Cartographie des complexes multiprotéiques
humains suite à la modification ciblée du génome
Mémoire
Jérémy Loehr
Maitrise en biologie cellulaire et moléculaire
Maitrise ès Sciences (M.Sc.)
Québec, Canada
© Jérémy Loehr, 2017
RÉSUMÉ
La purification par affinité couplée à l’analyse par spectrométrie de masse (AP-MS) est une méthode de choix pour l’étude des interactions protéines-protéines chez les cellules humaines. Par contre, cette technique est sensible aux perturbations causées par la surexpression ectopique des protéines cibles. Des effets anormaux, tels que la formation d’agrégats et la délocalisation des protéines cibles, peuvent mener à des conclusions erronées. Il est donc important de reproduire le plus précisément possible les niveaux physiologiques normaux des protéines à l’étude. Les travaux présentés dans ce mémoire décrivent le développement d’un système robuste et rapide couplant l’édition du génome et la protéomique permettant l’isolation de complexes protéiques natifs exprimés à des niveaux quasi physiologiques. L’approche a servie de tremplin afin d’atteindre l’objectif ultime qui est de caractériser les protéines exprimées à partir de leur contexte génomique naturel. À l’aide des outils d’édition génomique, nous avons introduit de façon ciblée au locus AAVS1 une cassette permettant l’expression de protéines d’intérêt étiquetées avec une séquence permettant la purification par affinité. Ainsi, nous avons purifié de nombreuses holoenzymes impliquées dans la réparation de l’ADN et la modification de la chromatine. Nous avons identifié de nouvelles sous-unités et interactions au sein de complexes déjà bien caractérisés et rapportons l’isolation de MCM8/9, soulignant ainsi l’efficacité et la robustesse de notre approche. La technique présentée dans ce mémoire améliore et simplifie l’exploration des interactions protéiques ainsi que l’étude de leur activité biochimique, structurelle et fonctionnelle.
ABSTRACT
Conventional affinity purification followed by mass spectrometry (AP-MS) analysis is a broadly applicable method to decipher molecular interaction networks and infer protein function. However, it is sensitive to perturbations induced by ectopically overexpressed target proteins and does not reflect multilevel physiological regulation in response to diverse stimuli. Here, we developed an interface between genome editing and proteomics to isolate native protein complexes produced from their natural genomic contexts. We used CRISPR/Cas9 and ZFNs to insert cDNA of interest in the endogenous genomic safe harbor locus AAVS1 and purified several DNA repair and chromatin modifying holoenzymes to near homogeneity. We uncovered novel subunits and interactions amongst well-characterized complexes and report the isolation of MCM8/9, highlighting the efficiency and robustness of the approach. These methods improve and simplify both small and large-scale explorations of protein interactions, as well as the study of biochemical activities and structure-function relationships.
TABLE DES MATIÈRES
RÉSUMÉ iii
ABSTRACT v
TABLE DES MATIÈRES vii
LISTE DES TABLEAUX xi
LISTE DES FIGURES xiii
LISTE DES ABRÉVIATIONS ET SIGLES xv
REMERCIEMENTS xvii
AVANT PROPOS xix
INTRODUCTION 1
1.0 Identification des complexes protéiques cellulaires ... 3
1.1 Techniques utilisées pour la purification des complexes protéiques ... 3
1.1.1 Purification d’affinité en tandem ... 3
Limites ... 5
1.1.1.1 1.1.2 Purification par étiquette d’affinité ... 6
L’étiquette FLAG ... 7
1.1.2.1 L’étiquette Strep ... 8
1.1.2.2 1.2 Les systèmes inductibles avec la tétracycline ... 9
1.2.1 Le système Tétracycline-Off ... 9
1.2.2 Le système Tétracycline-On ... 10
1.2.3 L’amélioration des systèmes ... 11
1.2.4 Limites ... 12
1.3 Limites des techniques présentement utilisées. ... 12
2.0 Concept de genomic safe harbour ... 13
2.1 Insertion au locus AAVS1 ... 14
3.0 L’édition du génome grâce aux nucléases d’ingénierie ... 14
3.1 Les endonucléases ... 15
Reconnaissance et clivage d’une séquence spécifique d’ADN.... 15
3.1.1.2 Utilité pour la modification du génome ... 16
3.1.1.3 Limites ... 17
3.1.1.4 3.1.2 Les nucléase effectrices de type activateur de transcription ... 17
Origines ... 17
3.1.2.1 Reconnaissance et clivage d’une séquence spécifique d’ADN.... 18
3.1.2.2 Utilisation pour la modification du génome ... 19
3.1.2.3 Limites ... 19 3.1.2.4 3.1.3 Le système CRISPR/Cas9 ... 19 Origines ... 19 3.1.3.1 Reconnaissance et clivage d’une séquence spécifique d’ADN.... 20
3.1.3.2 Protospacer Adjacent Motif ... 21
3.1.3.3 Utilisation pour la modification du génome ... 22
3.1.3.4 Limites ... 22
3.1.3.5 4.0 Mécanismes de réparation de l’ADN ... 23
4.1 Réparation des coupures simple brin ... 23
4.2 Réponse aux dommages à l’ADN ... 24
4.2.1 Jonction des extrémités non-homologues ... 26
Mécanisme ... 26
4.2.1.1 Protéines impliquées ... 26
4.2.1.2 Importance de ce mécanisme pour l’invalidation génique ... 28
4.2.1.3 4.2.2 Recombinaison homologue ... 28 Mécanisme ... 28 4.2.2.1 Protéines impliquées ... 28 4.2.2.2 Voies de réparation menant au HR ... 29
4.2.2.3 4.2.2.3.1 Synthesis Dependent Strand Annealing ... 29
4.2.2.3.2 Double-Strand Break Repair ... 30
4.2.2.3.3 Break-induced repair ... 31
4.2.2.3.4 Single-Strand Annealing... 31
Importance du processus de réparation pour l’édition du génome32 4.2.2.4 4.2.3 Utilisation temporelle des mécanismes de réparation de l’ADN ... 33
PROBLÉMATIQUE ET OBJECTIFS DES TRAVAUX 35 CHAPITRE 1 37 AVANT-PROPOS ... 39 RÉSUMÉ ... 41 ABSTRACT ... 43 INTRODUCTION ... 45 EXPERIMENTAL PROCEDURES... 47 RESULTS ... 50 DISCUSSION ... 68 REFERENCES ... 72
SUPPLEMENTAL EXPERIMENTAL PROCEDURES ... 79
SUPPLEMENTAL INFORMATION ... 83
DISCUSSION GÉNÉRALE 97
CONCLUSION 104
LISTE DES TABLEAUX
IntroductionTableau 1: Caractéristiques des étiquettes d’affinités communément utilisées ... 7 Tableau 2: Motif de liaison du PAM dans des orthologues de Cas9 ... 22
Chapitre 1
Table S1, Related to Figures 1 and 2. NuA4 Complex Subunits Identified by Mass
Spectrometry Analysis ... 54 Table S2, Related to Figure 3. PRC2 Complex Subunits Identified by Mass Spectrometry
Analysis ... 60 Table S1, Related to Figures 1 and 2. NuA4 Complex Subunits Identified by Mass
Spectrometry Analysis ... 93 Table S2, Related to Figure 3. PRC2 Complex Subunits Identified by Mass Spectrometry
Analysis ... 94 Table S3, Related to Figure 4. FA Core and Anchor Complexes Subunits Identified by
Mass Spectrometry Analysis ... 94 Table S4, Related to Figure 5. MCM8 Complex Subunits Identified by Mass Spectrometry
Analysis ... 95
Discussion
Tableau 3 : Avantages et désavantages des techniques de purification de complexes
protéine-protéine utilisées précédemment et de celle décrite dans ce mémoire ... 100 Tableau 4: Avantages et désavantages des systèmes Tet-On et notre système
LISTE DES FIGURES
IntroductionFigure 1 : Un survol de la stratégie de purification TAP ... 4
Figure 2 : Séquences d'acide aminé de l'étiquette FLAG et 3xFLAG ... 8
Figure 3 : Représentation schématique du principe de purification d’un strep-tag et d’un twin-strep-tag ... 9
Figure 4 : Tétracycline Off et Tétracycline On ... 11
Figure 5 : Le système Tet-On 3G permet l’induction de l’expression d’un gène en présence de la doxycycline. ... 12
Figure 6 : Schéma du mécanisme d’action et de la structure d’une nucléase à doigt de zinc ... 16
Figure 7 : Représentation schématique de la structure et de la fonction des TALENs ... 18
Figure 8 : Schéma de la nucléase Cas9 guidée par l’ARN ... 21
Figure 9 : Schéma de réparation d’une DSB par les voies NHEJ et HR ... 25
Figure 10 : Représentation d’une réparation par Synthesis Dependent Strand Annealing .. 30
