Identification de la base moléculaire de
l’antigène érythrocytaire de haute fréquence PEL
Mémoire
Corinne Nadeau Larochelle
Maîtrise en biochimie
Maître ès sciences (M.Sc)
Québec, Canada
iii
Résumé
En 2013, la Société internationale de médecine transfusionnelle (ISBT) reconnaît 33 groupes sanguins et 339 antigènes érythrocytaires différents, dont plus de 297 sont déjà associés à un groupe sanguin. Les autres antigènes sont classés dans des séries et des collections en attendant la découverte de leur base moléculaire. En 1996, Geoff Daniels et collaborateurs identifiaient l’antigène PEL. À la suite de cette découverte, des travaux ont démontré qu’il est présent chez plus de 99,9% de la population en général et que le phénotype PEL négatif semble être spécifique à la population québécoise. À la lumière des caractéristiques connues à propos de l’antigène PEL, des analyses génomiques et protéomiques ont été effectuées dans le but de découvrir sa base moléculaire. Cependant, l’analyse des ARN messagers séquencés, ainsi que les résultats obtenus lors des expériences en protéomique, n’ont pas permis d’identifier la base moléculaire de l’antigène PEL.
v
Avant-propos
Premièrement, je tiens à remercier ma directrice de recherche, Maryse St-Louis, d’abord pour m’avoir accueillie au sein de son équipe, mais aussi pour m’avoir fait bénéficier de son expérience et de sa grande disponibilité tout au long de mon projet.
Merci également à Josée Perreault, Élaine Deschênes, Danny Brouard et Josée Lavoie pour leur soutien et leur disponibilité. Merci aussi à Maureen Thompson pour son aide lors du stage qu’elle a effectué au sein de l’équipe.
Je veux aussi remercier les membres de mon comité d’encadrement : Manon Couture, Louis Thibault et Michel Vincent, pour le temps qu’ils m’ont accordé et pour tous leurs judicieux conseils.
Un merci particulier à Patrick Trépanier, Tony Tremblay et Annie Roy pour l’aide fournie pendant certaines parties de mes travaux et au Laboratoire de référence et des cellules souches (LRCS) d’Héma-Québec pour avoir fait toutes les expériences de cartes-gel.
Merci à tous les employés du département de Recherche et Développement d’Héma-Québec. Vous faites de la R&D un environnement enrichissant et stimulant, où il est agréable de travailler.
Un merci spécial à mes deux collègues et amies Dominique Chabot et Lee-Ann Mckinnon pour tous les fous rires et pour avoir contribué à rendre ces deux années de travail une vraie partie de plaisir!
Merci à mon amoureux, aux membres de ma famille et à mes ami(e)s pour leur appui inconditionnel et leurs encouragements.
Finalement, je veux remercier Héma-Québec, le CRSNG, ainsi que le FQRNT pour le soutien financier reçu tout au long de ce projet.
vii
Tables des matières
Résumé... iii
Avant-propos ... v
Tables des matières ... vii
Liste des tableaux ... x
Liste des figures ... xii
Liste des abréviations... xiv
1. Introduction ... 1
1.1. Héma-Québec ... 1
1.2. Le sang ... 1
1.3. Les globules rouges ... 3
1.4. Les groupes sanguins ... 4
1.5. Les antigènes érythrocytaires ... 9
1.6. La sérologie érythrocytaire ... 10
1.6.1. Les immunoglobulines ... 11
1.6.2. Les immunoglobulines de type G ... 12
1.6.3. La liaison antigène-anticorps ... 14
1.7. Le génotypage sanguin ... 16
1.8. L’antigène PEL ... 19
1.9. Contexte de recherche et hypothèse ... 20
1.10. Objectifs ... 21
2. Matériel et méthodes ... 23
2.1. Analyses génomiques ... 23
2.1.1. Échantillons ... 23
2.1.2. Extraction d’ARN ... 23
2.1.3. Design d’amorces pour les RT-PCR ... 23
2.1.4. Amplification par RT-PCR ... 24
2.1.5. Amplification des exons 1 à 5 du gène codant pour la glycoprotéine CD44 ... 26
2.1.6. Amplification de la séquence codante de SMIM1 par PCR classique ... 27
2.1.7. PCR nichée de l’amplicon de l’aquaporine-1 et PCR à demi nichée du deuxième fragment de l’ARN messager du CD238 ... 28
viii
2.1.9. Clonage de l’amplicon correspondant à l’ARNm de CD47 ... 29
2.1.10. Séquençage et analyse des résultats ... 30
2.2. Analyses protéomiques ... 31
2.2.1. Préparation de globules rouges fantômes ... 31
2.2.2. Traitement des globules rouges fantômes à la ficine ... 31
2.2.3. Électrophorèse SDS-PAGE et coloration des gels ... 32
2.2.4. Spectrométrie de masse ... 32
2.2.5. Adsorption-élution des anticorps anti-PEL ... 32
2.2.6. Purification des IgG totales du plasma PEL- par Fast Protein Liquid Chromatography (FPLC) ... 33
2.2.7. Fractionnement des IgG totales purifiées par focalisation isoélectrique .. 33
2.2.8. Tests d’agglutination en carte-gel ... 34
2.2.9. Immunobuvardages de type Western ... 36
2.2.10. Observation de l’expression des protéines CD55 et CD59 par immunobuvardage de type Western ... 37
2.2.11. Immunoprécipitation en conditions non-dénaturantes ... 37
2.2.12. Immunoprécipitation avec crosslinking ... 38
3. Résultats ... 39
3.1. Analyses génomiques ... 39
3.1.1. Amplification par RT-PCR des différents ARNm codant pour les protéines d’intérêt ... 39
3.1.2 Amplification des exons 2 à 5 du gène codant pour la glycoprotéine CD44 ... 39
3.2. Analyses protéomiques ... 40
3.2.1. Traitement des globules rouges fantômes à la ficine ... 40
3.2.2. Coloration de gels SDS-PAGE et envoi de bandes en spectrométrie de masse ... 41
3.2.3. Envoi des globules rouges fantômes PEL+ et PEL- en solution pour être analysés en spectrométrie de masse ... 44
3.2.4. Purification des IgG totales du plasma PEL- par FPLC ... 45
3.2.5. Séparation des IgG totales selon leur point isoélectrique par chromatofocalisation ... 47
3.2.6. Tests d’agglutination sur les fractions d’IgG purifiées séparées par chromatofocalisation ... 48
ix
3.2.7. Adsorption-élution des anticorps anti-PEL ... 49
3.2.8. Immunobuvardages de type Western ... 49
3.2.9. Observation de l’expression des protéines CD55 et CD59 par immunobuvardage de type Western ... 50
4. Discussion ... 51
4.1. Analyses génomiques ... 51
4.2. Analyses protéomiques ... 53
4.2.1. La purification des anticorps anti-PEL du plasma ... 53
4.2.2. Adsorption-élution des anticorps anti-PEL ... 53
4.2.3. Les immunobuvardages de type Western... 55
4.2.4. Observation de l’expression des protéines CD55 et CD59 par immunobuvardage de type Western ... 56
4.2.5. Les immunoprécipitations avec et sans crosslinking ... 57
4.2.6. Les analyses en spectrométrie de masse ... 58
4.2.7. Électrophorèses IEF ... 59
4.2.8. Électrophorèses en deux dimensions (2D) ... 60
5. Conclusion ... 61
Bibliographie ... 63
x
Liste des tableaux
Tableau 1.1. : Classement des différents groupes sanguins, du nombre d’antigènes
et du (des) gène(s) associé(s). ... 5
Tableau 2.2. : Description des protéines dont l’ARNm a été ciblé pour les analyses
génomiques. ... 24
Tableau 2.3. : Conditions expérimentales pour l'amplification des exons 1 à 5 de
l'ARNm de la glycoprotéine CD44 ... 27
Tableau A.1. : Paramètres utilisés pour l'amplification RT-PCR des ARNm des
protéines ciblées. ... 66
Tableau A.2. : SNP identifiés à la suite du séquençage des ARNm de différentes
protéines membranaires érythrocytaires. ... 67
xii
Liste des figures
Figure 1.1. : Visualisation des composantes du sang après centrifugation. ... 2 Figure 1.2. : Processus qui mène à la maladie hémolytique du nouveau-né causée
par des anticorps anti-D. ... 8
Figure 1.3. : Illustration de la disposition de certaines molécules porteuses
d'antigènes érythrocytaires. ... 9
Figure 1.4. : Structure d’une immunoglobuline. ... 12 Figure 1.5. : Fonctions et propriétés physiques des immunoglobulines humaines. . 13 Figure 1.6. : Concept d’affinité dans la liaison antigène-anticorps. ... 15 Figure 2.1. : Principe d'agglutination en carte-gel.. ... 35 Figure 2.2. : Interprétation des résultats d'agglutination obtenus en carte-gel. ... 36 Figure 3.1. : Résultat du traitement des globules rouges fantômes à la ficine en
immunobuvardage de type Western ... 40
Figure 3.2. : Premier gel de polyacrylamide (12%) qui a permis l'envoi de bandes en
spectrométrie de masse.. ... 42
Figure 3.3. : Deuxième gel de polyacrylamide (12%) qui a permis l'envoi de bandes
en spectrométrie de masse.. ... 43
Figure 3.4. : Test d'efficacité de la purification des IgG totales effectuée par FPLC.
