ROLE DE DIVERS ANTIGENES GRAND T MUTANTS DU VIRUS SIMIEN 40 (SV40) DANS L'ONCOGENESE
par
ALEXANDRE MADARNAS
.Département de microbiologie
Mémoire présenté à la Faculté de médecine en vue de l'obtention du grade de
maître es sciences (M. Sc.)
, '
11..L1
National Utxary Bibliothèque nationale D..,.... or Canada du Canadacaoacftan Theses Secvice Service des t~ canadierines Ottawa, Canada
K1AON4
The author has granted an irrevocabte non· exclusive licence allowing the National Ubrary
of Canada to reproduce, loan, <f&Sbibute or seU copies of his/her thesis by eny means end ln any foon or foonat, making th1s thesis aval1abte to interested persons.
The author retains ownership of the copyright in his/her thesis. Neither the thesis nor substantial extracts trom it may be printed or otherwise reproduced without hi~her
per-mission.
L'auteur a accordé une ncence Irrévocable et non exclusive permettant à la Btbliothèque natiOnale du Canada de reproduire, prêter, cfcstn"buer ou vendre des copies de sa thèse de quelque manière et sous quelque
tonne
que ce soit pour mettre des exemplaires de cette thèse à la disposition des personnes intéressées.L'auteur conserve la propriété du droit d'auteur qui protège sa thèse. Ni la thèse ni des extraits substantiels de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation.
ISBN ©-315-76187-3
TABLE DES MATIERES
TABLE DES MATIERES ...••...••.•...•.•...•...•••••.•....•.... I
LISTE DES FIGURES .•...•...•..•...•••....•.•.•••..•..•... III
LISTE DES TABLEAUX ...••.•...•..•.•....•... IV
LISTE DES ABREVIATIONS ..•••..•••.••••••.••.•... V
RESIJr..Œ • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • •••••••••••• VI
INTRODtJCTI ON . . . • . . . • . . . • . • . . . ••.•... 1
MATERIEL ET :METHODES .•...•..•...••...•..••....•..•... 15
I. Souche virale ....•....•... 15
II. Plasmides recombinants ••...•.•..•...••... 15
III. Méthodes de clonage •...••..• 16
IV. Mutagénèse dirigée avec oligonucléotide synthétique ... 17
1. Clonage dans le phage Ml3 .•.•.•....•.•..•....••... 17
2. Mutagénèse dirigée ...•.•...•.. 18
i. Phosphorylation de l'oligonucléotide ..••...•.••.. 18
ii. Hybridation de l'oligonucléotide à l'ADN rnonocaténaire ....•... 18
3. Purification de l'hétéroduplexe synthétisé par mutagénèse .19 4. Transformation de la souche E. coli JM109 avec la
farine hétéroduplexe ... . 19
i. Préparation des bactéries compétentes ••••••••••.•..•• 20 ii. Transformation des bactéries compétentes •....••...••• 20 V. Analyse des mutants par "dot blot" ..•••.••••••••.••••••••••.• 21 1. Préparation et hybridation de la sonde •••••.•••••••••... 22 2. Purification sur pétri •••••••.••.•.•••••.•.•.••••••.••••.• 23 3. séquençage de l'ADN des clones mutants •••••...••••... 23 VI. Transfection de cellules primaires et essai
d' irmnortalisation •••••••••.•..•..•.•.••...••••••••.•..••.•••• 24 1. Préparation des fibroblastes d'embryon de rat ••••....••.•• 24 2. Transfection au chlorure de calcium ••••••••..••.•••.•••••• 24 3. Sélection à l'antibiotique G-418 et irnmortalisation ..•••.• 25 VII. Transfection de lignées cellulaires établies •..•.••••••.•... 26 VIII. Immunoprécipitation des antigènes mutants .•••.•••..••.•... 26
RESIIl..TATS ••••..•••.••••.•••..••...•••••••••..••••.••••.••....••... 29
DISCUSSION •.•...•..••••...•••.•.•••••••••••••••••.••..••..••••..••• 42
REMERCifilŒN'rS •...•.••..•••....•.••••••••••.•••••.••••••••.••••.••...• 4 9
LISTE DES FIGORES
Figure 1. Carte physique du virus SV40 .••••...•••.••...••••.••••.••. 3
Figure 2. Localisation des domaines fonctionnels de l'antigène
grand T du virus SV 40 •.•...•...•••..••••••.••••.•...• 8
Figure 3. Homologie de séquence entre les antigènes grand T du groupe des virus du polyome et les oncoprotéines virales ElA d'adénovirus et E7 du virus du papillome
humain ... 11
Figure 4. Structure des recombinants du phage Ml3 utilisés lors
de la mutagénèse dirigée dans les régions 1 et 2 •.••.•••••• 30
Figure 5. Autoradiograrnme de la séquence des mutants de la
région conservée 1 et 2 de grand T ..•••.••...••••...••• 33
Figure 6. Clonage du mutant dll02 dans le plasmide pSV2néo ••••.•••••. 34
Figure 7. Analyse des antigènes grand T mutants par
LISl'E DES TABLEAIIX
Tableau 1. Capacité relative de certains mutants du grand T de SV40 et du grand T de polyorne à immortaliser
