gènes du collagène médiant aussi la fibrose (114). TGF-β permet également aux fibroblastes de patients atteints de l'EBDR de se comporter comme des fibroblastes associés au cancer (115). Les fibroblastes associés au cancer chez ces patients vont surexprimer le collagène de type XII et de type V, améliorant leur adhésion et nuisant à la migration cellulaire respectivement (115-117). Ainsi, la perte de fonction du COLVII entraîne des bris répétés de la peau augmentant l'activité de TGF-β menant vers une fibrose qui déclenche la voie de signalisation intégrine-FAK-AKT de l'intégrine β1 permettant la progression tumorale (108).
Les traitements pour l’épidermolyse bulleuse dystrophique récessive Thérapie pharmacologique
Diverses méthodes ont été développées pour traiter l’EBDR, dont la thérapie à partir de produits de synthèse chimique. Près de 30 % des patients atteints de l'EBDR expriment des CSP (118). L’administration de gentamicine ou d’amlexanox, des aminoglycosides, sur des kératinocytes et fibroblastes de patients atteints de l'EBDR permet selon la dose de supprimer l’effet des CSP et d’induire l’expression soutenue de COLVII qui ira se localiser à la JDE et d’ainsi renverser le phénotype EBDR (119-121). La gentamicine permet la traduction en se liant à un site spécifique sur l’ARN ribosomal de mammifère nuisant à la reconnaissance codon-anticodon au site accepteur d’acide ribonucléique de transfert (ARNt) et n’induit pas significativement la mauvaise lecture des codons stop normaux (122, 123). Toutefois, dépendamment du type de CSP (UGA, UAG ou UAA), et des nucléotides suivants, la capacité à surmonter le CSP sera variable (119, 124). L’administration systémique est reconnue pour causer de l’ototoxicité et des problèmes rénaux (125, 126). Ainsi l’approche développée prend la forme d’une crème pour l’application topique (119, 120). La limitation majeure de cette technique est sa restriction aux mutations non-sens.
Un autre moyen pharmacologique de traiter les patients atteints de l'EBDR est l’usage d’oligoribonucléotides anti-sense (AON) livrés par injection sous-cutanée dans un gel solidifiant à 37°C, suite à une greffe de peau, permettant leur diffusion lente et localisée (127). Ces ARN anti-sens vont se lier à des séquences spécifiques du pre-ARNm du COL7A1 et moduler l’épissage d’introns ou d’exons contenant des mutations permettant la réexpression du COLVII tronqué, mais fonctionnel se localisant à la JDE (127, 128). Ce genre de traitement est facile à manufacturer à grande échelle, permet de traiter plus d’une mutation (dans le même exon ou intron) et est non immunogène (127). Toutefois, les AON sont moins efficaces pour traiter des patients ayant des mutations hétérozygotes et tous les exons ne peuvent être retirés sans conséquence sur la fonctionnalité de la protéine (127).
La thérapie protéique permet de traiter l’EBDR par administration de la protéine sauvage aux patients. Trois traitements visant l’administration de COLVII ont été développés soit l'administration sous forme topique et l’injection localisée ou par voie systémique de la protéine. L’application topique de COLVII a montré que ce traitement permet de livrer le COLVII qui se localise à la JDE et permet d'accélérer la guérison des plaies (129). Toutefois, l’application de crème sur des régions non blessées ne permet pas au COLVII de traverser l’épiderme jusqu’à la membrane basale pour prévenir les bris éventuels de peau (129). Ainsi l’application topique de crème n’est efficace que pour les plaies à vif et la demi-vie in vivo du COLVII n’est que d’environ un mois et ne permet pas de guérir définitivement les patients (129, 130). De plus, ce genre de traitement se heurte à une mauvaise rétention du COLVII au niveau de la plaie et à l’instabilité de la protéine administrée dans un environnement riche en protéase (129). L'injection de COLVII a été tentée au niveau du derme et la protéine est alors capable de se localiser à la JDE et permet de restaurer l’adhésion de l’épiderme, tel que montré par des expériences chez la souris (131). Toutefois, ce traitement n’est pas idéal pour traiter de grandes surfaces de peau EBDR, car la diffusion du COLVII injecté est plutôt restreinte (92). L’injection intraveineuse dans la queue de souris a aussi été tentée (92). Le COLVII s’incorpore alors dans les plaies et n'a pas été détecté dans la peau d'organes non endommagés (92). Toutefois, la détection de réponse immunitaire contre la protéine complète injectée n'a pas été évaluée.