... 46
Figure 3.5. : Séparation des IgG totales préalablement purifiées par FPLC en huit
fractions selon leur point isoélectrique. ... 47
Figure 3.6. : Tests d’agglutination effectués en carte-gel effectués pour les huit
fractions obtenues lors de la chromatofocalisation. ... 48
Figure 3.7. : Exemple d'immunobuvardage de type Western effectué avec les
xiv
Liste des abréviations
ADN : Acide désoxyribonucléique ADNc : ADN complémentaire ARN : Acide ribonucléique ATP : Adénosine tri-phosphate CHUL : Centre hospitalier de
l’Université Laval
CH/RG : Système sanguin
Chido/Rodgers
CO2 : Dioxyde de carbone
COST : Collection sanguine Cost CROM : Système sanguin Cromer DTT : Dithiothréitol
ER : Collection sanguine Er ERMAP : Erythroid membrane-associated protein
FPLC : Fast Protein Liquid
Chromatography
FY : Système sanguin Duffy GE : Système sanguin Gerbich GLOB : Système sanguin Globoside Glut-1 : Transporteur de glucose 1 GPI : Glycosylphosphatidylinositol H : Système sanguin Hh
I : Système sanguin Ii IgG : Immunoglobuline G IN : Système sanguin Indian ISBT : La Société internationale de
médecine transfusionnelle
kDa : Kilodalton
KEL : Système sanguin Kell
KN : Système sanguin Knops LRCS : Laboratoire de référence et
des cellules souches
LU : Système sanguin Lutheran LW : Système sanguin
Landsteiner-Wiener
NCBI : National Center for
Biotechnology Information
ng/ml : Nanogramme par millilitre nm : Nanomètre
nmoles : Nanomoles pb : Paires de bases
PBS : Phosphate buffered saline PEL- : Individus dont les globules
rouges n’expriment pas l’antigène PEL
PEL+ : Individus dont les globules
rouges expriment l’antigène PEL
PCR : Réaction en chaîne par
polymérase
PCR-AS : PCR allèle-spécifique PCR-RFLP : Restriction fragment
length polymorphism PCR
RT-PCR : Reverse-Transcriptase PCR SC : Système sanguin Scianna
SDS-PAGE : Électrophorèse sur gel de polyacrylamide en présence de sodium dodécyl sulfate
SNP : Single Nucleotide Polymorphism
1
1. Introduction
1.1. Héma-Québec
Héma-Québec est responsable de la gestion et de l’entreposage des produits sanguins au Québec. Chaque année, l’entreprise livre environ 500 000 produits sanguins aux hôpitaux de la province[1]. Aussi, en plus des produits sanguins, Héma-Québec prépare et gère la
distribution des cellules souches de sang de cordon et des tissus humains. Plus récemment en 2013, le gouvernement québécois a donné son accord pour la mise sur pied, par Héma-Québec, de la première banque publique de lait maternel au Canada.
En constante évolution, cette entreprise a su s’adapter, au fil du temps, aux nouvelles réalités québécoises. Parmi celles-ci, il y a la présence grandissante des diverses communautés culturelles à travers la province. Le défi pour Héma-Québec a été de recruter des donneurs de sang au sein de ces communautés culturelles. Pour plusieurs raisons, certaines communautés culturelles ne voient pas d’un très bon œil le don de sang. Ainsi, Héma-Québec a dû redoubler d’ardeur pour recruter des donneurs, puisque les gens de ces communautés ont, comme les Québécois de souche, parfois besoin de transfusions.
Avant qu’il ne soit transfusé, le sang du donneur doit être compatible avec celui du receveur. Puisqu’il existe plusieurs variants au sein des diverses communautés culturelles et afin de fournir avec efficience et efficacité des produits sanguins à tous les Québécois, Héma-Québec a créé, au fil des années, une banque de sang rare.
1.2. Le sang
Le sang est indispensable au corps humain à plusieurs niveaux. Aussi, il permet de véhiculer tout ce qui est nécessaire à la survie des cellules : l’oxygène, les nutriments, les vitamines, les électrolytes, ainsi que plusieurs autres molécules indispensables au bon fonctionnement du corps humain[1]. Chez un adulte de taille moyenne, la quantité de sang
2
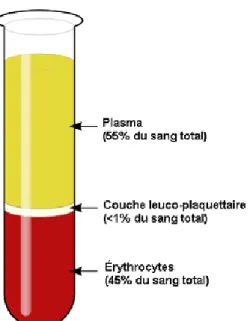
Figure 1.1. : Visualisation des composantes du sang après centrifugation.
Le sang est un liquide biologique homogène qui, après avoir été centrifugé, est séparé en trois phases distinctes : le plasma, la couche leuco-plaquettaire et le culot globulaire (Figure 1.1.). Puisqu’aucune composante synthétique n’existe encore pour le remplacer, les dons de sang sont d’une importance capitale en médecine. En fait, puisqu’un individu en santé ne peut donner du sang qu’une fois tous les 56 jours, le traitement distinct de chacune des trois phases du sang permet de traiter un plus grand nombre de patients. Ainsi, chaque patient reçoit la composante sanguine dont il a besoin.
En fait, après avoir été centrifugé, le sang total est séparé en trois composantes distinctes. À partir de la transformation d’un seul don de sang total, il est possible d’obtenir quatre composés : le plasma, les plaquettes, le culot globulaire et le cryoprécipité[1]. Ce dernier
n’est pas obtenu directement après la centrifugation du sang, mais plutôt en congelant et en décongelant le plasma[1]. Ensuite, ces quatre composés peuvent être donnés
séparément à quatre receveurs, d’où vient la possibilité de sauver quatre vies avec un seul don de sang total[1].
3 La phase supérieure correspond au plasma. Il s’agit du liquide jaunâtre dans lequel baignent les cellules sanguines : les globules rouges (érythrocytes), les globules blancs (leucocytes) et les plaquettes (thrombocytes). À lui seul, le plasma occupe de 50 à 60% du volume total du sang. En plus de servir à véhiculer les cellules sanguines, iI contient plusieurs molécules utiles à toutes les cellules, comme des immunoglobulines, des facteurs de coagulation, des lipides, des hormones et plusieurs protéines, dont la plus abondante est l’albumine.
Sous le plasma, se trouve la couche leuco-plaquettaire contenant toutes les cellules immunitaires appelées cellules mononuclées du sang périphérique (PBMC). Ces cellules jouent un rôle fondamental, autant dans l’immunité innée que dans l’immunité acquise. Elles englobent, entre autres : les lymphocytes, les macrophages, les neutrophiles, les cellules NK et les monocytes. La plupart du temps, à cause de leur pouvoir immunogène, ces cellules sont retirées du don de sang total par filtration. En fait, ces cellules expriment plusieurs antigènes, ce qui les empêche d’être transférables aisément d’une personne à une autre sans causer de réactions néfastes.
Finalement, le culot globulaire occupe la phase inférieure tout au fond du tube et contient, comme son nom l’indique, les globules rouges.
1.3. Les globules rouges
Les globules rouges sont des cellules de forme biconcave, qui mesurent de 8 à 9 μm et qui sont dépourvues de noyau. Ainsi, à maturité, les globules rouges ne contiennent pas de matériel génétique. Dans le corps humain, ils circulent pendant environ 120 jours avant d’être éliminés dans la rate. Par contre, in vitro, la durée de conservation des culots globulaires est de 42 jours lorsqu’ils sont conservés à 4ºC[2]. Après cette période, les
paramètres biochimiques au sein des culots globulaires sont trop altérés pour que ces derniers soient transfusés. La couleur conférée aux globules rouges est due à l’hémoglobine, une protéine composée de quatre chaînes polypeptidiques, deux chaînes α et deux chaînes β, qui contiennent chacune une molécule d’hème. Cette protéine permet aux globules rouges de remplir leur principale fonction au sein du corps humain, c’est-à-dire le transport de l’oxygène.