a.a. BSA q:m ddNTP DEAE DMEM Il-iF' DMSO dNTP D'IT EDTA IPm Kd Kb mM nt pb PVP rpm SDS Tris
u
µCi X-GalLISTE DES ABBREVIATIONS
acide aminé
albumine sérique bovine coup par minute
didéoxynucléotide 51 triphosphate
diéthylaminoethyl
Milieu Eagle modifié par Dulbecco N,N-diméthylformamide diméthylsulfoxyde déoxynucléotide 5' triphosphate dithiothréitol éthylène-diamine-tetraacétate de sodium isopropylthio-B-D-galactoside kilodalton kilobases milimolaire nucléotide paire de bases polyvinylpyrrolidone révolutions par minute sodium dodécyl sulfate
tris (hydroxyméthyl) amino méthane unité enzymatique internationale microcurie
L'objectif de ce travail est de mieux comprendre l'importance de la région N-terminale de l'antigène grand T du virus SV40 dans l'irnrnortalisation cellulaire. En particulier, nous nous sormnes intéressés à deux séquences d'acides aminés hautement conservées parmi les papovavirus, appelées régions conservées 1 et 2. Dans ce but, nous avons construit différents mutants de délétion et de substitution dans les régions 1 et 2 de la protéine grand T par mutagénèse dirigée. La région N-terminale de grand T, qui contient les régions 1 et 2, est impliquée dans l'i:rrmortalisation cellulaire et est essentielle à la liaison de la protéine cellulaire du retinoblastome pRB. pRB est impliquée dans la régulation négative de la croissance celulaire.
Les protéines ElA d'adénovirus et de grand T de polyome s'associent aussi à pRB par l'intermédiaire des deux régions conservees. Dans le , cas de grand T de SV40, il semblerait que seule la région 2 est importante dans l'interaction avec RB, le rôle de la région 1 étant encoore mal défini dans cette interaction. La région 2 contient le motif (N/D)LXCXE. Des études récentes ont démontré que la délétion de ce motif dans le grand T de polyome (mutant dll41) élimine sa liaison à RB dans un essai in vitro, ainsi que son pouvoir inrrnortalisant (Larose et al., 1990). Nous avons vérifié l'hypothèse selon laquelle la délétion du motif (N/D)LXCXE du mutant pSV24 de SV40 éliminerait son pouvoir inunortalisant en abolissant sa capacité de complexer la protéine RB. Le nouveau mutant (dll02) serait l'équivalent fonctionnel du mutant dll41 de polyome, c'est-à-dire, il serait négatif à la fois pour la
liaison à RB et à p53 (une autre protéine impliquée dans la régulation négative de la croissance cellulaire).
Des études de mutagénèse dans la région 1 de grand T du virus du polyome (mutants 13 val, 16 val et dll3) ont démontré l'importance de cette région dans 11immortalisation. Les trois mutants lient la
protéine RB mais les mutants 13 val et dll3 sont incapables d'immortaliser des cellules et de transactiver des promoteurs viraux. Les résultats obtenus avec le grand T de polyome démontrent également que même une simple substitution conservative (mutant 13 val) peut abolir la fonction d1immortalisation. Ces mêmes mutations ont été
faites dans le mutant SV24 afin de mieux comprendre le rôle de la région 1 dans le grand T de SV40.
Le virus SV40 fait partie de la famille des papovavirus et se retrouve plus particulièrement dans le groupe des virus du polyorne. Douze virus identifiés jusqu'à maintenant font partie du groupe des virus du polyome. Parmi ceux-ci on retrouve notamnent, le virus du polyome et les virus BK et JC. Les virus SV40 et polyorne sont les seuls à avoir été étudiés en détail. Ces études se sont avérées très utiles pour mieux comprendre les phénomènes moléculaires impliqués dans la régulation de l'expression et de la réplication de l'ADN des cellules eucaryotes. L'observation que ces virus sont turnorigènes chez certains animaux et peuvent transformer des cellules en culture a fourni aux chercheurs des outils pour étudier les mécanismes qui provoquent la turnorigenèse. L'intérêt renouvelé dans !'oncogenèse occasionné par SV40 et polyome découle des observations suivantes: les antigènes viraux s'associent à des protéines cellulaires (Lane et Crawford, 1979; Linzer et Levine, 1979; McCormick et Harlow, 1980) et les gènes immortalisants et transformants des virus oncogènes à ADN sont interchangeables avec des oncogènes cellulaires dans des expériences de cotransfection (Land et al., 1983; Ruley, 1983).