Figure 11 : Représentation de Double-Strand Break Repair ... 30
Figure 12 : Représentation du Break-Induced Repair ... 31
Figure 13 : Représentation de Single-Strand Annealing ... 32
Chapitre 1 Figure 1. ZFN-Driven Gene Addition to the AAVS1 Locus Simplifies Tandem Affinity Purification of Multisubunit Protein Complexes ... 51
Figure 2. CRISPR/Cas9-Driven Tagging of NuA4 Subunits Enables Reciprocal Tandem Affinity Purification of the Endogenous Native Complexes ... 57
Figure 3. Tandem Affinity Purification of the Native PRC2 Protein Complex ... 59
Figure 4. Tandem Affinity Purification of Endogenously Tagged Fanconi Anemia Core Complex... 63
Figure 5. Tandem Affinity Purification of Endogenously Tagged Minichromosome Maintenance Complex Component 8 ... 64
Figure 6. Efficient Complex Purification from Unselected Gene-Modified Cell Pools ... 66 Figure S1. Related to Figure 1. Determination of the Optimal Purification Steps for TAP
and Protein Expression in Single Cell-Derived Clones After ZFN / CRISPR-Driven Gene Addition to the AAVS1 Locus ... 83 Figure S2, Related to Figure 2. Strategy for CRISPR/Cas9-Driven Insertion of the TAP Tag to the C-Terminus of the EPC1, EP400 and MBTD1 Proteins ... 85 Figure S3, Related to Figure 2. CRISPR/Cas9-Driven Insertion of the TAP Tag to the
C-Terminus of the EPC1 and EP400 Proteins ... 86 Figure S4, Related to Figure 3. TALEDriven Insertion of the TAP Tag to the
N-Terminus of the EZH2 Protein ... 87 Figure S5, Related to Figures 4 and 5. Strategy for CRISPR/Cas9-Driven Insertion of the
TAP Tag to the N-Terminus of FANCF and to the C-Terminus of MCM8 ... 89 Figure S6, Related to Figure 1. Tandem-Affinity Purification (TAP) of JADE1L, JADE1S,
and the ATM kinase along with the Presentation of the Auto-Regulated Tet-On 3G System. ... 92
LISTE DES ABRÉVIATIONS ET SIGLES
AAVS1 Site d’intégration 1 du virus associé à l'adénovirus AAVS1 Virus associé à l'adénovirus
AP Apurinique ou apyrimidine ATM Ataxia telangiectasia mutated BER Réparation par excision de bases BIR Break-induced repair
Cas CRISPR-associé
CBP Peptide liant à la calmoduline
CRISPR Courtes répétitions palindromiques groupées et régulièrement espacées
crRNA CRISPR RNA
DBD Domaine de liaison à l'ADN DDR Réponse aux dommages à l'ADN D-loop Boucle de déplacement
DNA-PKcs DNA-dependent protein kinase catalytic subunit DSB Cassure double brin
DSBR Double-Strand Break Repair
EGTA Acide tétraacétique d’éthylène glycol
EK Entérokinase
ESCs Cellules souches embryonnaires
gRNA Guide RNA
GSH Genomic safe harbor HJ Jonction d'Holliday
HR Recombinaison homologue
HSPC Cellules souches et progéniteurs hématopoïétique
IgG Immunoglobulin G
Indels Insertions ou délétions
iPSCs Cellules souches pluripotentes induites KO Invalidation génique
MMR Réparation des mésappariements NER Réparation par excision de nucléotides NHEJ Jonction d'extrémités non homologues PAM Protospacer Adjacent Motif
PPP1R12C Phosphatase 1 Regulatory Subunit 12C ProtA Protéine A
pTRE Promoteur Tetracycline-Response Element pTRE-BI pTRE-bidirectionel
rTetR Représseur Tet inversé
rtTA Transactivateur contrôlé. par la tétracycline inversé SDSA Synthesis Dependent Strand Annealing
sgRNA Single guide RNA
SpCas9 Streptococcus pyogenes Cas9 SSA Single-Strand Annealing
TALEN Nucléases effectrices de type activateur de transcription TALEs L'effecteur transcriptionel activator-like
TAP Purification d'affinité en Tandem tetO Opéron Tétracycline
tet-Off Tétracycline - Off tet-On Tétracycline - On
tetR Répresseur de la tétracycline TEV Tobacco Etch Virus
tracrRNA Trans-activating crRNA
tTA Transactivateur contrôlé par la tétracycline ZFN Nucléase à doigt de zinc
REMERCIEMENTS
Ce mémoire est le résultat d’un travail de recherche de près de deux ans. Je veux adresser tous mes remerciements aux personnes qui m’ont permis de l’accomplir et qui m’ont aidé pour la rédaction de ce mémoire.
Je souhaite tout d’abord remercier mon directeur de recherche Dr Yannick Doyon, qui m’a accompagné tout au long de ma maitrise. Merci d’avoir pris un risque en m’acceptant comme ton premier étudiant, d’avoir pris le temps nécessaire pour me montrer des techniques de laboratoire ainsi que de m’avoir permis de développer une rigueur scientifique dans mon travail.
Je tiens à remercier les gens avec qui j’ai eu la chance de travailler pendant mes études graduées. En premier lieu, je veux remercier les gens de l’équipe Doyon. Je souhaite particulièrement remercier Caroline pour avoir répondu à mes questions et avoir établi une atmosphère de travail très agréable, Sophie pour ton aide qui a été indispensable dans la rédaction de ce mémoire, et Alexandre pour ta facilité avec les mots.
Je souhaite remercier les membres des nombreuses équipes de recherche de l’étage d’avoir permis aux membres de l’équipe Doyon (moi) d’emprunter des ressources de laboratoire lorsque nous étions dans le stade embryonnaire du laboratoire. Et surtout d’avoir pris le temps pour répondre à mes questions, merci à Christine, Francis, Nicolas, Olivier, Philippe, et Suzanne.
Je souhaite remercier Jacques Côté pour m’avoir accueilli dans son laboratoire de recherche et de m’avoir permis d’entamer les premières expériences de ma maitrise. Merci à Valérie et à Céline pour vos conseils et votre support technique professionnel.
Je tiens à remercier tous les gens faisant partie de mon quotidien. Entre autres, Ginette, Annie et Bertrand pour tous les soupers du dimanche et surtout pour les lunchs du lendemain. Guillaume, Mihnea et Roccio lorsque nous réussissons à trouver du temps, nos
Un grand merci à ma famille. Merci à mes parents pour essayer de comprendre ce que je fais, et toujours être de mon côté. Merci à mon frère, tu es mon grand frère préféré.
J’adresse mes plus sincères remerciements ma conjointe Katrine. Sans toi je serais perdu, je t’aime.
AVANT PROPOS
Le présent ouvrage est déposé à la Faculté des Études Supérieures de l’Université Laval pour l’obtention du diplôme de Master ès Sciences (MSc.). Ce mémoire porte sur le développement d’une technique simple et rapide permettant la purification de complexes protéiques dans leur contexte génomique natif. L’étude décrite dans cet ouvrage s’attarde spécifiquement au développement d’une méthode à l’interface entre l’édition génique et la protéomique permettant la purification de complexes protéiques. Ce mémoire contient une introduction générale rédigée en français portant sur la réparation de l’ADN et l’édition génique. Le chapitre 1 constitue le corps de l’ouvrage et relate en détail les travaux de recherche réalisés pour ce mémoire. Ce chapitre est rédigé en anglais sous forme d’un article scientifique tel que présenté en vue de sa publication, selon les exigences éditoriales. Finalement, une conclusion termine ce mémoire en résumant les principaux résultats obtenus, en analysant leurs liens respectifs et en discutant des voies futures à explorer.
L’article présenté dans ce mémoire est le fruit de mes travaux de maitrise qui ont été réalisés au cours des deux dernières années dans le laboratoire du Dr Yannick Doyon. L’étude présentée implique mon entière participation dans les techniques de clonage, de transfection, de culture cellulaire, d’immuno-buvardage. Ce travail exécuté sous la supervision du Dr Yannick Doyon a été réalisé grâce à la collaboration de Mathieu Dalvai (étudiant postdoctoral de Dr Jacques Côté, Université Laval), Karine Jacquet (Doctorante de Jacques Côté, Université Laval), Dr Caroline Huard (professionnelle de recherche du Dr Yannick Doyon, Université Laval), de Céline Roques (professionnelle de recherche du Dr Jacques Côté, Université Laval), de Dr Pauline Herst (étudiante postdoctorale de Dr Jacques Côté, Université Laval) et du Dr Jacques Côté.