4
Du point de vue structural, les globules rouges sont entourés d’une membrane plasmique qui contient un nombre impressionnant de protéines et de molécules diverses. À ce jour, le nombre de protéines que contiendrait le globule rouge est évalué à 2289[3]. Notamment, les dernières avancées en spectrométrie de masse ont permis d’atteindre ce nombre au fil des ans. Bien que les techniques d’analyses soient de plus en plus sensibles, l’hémoglobine, qui forme à elle seule 98% du protéome érythrocytaire, reste un contaminant majeur lors des études protéomiques[4]. Comme l’ont décrit Singer et
Nicolson en 1972, la membrane plasmique des globules rouges est une mosaïque fluide où toutes les composantes sont en mouvement constant[5]. Ces diverses protéines et
molécules sont à la base des groupes sanguins et, sont d’une importance capitale en médecine transfusionnelle.
1.4. Les groupes sanguins
La médecine a été révolutionnée par la découverte du groupe sanguin ABO par Karl Landsteiner en 1900[6]. Depuis cette découverte, la Société internationale de
médecine transfusionnelle (ISBT) reconnaît 33 groupes sanguins distincts, qui sont tous codés par un (des) gène(s) différent(s)[6]. Les groupes sanguins sont définis par
l’expression et l’exposition des antigènes érythrocytaires à la surface des globules rouges de chaque individu. Bien que les antigènes érythrocytaires soient les mêmes pour tous et que leur expression varie au sein des différentes populations, l’expression de certains d’entre eux est spécifique à l’ethnicité des individus[7]. Parmi tous les groupes sanguins,
5
Tableau 1.1. : Classement des différents groupes sanguins, du nombre d’antigènes et du (des) gène(s) associé(s). Information tirée de[6].
En ce qui concerne le groupe sanguin ABO, une personne est dite de groupe sanguin A lorsque ses globules rouges expriment l’antigène A. Pareillement, les individus de groupe sanguin B expriment l’antigène B et ceux de groupe AB expriment à la fois les antigènes A et B. Pour leur part, les globules rouges des individus de groupe O n’expriment aucun des deux antigènes A et B. Les antigènes A et B ne sont pas produits directement par l’expression d’un gène, mais sont plutôt définis par des sucres (GalNAc pour l’antigène A et Gal pour l’antigène B), attachés à un ou jusqu’à quatre différents types de chaînes d’oligosaccharides, qui elles sont portées par des glycosphingolipides et des glycoprotéines[7].
Pour chaque antigène érythrocytaire, il existe un anticorps correspondant. Les anticorps du groupe sanguin ABO ont la particularité d’être des anticorps naturels réguliers; c’est-à-dire que les cellules immunitaires de chaque individu produisent des anticorps dirigés contre le ou les antigènes qui ne sont pas exprimés à la surface des globules rouges. Par exemple, le plasma d’une personne de groupe O contiendra à la fois des anti-A et des anti-B et à l’opposé, celui d’une personne de groupe AB ne contiendra aucun anticorps dirigé contre les antigènes du groupe ABO. Bien que les anticorps du groupe ABO soient présents systématiquement dans l’organisme, ce n’est pas le cas pour la plupart des anticorps de groupes sanguins.
6
Le Rh est le deuxième groupe sanguin le plus connu, mais a été le quatrième à être découvert. Contrairement à l’ABO qui n’a que deux antigènes (A et B), le Rh en possède jusqu’à 54. Lorsqu’une personne est dite « positive » ou « négative » à la suite de l’énonciation de son groupe sanguin ABO, il s’agit en fait de la présence ou non de l’antigène D à la surface de ses globules rouges. Environ 85% de la population caucasienne exprime l’antigène D et ces personnes sont dites D positif[1]. Contrairement à
celle d’autres antigènes érythrocytaires, l’expression de l’antigène D ne résulte pas de la mutation d’un seul acide aminé au sein de la protéine (SNP) qui porte cet antigène, mais bien de la présence ou de l’absence du gène RHD[7]. Donc, l’absence du gène RHD chez
l’autre 15% de la population est responsable du phénotype D négatif. Aussi, il est à noter que l’antigène D est une mosaïque de différents épitopes et que l’absence de certains de ces épitopes peut mener au phénotype D partiel[7]. Quant à lui, le phénotype D faible est
caractérisé par une expression moins élevée de tous les épitopes de l’antigène D[7].
Tout comme les antigènes A et B, l’antigène D est très immunogène[7]. Les antigènes de
groupes sanguins immunogènes ont la capacité de déclencher une réaction immunitaire lorsqu’ils sont reconnus par des anticorps spécifiques (ex : l’antigène D qui est reconnu par des anticorps anti-D). Cette mise en branle du système immunitaire amène des réactions post-transfusionnelles. Ces dernières sont, la plupart du temps, causées par la reconnaissance d’un ou de plusieurs antigènes présents sur les globules rouges du donneur par les anticorps présents chez le receveur. La gravité des réactions post-transfusionnelles immunes, c’est-à-dire celles qui sont causées par une incompatibilité antigène-anticorps, est variable et elle dépend de plusieurs facteurs. La réaction hémolytique immédiate, par exemple, se caractérise par la destruction rapide des globules rouges transfusés et elle est la forme la plus grave de réaction transfusionnelle[8]. Elle peut
se produire lorsqu’il y a une incompatibilité antigène-anticorps pour certains groupes sanguins (ABO, Kidd, Kell, Duffy et Lewis), ce qui entraîne, entre autres, l’activation du complément[8]. Dans le cas d’une incompatibilité ABO par exemple, qui correspond à 90%
des cas, cela peut entraîner la mort du receveur[8, 9].
Heureusement, tous les groupes sanguins n’ont pas la même importance clinique en médecine transfusionnelle. Les plus immunogènes sont l’ABO, le Rh, le Kell, le Kidd et le Duffy[8]. La raison qui explique leur importance clinique est que certains de leurs antigènes
7 respectifs peuvent mener à l’immunisation du receveur, c’est-à-dire à la production d’anticorps chez ce dernier. Exceptionnellement, l’immunisation se fait naturellement chez un individu qui développe des anticorps contre ses propres antigènes[10]. Ces anticorps
sont appelés autoanticorps. Ce phénomène d’immunisation est plus souvent observé chez les personnes âgées, mais la cause de celui-ci est toujours inconnue à ce jour. Dans la plupart des cas, l’immunisation se produit lors de transfusions sanguines ou de grossesses[11, 12].
L’immunisation d’un individu peut compliquer de manière significative le processus de transfusion et entraîner certaines complications, notamment dans le cas où la personne immunisée est une femme enceinte[13]. Tout comme ceux du système sanguin Rh, les
anticorps dirigés contre les antigènes du système sanguin Kell peuvent causer la maladie hémolytique du nouveau-né (Figure 1.2.)[7]. Cependant, outre ceux-ci, d’autres antigènes
8
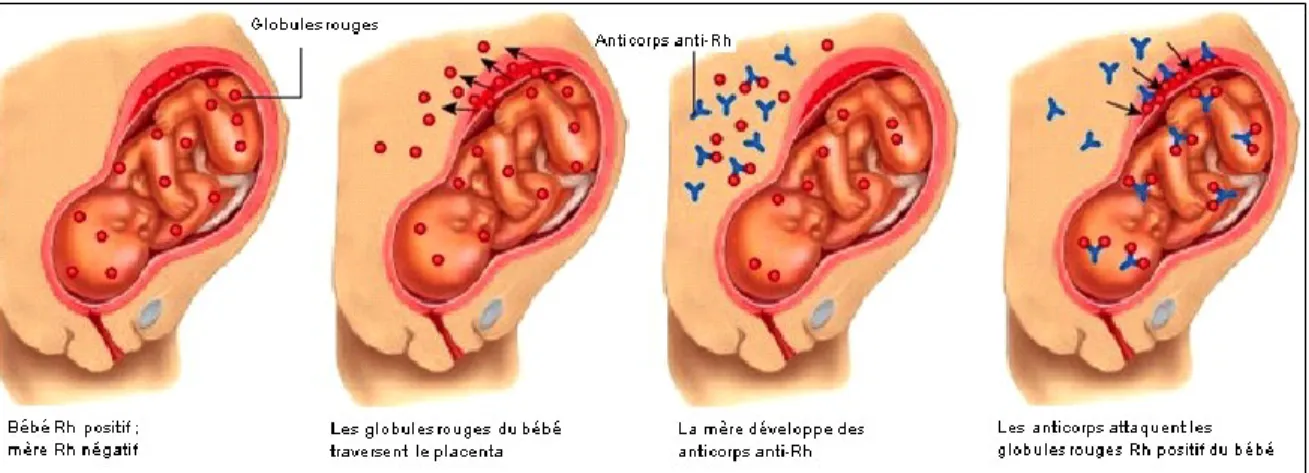
Figure 1.2. : Processus qui mène à la maladie hémolytique du nouveau-né causée par des anticorps anti-D. Adaptée de [14].