Thérapie cellulaire
Des essais ont été menés pour traiter l’EBDR par l’injection de différentes cellules allogéniques. L’administration intradermique ou systémique de MSC (acronyme de la terminologie anglaise pour Mesenchymal Stem Cell; cellules souches mésenchymateuses) allogéniques issues de la moelle osseuse ou l'administration intradermique de fibroblastes allogéniques chez des patients atteints de l'EBDR permet la production de COLVII qui localise à la JDE, peut prévenir l’érosion de la peau et accélère la guérison des plaies (132-135). En effet, les MSC permettent la production de facteurs trophiques qui stimulent la migration de cellules progénitrices endogènes, leur prolifération et leur différenciation ainsi que la néo-vascularisation (136, 137). Toutefois, ces approches ne permettent qu'une production transitoire de la protéine, car les cellules seront rejetées par l'organisme (138).
Il a également été rapporté que l'allogreffe de cellules souches de la moelle osseuse pouvait améliorer les symptômes de la maladie chez les patients atteints de l'EBDR. Dans ce cas-ci, ce genre d’approche est intéressante puisqu’elle permet de traiter l'épiderme ainsi que les muqueuses (139). Toutefois, ce traitement n'est pas très efficace, car seulement une correction légère des symptômes a été obtenue chez 50 % des patients (139). De plus cette approche est associée à un risque élevé de mortalité (139).
Un autre moyen de traiter l’EBDR est par la transplantation de cellules révertantes autologues (140). Dans de rares cas, les cellules somatiques mutées peuvent acquérir une seconde mutation dans le même gène restaurant l’expression d’une protéine fonctionnelle comme une délétion ou une insertion permettant le réalignement du cadre de lecture ou la mutation faux sens d’un CSP (140, 141). Dans ce genre de thérapie, des biopsies de peau sont prélevées chez le patient à des régions saines, puis les cellules sont amplifiées in vitro et ensuite greffées sur les plaies permettant leur fermeture (140). Cette méthode protège contre d’éventuelles érosions et est davantage sécuritaire puisqu'elle amoindrit le risque de rejet de greffe comparativement aux allogreffes, et ne nécessite pas la manipulation du génome in vitro (140). Cette approche serait à privilégier si la fréquence de génération de révertants était plus élevée (142).
L’édition du génome
L’approche par édition du génome permet de corriger certaines mutations dans le COL7A1. Le traitement de l'EBDR pourrait se faire par l’emploi de ZFN (acronyme de la terminologie anglaise pour Zinc Finger Nuclease; nucléases à doigts de zinc) en générant une coupure double-brin de l'acide désoxyribonucléique (ADN) à un site spécifique dans le génome, suivi d'une recombinaison homologue (RH) à partir d'un ADN donneur (143). Bien que cette technique soit très complexe pour les non-initiés, elle demande beaucoup de temps, est plus dispendieuse que les CRISPR (acronyme de la terminologie anglaise pour Clustered Regularly Interspaced Short Palindromic Repeats; courtes répétitions palindromiques groupées et régulièrement espacées) est sous-optimale pour cibler les gènes, peut cliver le génome à des sites non-spécifiques et a un faible taux de succès lorsque la RH est nécessaire ce qui peut expliquer pourquoi elle est peu utilisée pour traiter l’EBDR (143-146).