Dalvai M*, Loehr J*, Jacquet K, Huard CC, Roques C, Herst P, Côté J, and Doyon Y, A Scalable Genome-Editing-Based Approach for Mapping
Multiprotein Complexes in Human Cells. Cell Reports 13, 621–633, October 20,
1.0 Identification des complexes protéiques cellulaires
Les machineries protéiques de la cellule sont responsables de la coordination et de l’exécution des fonctions cellulaires [1, 2]. Il est donc impératif d’identifier et de caractériser les composantes de ces complexes de façon à mieux comprendre les voies perturbées lorsque l’organisme est malade.
1.1 Techniques utilisées pour la purification des complexes protéiques
La purification des protéines suivie d’analyses par spectrométrie de masse demeure la méthode la plus couramment utilisée pour l’identification des complexes protéiques [3, 4]. Il est important d’avoir une quantité suffisante de la protéine à l’étude pour la réalisation de cette technique [4]. L’étape limitante de cette caractérisation demeure donc la purification du complexe et non l’identification des protéines [4]. Lors de l’étude de protéines faiblement exprimées, cette limite peut être contournée par une surexpression de la protéine cible [5]. Toutefois, cette approche couramment utilisée peut mener à la formation d’interactions non spécifique et mener à des conclusions erronées [5].
1.1.1 Purification d’affinité en tandem
La méthode de purification des complexes protéiques utilisée chez la levure est la purification d’affinité en tandem (TAP), décrite par Puig et al. en 2001 [6]. Cette technique répond à la problématique précédemment décrite, car elle est optimisée de façon à obtenir des complexes protéiques dans des conditions natives [6]. La méthode de TAP consiste en la fusion de l’étiquette-TAP à la protéine cible par recombinaison homologue (HR). L’étiquette-TAP est composée de trois modules : i) deux domaines de reconnaissance des IgG de la protéine A (ProtA) provenant de Staphylococcus aureus, ii) un peptide qui se lie à la calmoduline (CBP, Calmoduline binding protein) séparé par iii) un site de clivage de la protéase du Tobacco Etch Virus (TEV) [6]. Cette construction, CBP-TEV-ProtA, est conçue pour l’étiquetage en C-terminal d’une protéine cible (Figure 1A) [6]. Dans une
fonction native de la protéine, produire des déficiences dans la croissance ou induire la mort cellulaire [6]. Pour pallier à cette problématique, le même groupe de recherche a développé une stratégie visant à placer l’étiquette-TAP en N-terminal de la protéine en inversant l’ordre des modules, ProtA-TEV-CBP-EK (Figure 1A) [6]. Un site de clivage (DDDDK) de l’entérokinase (EK) a aussi été ajouté, permettant ainsi de retirer complètement les résidus de l’étiquette suite à la purification [6].
Figure 1 : Un survol de la stratégie de purification TAP
A) Représentation schématique des étiquettes TAP en C- et N-terminal. B) Survol de la stratégie de
purification TAP. [6]
À partir des extraits cellulaires préparés, les protéines fusionnées ainsi que les protéines du même complexe sont purifiées [7]. L’extrait cellulaire est incubé dans une première colonne de bille Sepharose IgG où le module ProtA de l’étiquette-TAP se lie
fortement à la matrice [6]. La protéase TEV est ensuite utilisée pour couper la protéine chimère au site de clivage TEV permettant à la protéine cible de se dissocier de la matrice IgG [6]. L’éluat, contenant la protéine cible fusionnée avec le module CBP, est incubé dans une deuxième colonne contenant des microbilles liées à la calmoduline. Suite aux étapes de lavage visant à enlever les protéases TEV et les contaminants, le complexe protéique est relâché via l’ajout d’EGTA (Acide Tétraacétique d’éthylène glycol) [6]. Lors de l’utilisation de la stratégie d’étiquetage-TAP en N-terminal, un traitement additionnel à l’entérokinase peut être réalisé de façon à retirer complètement le résidu de l’étiquette (Figure 1B) [6].
Suite à la purification, les complexes protéiques sont séparés sur un gel de SDS-polyacrylamide, puis colorés à l’argent. Les bandes sont par la suite analysées par spectrométrie de masse, permettant ainsi l’identification des protéines formant le complexe purifié. Comme la purification TAP est faite sous conditions douces, des essais d’activité in
vitro peuvent être réalisés avec les complexes purifiés [6-8]. Cette technique est conçue
pour permettre la purification rapide de complexes à partir d’une quantité relativement faible de cellules. De plus, aucune notion antérieure de la composition, l’activité, ou même la fonction de la protéine cible et de son complexe n’est nécessaire pour l’utilisation de cette technique [7].
Limites 1.1.1.1
Bien que la purification par étiquette-TAP soit largement utilisée, il existe néanmoins certaines limites à son usage. La localisation de l’étiquette dans la structure 3D de la protéine peut diminuer son exposition et ainsi réduire sa liaison aux billes d’affinité. La présence de l’étiquette peut aussi affecter la fonction et les niveaux d’expression de la protéine cible [6]. Changer la position de l’étiquette-TAP de C- à N-terminal peut remédier à ces problématiques [6].
L’utilisation de la purification TAP a permis la purification de plus de 589 complexes protéiques, permettant ainsi l’étude systématique du protéome de la levure [9]. Malgré les succès obtenus dans la levure, cette technique se transpose difficilement dans les eucaryotes supérieurs, car la HR est moins efficace dans ces organismes [9]. L’expression dans la levure de gènes humains fusionnés à l’étiquette-TAP a permis l’étude fonctionnelle de nombreuses protéines [9]. Toutefois, les conclusions tirées de ces études demeurent des inférences, puisque ces dernières n’ont pas été réalisées en conditions natives chez l’humain [10-18]. Malheureusement, cette technique est difficilement applicable dans les cellules humaines [9]. La présence de l’étiquette-TAP peut mener à de l’interférence dans la formation de la structure tridimensionnelle de la protéine et lors des interactions avec d’autres protéines [19]. Aussi, certaines protéines de mammifère peuvent interagir avec la calmoduline créant beaucoup de bruit de fond annulant ainsi la fonction principale de l’étiquette-TAP [20, 21]. Des modifications visant à diminuer les perturbations dans la stœchiométrie des interactions protéiques et à prévenir la localisation aberrante, l’agrégation, l’effet dominant-négatif, ainsi que la toxicité devraient être apportées de façon à permettre une meilleure caractérisation des complexes multiprotéiques [22, 23].
1.1.2 Purification par étiquette d’affinité
L’étiquette d’affinité est une séquence polypeptidique fusionnée à une protéine d’intérêt de façon à en faciliter la purification et la détection. Les étiquettes d’affinités peuvent être composées de protéines entières, de domaines protéiques ou de petites séquences peptidiques. Les étiquettes de ce groupe partagent toutes certaines caractéristiques : i) une procédure de purification simple; ii) un effet minimal sur la structure tertiaire et l’activité biologique et iii) une application simple [24]. Tel que décrit précédemment, l’utilisation de larges étiquettes constituées de domaines protéiques peut impacter de façon négative la solubilité et la fonction de certaines protéines; il est donc important de les retirer lors d’études fonctionnelles [24]. Une approche plus simple a été développée et consiste en l’utilisation de très petites séquences peptidiques [25]. Il n’est toutefois pas nécessaire de retirer ces étiquettes, car considérant leur petite taille, ces dernières n’affectent généralement pas la fonction ou les interactions de la protéine à
l’étude [25]. Les étiquettes d’affinités les plus communément utilisées sont : poly-Arg-, FLAG, poly-His-, c-myc-, S-, et Strep II-tag [25]. De façon à optimiser l’étude d’une protéine d’intérêt, les caractéristiques de l’étiquette d’affinité utilisée doivent être prises en compte (Tableau 1) [25]. À titre d’exemple, les étiquettes d’affinité FLAG et Strep seront décrites plus en détail.
Tableau 1: Caractéristiques des étiquettes d’affinités communément utilisées
Tableau tiré de Terpe et al. 2003 [25]
L’étiquette FLAG 1.1.2.1
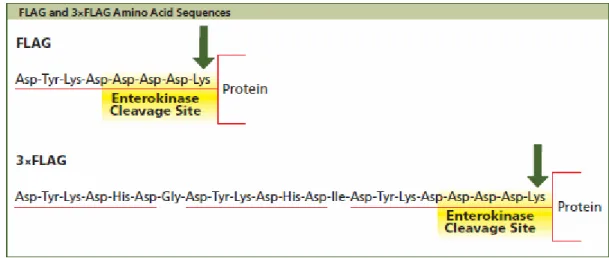
L’étiquette d’affinité FLAG est une petite séquence peptidique de 8 acides aminés (DYKDDDDK)[26]. L’anticorps anti-FLAG monoclonal M2 se lie à la partie N-terminale du peptide FLAGTM permettant ainsi l’immunoprécipitation des protéines liées à l’étiquette FLAG et de leurs complexes [27]. Cette étiquette peut être placée en C- ou en N-terminal de la protéine cible [27]. Ce système est efficace dans plusieurs types cellulaires différents, tels que les bactéries, la levure, et les cellules de mammifères [26, 28-32]. Les conditions de purification du système sont généralement non dénaturantes ce qui permet de purifier une protéine de fusion active [27]. Une version améliorée du système FLAG est le système 3xFLAG, constitué de trois épitopes FLAG regroupés ensemble (Figure 2). Il s’agit d’une étiquette de 22 acides aminés (N-MDYKDHD-G-DYKDHD-I-DYKDDDDK-C), hydrophile qui contient un site de clivage entérokinase [27]. La purification basée sur le système
3xFLAG est en mesure de détecter des quantités aussi petites que 10 fmol d’une protéine cible tandis que le système FLAG est capable d’en détecter 100 fmol [25].