La maladie hémolytique du nouveau-né est caractérisée par la reconnaissance des antigènes présents à la surface des globules rouges du fœtus par les anticorps présents dans le sang de la mère. Pour causer cette maladie, les anticorps produits par la mère doivent être de type IgG, puisque seul cet isotype a la capacité de traverser la barrière placentaire[15]. En fait, le transport sélectif des IgG de la mère vers le fœtus se fait grâce à
une protéine de transport des IgG du placenta, FcRn, qui reconnaît la partie Fc des IgG[15].
Lorsque la mère est D négatif et que le père est D positif, le fœtus a 50% ou 100% de chances d’être D positif selon le génotype du père (Dd ou DD). Dans ces cas, la mère peut produire des anticorps dirigés contre l’antigène D. Ce phénomène survient, dans la plupart des cas, lorsqu’il y a contact entre le sang de la mère et celui du bébé. Lorsque cela se produit, c’est le plus souvent lors de l’accouchement. Ainsi, les grossesses subséquentes d’un fœtus D positif sont plus à risques d’être problématiques pour une femme D négatif, puisque les anti-D sont encore la cause principale de la manifestation de la maladie hémolytique du nouveau-né[16].
En résumé, lorsqu’il y a eu un premier contact entre le sang de la mère D négatif et celui du fœtus D positif, les cellules sécrétrices d’anticorps anti-D de la mère peuvent être activées pour en produire lors des grossesses subséquentes. Donc, ces anticorps en circulation chez la mère peuvent traverser la barrière placentaire et détruire les globules rouges D positif du fœtus. Puisque les femmes D négatif qui possèdent des anti-D ont un
9 suivi de grossesse à fréquence élevée, l’apparition de cette maladie est plutôt rare de nos jours[16]. Cependant, dans certains cas graves, les médecins doivent procéder à une
plasmaphérèse chez la mère ou, dans les cas extrêmes, transfuser le fœtus de manière intra-utérine.
1.5. Les antigènes érythrocytaires
L’ISBT répertorie tous les antigènes érythrocytaires et ce nombre est de 339 à ce jour[6].
D’une part, environ 297 antigènes sont déjà associés à un groupe sanguin. D’autre part, les autres sont classés dans des séries et des collections en attendant la découverte de leur base moléculaire. D’un côté, la série 901 regroupe les antigènes de haute fréquence exprimés par plus de 90% de la population et dont le phénotype négatif a été déterminé comme étant hérité génétiquement[6]. À l’inverse, la série 700 regroupe les antigènes de
faible fréquence exprimés par moins de 1% de la population et dont la transmission héréditaire a été démontrée et ce, pour au moins deux générations[6]. Finalement, les
différentes collections regroupent chacune au moins deux antigènes, qui sont liés de manière sérologique, biochimique ou génétique et qui ne peuvent pas être classés dans l’une ou l’autre des séries[6].
Figure 1.3. : Illustration de la disposition de certaines molécules porteuses d'antigènes érythrocytaires[17].
Les antigènes érythrocytaires sont portés par différentes molécules présentes au sein de la membrane plasmique des globules rouges (Figure 1.3.). Ces molécules peuvent être des chaînes glucidiques, attachées ou non à un lipide ou à une protéine, des protéines
10
transmembranaires ou des protéines liées à un ancrage GPI[18]. En fait, les protéines
codées par les gènes se combinent à des polypeptides, à des lipides ou à des oligosaccharides pour former les molécules qui seront porteuses de ces différents antigènes érythrocytaires[8]. Bien que la majorité des antigènes érythrocytaires soient
portés par des molécules membranaires, certains ont, en plus, des composantes hydrosolubles. Par exemple, les antigènes du groupe sanguin Lewis ne sont pas intrinsèques à la membrane des globules rouges, mais ils sont plutôt localisés sur des glycosphingolipides, qui eux sont adsorbés par les globules rouges à partir du plasma[7].
Ces dernières sont en circulation dans l’organisme et elles possèdent des déterminants antigéniques semblables à ceux retrouvés dans la membrane plasmique.
Les protéines membranaires peuvent avoir plusieurs fonctions. Notamment, celles qui sont liées à un groupe sanguin et qui sont les plus abondantes (>200 000 copies par cellule) contribuent aux fonctions essentielles de la cellule, soit en étant des transporteurs membranaires (Bande 3, RhAG, aquaporine-1, SLC14A1, Kx), soit en fournissant une structure qui permet l’ancrage au cytosquelette, ou encore en facilitant l’assemblage des complexes protéiques dans la membrane (RhD, RhCE, glycophorines A et B, CD151)[17].
Ces complexes membranaires ont la fonction de lier la membrane plasmique au cytosquelette, aidant ainsi à garder la structure des globules rouges intacte[17]. Les
protéines moins abondantes à la surface des globules rouges, mais dont le lien avec un groupe sanguin a déjà été démontré, ont des fonctions qui ne sont pas très bien connues à ce jour. Cependant, bien que les protéines qui servent de base moléculaire aux groupes sanguins remplissent diverses fonctions, seule l’absence de la protéine Kx, du groupe sanguin du même nom, a été identifiée comme étant la cause d’une pathologie[17].
1.6. La sérologie érythrocytaire
En sérologie érythrocytaire, les antigènes présents à la surface des globules rouges doivent être reconnus par un anticorps spécifique[6]. Cette liaison antigène-anticorps
permet l’agglutination des globules rouges et ainsi, il est possible de détecter la plupart des antigènes érythrocytaires. Les anticorps spécifiques aux antigènes érythrocytaires sont de type immunoglobuline G (IgG), IgM et dans de rares cas, IgA[19]. Particulièrement,
11 antigéniques. Ainsi, les IgM ont la capacité de faire agglutiner les globules rouges dans les conditions sérologiques standards, c’est-à-dire dans une solution saline 0,85%[8]. À
l’opposé, les IgG et les IgA sont le plus souvent retrouvées sous forme de monomères dans le plasma, mais les IgA peuvent aussi s’associer sous forme de dimères[8]. Ainsi,
l’agglutination des globules rouges dans les conditions sérologiques standards par les isotypes IgG et IgA nécessite un nombre important de sites antigéniques. Dans les autres cas, l’ajout d’un anti-IgG commercial est nécessaire pour visualiser l’agglutination. Ce réactif permet le pontage des IgG fixées spécifiquement à la surface des globules rouges pour créer l’agglutination. La sérologie érythrocytaire permet de détecter, de manière rapide et spécifique, quel(s) antigène(s) est ou sont présent(s) sur les globules rouges et quel(s) anticorps est ou sont présent(s) dans le sang des individus.
1.6.1. Les immunoglobulines
Un des outils grandement utilisé en sérologie demeure les anticorps ou immunoglobulines (Ig). Dans le corps humain, elles sont sécrétées par les plasmocytes et elles sont à la base de l’immunité humorale. Chaque Ig possède au moins un site de liaison spécifique à un antigène. Le nombre de sites de liaison antigénique dépend, entre autres, du type d’Ig. Chez l’humain, il existe cinq classes d’Ig : les IgM, IgG, IgD, IgE et IgA. La structure de chaque classe d’Ig est très semblable et elle se définit par la structure de sa chaîne lourde[15]. Il s’agit d’une molécule en forme de « Y », formée de deux chaînes lourdes (H
pour heavy) et de deux chaînes légères (L pour light). Chacune de ces chaînes est formée de peptides dont les séquences en acides aminés sont plus ou moins variables. Les régions déterminant la complémentarité (CDR) des domaines variables des chaînes lourdes et légères contribuent au site de liaison antigénique, c’est-à-dire que c’est l’association de la chaîne légère et de la chaîne lourde, et non une seule des deux, qui détermine la spécificité antigénique[15].
12
Figure 1.4. : Structure d’une immunoglobuline[20]. Structure type en «Y», où les deux chaînes lourdes sont liées par deux ponts disulfures et où chaque chaîne lourde lie une chaîne légère avec un seul pont disulfure.
Chaque classe d’Ig est conçue pour assurer une fonction principale et leur quantité varie considérablement en fonction du tissu ou du liquide biologique étudié. Chez l’humain, les IgG sont distribuées également dans le sang et les tissus et ce sont les plus abondantes dans le sérum[21]. Ainsi, elles sont souvent impliquées dans les interactions avec les
antigènes érythrocytaires. Les anticorps dirigés contre des antigènes de haute fréquence peuvent parfois causer des problèmes aux gens qui les développent puisqu’il devient parfois très difficile de trouver du sang compatible à leur transfuser[22].