Le virus SV40 est retrouvé dans les cellules rénales de son hôte naturel, le singe rhésus. L'infection de celui-ci par le virus est sans conséquence et ne présente pas d'effets cytopathogènes facilement détectables. Le virus SV40 a été découvert indirectement suite à des travaux sur le vaccin contre le virus de la poliomyélite. Le vaccin de la polio, qui était produit dans les cellules rénales du singe rhésus,
\
contenait un certain nombre de particules du virus SV40. Ces particules contaminantes causaient des effets cytopathogènes facilement observables lorsque les cellules d'autres espèces de singe étaient utilisées dans les essais (SWeet et Hilleman, 1960). Le virus SV40 est un virus non enveloppé avec une capside icosahédrique de 45 nrn de diamètre (Tooze, 1981). Le génome viral est constitué d'une molécule d'ADN bicaténaire circulaire de 5243 pb, associée à des histones cellulaires. La figure 1 illustre l'organisation du génome viral qui est composé de deux régions codantes, l'une dite région précoce et l'autre région tardive, séparées par une région non-codante régulatrice qui comprend l'origine de réplication, les promoteurs précoce et tardif et les activateurs de transcription (Reddy et al., 1978; Benoist et Chambon, 1981; Cereghini et al., 1983; Gruss et al., 1981; Hartzell et al., 1984a; 1984b).
La région précoce de SV40 est transcrite très tôt lors d'une infection lytique, avant le début de la réplication de l'ADN viral. Cette région encode deux protéines différentes appelées antigènes grand T (96 Kd) et petit T (17 Kd) qui ont d'abord été détectés par la présence d'anticorps anti-T dans le sérum d'animaux porteurs de tumeurs et ensuite dans les immunoprécipitations de cellules infectées par le virus (Black et al., 1963; Prives et al., 1975; 1977; Tegtmeyer et al., 1975). Les deux antigènes sont générés à partir d'un même ARN prémessager suite à des événements d'épissage différents. En raison de l'épissage différentiel les 82 premiers acides aminés de la région N-terminale des deux protéines sont identiques. L'antigène petit T possède en plus 92 acides aminés uniques de l'intron de grand T (Tooze,
Figure 1. carte physique du virus SV40.
Représentation schématique du génome viral de SV40 indiquant les unités de transcription précoce et tardive codant respectivement pour les antigènes grand T, petit T et les protéines de la capside VPl, VP2 et VP3. La numérotation des nucléotides commence au site ~I qui se
retrouve à l'intérieur de l'origine de réplication et procède dans le sens horaire. Les chiffres présents à l'intérieur des régions codantes indiquent les positions des sites d'initiation, d'épissage et de terminaison (Tooze, 1981).
\
1981). De nombreuses expériences biochimiques, génétiques et imrnunologiques ont permis la distinction entre ces deux antigènes (Shenk et al., 1976; Prives et al., 1977; Paucha et al., 1978). La région tardive de SV40, qui est transcrite après le début de la réplication de l'ADN viral, encode quatre protéines distinctes. Les protéines VPl (45 Kd), VP2 (42 Kd) et VP3 (30 Kd) sont des protéines structurales qui constituent la capside virale (Tooze, 1981). L'agnoprotéine, une petite protéine de 61 acides aminés, est également encodée par les séquences tardives (Jay et al., 1981). Certaines évidences génétiques et biochimiques suggèrent que celle-ci joue un rôle tardif dans le cycle lytique, possiblement dans l'assemblage de virions (Ng et al., 1985).
Les cellules de singe sont les seules cellules permissives pour le virus SV40, c'est-à-dire qu'elles pennettent la réplication et la transcription de l'ADN viral menant à la production de virions. Les cellules de rongeur, particulièrement celles de souris et de rat, se retrouvent à l'autre extrême et sont non-permissives pour le virus SV40 puisqu'elles ne pennettent pas la réplication virale. Les cellules humaines sont semi-permissives pour SV40 car il est possible de détecter un faible niveau de réplication virale. Un faible pourcentage de cellules non-permissives peut être transformé par le virus SV40 lorsque celui-ci est intégré de façon stable dans le génome cellulaire et ses fonctions virales précoces sont exprimées efficacement. Les cellules en culture transformées par SV40 ou le virus du polyome acquièrent un certain nombre de nouvelles propriétés qui les distinguent des cellules primaires. Parmi ces propriétés, on retrouve l'immortalisation ou la capacité de croître en culture de façon continue sans entrer en crise,
la perte d'inhibition de contact, la croissance à faible concentration de sérum, une forte densité de saturation, l'indépendance d'ancrage, des changements morphologiques ainsi que la capacité d'induire des tumeurs chez les nouveau-nés de certains rongeurs. L'utilisation de mutants thermosensibles ainsi que des expériences de transfection dans des cellules en culture ont démontré que la région précoce du virus SV40 est suffisante pour induire des tumeurs in vivo et pour transformar les cellules in vitro (Tegtrneyer, 1975; Van der Eb et al., 1979).