Figure 2 : Séquences d'acide aminé de l'étiquette FLAG et 3xFLAG
Figure tirée de Sigma-Aldrich [33]
L’étiquette Strep 1.1.2.2
Développé, en 1993, comme outil d’affinité pour la purification, le Strep-tag est une petite séquence peptidique de 8 acides aminés (WSHPQFEK) qui se lie fortement dans la pochette de liaison à la biotine de la streptavidine [34]. La Strep-Tactine est une variation de la streptavidine qui est mutée aux positions 44, 45, et 47 [35]. Le Strep-tag a une affinité près de 100 fois plus forte pour la Strep-tactine que pour la streptavidine [35-37]. L’élution de la protéine fusionnée Strep-tag est faite sous condition douce avec une concentration faible en d-desthiobiotin, résultant en un produit purifié se rapprochant de l’état natif [36]. Ceci permet donc l’étude des fonctions des complexes protéiques suite à leur purification [36]. Une version améliorée du Strep-tag II est le Twin-Strep-tag (WSHPQFEK-GGGSGGGSGG-SAWSHPQFEK) [38]. Cette version permet une plus grande affinité pour la Strep-Tactine (Figure 3) [38]. Le Twin-Strep-tag consiste en deux Strep-tag II en tandem avec une région de liaison [38].
Figure 3 : Représentation schématique du principe de purification d’un strep-tag et d’un twin-strep-tag
Figure tirée de Schmidt et al. 2007 [38].
1.2 Les systèmes inductibles avec la tétracycline
Malgré l’éventail de techniques disponibles, il est dans certains cas impossible d’utiliser une des méthodes précédemment décrites pour étudier la fonction d’une protéine cible. Par exemple, il est impossible de surexprimer de façon constitutive une protéine cytotoxique pour en étudier les fonctions. Une approche alternative vise à induire de façon temporelle l’expression du gène d’intérêt. Dans un premier temps, le système inductible LacR/O provenant de E.coli a été utilisé dans les cellules de mammifères, toutefois l’agent inductible β-D-thiogalactopyranoside réagissait trop lentement pour une induction adéquate [39]. Pour répondre à cette problématique, deux systèmes inductibles, provenant de E. coli., ont été développés [40, 41]. Les systèmes tétracycline-Off Off) et tétracycline-On (tet-On) sont fonctionnels dans les cellules humaines HeLa et démontrent une induction rapide de l’expression du gène d’intérêt [40, 41]. Il existe cependant deux éléments clés nécessaires à une induction efficace dans les cellules de mammifères: i) un plasmide exprimant une protéine régulatrice, le transactivateur, et ii) un plasmide comprenant le promoteur inductible.
1.2.1 Le système Tétracycline-Off
Dans le système tet-Off, le transactivateur contrôlé par la tétracycline (tTA) est exprimé constitutivement et se lie au promoteur Tetracycline-Responce Element (pTRE)
activant ainsi le gène d’intérêt (Figure 4) [40]. Le pTRE est composé de 7 répétitions de 19pb de la séquence de l’opéron tétracycline (tetO) suivi d’un promoteur minimal CMV et est reconnu par le répresseur de la tétracycline (tetR) [42]. Le tTA a été créé en fusionnant tetR avec le domaine activateur de la protéine virale 16 (VP16). Lorsque la tétracycline est présente, le tTA se lie préférentiellement à la tétracycline et non au pTRE, donc, inactive le système [40, 41]. L’absence de tétracycline permet la liaison du tTA au pTRE, ce qui induit l’expression du gène d’intérêt.
1.2.2 Le système Tétracycline-On
Le système Tet-On a été développé à l’inverse du système tet-OFF (Figure 4) [40]. Il s’agit donc d’un système inactif en conditions natives dans lequel l’expression du gène d’intérêt est induite par l’ajout de tétracycline [41]. Par mutagenèse aléatoire, Gossen et al. sont parvenus à identifier les acides aminés présents dans tetR responsables de la répression par la tétracycline [41]. Cette découverte a permis le développement d’un répresseur tet inversé (rTetR, reverse Tet repressor). Le transactivateur inversé de tTA (rtTA) a été créé en fusionnant rTetR avec le domaine C-terminal de VP16, permettant ainsi d’activer le système par l’ajout de la tétracycline au milieu.
Figure 4 : Tétracycline-Off et Tétracycline-On
Figure tirée de Gossen et al. 1992 [40].
1.2.3 L’amélioration des systèmes
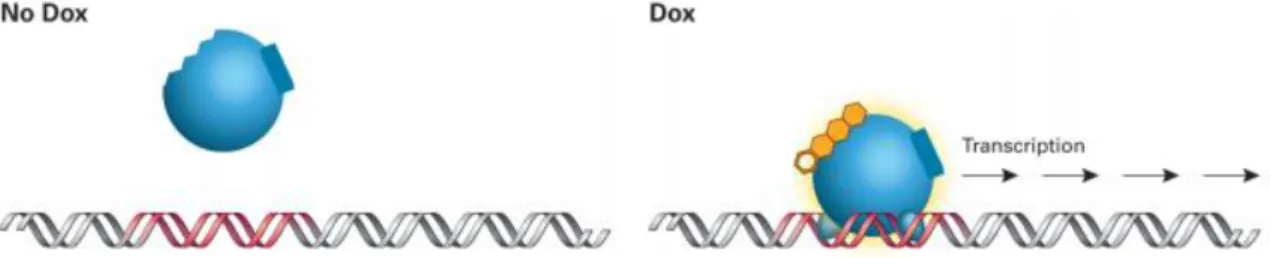
Au cours des années, plusieurs versions améliorées des systèmes ont été conçues, tel le système Tet-On 3G Bidirectionnel [42, 43]. Dans le Tet-On 3G, la doxycycline, un dérivé synthétique de la tétracycline, remplace la tétracycline (Figure 5). La concentration de doxycycline nécessaire pour induire le système est inférieure à celle de tétracycline. Cette concentration inférieure diminue les effets cytotoxiques, ce qui est un net avantage pour les études in vivo [44]. La doxycycline possède une demi-vie de 24 heures, donc pour maintenir l’induction, il est nécessaire d’ajouter de la doxycycline fréquemment au milieu de culture. Il est aussi important de noter que le nouveau système Tet-On 3G n’est inductible que par la doxycycline et non la tétracycline comme l’était son prédécesseur [41]. Le pTRE-bidirectionel (pTRE-BI) permet l’induction des gènes en 5’ et en 3’ [42]. Cette caractéristique permet l’expression inductible en simultané en amont et en aval des deux transgènes d’intérêt. Comme le contrôle de l’expression du gène en 5’ est considéré comme «fuyant», le gène placé en amont est souvent un marqueur visuel, tel mCherry ou
ZsGreen1, permettant la sélection des cellules ayant intégré le système. Le pTRE-BI ne contient aucun site pour la liaison de facteurs de transcription endogènes chez les mammifères, rendant ainsi impossible l’étude en condition d’expression native.
Figure 5 : Le système Tet-On 3G permet l’induction de l’expression d’un gène en présence de la doxycycline.
Figure tirée de clontech [45]
1.2.4 Limites
Les techniques décrites précédemment comportent certaines limitations. Tel que conçus, ces systèmes artificiels nécessitent une transfection simultanée de deux plasmides et fonctionnent par intégration aléatoire dans le génome. La localisation aberrante du transgène par l’intégration aléatoire peut mener à des effets indésirables dans la cellule [22] Ces effets peuvent mener à des conclusions erronées en modifiant le contrôle transcriptionnel réel, en invalidant ou surexprimant des gènes endogènes via la modification de certaines régions régulatrices ou en ayant d’autres effets négatifs via l’agrégation de faux complexes ou la création d’un effet dominant-négatif [22, 23].
1.3 Limites des techniques présentement utilisées.
La purification en tandem suivi de la spectrométrie de masse demeure une technique utile pour l’étude des complexes protéiques, mais cette technique se transpose difficilement dans les cellules humaines [6, 9]. Notamment, l’étiquette-TAP peut mener à l’interférence dans la formation de la structure de la protéine ainsi que des interactions protéiques non retrouvées à l’état naturel dans les cellules de mammifères [19-21]. Les études
fonctionnelles des protéines humaines ont été obtenues par l’expression de gènes humains fusionnés à l’étiquette-TAP dans la levure [9]. Toutefois, les conclusions tirées de ces études sont inférées, puisque les études n’ont pas été réalisées en conditions natives chez l’humain [10-18].