1.6.2. Les immunoglobulines de type G
Les IgG représentent 75% des anticorps du sérum humain. Elles sont formées de quatre chaînes de peptides : deux chaînes lourdes dites gamma (γ) et deux chaînes légères dites
kappa (κ) ou lamba (λ). Les chaînes légères sont κ ou λ, jamais les deux à la fois. La
fonction de chaque type, κ ou λ, est toujours inconnue à ce jour et le ratio κ sur λ est de deux pour un chez l’humain[21]. La masse moléculaire d’une IgG est d’environ 150 kDa
(50 kDa par chaîne lourde et 25 kDa par chaîne légère). Du point de vue structural, les deux chaînes lourdes sont liées entre elles par des ponts disulfures. Chaque chaîne lourde lie une chaîne légère par un seul pont disulfure.
13 Dans le sérum, les IgG sont retrouvées majoritairement sous forme de monomère et chacune possède deux sites de liaison à l’antigène. Ces sites se trouvent aux extrémités supérieures du « Y », là où se trouvent les régions hypervariables des chaînes lourdes et légères. La région constante est composée d’une partie des deux chaînes lourdes et ce fragment interagit avec les molécules effectrices et les autres cellules[21].
Il existe quatre sous-classes d’IgG et elles sont nommées ainsi en fonction de leur concentration relative dans le sérum humain : IgG1, IgG2, IgG3 et IgG4[23]. Les
sous-classes diffèrent aussi selon leur fonction (Figure 1.5.), mais aussi selon d’autres propriétés biologiques, comme leur temps de demi-vie. La figure 1.5. démontre aussi que plusieurs sous-classes d’IgG peuvent remplir la même fonction. En général, les IgG agissent, d’une part, directement sur la réponse immunitaire en neutralisant les virus et les toxines et d’autre part, indirectement en activant le complément ou d’autres cellules impliquées dans l’immunité.
14
1.6.3. La liaison antigène-anticorps
La liaison entre l’anticorps et l’antigène peut être comparée à une clé qui permet d’ouvrir spécifiquement une serrure. Chaque anticorps est conçu pour lier spécifiquement un seul épitope. En fait, certains anticorps reconnaissent un épitope unique, mais qui est formé par l’association de plusieurs molécules sous forme de complexe. Étant donné que chaque anticorps est conçu pour reconnaître et lier de manière spécifique un seul épitope antigénique, plusieurs anticorps différents peuvent reconnaître le même antigène en liant des épitopes différents. La spécificité de liaison d’un anticorps à un seul ou à plusieurs épitope(s) sur un antigène permet de différencier les anticorps monoclonaux des anticorps polyclonaux. Les anticorps monoclonaux vont reconnaître tous le même épitope d’un antigène, alors que les anticorps polyclonaux vont reconnaître le même antigène, mais en ne se liant pas tous au même épitope, donc au même endroit sur l’antigène. Le sérum humain est un mélange d’anticorps polyclonaux. Ainsi, la purification biochimique d’un seul type d’anticorps monoclonal est très difficile, en partie parce que la concentration de n’importe quel anticorps dans l’organisme est très faible[24]. La préparation d’anticorps
monoclonaux nécessite, dans un premier temps, l’isolement de plasmocytes qui sécrètent spécifiquement cet anticorps[25]. Cette cellule doit ensuite être mise en culture, puis
fusionnée avec une cellule immortelle, dérivée d’un myélome par exemple. Ensuite, les anticorps sécrétés sont récoltés du milieu de culture, puis purifiés. Plusieurs réactifs utilisés en sérologie sont des anticorps monoclonaux fabriqués en laboratoire.
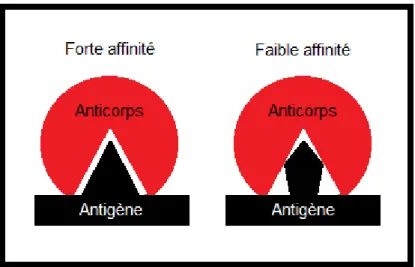
Les réactions antigène-anticorps peuvent être caractérisées selon deux facteurs : l’affinité et l’avidité. D’une part, l’affinité représente la force de liaison entre un site antigénique et un seul site de liaison sur l’anticorps (Figure 1.6.). Cette force de liaison est dépendante de la somme des forces attractives et répulsives qui sévissent entre l’anticorps et l’antigène[8].
15
Figure 1.6. : Concept d’affinité dans la liaison antigène-anticorps.
Puisque la liaison antigène-anticorps est non-covalente, les forces impliquées sont celles de Van der Waals, les liaisons électrostatiques et hydrophobes, ainsi que les liaisons hydrogènes.
D’autre part, l’avidité représente la force totale de la liaison entre un anticorps et un antigène. L’avidité d’un anticorps pour un antigène dépend de l’affinité de chacun des sites de fixation de l’anticorps aux différents déterminants antigéniques[26]. Par exemple, une
affinité moindre entre une IgM et un site de liaison antigénique pourrait être compensée par le fait que les IgM, lorsqu’elles sont associées en pentamères, offrent dix sites identiques de liaison à l’antigène. Ce phénomène permet d’augmenter considérablement l’avidité. Les concepts d’affinité et d’avidité dans la réaction antigène-anticorps sont à la base des tests sérologiques effectués dans les banques de sang.
Héma-Québec teste et répertorie le phénotype sanguin de chacun des donneurs de sang. Ce registre s’avère essentiel dans la recherche de sang compatible à chaque receveur. Dans la plupart des cas, les tests effectués en sérologie érythrocytaire permettent le typage sanguin d’un individu. Cependant, comme chaque technique d’analyse, elle a ses limites. Par exemple, lorsqu’un donneur ne peut pas être phénotypé avec les tests sérologiques standards ou que des résultats divergents sont observés à la suite de ces
16
tests, d’autres méthodes doivent être envisagées. Le génotypage sanguin en est un exemple.
1.7. Le génotypage sanguin
Dans le cas des patients récemment transfusés ou greffés de moelle osseuse, le phénotype sanguin est impossible à établir de manière traditionnelle. En fait, les individus récemment transfusés ont en circulation, en plus de leurs propres globules rouges, ceux qui proviennent du (des) donneur(s). À cause de la présence simultanée de ces deux populations cellulaires dans l’organisme, il est impossible de procéder aux tests sérologiques standards. En fait, il n’est pas possible d’isoler les globules rouges intrinsèques du receveur. Dans le même sens, avant d’être greffés de moelle osseuse, les patients sont tous irradiés. Cette irradiation altère de façon majeure les fonctions des cellules souches, lesquelles sont les cellules progénitrices des cellules sanguines[27]. Une
fois irradié, le receveur est greffé des cellules souches saines du donneur dans le but de reconstruire son système immunitaire et sanguin. Lorsque la greffe est un succès, les cellules sanguines du receveur deviennent de manière intrinsèque celles du donneur, et ce, pour le restant de sa vie. Ainsi, l’ADN retrouvé dans les cellules sanguines du receveur sera différent de celui retrouvé dans ses autres types de cellules. Cependant, pendant la prise de greffe, le receveur exprime une mosaïque hétérogène d’antigènes sanguins, c’est-à-dire qu’il exprime à la fois les siens et ceux du donneur. Ainsi, à cause de la présence des deux populations de cellules sanguines, les tests d’agglutination sérologiques standards sont très difficiles à effectuer.
Ces deux exemples démontrent bien l’utilité du génotypage sanguin. Il existe cependant un bémol au génotypage sanguin dans les cas de prise de greffe. En fait, pendant celle-ci, il est difficile de génotyper les patients puisque les cellules du donneur sont encore présentes dans leur organisme. À mesure que la greffe s’implante, de moins en moins de cellules sanguines vont exprimer le génotype intrinsèque du receveur, mais elles vont plutôt exprimer celui du donneur. Donc, lorsque la prise de greffe est terminée, le génotypage sanguin permet de confirmer le fait que le receveur exprime bel et bien les antigènes érythrocytaires du donneur.
17 Le génotypage sanguin est apparu au début des années 1990[6]. Cette technique
d’analyse permet de prédire le phénotype par l’analyse du génotype. En fait, la majorité des antigènes érythrocytaires sont définis par des polymorphismes observables dans l’ADN génomique, qui affectent la plupart du temps un seul nucléotide (SNP)[28]. Ces SNP
codent spécifiquement certains acides aminés dans la portion extracellulaire des protéines membranaires érythrocytaires.