L'antigène grand T, une phosphoprotéine de 708 a.a., possède plusieurs activités biochimiques qui agissent seules ou ensemble pour contrôler divers aspects de l'infection virale et du comportement cellulaire. Des expériences d'immunofluorescence ont démontré que la protéine grand T est localisée principalement dans le noyau de la cellule et qu'environ 5% des molécules sont fortement associées à la matrice nucléaire (Verderarne et al., 1983). Dans les cellules permissives infectées par SV40, l'antigène grand T dirige toute une série d'événements et contrôle le passage de la phase précoce de l'infection, avant la réplication de l'ADN viral, à la phase tardive durant laquelle les virions sont produits. Lors de l'infection lytique chez le singe, la protéine grand T altère les patrons de transcription cellulaire, stimule la synthèse de l'ADN cellulaire et initie la réplication de l'ADN viral (Oda et Dulbecco, 1968; Tegtmeyer 1972; Tjian et al., 1978). Au moins trois fonctions biochimiques de l'antigène grand T notaiment, son activité ATPase (Giacherio et al., 1979; Tjian et Robbins, 1979) son activité hélicase (Stahl et al., 1986) et son affinité pour l'origine de réplication viral (Reed et al., 1978; Tjian,
1978) sont nécessaires dans l'initiation de la réplication de l'ADN viral. Pour passer de la phase précoce de l'infection à la phase tardive, grand T réprime la transcription de la région précoce et transactive la transcription de la région tardive (Reed et al., 1976; Alwine et al., 1977; Brady et al., 1984; 1985; Keller et Alwine, 1985). De plus, l'antigène grand T peut également transactiver toute une série de promoteurs viraux et cellulaires mais l'irrtIX>rtance biologique des ces activations est encore inconnue (Loeken et al., 1986; Saffer et al., 1990). La protéine grand Test aussi impliquée dans l'intégration, l'amplification et l'excision du génome viral dans l'ADN cellulaire
(Botchan et al.,1979).
L'antigène grand T de SV40 se distingue principalement de son homologue, le grand T du virus du polyome, par le fait qu'il peut à lui seul immortaliser et transformer les cellules primaires. Le grand T du virus du polyome ne possède que la capacité d'innnortaliser les cellules primaires, telles que les cellules embryonnaires de rat et d'humain (Strauss et al., 1990). L'antigène petit T n'est pas essentiel dans la transformation in vitro par SV40 si les cellules sont infectées par un rétrovirus recombinant (MV40) qui assure une expression très élevée de l'antigène grand T (Kriegler et al., 1984). Petit T semble plutôt jouer un rôle promoteur ou auxiliaire dans les cellules qui ne sont pas en phase de croissance active ou lorsque le niveau d'expression de l'antigène grand T est limitant (Frisque et al., 1979; Bikel et al., 1987). En dépit du fait que le grand T de SV40 peut à la fois innnortaliser et transformer des cellules de rongeur, il le fait moins efficacement que lorsque le grand T de polyome complémente l'antigène
\
moyen T (Salzman, 1986). Les activités ATPase, hélicase et l'affinité pour !'ADN viral de l'antigène grand T ne sont pas requises dans l'irnmortalisation ou la transformation (Cole et al., 1986; Manas et Gluzman 1984; 1985). Les mécanismes moléculaires par lesquels l'antigène grand T occasionne la transformation cellulaire ne sont pas bien connus. Cependant, il semblerait que la liaison à deux protéines cellulaires, p53 et pRB, joue un rôle essentielle dans la production du phénotype transformé car des antigènes grand T mutants, qui sont incapables de lier l'une ou l'autre de ces protéines, sont au moins partiellement défectifs dans la transformation (DeCaprio et al., 1988; Peden et al., 1989). L'antigène grand T forme des complexes avec au moins sept protéines cellulaires: !'ADN polymérase a (Gannon et Lane,
1987; Smale et Tjian, 1986); la protéine de choc thermique hsp73 (Sawai et Bute!, 1989); le facteur de transcription AP-2 (Mitchell et al., 1987); deux protéines impliquées dans la régulation négative de la croissance cellulaire, p53 et pRB (DeCaprio et al., 1988; Linzer et Levine, 1979); la protéine p107/120 qui possède une structure semblable à pRB et qui pourrait aussi être impliquée dans le contrôle du cycle cellulaire (Dyson et al., 1989; Ewen et al., 1989; 1991), et la protéine p185 (Kohrman et Imperiale, 1992). La figure 2 indique la localisation des domaines fonctionnels de l'antigène grand T ainsi que les résidus de la protéine qui sont phosphorylés.
La protéine RB est le produit du gène de la susceptibilité au rétinoblastome (RBl). C'est une phosphoprotéine nucléaire de 110 Kd
(pllO-RB) qui possède une affinité pour !'ADN (Lee et al., 1987). La perte ou l'inactivation du gène RBl chez les enfants en bas âge les
NLS
(126·132)
Finger
(302·320)Host Range
(682-708)Pol ex
Pola, p53
ATPase, ATP bindin
l,oosroAP1IHDHNP-11-KK
S~ S~6& SU7 p S671
K
1!0517
Figure 2. Local.isation des dœeines fonctionnels de 1' antigène grand T du virus SV40.