Les étiquettes d’affinités sont de puissants outils permettant la purification de protéines cibles. Par contre, l’utilisation de larges étiquettes constituées de domaines protéiques peut impacter de façon négative la solubilité et la fonction de certaines protéines [24]. L’utilisation d’étiquettes formées de très petites séquences peptidiques ne nécessite pas de retirer ces étiquettes et n’affecte généralement pas la fonction ou les interactions de la protéine à l’étude [25]. Par contre, les caractéristiques de l’étiquette d’affinité utilisée doivent être prises en compte [25].
Les systèmes tétracycline permettent un contrôle sur l’expression de la protéine cible. Toutefois, l’intégration aléatoire des deux vecteurs demeure une limite importante, causant beaucoup de problématique potentielle dans la cellule et pouvant erroner les conclusions ressorties [22, 23].
Des modifications visant à diminuer les perturbations dans la stœchiométrie des interactions protéiques et à prévenir la localisation aberrante, l’agrégation, l’effet dominant-négatif, ainsi que la toxicité devraient être apportées de façon à permettre une meilleure caractérisation des complexes multiprotéiques [22, 23].
2.0 Concept de genomic safe harbour
Un genomic safe harbor (GSH) est une région du génome où il est possible d’intégrer du matériel génétique sans perturber la fonction, la transcription et la régulation des séquences codantes natives, ainsi que la structure génique [46]. Un GSH permet l’expression suffisante d’un transgène sans prédisposer la cellule à une transformation maligne ou en altérer la fonction [46, 47]. Aussi, l’expression du transgène par le GSH doit être possible chez différents types cellulaires [46].
2.1 Insertion au locus AAVS1
Le locus Adeno-Associated Virus Integration Site 1 (AAVS1) aussi sous le nom de Phosphatase 1 Regulatory Subunit 12C (PPP1R12C) est un gène qui encode une protéine dont la fonction n’est pas encore connue. Chez l’humain, ce dernier se situe dans le chromosome 19 à la position 19q13.42 et est le site d’intégration préférentiel du virus associé à l’adénovirus (AAV) [48-50]. Comme l’infection par le AAV n’est associée à aucune pathologie connue et que son intégration dans le locus AAVS1 ne pose aucun effet néfaste, l’intégration au site AAVS1 est considérée inoffensive [51, 52]. Les cellules souches embryonnaires (ESCs) et les cellules souches pluripotentes induites (iPSCs) conservent leur pluripotence suite à l’intégration d’un transgène à AAVS1 [52-56]. De plus, l’expression du transgène est maintenue suite à leur différenciation [52-56]. L’intégration à
AAVS1 a été démontrée comme étant bénigne dans les cellules T humaines en culture [57].
Le gène intégré est transcrit dans les lignées cellulaires communément utilisées : K562, HeLa, HEK293, DU-145, et Hep3B, ce qui facilite son utilisation [47]. Le locus AAVS1 répond aux deux critères pour être considéré un GSH : i) l’intégration au site ne résulte pas en effets néfastes et ii) la transcription du transgène est compétente dans plusieurs types cellulaires [47].
3.0 L’édition du génome grâce aux nucléases d’ingénierie
L’édition du génome est une méthode qui permet l’introduction de modifications désirées à un endroit précis du génome. Ces modifications peuvent prendre la forme d’invalidation génique (KO, Knock-out), d’introduction de séquence exogène, ou d’altération d’un ou plusieurs nucléotides. Cette technique nous permet de repousser les limites des études fonctionnelles en protéomique en facilitant la modification génique. L’édition du génome se fonde sur la création de cassures double brin (DSB) à un site spécifique dans l’ADN à l’aide d’endonucléases, telles que les nucléases à doigt de zinc (ZFN), les nucléases effectrices de type activateur de transcription (TALEN) et CRISPR (Courtes répétitions palindromiques groupées et régulièrement espacées)/Cas
(CRISPR-associé). Ces systèmes, développés en 2005, 2011 et 2013 respectivement, possèdent une facilité d’utilisation ouvrant la porte à la compréhension de certains phénomènes biologiques jusqu'à présent impossible à caractériser.
3.1 Les endonucléases
3.1.1 Les nucléases à doigt de zinc Origines
3.1.1.1
Les ZFN sont des endonucléases chimériques composées d’un domaine de liaison à l’ADN (DBD) fusionné à un domaine de clivage [58]. Le DBD est composé de modules à doigts de zinc Cys2His2 en tandem, modifiés pour reconnaître environ 3 nucléotides spécifiques. Chacun de ces modules dérive de facteur de transcription eucaryote [59-61]. Le domaine de clivage utilisé dans la ZFN est le domaine de clivage non spécifique de l’endonucléase FokI [58].
Reconnaissance et clivage d’une séquence spécifique d’ADN 3.1.1.2
Typiquement, 3 à 6 modules sont utilisés de façon à créer le DBD d’une ZFN. Chacun des modules est formé d’environ 30 acides aminés d’une configuration typique de ββα [62]. Certains acides aminés situés sur la surface de l’hélice-α interagissent avec environ 3 paires de bases se trouvant dans le grand sillon de l’ADN [62]. L’assemblage de plusieurs doigts de zinc permet donc la reconnaissance d’une séquence spécifique d’ADN [62].
La fonction endonucléase de la ZFN est assurée par la fusion du domaine de clivage non spécifique de l’enzyme de restriction Fok1 aux modules de protéines à doigt de zinc. Cette dernière doit dimériser pour être active [63]. Une paire de ZFN dans une orientation adéquate et espacée de 4-7 pb est nécessaire pour créer une DSB à un endroit précis dans le génome (Figure 6) [63]. L’obligation des ZFN à travailler en paire augmente la longueur de
la séquence de reconnaissance à l’ADN de 18 à 36 pb, accentuant considérablement le potentiel d’obtenir un site de reconnaissance unique et limitant ainsi les sites de clivages potentiels hors cible [64, 65].
Figure 6 : Schéma du mécanisme d’action et de la structure d’une nucléase à doigt de zinc
Figure tirée de Urnov et al. 2010 [64]
a) Schéma d’une ZFN dimérisée et liée à sa cible. Chaque ZFN contient le domaine de clivage FokI
lié à 3 à 6 modules à doigt de zinc. Ces derniers sont conçus pour reconnaître une séquence spécifique (boîtes bleues et rouges) sur chacun des brins de l’ADN. Un petit nombre de bases (typiquement 5 à 6) sépare les séquences cibles de la ZFN (Miller et al 2007 ref 58). b) L’assemblage des modules d’une ZFN. Pour générer une protéine à doigt de zinc avec une spécificité à la séquence GGGGGTGAC, trois modules liant spécifiquement un triplet de bases sont identifiés, puis liés. [63]
Utilité pour la modification du génome 3.1.1.3
Les ZFN furent les premières nucléases d’ingénierie à être développées et ont servi à faire des études pionnières dans les plantes, les poissons et les mammifères [64, 66-68]. La ZFN est actuellement la seule nucléase à être utilisée lors d’études cliniques chez l’humain. Sangamo Biosciences utilise une ZFN à la base de leurs stratégies de correction
génique pour différentes maladies (clinicaltrials.gov). Notamment, ils ont un projet en Phase 2 visant à modifier le gène CCR5, un corécepteur utilisé par le VIH pour infecter les cellules T. Ils ont aussi actuellement deux projets en Phase 1, l’un ciblant les cellules souches et progéniteurs hématopoïétique (HSPC) des patients VIH, et l’autre ciblant les HSPC dans les patients souffrant d’anémie à cellules falciformes.
Limites 3.1.1.4
L’utilisation des ZFN comporte toutefois certaines limites. Il faut synthétiser les protéines spécifiques pour chaque site de coupure, ce processus peut être ardu à exécuter et nécessite beaucoup de temps et d’argent. Seul un certain nombre de combinaisons de reconnaissance de nucléotide sont possibles [69]. En effet, les modules protéiques adjacents peuvent exercer une influence les uns sur les autres, ce qui complique la synthèse de ZFN [69]. Pour fonctionner, les ZFN doivent être utilisés en paires ce qui permet de réduire les DSB hors cible, mais ces derniers ne sont pas complètement éliminés [69].