Les protéines membranaires, comme toutes les autres protéines, sont synthétisées à la suite de la traduction des codons des ARNm en acides aminés. Les ARNm sont transcrits par l’ADN polymérase II et correspondent à une version simple brin de l’ADN qui sert de matrice. Chaque codon est composé de trois bases azotées et l’enchaînement de ces trois bases code pour un seul acide aminé. En fait, l’ARNm mature contient seulement les séquences codantes de l’ADN, c’est-à-dire les exons. Les introns sont transcrits dans le pré-ARNm, mais sont ensuite retirés par un processus nommé épissage. Chez les eucaryotes, les ARNm contiennent une coiffe en 5’ et une queue de poly-A en 3’. Contrairement à la transcription de l’ADN en ARNm qui a lieu dans le noyau, le processus de traduction des ARNm en protéines a lieu dans le réticulum endoplasmique. En fait, les ribosomes enchaînent les acides aminés l’un à la suite de l’autre jusqu’à ce qu’ils aient formé une protéine complète.
Selon la structure des atomes qui composent les acides aminés, ces derniers forment des chaînes qui se replient pour former d’abord des structures secondaires en hélices α et en feuillets β, puis des structures tridimensionnelles. Dans la plupart des cas, la protéine sera fonctionnelle seulement dans sa structure tridimensionnelle. Cependant, il arrive que certaines protéines se regroupent entre elles pour former des structures quaternaires et ainsi, elles peuvent former une nouvelle protéine avec une tout autre fonction. En général, la structure des protéines renseigne beaucoup à propos de leur(s) fonction(s). Par exemple, celles qui traversent de part et d’autre la membrane plasmique des cellules peuvent avoir une fonction de canal ou de pompe, ou encore avoir un rôle purement structural.
Une quantité importante de maladies sont causées par une ou des mutations dans une protéine en particulier. C’est notamment le cas de la fibrose kystique[29]. Dans l’ADN, une
18
quantité importante de mutations s’accumulent au fil du temps. La substitution des bases, surtout des transitions de cytosine à thymine, est la mutation somatique la plus fréquente, mais des insertions/délétions, des duplications, des réarrangements chromosomiques et des translocations peuvent aussi avoir lieu[30].
De la même manière, les mutations dans les protéines membranaires des globules rouges définissent les antigènes érythrocytaires qui eux, sont à la base des groupes sanguins. En fait, plus de 90% des antigènes de groupes sanguins sont codés par un seul polymorphisme (SNP), qui n’altère pas la fonction de la protéine porteuse de ces antigènes[31]. Cependant, bien que la plupart des SNP n’affectent pas la fonction première
des protéines, certains peuvent avoir un effet majeur sur la structure de celles-ci, selon l’acide aminé touché et surtout de sa position au sein des protéines. Donc, comme structure rime avec fonction, la mutation d’un seul acide aminé dans une protéine peut avoir comme effet de l’inactiver complètement.
À défaut d’éliminer complètement la fonction d’une protéine, certaines mutations vont seulement l’altérer. Au fil des ans, plusieurs SNP ont été identifiés et associés à des maladies. Par exemple, l’anémie falciforme est une maladie qui atteint deux individus sur mille de souche africaine et elle est causée par une mutation faux-sens dans le codon 6 de la protéine normale par un résidu valine dans la protéine mutée[24]. Ce changement
d’acide aminé altère profondément la structure de l’hémoglobine, ce qui entraîne une rigidité plus importante des globules rouges et ultimement, des lésions tissulaires[24].
Cependant, l’effet réel de ces SNP sur la transcription génique est difficile à connaître puisqu’un seul SNP peut modifier la transcription d’autres gènes[32]. Il a été observé
maintes fois dans la littérature que plusieurs des fonctions vitales au sein des cellules peuvent être gérées par plus d’une protéine. Ceci serait le résultat de l’évolution, qui avait pour but de garder en vie les cellules, même dans le cas de l’apparition de certaines mutations[33]. En fait, ensemble, les mutations épigénétiques et génétiques amènent un
nombre élevé de variations au sein des cellules, des variations sur lesquelles une sélection peut avoir lieu. C’est la raison pour laquelle la majorité des mutations délétères, celles qui affectent la survie de la cellule, vont être éliminées par le principe de la sélection naturelle[30]. Donc, chez les eucaryotes, chaque protéine est conçue pour remplir un ou
19 plusieurs rôle(s) et à ce jour, ce(s) rôle(s) est (sont) inconnu(s) pour une grande quantité d’entre elles. Heureusement, plusieurs méthodes de protéomique et de biologie moléculaire sont utilisées afin d’élucider la fonction des protéines.
Les méthodes de biologie moléculaire utilisées pour génotyper les groupes sanguins peuvent être classées en deux catégories : celles à faible-moyen débit (PCR, PCR-RFLP, PCR-SSP ou PCR-ASP, PCR en temps réel, séquençage de l’ADN et le pyroséquençage) et celles à haut-débit, qui regroupent tous les systèmes basés sur les puces[28]. En plus de
permettre la résolution des cas complexes en sérologie érythrocytaire traditionnelle, l’apparition des plates-formes automatisées de génotypage sanguin a permis d’augmenter considérablement l’efficacité à créer les registres de donneurs de sang.
1.8. L’antigène PEL
L’antigène érythrocytaire PEL a été identifié pour la première fois en 1996 par l’équipe du chercheur Geoff L. Daniels en Angleterre[22]. Depuis ce jour, il a été classé dans la série
901 par l’ISBT, celle qui regroupe tous les antigènes de haute fréquence[6]. Il a été nommé
PEL en l’honneur de la femme dénommée Pelletier chez qui les anticorps anti-PEL ont été découverts. Cette femme de 29 ans passait des tests sérologiques lors de sa deuxième grossesse lorsqu’ils ont identifiés les anticorps. À la suite de différents tests sérologiques, ils sont parvenus à la conclusion que ces anticorps étaient spécifiques à un nouvel antigène, inconnu jusque-là. Les mêmes anticorps anti-PEL ont été identifiés chez une autre femme, elle aussi d’origine québécoise. Aussi, ils ont identifié, chez deux autres québécoises, des globules rouges qui exprimaient l’antigène PEL, mais en très faible quantité. Ces deux femmes avaient développé des anticorps, mais contrairement à ceux qui avaient été identifiés précédemment, ceux-ci ne réagissaient pas avec les cellules PEL-. Pour cette raison, ils ont été nommés anti-MTP[22].
Même si l’antigène PEL a une importance clinique, il n’a jamais été démontré que ses anticorps spécifiques peuvent causer la maladie hémolytique du nouveau-né[22]. Le sérum
des deux femmes identifiées comme étant PEL- a réagi avec des antiglobulines polyspécifiques et avec des anti-IgG. De plus, l’analyse sérologique des enfants de ces deux femmes a démontré que le phénotype PEL- était hérité de manière récessive.
20
Puisque la base moléculaire de l’antigène PEL n’a pas été découverte, très peu de caractéristiques sont connues à son sujet. Outre le fait qu’il s’agit d’un antigène de haute fréquence présent chez plus de 99% de la population, le phénotype PEL- semble être spécifique à la population québécoise. Aussi, il a été démontré qu’il résiste au traitement des globules rouges par ces enzymes protéolytiques : ficine, papaïne, trypsine, alpha-chymotrypsine, pronase, sialidase ainsi que par le DTT[7]. De plus, des tests
d’agglutination en cartes-gel effectués en sérologie ont permis de déterminer que les anticorps anti-PEL étaient des IgG.
1.9. Contexte de recherche et hypothèse
En 1996, Goeff Daniels et son équipe ont fait la découverte d’anticorps anti-PEL dans le sérum de deux femmes d’origine québécoise[22]. Bien qu’ils soient parvenus à déterminer
que ces anticorps reconnaissent un antigène qui diffère de tous ceux qui ont été identifiés jusqu’à maintenant, la base moléculaire de l’antigène PEL demeure inconnue à ce jour. La découverte de la base moléculaire des antigènes érythrocytaires permettrait de les associer à un groupe sanguin déjà existant ou d’en créer un nouveau. En fait, pour qu’un nouveau groupe sanguin soit nommé, la base moléculaire de l’antigène érythrocytaire doit être codée par un gène qui n’est pas déjà associé à un autre groupe sanguin[6]. Lorsque la
base moléculaire est connue, il devient possible de connaître le génotype de ces individus concernant cet antigène, en complément du phénotype qui a été préalablement identifié en sérologie. Récemment, au mois de février 2012, le groupe de Lionel Arnaud publiait la découverte de la base moléculaire des antigènes Lan et Jra, qui sont deux antigènes de
haute fréquence, tout comme l’antigène PEL[34, 35]. Par exemple, dans le cas de l’antigène
Lan, la création d’un anticorps monoclonal spécifique a permis d’identifier un complexe immun par immunobuvardage. La bande correspondant au complexe immun a été envoyée en spectrométrie de masse et c’est ainsi qu’ils ont lié l’antigène Lan au transporteur d’ATP ABCB6. En sachant que la protéine ABCB6 est la base moléculaire de l’antigène Lan, l’analyse du gène ABCB6 de certains individus de phénotype Lan+ versus ceux de phénotype Lan-, a permis d’identifier divers SNP responsables de ce phénotype[34].