Les TALENs (acronyme de la terminologie anglaise pour Transcription Activator-Like Effector
Nuclease; nucléases effectrices ressemblant aux activateurs de transcription) peuvent être utilisées
pour corriger des mutations dans les fibroblastes et kératinocytes de patients atteints de l'EBDR en effectuant une coupure double-brin de l’ADN, suivi d'une RH à partir d'un ADN donneur (147, 148). Toutefois, cette technique peut cliver le génome à des endroits non désirés (147). De plus, cela reste un défi d’effectuer la RH dans des kératinocytes souches et de préserver leur état (148). Or, une équipe a contourné ce problème en utilisant les TALENs pour corriger des fibroblastes de patients atteints de l'EBDR, puis les a reprogrammés en cellules souches pluripotentes induites qui ont montré un potentiel de kératinocytes lors de la formation de tératomes (147). Les TALENs ont l’avantage d’être plus rapides à générer que les ZFN et peuvent cibler des régions du génome à environ toutes les 35 pb ce qui est optimal considérant que la RH nécessite que de l’ADN soit clivé à moins de 200 pb de la région à corriger les rendant plus flexibles que les CRISPR ou les ZFN (147, 149, 150).
Le système CRISPR/Cas9, couplé à un ADN donneur pour la RH, permet de générer des clones de kératinocytes de patients atteints de l'EBDR exprimant et sécrétant le COLVII à des niveaux similaires à des kératinocytes sains tel que montré par des greffes d’épidermes humains corrigées sur des souris immunodéficientes (151). De plus, l’usage de la version mutante de Cas9, Cas9 D10A nickase (Cas9n), améliore la fréquence de RH et sa spécificité, donc la sécurité de cette
technologie (152). Toutefois, cette technique n'est pas à l'abri de coupures dans le génome à des endroits non-spécifiques. Le principal défaut de l’usage de CRISPR/Cas9 basé sur la RH est la faible efficacité de RH, qui nécessite l’emploi d’une cassette de sélection pour isoler les cellules ayant acquis la séquence d'ADN donneur et suite à l'excision de cette cassette par le système CRE-LOX, la séquence restante du vecteur peut influencer le patron d’épissage en cis de la région ciblée (151). Cependant, l’usage de plasmides en mini-cercles contenant des sites de restriction situés entre les séquences d'homologie pour la RH permet, lorsqu'intégré dans le génome, de modifier les gènes sans laisser de traces (151). D'autre part, dans certains cas, l’utilisation d’un ADN donneur pour la RH n’est pas essentielle, car la réparation NHEJ (Non-Homologous End-Joining) peut produire une protéine fonctionnelle mutée (148). Comme chaque patient a des mutations différentes, différents ARN guides devront être utilisé et n'auront pas tous la même spécificité dans le génome ce qui augmente le travail nécessaire pour traiter plusieurs patients avec le système CRISPR/Cas9 et les probabilités qu'un ARN guide soit non-spécifique.
En conclusion, l’édition du génome vise à corriger le gène muté et peut rétablir le phénotype EBDR même si un seul allèle est ciblé à corriger puisqu'environ 30 % d’expression de COLVII comparativement au niveau normal est suffisant pour obtenir un phénotype normal (106, 147). De plus, il est possible de séquencer le génome des cellules traitées avant de les greffer aux patients afin de confirmer la spécificité de la coupure. Toutefois, il est également possible de livrer une copie fonctionnelle du gène muté avec les vecteurs viraux.