3.1.2 Les nucléase effectrices de type activateur de transcription Origines
3.1.2.1
Les TALENs ont été développées comme une alternative aux ZFN, car elles sont plus simples à générer. Tout comme les ZFN, ces dernières utilisent un code protéine-ADN simple d’utilisation et sont composées d’un DBD et d’un domaine de clivage (Figure 7) [70]. Le DBD est composé de séquences répétées provenant des nucléases effectrices de type activateur de transcription (TALEs), une protéine sécrétée par la bactérie
Xanthomonas qui altère la transcription de gène dans la plante hôte, favorisant ainsi son
infection [71]. Les TALENs clivent l’ADN grâce à leur fusion au domaine endonucléase de l’enzyme de restriction Fok1.
Figure 7 : Représentation schématique de la structure et de la fonction des TALENs
a) Structure d’un TALEN. b) Assemblage de deux TALENs pour créer une DSB. Le clivage par le
domaine FokI s’effectue dans la séquence qui se situe entre les deux régions de l’ADN lié par les deux monomères TALEN. c) Chacune des protéines TALE reconnaît une paire de bases spécifiques grâce à deux résidus. d) Liaison d’un TALEN à l’ADN. Chaque domaine répété se lie à une paire de bases, notons la présence d’une thymine en 5’ de la première base liée par une répétition TALE. [70]
Reconnaissance et clivage d’une séquence spécifique d’ADN 3.1.2.2
Le domaine de répétition TALEs est composé de plusieurs séquences répétitives qui se lient spécifiquement à une seule base d’ADN. Les séquences répétitives sont composées de 33-35 acides aminés. Parmi ceux-ci, les résidus retrouvés aux positions 12-13 confèrent l’interaction spécifique avec une base de l’ADN [72]. Les résidus principalement utilisés à ces positions sont NN, NI, HD et NG, pour reconnaître les bases G, A, C et T, respectivement [70]. La co-crystalisation, des domaines de répétition TALEs liés à la séquence d’ADN cible, a démontré que les séquences répétitives individuelles sont constituées de deux hélices-α en forme de V qui se chevauchent. Les résidus aux positions 8 et 12 interagissent ensemble de façon à stabiliser la structure [2, 73]. Ces dernières
forment une super-hélice autour de l’ADN plaçant ainsi les résidus en position 12 et 13 dans le grand sillon de l’ADN [2, 73].
Utilisation pour la modification du génome 3.1.2.3
Plusieurs études ont démontré que l’efficacité des TALEN à créer des DSB est similaire à celle des ZFN [74-77]. Les répétitions TALE peuvent être assemblées facilement. Notamment, une libraire de TALENs ciblant 18740 gène humains codant des protéines a été développée [78]. Une étude préclinique a utilisé des TALEN ciblant le gène CCR5 pour y induire des petites insertions ou délétions (Indels) et, en conséquence, induire une protection contre l’infection du VIH-1 in vitro [79].
Limites 3.1.2.4
Le site de liaison à l’ADN reconnu par les TALENs doit débuter par une thymine (T) ce qui limite l’éventail des séquences pouvant être ciblées [78]. La livraison des composantes du système TALEN pose aussi un problème puisque leur grande taille rend impossible leur empaquetage dans un virus adéno-associé, le vecteur de choix pour la thérapie génique. [80].
3.1.3 Le système CRISPR/Cas9 Origines
3.1.3.1
Le système CRISPR/Cas est le système d’édition génique le plus récent et démontre une grande facilité d’utilisation [81]. Les séquences répétées, maintenant nommées CRISPR, ont été initialement identifiées en observant cinq répétitions de 29 paires de bases espacées par 32 paires de bases à proximité du gène iap chez la bactérie Escherichia coli [82]. En 2007, Barrangou, Horval et Moineau, ont lié les CRISPR à une réponse immunitaire dans les procaryotes lorsqu’ils ont démontré l’acquisition d’une résistance aux
attaques de phages en introduisant les séquences de CRISPR chez Streptococcus
thermophilus [82]. L’immunité est possible, car un ARN produit par la séquence CRISPR
guide la nucléase du système CRISPR/Cas aux séquences spécifiques du phage invasif, permettant son clivage et sa désintégration [83]. Le système CRISPR/Cas et ses variantes ont été retrouvés dans nombreuses espèces de bactérie et archaea [83]. Les systèmes CRISPR/Cas ont été classifiés en trois groupes : type I, type II et type III, ainsi que dans des sous-types respectifs [84]. En 2012, le système CRISPR/Cas9 appartenant au type II a été démontré comme étant un puissant outil d’ingénierie génétique, ce système est capable de produire une DSB ciblée [83]. Ce système a été utilisé avec succès dans plusieurs organismes différents [85-89].
Reconnaissance et clivage d’une séquence spécifique d’ADN 3.1.3.2
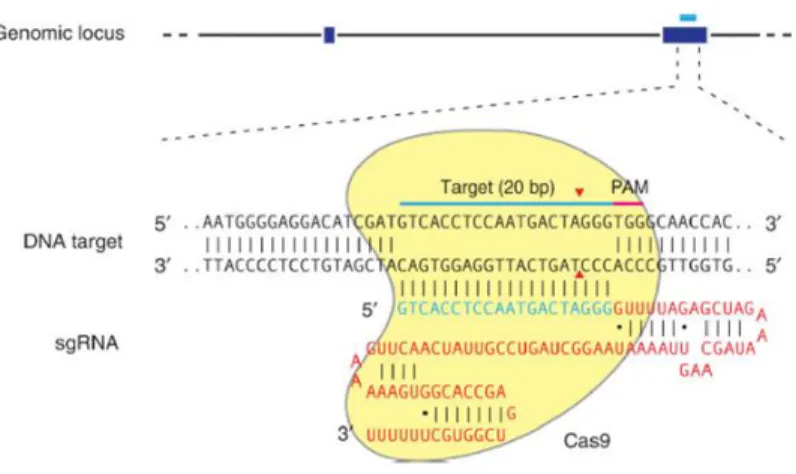
Le système CRISPR/Cas9 à son état naturel consiste en trois éléments : crRNA (CRISPR RNA), tracrRNA (trans-activating crRNA) et Cas. L’hybridation du crRNA avec le tracrRNA permet leur association avec la nucléase Cas9 [90-93]. Une fusion chimérique de l’ARN tracrRNA :crRNA, nommée sgRNA (single guide RNA) ou gRNA (guide RNA), est plus fréquemment utilisée pour l’édition de génome (Figure 8) [83]. La séquence de 20 nucléotides guide faisant partie du crRNA, permet de diriger avec précision la nucléase à la séquence homologue cible dans le génome [83, 90, 93]. Cette séquence cible doit être située directement en amont d’une séquence génomique appelée Protospacer Adjacent Motif (PAM) [83, 90, 93]. Cas9 subit un changement de conformation plaçant les deux domaines endonucléiques, RuvC et HNH sur les brins opposés de l’ADN ciblé et le clive à environ 3 à 4 nucléotides en amont du PAM [83, 90, 93].
Figure 8 : Schéma de la nucléase Cas9 guidée par l’ARN
La nucléase Cas9 de S. pyogenes (jaune) est ciblée pour l’ADN génomique (en exemple dans le locus EMX1 humain) par un ARN-guide consistant en une séquence guide de 20 nucléotides (bleu) et un échafaud (rouge). La séquence guide se lie avec l’ADN cible (ligne bleue), directement en aval du 5’-NGG
PAM (rose). Cas9 crée une DSB à ̴ 3 pb en amont du PAM (triangle rouge).Tirée deJinek et al.[83].
Protospacer Adjacent Motif 3.1.3.3
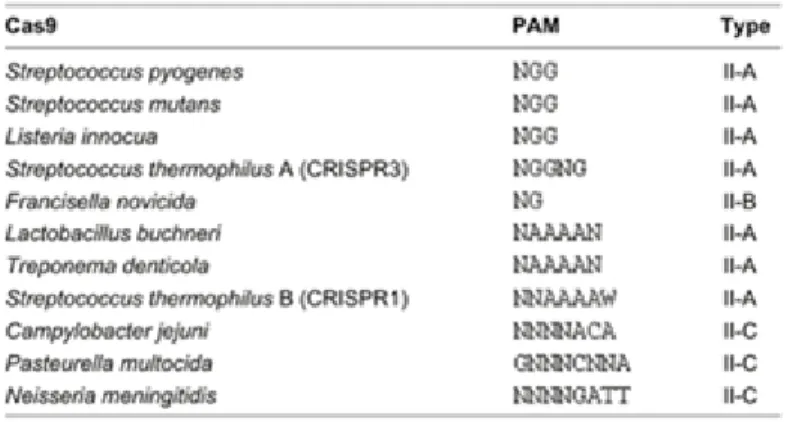
Le PAM est une petite séquence dans l’ADN génomique obligatoire pour la reconnaissance d’une séquence génique par le sgRNA [94]. Toutefois, cette nécessité ne limite pas significativement les possibilités de séquence guide considérant sa petite taille [94]. Par exemple, la séquence PAM de SpCas9 (Streptococcus pyogenes Cas9) est 5’-NGG-3’, une séquence qui est présente à environ chaque 8 à 12 pb dans le génome humain [94, 95]. Certaines nucléases provenant d’espèces différentes peuvent avoir un PAM différent, ce qui permet plus de flexibilité pour la sélection d’un site de coupure dans le génome (Tableau 2) [96]. Le PAM et les 8 à 12 premières bases du guide sont les plus importants pour la reconnaissance du site [83]. Cette séquence appelée ‘seed’ est primordiale à la liaison du guide ARN [83]. Cas9 reconnait tout d’abord un PAM dans le génome. Elle ouvre la double hélice d’ADN et teste l’appariement systématique de chaque base du sgRNA allant du 3’ jusqu’au 5’ [83]. Un mésappariement dans la région ‘seed’ va induire un relâchement du système tandis qu’un mésappariement après le ‘seed’ peut être toléré [83].