21 Les résultats obtenus par l’équipe de L. Arnaud démontrent bien que la découverte de la base moléculaire d’un antigène de haute fréquence peut se faire selon deux types d’analyses, soient par des analyses génomiques, soient par des analyses protéomiques.
Puisqu’il n’y a qu’un seul article publié à ce jour à propos de l’antigène PEL et qu’il a été démontré que les anticorps anti-PEL sont des IgG, l’hypothèse émise est que la base moléculaire de cet antigène est de nature protéique. Des analyses génomiques et protéomiques ont été effectuées sur des échantillons PEL+ et PEL- pour tenter de découvrir la base moléculaire de l’antigène PEL.
1.10. Objectifs
Ce projet de maîtrise avait pour but d’identifier la base moléculaire de l’antigène PEL. Pour tenter d’y parvenir, plusieurs analyses génomiques et protéomiques ont été envisagées en fonction des caractéristiques déjà connues à propos de cet antigène. Tout d’abord, les analyses génomiques consistaient en l’analyse des séquences codantes des ARNm de certaines protéines déjà connues comme étant la base moléculaire de certains groupes sanguins. En fait, bien qu’il soit possible que l’antigène PEL soit porté par une protéine qui n’est pas déjà associée à un groupe sanguin, il se peut aussi qu’il soit la manifestation d’une ou de certaine(s) modification(s) transcriptionnelle(s) au sein d’une protéine déjà associée à un groupe sanguin.
Les protéines à analyser ont été sélectionnées selon le fait que leur(s) antigène(s) réagissait(ent) de la même manière que l’antigène PEL aux enzymes protéolytiques[23].
Aussi, en sachant que les anticorps anti-PEL sont des IgG et que cet isotype reconnaît souvent des antigènes protéiques, l’hypothèse du projet était que la base moléculaire de l’antigène PEL est de nature protéique. Ainsi, il a été envisagé d’isoler l’antigène par des méthodes de capture et de reconnaissance des protéines.
L’objectif principal de la recherche était d’identifier la base moléculaire de l’antigène érythrocytaire de haute fréquence PEL.
22
Pour arriver à atteindre l’objectif principal de la recherche et dans le but de vérifier l’hypothèse, voici l’objectif de la recherche en ce qui concerne les analyses génomiques :
Séquencer la partie codante de différents ARNm de plusieurs protéines, associées ou non à un groupe sanguin, dans des échantillons PEL+ et PEL- afin de les comparer
Parallèlement, voici les objectifs de la recherche en ce qui concerne les analyses protéomiques :
Faire des électrophorèses SDS-PAGE avec des échantillons PEL+ et PEL- pour en comparer le motif de migration
Envoyer des échantillons PEL+ et PEL- en spectrométrie de masse pour en comparer le protéome érythrocytaire respectif
Isoler les anticorps anti-PEL du plasma par diverses méthodes de purification Identifier un complexe immun par immunobuvardage de type Western en utilisant
diverses préparations biologiques contenant des anticorps anti-PEL comme anticorps primaires
23
2. Matériel et méthodes
2.1. Analyses génomiques
2.1.1. ÉchantillonsTous les échantillons PEL+ (ceux dont les globules rouges expriment l’antigène PEL) et tous les échantillons PEL- (ceux dont les globules rouges n’expriment pas l’antigène PEL) ont été obtenus après la signature d’un formulaire de consentement éclairé et leur utilisation a été approuvée par le comité d’éthique de la recherche d’Héma-Québec. Les deux échantillons sanguins PEL- et les deux échantillons contrôles PEL+ provenaient tous deux d’individus caucasiens et d’origine québécoise. Préalablement, le Laboratoire de référence et cellules souches (LRCS) d’Héma-Québec a confirmé, en sérologie, les phénotypes PEL+ et PEL- des échantillons. De plus, de précédentes analyses sérologiques ont démontré la présence d’anticorps anti-PEL dans le plasma des deux individus PEL-.
2.1.2. Extraction d’ARN
L’ARN des échantillons PEL+ et PEL- a été extrait à partir du sang total préalablement congelé à -20ºC dans du RNA Later (Life Technologies Inc., Burlington, Ontario). Le protocole d’extraction d’ARN provenait de la trousse commerciale RiboPure™-Blood Kit (Life Technologies Inc.) et il a été suivi à la lettre selon les instructions du fabricant. L’ARN purifié était dosé au NanoDrop® (Thermo Scientific, Delaware, États-Unis), un spectrophotomètre qui mesure l’absorbance à 260 nm. Une fois dosé, l’ARN était aliquoté, puis congelé à -80ºC pour toute la durée du projet.
2.1.3. Design d’amorces pour les RT-PCR
Pour les ARNm dont le laboratoire ne possédait pas encore d’amorces spécifiques à utiliser en RT-PCR, des amorces ont dû être sélectionnées. La sélection des amorces s’est faite avec l’application Primer3 intégrée dans le logiciel Geneious (Biomatters Ltd, Nouvelle-Zélande) à partir de la séquence complète des ARNm. Une fois sélectionnées, les amorces étaient recherchées dans une base de données pour s’assurer de leur
24
spécificité. Un total de 25 nmoles de chaque amorce a été commandé chez Integrated
DNA Technologies Inc. (San José, Californie).
2.1.4. Amplification par RT-PCR
La majorité des amplifications par RT-PCR ont été faites avec la trousse commerciale
OneStep RT-PCR kit (Qiagen, Toronto, Canada). Les réactions PCR étaient effectuées
dans un volume final de 50 µl qui contenait de 75 ng à 170 ng d’ARN, 10 µl de tampon 5X, 0,4 mM de dNTP (Qiagen), 40 U d’inhibiteur de RNAse (Life Technologies Inc.), 0,6 µM de chacune des amorces et 2 µl du mélange d’enzymes (Qiagen) provenant de la trousse. L’amplification des ARNm avait lieu dans les thermocycleurs GeneAmp® PCR system
9700 (Life Technologies Inc.). Les protéines ciblées pour l’amplification de leur ARNm sont
présentées dans le tableau 2.2..
25 Les programmes des thermocycleurs variaient selon les ARNm amplifiés. Les ARNm des molécules : aquaporine-1, CD55, CD59, RhD, ERMAP, FLOT-1, GBGT1, Kx et Oka ont été
amplifiés dans les conditions suivantes :
}
Pour leur part, les ARNm des molécules : bande 3, CD47, GLUT-1, SLC14A1 et RhAG étaient amplifié dans les conditions suivantes :
}
L’ARNm de RhCE était amplifié dans les conditions suivantes :
}
}
L’ARNm de la glycophorine A était amplifié dans les conditions suivantes :
}
L’ARNm de la glycophorine B était amplifié dans les conditions suivantes :
}
26
Finalement, l’amplification de l’ARNm de la protéine CD238 était faite dans les conditions suivantes :
}
Puis, pour le séquençage, une PCR semi-nichée a été nécessaire à la suite de la RT-PCR pour la protéine CD238. La PCR semi-nichée, tout comme la PCR nichée, consiste à faire deux amplifications PCR successives, en utilisant, lors de la deuxième amplification PCR, une ou deux amorce(s) différente(s) de celle(s) utilisée(s) lors de la première amplification. Donc, la PCR semi-nichée, dans ce cas, s’est faite dans les conditions suivantes :
}
Les détails concernant ces amplifications, notamment la séquence des amorces utilisées, sont présentés en annexe (Tableau A.1.).
2.1.5. Amplification des exons 1 à 5 du gène codant pour la glycoprotéine CD44
Puisque l’amplification en une seule étape de la séquence codante de l’ARNm de la protéine CD44 s’est avérée infructueuse, les exons 2 à 5 ont été amplifiés par PCR classique selon le protocole décrit dans l’article de Poole et collaborateurs à partir de l’ADN génomique[37]. Étant donné l’échec de l’amplification de l’exon 1 basée sur le
protocole décrit dans l’article, d’autres amorces ont été sélectionnées selon la même méthode que celle décrite précédemment. Seuls les exons 1 à 5 ont été ciblés pour être amplifiés et séquencés, puisque ceux-ci codent pour les parties extramembranaires de la glycoprotéine CD44.