Thérapie génique
Historique de la thérapie génique
La thérapie génique est un traitement visant à livrer un gène thérapeutique dans les cellules de manière in vivo, ou in vitro suivi de greffes des cellules corrigées au patient, permettant l’expression d’une protéine supplémentaire dans les cellules traitées de l’organisme. Pour introduire le gène dans les cellules cibles, des approches physiques, chimiques et biologiques ont été développées dont : l’électroporation, des fusils à gènes, la microporation au laser, des liposomes, des peptides, des tensioactifs, des vecteurs viraux, etc. (153). Les vecteurs viraux sont des virus
dont les gènes de réplication ont été retirés et remplacés par un transgène thérapeutique. Les rétrovirus sont fréquemment utilisés en thérapie génique pour leur capacité à injecter leur génome intact (contenant le transgène) dans les cellules et leur capacité à s’intégrer de façon permanente dans leur génome procurant un effet thérapeutique à long terme.
Le premier essai clinique de thérapie génique qui a montré une efficacité chez l’homme a été publié en 2000 dans lequel un rétrovirus a été utilisé pour traiter des patients atteints de X-SCID (acronyme de la terminologie anglaise pour X-Linked Severe Combined Immunodeficiency; déficit immunitaire combiné sévère lié à l’X) ce qui a montré la faisabilité de cette approche et ouvert la porte pour le traitement de diverses maladies (154). Beaucoup d’efforts ont ensuite été déployés pour traiter des patients atteints de déficit en adénosine-désaminase (155-159). Depuis lors, les rétrovirus et certains AAV (acronyme de la terminologie anglaise pour Adeno-Associated Virus; virus associés-aux-adénovirus) ont été utilisés en thérapie génique pour traiter avec succès diverses maladies, dont l’adrénoleukodystrophie, le syndrome de Wiskott-Aldrich, l’hémophilie A et B, l’amyotrophie spinale et l’anémie falciforme (160-165). Des récepteurs antigéniques chimériques de cellules T ont aussi été générés et sont utilisés pour le traitement de divers cancers (166). Les trois premiers produits de thérapie génique ont tout juste été approuvés par la FDA en 2017 pour le traitement des tumeurs de lymphocytes B (Kymriah et Yescarta) et de l’amaurose congénitale de Leber (Luxturna) alors que la Chine et l’Europe approuvaient déjà des produits comme le Gendicine et le Macugen en 2003 et 2006 respectivement (167, 168). De plus, un rapport publié par le MIT NEWDIGS (Massachusetts Institute of Technology New Drug Development Paradigms) prévoit que 40 produits de thérapie génique seront approuvés d’ici 2022 (169). Ces essais montrent bien que la thérapie génique est fonctionnelle et en plein essor.
Virologie
Différents vecteurs viraux disponibles pour la thérapie génique
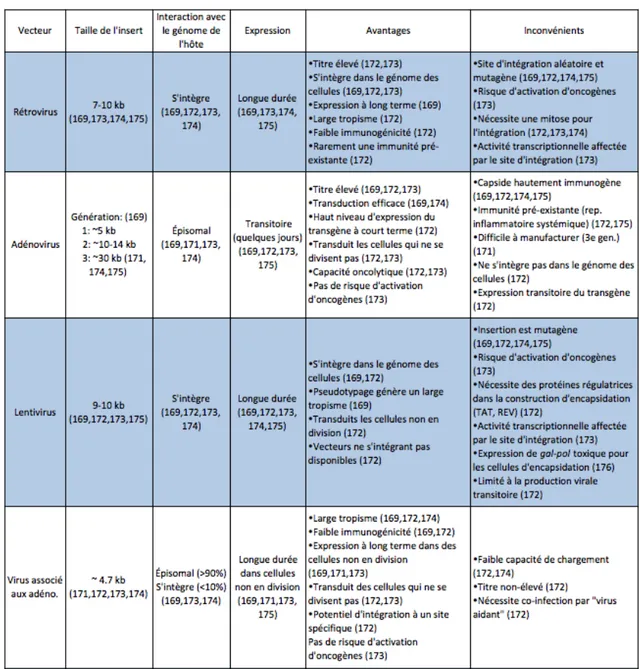
Plusieurs vecteurs viraux peuvent être utilisés en thérapie génique. Les plus fréquemment utilisés sont: les rétrovirus, les lentivirus, les adénovirus et les AVV. Leurs principales caractéristiques sont résumées au tableau 1.(168, 170-175)
Tableau 1. Principaux virus utilisés en thérapie génique. Les caractéristiques essentielles des virus utilisés en thérapie génique sont présentées comme la taille de l’insert pouvant être inséré, le type
d’interaction du virus avec le génome de l’hôte, la durée d’expression du transgène ainsi que les avantages et les inconvénients rattachés à l'utilisation des vecteurs.