Localisation des principales fonctions biochimiques de l'antigène grand T. Les séquences impliquées dans les activités ATPase et hélicase, la liaison au protéines cellulaires pRB, p107, p53 et l'ADN polymerase a
(Pol.Œ), ainsi que les séquences nécessaires à la liaison à l'ADN, à la localisation dans le noyau (NLS), le motif doigt de zinc et un domaine impliqué dans l'assemblage des virions (Host Range function) sont indiquées. Les résidus sérines et thréonines de la protéine qui sont phosphorylés dans les cellules de mammifères sont indiquées par une lettre P encerclée et ceux qui le sont dans les cellules d'insecte sont indiquées par une lettre P encadrée. Les astérisques indiquent les endroits de la protéine qui sont sensibles aux protéases (Fanning,
prédispose à développer le cancer du rétinoblastome (Friend et al., 1986; Fung et al., 1987; Lee et al., 1987). Les altérations du gène RBl sont aussi associées à plusieurs autres tumeurs humaines telles que le cancer du sein, certains ostéosarcomes, certains sarcomes de tissu mou, les carcinomes de poumon et de vessie et les leucémies (Friend et al., 1987; Mendoza et al., 1988; Lee et al., 1988; Cheng et al., 1990). La réintroduction d'un gène RB fonctionnel dans des cellules tumorales ayant perdu les deux allèles de ce gène supprime le phénotype transformé (Huang et al., 1988). Toutes ces observations viennent appuyer l'hypothèse selon laquelle la protéine RB serait impliquée dans la régulation négative du cycle cellulaire. Ainsi lorsque la protéine RB est mutée ou absente, il n'y a plus de contrôle de la prolifération cellulaire et ceci amène indirectement la formation de tumeurs (Green, 1989). La protéine RB oscille entre la forme déphosphorylée et la forme phosphorylée au cours du cycle cellulaire. Dans les phases GO/Gl du cycle cellulaire c'est la forme déphosphorylée de la protéine qui est majoritaire alors qu'elle est présente sous forme phosphorylée dans les cellules en phase S et G2 (Buchkovich et al., 1989). L'observation que RB lie spécifiquement les régions transf orrnantes des oncoprotéines virales ElA d'adénovirus, E7 des HPV-16 et 18, et grand T de SV40, suggère que celle-ci est impliquée dans la transformation par ces virus
(Dyson et al., 1989a; Whyte et al., 1988a; 1989). En effet, toutes les oncoprotéines virales qui lient la protéine RB possèdent deux courtes séquences non contigues d'acides aminés hautement conservées appelées régions conservées 1 et 2 (crl et cr2) (Dyson et al., 1990; necaprio et al., 1988; Whyte et al., 1988a et b; Munger et al., 1989) (figure 3). L'intégrité des sites de liaison à RB localisés dans ces oncoprotéines
est importante à leur activité transfonnante car l'introduction de mutations réduisant la liaison à RB réduit ou abolit cette activité (Edmonds et Vousden, 1989; Lillie et al., 1986; 1987}. L'hypothèse expliquant la corrélation entre la liaison à RB et la transformation présume que les oncoprotéines virales peuvent empêcher la protéine RB d'exercer son rôle dans le cycle cellulaire en la complexant. L'effet final de cette interaction serait donc l'équivalent d'une délétion génétique du gène RBl. Plusieurs études on démontré que les 130 premiers résidus de la région N-terminale de grand T possèdent une activité transfonnante (Colby et Shenk, 1982; Clayton et al., 1982; Sompayrac et Danna, 1984; 1985; 1988; Asselin et Bastin, 1985; Pan et al., 1985). Ce fragment N-terminal comprend les régions crl et cr2. La région cr2 possède comme séquence consensus le motif (D/N}LXCXE qui est présent dans tout les antigènes grand T des virus du polyome à l'exception du virus BDF {un virus aviaire} (Pipas, 1992). Dans SV40 le motif représente les acides aminés 102 à 107 de la protéine et fait partie du site de liaison à RB et à p107/120 (Decaprio et al., 1988; Dyson et al., 1989; Ewen et al., 1989). Des mutations entre les résidus
105 et 115 rendent défectif l'antigène grand T dans la transformation cellulaire (Kalderon et Smith, 1984). Par ailleurs, la délétion du motif DLXCXE de l'antigène grand T du virus du polyome (mutant dll41} abolit sa capacité de lier RB in vitro ainsi que sa capacité d'inmortaliser les cellules primaires (Larose et al., 1990).