Tableau 2: Motif de liaison du PAM dans des orthologues de Cas9
Des orthologues de Cas9 avec les séquences de PAM connu. Le PAM du Cas9 de Lactobacillus
buchneri a été inféré à partir de d’autres séquences connues, mais n’a pas été validé expérimentalement. [96]
Utilisation pour la modification du génome 3.1.3.4
CRISPR/Cas9 est actuellement l’outil le plus puissant pour l’édition du génome, sa facilité de synthèse et le faible coût y étant associé jouent un grand rôle dans sa popularité. Plusieurs domaines de recherche ont été facilités grâce au CRISPR, dont la génération d’animaux transgéniques. Le groupe de recherche de Rudolf Jaenisch a démontré qu’il est possible de créer plusieurs mutations chez la souris en une seule génération [97]. Les premières études cliniques chez l’humain viennent d’être approuvées en juin aux États-Unis. CRISPR/Cas9 sera utilisé pour la thérapie génique ex vivo dans des cellules T de patients souffrant de cancer [98].
Limites 3.1.3.5
Le PAM est la limite majeure pour la construction des gRNAs, car elle est invariable. Toutefois, de plus en plus d’orthologues de Cas9 qui sont découvertes possèdent des PAM différents de celui de SpCas9, ce qui permet d’augmenter les possibilités de cibler une séquence précise [92, 99-102]. D’autres types de nucléases peuvent aussi avoir des PAM différents. Cpf1, une endonucléase de Class 2 du système CRISPR-Cas, possède un PAM riche en thymine : 5’-TTN-3’ [103].
Une autre limite identifiée est la tolérance du système CRISPR/Cas aux mésappariements [47, 83, 95]. Les 10-12 pb en 5’ du gRNA sont généralement les plus tolérantes au mésappariement [83, 95]. Cette caractéristique mène à une problématique de DSB hors cible, qui peut mener à des mutations inattendues [104]. Des stratégies ont été apportées pour diminuer les DSB hors cible, mais elles ne réussissent pas à complètement les éliminer. En diminuant à 17 nucléotides le nombre de nucléotides pour former un ARN guide, les DSB hors cible sont réduits dans certains cas de 5000 fois [47]. Ce processus ne semble toutefois pas affecter l’efficacité des gRNA à atteindre leur cible [47]. Aussi, Cas9 D10A, une nickase créée en mutant et en désactivant le domaine endonucléique RuvC, nécessite deux CRISPR-Cas9-D10A pour faire une DSB. Cette technique, rappelant le fonctionnement des ZFN et des TALENs, augmente ainsi la spécificité du système [105]. De même, la fusion Fok1-dCas9 nécessite aussi d’être utilisée en paire. Ce système utilise la nucléase Fok1 qui doit dimériser pour couper l’ADN et une Cas9 désactivée [106].
4.0 Mécanismes de réparation de l’ADN 4.1 Réparation des coupures simple brin
Lorsque seul un brin d’ADN est coupé, l’autre brin agi comme gabarit pour permettre la correction. La réparation par excision de base (BER) permet de réparer un nucléotide grâce à la glycosylase d’ADN, qui reconnaît et enlève le nucléotide en question, créant un site apurinique ou apyrimidique (AP) [107]. Des endonucléases AP créent une coupure simple brin dans le site AP pour permettre la réparation en utilisant le brin complémentaire comme gabarit [107].
La réparation par excision de nucléotides (NER) répare le dommage à l’ADN provenant de rayons UV [108]. La région endommagée est enlevée en une procédure de trois étapes : i) reconnaissance du dommage, ii) excision du dommage à l’ADN en amont et en aval, iii) et resynthèse de la région d’ADN excisée en utilisant le brin complémentaire comme gabarit [108]. La réparation des mésappariements (MMR) permet la reconnaissance des mésappariements base-base, ainsi que des petites Indels de nucléotides pendant la
réplication et la recombinaison de l’ADN, évitant ainsi que la mutation ne devienne permanente dans la cellule [109-111].
4.2 Réponse aux dommages à l’ADN
Une DSB de l’ADN est potentiellement létale pour la cellule. La reconnaissance rapide des dommages à l’ADN et la réparation avec précision de l’ADN sont les aspects les plus importants de la réponse aux dommages à l’ADN (DDR) dans la cellule [57]. La réparation des DSB se fait principalement par deux voies de réparation de l’ADN, soit par jonction d’extrémités non homologues (NHEJ) ou par HR [112].
La réponse la plus précoce à une DSB est le recrutement du complexe MRN (Mre11, Rad50, Nbs1) suivi rapidement du recrutement de la kinase ataxia telangiectasia mutated (ATM) qui à son tour phosphoryle l’histone H2AX à proximité de la DSB [113-115]. MRE11, quant à elle, est une 3’ à 5’exonuclease double brin et une endonucléase d’ADN simple brin qui assure l’alignement de l’ADN brisé pour permettre sa liaison [116]. Rad50 est une protéine superhélice possédant une activité ATP-dépendante pour la liaison de l’ADN [116]. Le complexe MRN clive les extrémités d’ADN en guise de préparation pour la HR ou la NHEJ [116-118]. Le complexe NuA4 est le complexe protéique clé dans l’acétylation d’histone et la réparation des DSB chez les mammifères. Il contient au moins 16 sous-unités dont trois sous-unités possèdent une activité catalytique : Tip60 l’acétyltransférase, le moteur ATPase p400 et les hélicases Ruvbl1 et Ruvbl2 [119-122]. Tip60 (aussi connue sous le nom d’acétyltransférase KAT5) est recrutée au DSB, s’autophosphoryle et active l’ATM kinase [123-125]. De nombreuses études démontrent que l’ATM phosphoryle Kap1 ce qui est une étape critique pour la réparation des DSB dans l’hétérochromatine [126, 127]. Le recrutement du complexe NuA4 à la DSB permet l’acétylation de l’histone 4, ce qui, en combinaison avec l’activité de l’ATPase SWI/SNF DNA-dépendant de p400, permet de diminuer les interactions histone-histone [128]. Ce relâchement des histones permet une plus grande accessibilité pour la réparation de l’ADN au site de la cassure [128]. Les processus cellulaires menant au choix d’utiliser une voie de réparation ou l’autre ne sont pas encore bien compris, par contre, les protéines 53BP1 et
BRCA1 sont connues comme ayant un rôle clé à cet égard [129, 130]. Il a toutefois été démontré que CtIP interagit avec le complexe MRN et régule la résection, une étape clé dans la décision d’utiliser la réparation par HR dans les cellules mammifères [131].
Figure 9 : Schéma de réparation d’une DSB par les voies NHEJ et HR.
L’ADN ayant subi une DSB et le gabarit homologue utilisé pour la réparation sont respectivement représentés par une paire de lignes noires et grises. A) Les extrémités de la DSB sont liées par MR(X)N et le complexe Ku/DNA-PK. B) Dans la réparation NHEJ, les extrémités de la DSB sont stabilisées par MR(X)N et Ku/DNA-PK. C) MR(X)N et Ku/DNA-PK recrutent le complexe ligase et alignent les extrémités de la DSB. D) Les extrémités de la DSB sont ligaturées ou sont traitées pour d’autre ligation (repair). E) Dans la HR, les extrémités 5’ de la DSB sont réséquées par MR(X)N et d’autres nucléases. F) RPA se lie à l’ADN simple brin généré par la résection. G) L’ADN simple brin lié par la RPA est substitué par la formation du
filament-Rad51, impliquant Rad52, Rad55-Rad57 et Rad54. H) La recherche d’homologie et l’invasion du brin du filament-Rad51 mènent à la formation de la D-loop. I) À partir de la D-loop, différentes voies de HR
peuvent mener à la réparation de DSB. Tirée de Pardo et al. [132]
4.2.1 Jonction des extrémités non homologues Mécanisme
4.2.1.1
La NHEJ est l’une des voies majeures de réparation de l’ADN suite à une DSB. Cette méthode de réparation est active en tout temps lors du cycle cellulaire, mais elle prédomine lors des phases G0/G1 et G2 [133-136].