27
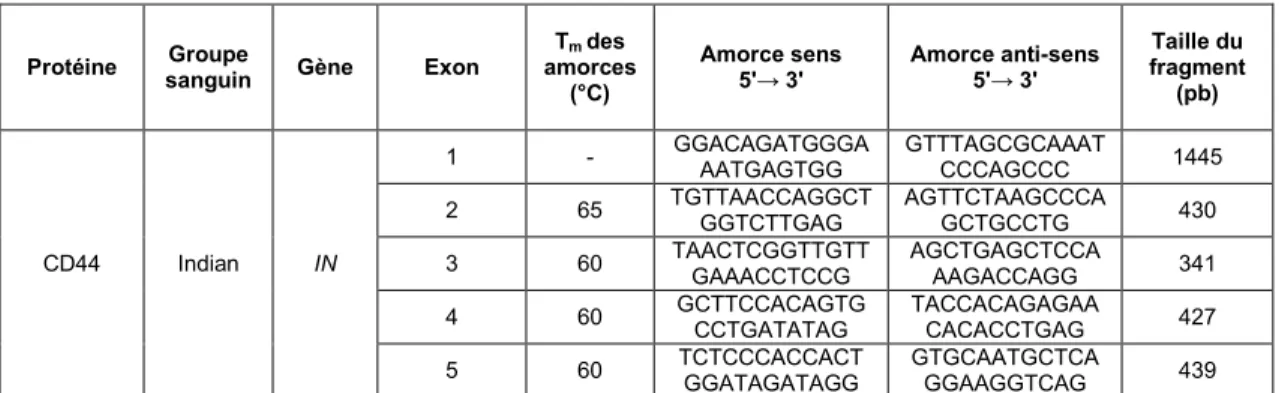
Tableau 2.3. : Conditions expérimentales pour l'amplification des exons 1 à 5 de l'ARNm de la glycoprotéine CD44.
Protéine sanguin Groupe Gène Exon amorces Tm des (°C) Amorce sens 5'→ 3' Amorce anti-sens 5'→ 3' Taille du fragment (pb) CD44 Indian IN
1 - GGACAGATGGGAAATGAGTGG GTTTAGCGCAAATCCCAGCCC 1445 2 65 TGTTAACCAGGCTGGTCTTGAG AGTTCTAAGCCCAGCTGCCTG 430 3 60 TAACTCGGTTGTTGAAACCTCCG AGCTGAGCTCCAAAGACCAGG 341 4 60 GCTTCCACAGTGCCTGATATAG TACCACAGAGAACACACCTGAG 427 5 60 TCTCCCACCACTGGATAGATAGG GTGCAATGCTCAGGAAGGTCAG 439
Sauf pour la Tm (qui correspond à X°C dans l’équation ci-dessous) qui change selon l’exon
amplifié, l’amplification PCR de chaque exon était faite selon la méthode suivante :
}
Pour des raisons techniques, l’exon 1 n’a pas été amplifié.
2.1.6. Amplification de la séquence codante de SMIM1 par PCR classique
Étant donné que la séquence codante de l’ARNm de la protéine SMIM1 du groupe sanguin Vel était de petite taille (237 pb), il était possible de l’amplifier par PCR classique en utilisant l’ADN génomique. Les réactions PCR étaient effectuées dans un volume final de 50 µl qui contenait de 75 ng à 170 ng d’ADN, 5 µl de tampon PCR 10X (Roche, Mississauga, Ontario), 0,2 mM de dNTP (Life Technologies Inc.), 0,6 µM de chacune des amorces et 2,5 U d’enzyme AmpliTaq® Gold (Celera, Alameda, Califonie). Le programme d’amplification PCR était le suivant :
28
2.1.7. PCR nichée de l’amplicon de l’aquaporine-1 et PCR à demi nichée du deuxième fragment de l’ARN messager du CD238
Dans deux cas, parce qu’il s’agissait de plus longs fragments (>1000 pb), une autre PCR était faite à partir de l’ADN complémentaire provenant de l’amplification précédente de l’ARNm. Dans le cas de l’ARNm de l’aquaporine-1 du groupe sanguin Colton, la PCR
nichée était faite selon la méthode suivante :
}
Pour ce qui est du deuxième fragment de l’ARNm de la protéine CD238, la PCR à demi nichée était faite selon la méthode suivante :
}
Ces amplifications permettaient d’obtenir assez d’ADN pour la purification et l’envoi au séquençage.
2.1.8. Purification et dosage des amplicons
À la suite des amplifications RT-PCR et PCR, les divers amplicons devaient être purifiés avant d’être envoyés au séquençage. Lorsqu’il n’y avait qu’une seule bande sur le gel d’agarose, les amplicons étaient purifiés par RapidTip® (D-Mark Biosciences, Toronto, Canada) selon les instructions du fabricant. Dans les cas où les amplifications ne se traduisaient pas en une seule bande sur le gel, les amplicons étaient purifiés à l’aide du
MinElute Gel Extraction Kit (Qiagen) selon les instructions du fabricant. En résumé, les
bandes correspondant aux fragments d’intérêt étaient découpées au scalpel, puis l’ADN était libéré lors de la fonte de l’agarose. Ensuite, l’ADN était purifié dans un système de colonne. La colonne est conçue pour retenir l’ADN, alors que ce dernier est purifié lors d’une étape de lavage et finalement recueilli lors d’une étape finale d’élution. Ces deux étapes sont effectuées par centrifugation.
29 Le dosage des amplicons non purifiés était effectué avec le marqueur de masses moléculaires Low DNA mass ladder (Life Technologies Inc.). Une quantité de 235 ng de ce marqueur était déposée sur un gel d’agarose 1%. La création d’une courbe standard à partir du marqueur permettait de doser 1 µl de chaque amplicon et d’obtenir la concentration de chaque échantillon. Le dosage sur gel d’agarose avait comme avantage de prouver la présence d’un seul fragment d’ADN et de s’assurer que ce dernier correspondait bel et bien au fragment d’intérêt.
Par souci d’économie de temps, les amplicons précédemment purifiés par RapidTip® étaient dosés au NanoDrop®.
2.1.9. Clonage de l’amplicon correspondant à l’ARNm de CD47
Afin d’améliorer la qualité des séquences obtenues à la suite de la RT-PCR, la séquence de l’ARNm du CD47 a dû être clonée. Le vecteur utilisé, le pDrive Cloning Vector provenait de la trousse commerciale PCR CloningPlus kit (Qiagen).
Tout d’abord, le clonage était précédé d’une amplification de la séquence à cloner par RT-PCR dans les conditions énoncées précédemment. Le programme du thermocycleur était le suivant :
}
Ensuite, les produits PCR étaient dosés sur gel d’agarose 1% avec le marqueur de masses moléculaires Low DNA mass ladder afin de s’assurer que la quantité d’amplicons soit suffisante pour procéder au clonage. Une fois dosés, les amplicons étaient purifiés avec les RapidTip®. Pour faciliter la réaction de ligation, il y avait ajout de queues poly-A aux produits PCR selon la méthode suivante : 4 µl de produits PCR purifiés (environ 250 ng d’ADN), auxquels sont ajoutés 0,5 µl de tampon Taq 10X (Life Technologies Inc.), 1 mM de dATP (Life Technologies Inc.) et 2,5 U de Taq polymérase (Life Technologies Inc.). Le volume total de la réaction était de 5,5 µl. L’ajout de queues poly-A se faisait à 72°C pendant 10 minutes, suivi d’un refroidissement à 4°C.
![Figure 1.3. : Illustration de la disposition de certaines molécules porteuses d'antigènes érythrocytaires [17]](https://thumb-eu.123doks.com/thumbv2/123doknet/7266447.206101/23.918.150.789.698.897/figure-illustration-disposition-molécules-porteuses-antigènes-érythrocytaires.webp)
![Figure 1.4. : Structure d’une immunoglobuline [20] . Structure type en «Y», où les deux chaînes lourdes sont liées par deux ponts disulfures et où chaque chaîne lourde lie une chaîne légère avec un seul pont disulfure](https://thumb-eu.123doks.com/thumbv2/123doknet/7266447.206101/26.918.275.601.149.473/figure-structure-immunoglobuline-structure-chaînes-disulfures-légère-disulfure.webp)
![Figure 1.5. : Fonctions et propriétés physiques des immunoglobulines humaines [15] .](https://thumb-eu.123doks.com/thumbv2/123doknet/7266447.206101/27.918.265.665.613.949/figure-fonctions-propriétés-physiques-immunoglobulines-humaines.webp)
![Tableau 2.2. : Description des protéines dont l’ARNm a été ciblé pour les analyses génomiques [6, 36]](https://thumb-eu.123doks.com/thumbv2/123doknet/7266447.206101/38.918.246.631.586.894/tableau-description-protéines-l-arnm-ciblé-analyses-génomiques.webp)
![Figure 2.1. : Principe d'agglutination en carte-gel. Adaptée de [8]. D’abord, les globules rouges sont placés dans la chambre de réaction en présence du liquide qui doit être testé (plasma ou autre)](https://thumb-eu.123doks.com/thumbv2/123doknet/7266447.206101/49.918.318.640.191.488/figure-principe-agglutination-adaptée-globules-placés-réaction-présence.webp)