La protéine p53 est une phosphoprotéine nucléaire de 53 Kd (Linzer et Levine, 1979; Dippold et al., 1981}. Elle a été mise en évidence initialement par son association avec l'antigène grand T de SV40 dans
A. cr1 reglon. El.A 3 E7 D T P T L H E Y H L D LU SV40 E S L Q L H D L L GœE R S A Il G N I P L Hlo BICV E S H E L H D L L G L E R A A Il G N L P L HlO JCV E S H E L H D L L G L D R S A Il G N I P V HlO LPV E R N E L H D L L Q I T R A A Il G N L S H Hlo HaPV E K Q A L I S L L D œE P Q Y 11 G D Y G R Hlo PyV D K E R L L E L L K L P R Q L Il G D F G R HlO !CV S Q R L H H L L K L P H E Q Y G N F P L HlO BPyV Y E E L R G L L G - - T P D I G N A D T L21 BFDV lS L R R L T C(!JP V T A T A - - A D 122 E X X X L X E L X X L D D I B. a2 re~on • • • SV40 101E N L F C S E E H P S S D E A T117
BICV 103E D i'.. F C H E 0 H F A S D E A T11t
JCV 103E D L F c H E <: H F A s D E N
T11'
LPV 1290 D L F C S E T M S S S S E D T10 HaPV 121E D L T C Q E E L S S S E E F T144 PyV t4op D L F C Y. E E P L L S p N P S 5u1 !CV 102r D L F C N E A F D R S O D E Q Elll BPyV 510 D L H D E E L E P S D N E E E74 BFDV 51E G L R A D E T L E D S D F E p E'' D L X C X E X X X X S N PFigure 3. Homologie de séquence entre 1es antigènes grand T du groupe des virus du po1yome et 1es oncoprotéines virales ElA d'adénovirus et E7 du virus du papi11ome humain.
Alignement des séquences d'acides aminés faisant partie des régions conservées 1 et 2 (crl et cr2) des antigènes grand T du virus SV40, BK et JC (BKV et JCV), du virus lymphotropique de singe (LPV), des virus du polyome humain, bovin et de hamster (PyV, BPyV et HaPV), du virus K de souris (KV), et du virus de la maladie budgerigar des jeunes oiseaux (BFDV) ainsi que les protéines virales ElA d'adenovirus et E7 du virus du papillome humain. La région conservée 1 est caracterisée par la séquence (E/D)XXXLX(E/D)LXX(L/I). La séquence consensus de la région 2 est le motif (D/N)LXCXE. Ce motif fait partie du site de liaison à RB dans le grand T de SV40, dans la protéine ElA d'adenovirus de type 5 et dans la protéine E7 du virus du papillome humain (Dyson, 1989a; MÜnger et al., 1989). Toutes les séquences identiques ou semblables des deux régions conservées sont encadrées (Pipas, 1992).
les cellules transfonnées (Linzer et Levine, 1979; I.ane et CraWford, 1979). Cette association a été aussi dénnntrée plus tard dans le cas des oncoprotéines ElB d'adénovirus et E6 du virus du papillome (Sarnow et al. , 1982; Werness et al. , 1990) • Les premiers travaux sur p53 suggèraient que celui-ci se classait parmi les oncogènes cellulaires car un clone génomique de p53, derivé de cellules hépatiques normales de souris, pouvait coopérer avec l'oncogène ras pour transformer des cellules de rongeur (Eliyahu et al., 1984; Jenkins et al., 1984; Parada et al., 1984), augmenter l'efficacité de transformation par SV40 (Michalovitz et al., 1986) et immortaliser les cellules primaires (Jenkins et al, 1985; Rovinski et Benchirool, 1988). La comparaison de ce clone avec d'autres clones du gène p53 isolés de cellules normales de souris ont déroontré que le clone oncogénique était mutant, et comportant une substitution de valine pour alanine au résidu 135 (Finlay et al., 1988). Le gène p53 de type sauvage possède plutôt une activité antitransformante et lorsqu'il est cotransfecté avec ras plus ElA, ras plus mye ou ElA plus ElB, il empêche la transformation par ces derniers (Eliyahu et al., 1989). Le gène p53, tout comme le gène RBl, subit des réarrangements ou délétions dans un nombre considérable de tumeurs humaines telles le cancer du pouroon, du colon et du sein ainsi que les sarcomes ostéogeniques (Bartek et al., 1990; Baker et al., 1989; Takahashi et al., 1989). Lorsque le gène p53 de type sauvage est transfecté dans des cellules transformées, il supprime leur phénotype (Mercer et al., 1990; Fukasawa et al., 1991). Par ailleurs, des cellules primaires et établies de souris qui exprinent un gène p53 exogène sont résistants à la transformation par SV40 (Fukasawa et al., 1991). Tous ces résultats viennent appuyer la théorie snJ on lnquelle lr.·
protéine p53 est impliquée dans le contrôle négatif de la prolifération cellulaire et qu'elle n'est plus capable d'exercer sa fonction régulatrice lorqu'elle est complexée par l'antigène grand T. Le site de liaison à p53 est localisée dans la région C-terminale (résidus 347 à
628) de l'antigène grand T (Zhu et al., 1991). Certaines études indiquent que des mutants de grand T affectés dans leur liaison à p53 sont incapables de transfonner des lignées établies de cellules de rat (REF52) mais gardent toutefois leur capacité à transfonner des lignées établies de cellules de souris (C3H1Drl/2)(Peden et al., 1989; Srinivasan et al., 1989). Ceci suggère que la liaison de grand T à p53 est requise dans la transformation des cellules REF52 mais serait non-essentielle pour la transformation des cellules C3H1Drl/2. D'autres chercheurs suggèrent que la liaison à RB par grand T n'est pas nécessaire pour l'irrmortalisation des fibroblastes de souris ou de rat (Chenet Paucha, 1990; Zhu et al., 1991). Ces résultats contrastent avec les études d'autres laboratoires qui proposent qu'un petit fragment N-terminal de grand T qui peut lier RB et p107, mais non pas p53, soit suffisant pour irrmortaliser les cellules de rat (Asselin et Bastin, 1985; Sompayrac et Danna, 1988). Tous ces résultats apparemment contradictoires soulèvent la possibilité que la protéine grand T pourrait peut-être irrmortaliser les cellules par deux mecan1smes ,
.