La NHEJ relie directement les extrémités d’ADN coupées, ce qui peut introduire de petits Indels de nucléotides pouvant mener à une invalidation génique. C’est pour cette raison que la NHEJ est considérée propice à l’erreur, malgré le fait que ce ne soit pas toujours le cas. Il existe trois étapes majeures dans la voie de réparation des DSB par NHEJ : a) retrait de l’ADN endommagé par des nucléases, b) la réparation par les polymérases, et c) la ligation des deux extrémités d’ADN par des ligases.
Protéines impliquées 4.2.1.2
Chez les mammifères, les protéines principalement impliquées dans la réparation par NHEJ sont l’hétéroduplexe Ku70/80, la sous-unité catalytique de la protéine kinase ADN-dépendent (DNA-dependent protein kinase catalytic subunit, DNA-PKcs) et les protéines du complexe de ligation : X-ray cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF, aussi connue sous le nom Cernunnos) ainsi que la DNA ligase IV [137-140].
Les protéines Ku70 et Ku80, nommées en fonction de leurs poids moléculaires en kDa, forment un hétéroduplexe en se liant aux extrémités coupées de l’ADN et recrutent l’ADN-PKcs une sérine/thionine kinase [141]. Une fois recrutée à la DSB, l’ADN-PKcs s’autophosphoryle, et s’autoactive. Par son activation, elle recrute et active Artemis en la
phosphorylant [134, 135]. Le complexe Artemis : ADN-PKcs possède une activité endonucléase 5’ qui a une préférence à cliver l’ADN simple brin 5’ pour y laisser des bouts francs, ainsi qu’une activité endonucléase 3’ avec une préférence pour l’ADNsb 3’ laissant 4 nt en surplomb [142]. De plus, le complexe Artemis : DNA-PKcs possède la capacité de cliver les boucles d’ADN [142]. Ces activités préparent les extrémités d’ADN pour la ligation par le complexe de ligation [142, 143].
La polymérase mu est impliquée dans la préparation d’ADN pré-ligation, car elle possède de multiples activités enzymatiques. Toutefois, d’autres polymérases peuvent contribuer à la réparation par NHEJ lorsque mu est absente, telle la polymérase lamdba [144, 145]. La polymérase mu se lie à la DSB via Ku par les domaines BRCT situés en N-terminal des polymérases [146]. Lorsque Ku et XRCC4 : ADN ligase IV sont présents avec ces polymérases, elles acquièrent la capacité de synthétiser l’ADN après les extrémités coupées, et ce, sans avoir besoin d’un gabarit [147-150]. Comme d’autres polymérases, la polymérase mu peut glisser sur le brin guide ce qui la rend propice à l’erreur [151-153]. La polymérase mu peut aussi synthétiser de l’ADN indépendamment d’un gabarit lorsqu’elle est seule ou en présence de XRCC4 : ADN ligase IV [154].
La ligation des extrémités d’ADN est réalisée par la ligase IV qui est liée par les protéines XRCC4 et XFL ancrées sur l’hétéroduplexe Ku70/80 [146, 155]. XRCC4 et XLF ont des propriétés similaires; elles ne possèdent pas d’activité enzymatique, mais stimulent l’activité de la ligase IV ainsi que sa readénylation lorsqu’elles y sont liées [155]. La ligase IV est capable de ligaturer des extrémités compatibles d’une longueur de 4 nt [156]. L’interaction de la ligase IV avec XRCC4 permet la ligation des extrémités possédant une microhomologie de 2 pb [155, 157, 158]. Si la ligase IV se trouve ancrée au site de la DSB avec Ku, l’activité de ligation est améliorée de 10 fois [154]. Lorsque XLF s’ajoute, le complexe XLF:XRCC4:DNA ligase IV est capable de ligaturer des extrémités d’ADN incompatible avec plus d’efficacité [159, 160]. La NHEJ est un mécanisme qui, malgré son potentiel d’introduire des indels, est nécessaire pour permettre la survie de la cellule dans une situation autrement mortelle [136].
Importance de ce mécanisme pour l’invalidation génique 4.2.1.3
L’invalidation génique est nécessaire pour étudier la fonction d’un gène dans un organisme spécifique. Une fois le gène invalidé, il suffit de comparer les fonctions de la cellule ou de l’organisme invalidé à celles d’un contrôle pour inférer les fonctions du gène. En utilisant une endonucléase ciblant spécifiquement le gène d’intérêt, il est possible d’invalider rapidement et efficacement un gène et étudier ses effets dans la cellule [161]. Récemment, le groupe de recherche de Feng Zhang a développé une stratégie pour induire multiples invalidations de gène en simultané dans les cellules de mammifère utilisant le système CRISPR-Cpf1 avec 4 ARN guides [161].
4.2.2 Recombinaison homologue Mécanisme
4.2.2.1
La HR est une des voies de réparation des DSB qui est considérée sans erreur, car elle utilise la séquence homologue de la chromatine sœur pour faire une correction parfaite [132]. Comme les chromatides sœurs ne sont présentes que lors des phases S et G2/M, la réparation par HR est restreinte à ces phases du cycle cellulaire [162]. La HR classique se caractérise principalement par trois étapes successives : 1) la résection de l’extrémité 5’ à la DSB, suivi de 2) l’invasion de brin de la chromatine sœur et la recherche d’une séquence d’homologie, puis 3) la résolution intermédiaire [132].
Protéines impliquées 4.2.2.2
La première étape de la réparation par HR est la dégradation de l’extrémité 5’ d’ADN. Cette étape est régie par les protéines MRE11, RAD50, NBS1, CtIP and EXO1, ainsi que par d’autres complexes qui demeurent à être identifiés [132]. Cette résection résulte en la production d’un ADN simple brin en 3’ qui est rapidement lié par la protéine de réplication A (RPA), un complexe hétérotrimérique qui se lie à l’ADNss avec haute affinité [163-165]. La liaison de RPA permet d’éviter la formation de structures secondaires
de l’ADNss ainsi que de recruter Rad52 et les protéines encodées par les gènes du groupe épistatique de RAD52 : RAD50, RAD51, RAD52, RAD54, RAD55, RAD57, RAD59,
RDH54, MRE11 et XRS2. Ces dernières forment le filament Rad51 qui remplace RPA sur
l’ADNss 3’ [163-167]. L’ADNss 3’ enrobé par le filament Rad51 peut donc amorcer une invasion du brin de la chromatine sœur en créant une boucle de déplacement, (D-loop) suivi d’une recherche de séquences homologues pour permettre la synthèse de l’ADN [168-170]. Rad54 jouerait un rôle important dans le processus de recherche de séquences d’homologie et dans la maturation d’intermédiaires de la recombinaison suite à la formation de la D-loop [171-173].
Voies de réparation menant au HR 4.2.2.3
Différentes voies de réparation mènent à une réparation par HR. Le processus associé au choix de la cellule d’utiliser l’une ou l’autre de ces voies est encore peu connu. Toutefois, chacune des voies présentées ci-dessous résulte en un échange de matériel génétique entre deux chromosomes homologues.
4.2.2.3.1 Synthesis Dependent Strand Annealing
Dans la voie de Synthesis Dependent Strand Annealing (SDSA), le brin invasif identifie une séquence homologue dans la chromatine sœur. Cette dernière sert de gabarit pour l’élongation [174]. La nouvelle séquence synthétisée se déplace à l’autre bout de la DSB et permet la synthèse d’une nouvelle région complémentaire [174]. Cette voie de réparation ne produit pas d’échange génétique par enjambement [175].
Figure 10 : Représentation d’une réparation par Synthesis Dependent Strand Annealing
A) Déplacement des brins par le brin invasif et liaison à l’autre extrémité du DSB. B) Clivage des
séquences non homologues, élongation et ligation produisent un enjambement. Figure tirée de Pardo et al. [132]
4.2.2.3.2 Double-Strand Break Repair
Lors d’une réparation par la voie classique du Double-Strand Break Repair (DSBR), l’élongation a lieu sur les deux brins d’invasion simultanément en utilisant le gabarit provenant de la même D-loop [174]. La résultante est la formation de jonctions d’Holliday (HJ) qui peuvent produire ou non des événements d’enjambement dépendamment du type de clivage utilisé pour résoudre la jonction [174].
Figure 11 : Représentation de Double-Strand Break Repair
Élongation et ligation du brin invasif : formation de double-jonction d’Holliday. Figure tirée de Pardo et al.[132]


![Figure 4 : Tétracycline-Off et Tétracycline-On Figure tirée de Gossen et al. 1992 [40]](https://thumb-eu.123doks.com/thumbv2/123doknet/5542677.132578/31.918.284.633.145.460/figure-tétracycline-off-tétracycline-figure-tirée-gossen-al.webp)