Génome des rétrovirus
Le génome des rétrovirus est composé des gènes gag, pro-pol et env contenus dans cet ordre entre deux LTRs (acronyme de la terminologie anglaise pour Long Terminal Repeat; séquence
terminale longue répétée): le LTR 5’ et le LTR 3’ (Figure 5) (176). Gag code pour la matrice (MA), la capside (CA) et les protéines de la nucléocapside (NC) ; pro-pol code pour une protéase virale (PR), la transcriptase inverse (RT) (acronyme de la terminologie anglaise pour Reverse Transcriptase; transcriptase inverse), une intégrase (IN) et la RNAse H ; env code pour les sous- unités de surface (SU) et transmembranaires (TM) (176). Chaque LTR est sous-divisé en 3 régions : U3, R et U5 (177). La région U3 (acronyme de la terminologie anglaise pour Unique Sequence at 3’ End of
Genome; séquence unique à l'extrémité 3' du génome) contient un enhancer et un
promoteur permettant le début de la transcription à la jonction U3-R du LTR 5’ ainsi que divers éléments de contrôle se chevauchant et qui régulent de manière positive ou négative la transcription du génome viral selon les facteurs de transcription présents dans la cellule (176, 177). De plus, dans la région U3 du Mo-MLV (acronyme de la terminologie anglaise pour Moloney Murine Leukemia
Virus; virus de la leucémie murine de Moloney) se trouve une séquence agissant en cis essentielle à
l’exportation de l’acide ribonucléique (ARN) viral hors du noyau (178). La région R (acronyme de la terminologie anglaise pour Repeat Sequence; séquence répétée) contient un signal de polyadénylation relativement faible, afin de prévenir la terminaison prématurée dans le LTR 5’, et elle permet un saut de la transcriptase inverse du site 5’ vers le site 3’ lors de la synthèse de l’ADN de polarité négative (176, 177, 179). La région U5 (acronyme de la terminologie anglaise pour Unique
Sequence at 5’ End of Genome; séquence unique à l'extrémité 5' du génome) adopte une structure
secondaire particulière dans laquelle, à la fin de cette région dans le LTR 5', est exposé le PBS (acronyme de la terminologie anglaise pour Primer Binding Site; site de liaison de l’amorce) qui est nécessaire au début de la transcription inverse (180). La jonction R-U5 du LTR 3’ est le lieu où se termine la transcription du génome viral (177). De plus, une séquence d’environ 300 pb composant la région R-U5 du LTR 5’ permet d’améliorer l’efficacité d’épissage du génome des rétrovirus résultant en une augmentation de la quantité d’enveloppes produites (181). À l’extrémité 5’ de U3 et à l’extrémité 3’ de U5 se trouve une répétition inversée composée de 13 pb essentielle pour l’intégration de l’ADN viral dans la cellule infectée (182, 183). En aval du LTR 5’ se trouve la séquence d’encapsidation ψ qui permet la dimérisation de l’acide ribonucléique génomique (ARNg) des rétrovirus, qui encapsident deux copies de leur génome par particule virale, et qui aurait un rôle à jouer dans la régulation de l’épissage et l’export nucléaire des transcrits viraux (184-186). En aval du LTR 5’, se trouve un site SD (acronyme de la terminologie anglaise pour Splice-Donor; site donneur d’épissage) et dans le gène env se trouve un site SA (acronyme de la terminologie anglaise pour