différents, l'un impliquant la liaison à p53, l'autre pas.Dans le but de préciser l'importance de certaines séquences faisant partie des régions conservées 1 et 2 de l'antigène grand T dans l'i:mmortalisation cellulaire et la liaison probable à RB, nous nous sommes intéressés aux motifs (E/D)XXXL(E/D)LXX(L/I) et (D/N)LXCXE
présents dans ces régions. Nous avons produit par mutagénèse dirigée un mutant, dll02, ou le motif NLXCXE (région 2) a été délété d'un antigène grand T tronqué (SV24) qui immortalise les cellules primaires de rat avec une grande efficacité (Asselin et Bastin, 1985) mais qui est négatif pour la liaison à p53. Corrme le mutant SV24 possède encore les séquences de la région 2 jugées nécessaires pour la liaison à RB, nous avons donc voulu vérifier si la délétion du petit motif NLXCXE de la protéine était suffisante pour abolir sa fonction immortalisante en éliminant son site de liaison à RB. Un mutant fonctionnel équivalent dans l'antigène grand T du virus du polyo:rœ (dll41) ou ce même motif a été délété s'est révélé négatif dans l'inmortalisation et n'est plus capable de lier la protéine RB in vitro. On démontre ici que le mutant dll02 est bel et bien exprimé dans les cellules transfectées mais n'immortalise pas. Dans la région 1, trois mutations on été produites. Le mutant dll3 représente une délétion de 5 acides aminés (résidus 13 à 17) dont trois résidus valine (aux positions 13, 16 et 17) hautement conservés chez les papovavirus. Les mutants 13 val et 16 val sont des substitutions de résidus valine pour résidus leucine aux positions 13 et 16 de la protéine. On sait que cette région fait partie du site de liaison à RB dans le cas de la protéine ElA d'adénovirus mais son rôle dans le grand T de SV40 reste mal compris. Les mêmes mutations dans la région 1 du grand T du virus du polyome (Larose et al., 1991) révèlent que la liaison à RB n'est pas trop affectée mais seul le mutant 16 val conserve sa capacité d'immortaliser.
MATERIELS ET ME'IHOD~
I. Souche virale
Le plasmide p777 contient le génome viral complet de SV40 cloné au site BamHI de pBR322. Les séquences virales proviennent de la souche 777.
II. Plasmides recanbinants
pSV2neo contient le gène de la résistance à la néomycine. Le gène néo, codant pour une aminoglycoside phosphotransférase de l'élément Tn5 de E. coli, est sous le contrôle du promoteur précoce de SV40 (Southern et Berg, 1982). Les cellules eucaryotes qui expriment ce plasmide peuvent être sélectionnées au G-418.
pSV2neoà2005 résulte du clonage du mutant à2005 de SV40, n'encodant que l'antigène grand T, au site BamHI de pSV2neo. à2005 est caracterisé par une délétion de 230 pb dans l'intron entre les nucléotides 4917 et 4572 qui inactive l'expression de l'antigène petit T mais n'affecte pas la région codante de l'antigène grand T (Sleigh et al., 1978).
pSV2neoSV24 résulte du clonage du mutant pSV24 au site BarnHI de pSV2neo. pSV24 est un mutant de ll 2005 comportant une délétion de 1227
pb entre les nucléotides 4407 et 3180. Ce mutant code pour un antigène grand T tronqué et provoque un changement dans le cadre de lecture de façon à ajouter 6 nouveaux acides aminés à la protéine (Asselin et Bas tin, 1985) •
III . Méthodes de clonage
Les enzymes de restriction utilisées pour digérer le vecteur et le fragment à cloner proviennent des compagnies New England Biolabs et Pharrnacia. Les digestions enzymatiques d'ADN sont effectuées selon les recorrmandations des manufacturiers. Les fragments d'ADN à purifier sont déposés sur un gel d'agarose puis extraits par la technique d'électroélution à cuvettes ou à l'aide d'une membrane de DEAE
(DEAE-NA45, Schleicher et Schuell).
Les ligations intramoléculaires et intermoléculaires se font à la température de la pièce pendant 2 à 4 heures dans 20 µl de tampon de ligation (50rrM Tris-HCl pH 7.4, lOrrM MgCl2, 10 rrM D'IT, lrrM ATP) contenant 2U de T4 ADN ligase. Les transformations bactériennes sont éffectuées dans les souches E. coli JM109 et DH5~ selon les méthodes
décrites par Messing (1983) et la compagnie BRL. La préparation des plasmides recombinants se fait selon les méthodes décrites par Maniatis et al., (1982) et celle des phages recombinants selon les méthodes de Messing (1983) et de la compagnie Amersham.
IV. Mutagénèse dirigée avec oligonuctéotide synthétique
La mutagénèse dirigée avec oligonucléotide synthétique permet de produire des mutations ponctuelles, des délétions ou des insertions. Le fragment d'ADN qui porte la séquence à mutagéniser est tout d'abord inséré dans le site de multiclonage du phage M13mp18 ou M13mp19. L'ADN monocaténaire du phage M13 recombinant sert de matrice à la réaction de mutagenèse. Un oligonucléotide synthétique, qui possède la mutation désirée et qui est complémentaire à la région voulue, est hybridé à la matrice. Cet oligonucléotide, à son tour, sert d'amorce pour la polymérisation du second brin par le fragment Klenow de l'ADN polymérase de E. coli. L'utilisation de l'enzyrre T4 ADN ligase, par la suite, permet la ligation du brin nouvellement synthétisé et la production d'une molécule bicaténaire fennée.
1. Clonage dans le phage Ml.3
Le fragment porteur de la séquence à mutagéniser est cloné dans le site de multiclonage du phage M13mp18 ou M13mp19 (Messing, 1983). Dans le présent travail, un fragment KpnI-BamHI (1547 pb) à été cloné aux sites KpnI-BamHI dans la region de multiclonage du phage M13mp18. Un autre fragment entre KpnI et TaqI (798 pb) à été cloné aux sites KpnI-AccI dans la région de multiclonage du phage M13mpl9. L'enzyrre de restriction Ace! peut également couper à l'intérieure de la séquence nucléotidique que TaqI. Après la mutagénèse, les fragments sont sousclonés dans différents vecteurs.
2. Mutagénèse dirigée
i. Phosphorylation de l'oligonucléotide
Les oligonucléotides requis pour la mutagenèse ont été synthétisés par le laboratoire de K. Deugau (Queen's University). L'oligonucléotide n'est pas phosphorylé à son extrérni té 5' . La phosphorylation est nécessaire pour permettre la ligation du brin nouvellement synthétisé.
La phosphorylation de l'oligonucléotide se fait selon la technique décrite par Aiœrsham dans: "Oligonucleotide-directed in vitro mutagenesis system version 211• Cinquante pmoles d'oligonucléotide
contenues dans 2.0 µl sont mélangées avec 25 µ1 d'eau bidistillée, 3 µl de tampon kinase (lM Tris-HC!, pH 8. 0, lOOrrM Mgc12 , 70 rrM IY.rl', 10 rrM ATP), et 2 U de T4 polynucléotide kinase. Après avoir mélangé doucement en pipettant, le tube est incubé à 3-Pc pendant 45 minutes. Pour arrêter la réaction, le tube est incubé à 7cf>C pendant 10 minutes.
ii. Hybridation de l'oligonucléotide à !'ADN monocaténaire
L'hybridation de l'oligonucléotide phosphorylé à la matrice monocaténaire se fait aussi selon les techniques décrites dans le livret d'Alœrsham. Il faut s'assurer d'un excès molaire de l'oligonucléotide sur celui de la matrice de l'ordre de lOx. Le mélange réactionnel contient 4 µg du phage M13 recombinant, 2 µ! de tampon d'hybridation (lM NaCl, lM Tris-HC!, pH 8.0) et suffisamment d'eau bidistillée pour que le volume total soit égal à 17 µ.!. Le mélange est incubé dans un bain à 65° C pendant 10 minutes pour dénaturer toute structure secondaire de !'ADN
\
et pour faciliter l'hybridation de l'oligonucléotide avec la rratrice. Le bain est ensuite refroidi graduellement pendant 40 minutes jusqu'à ce que la température atteigne 4CPC. Le mélange est ensuite incubé à la température de la pièce pendant 2 minutes puis placé sur glace.
iii. Synthèse et ligation du brin mutant
La synthèse et la ligation du brin mutant se fait en ajoutant aux 17 µl du mélange d'hybridation: 5 µl MgCl2 100 :rrM, 1 µl d'ATP 50 rrM, 7.5 µl d'un mélange de nucleotides (2.5 :rrM dATP, 2.5 :rrM dCTP, 2.5 rrM dGTP, 2.5 rrM d'I'l'P), 6U du fragment Klenow d'ADN polymérase I d'E. coli, 6U de T4 ADN ligase et suffisanunent d'eau bidistillée pour obtenir un -volume final de 55 µ 1. Le mélange est ensui te incubé à 16oc pendant 24 heures.
3. Purification de 1 'hétérodup1ex: synthétisé par 1111tagenèse
L'hétéroduplex synthétisé par mutagenèse est isolé des autres intermédiaires non-complétés d'ADN bicatenaire par électrophorèse sur un gel d' agarose de 0. 7% contenant 1 µ g/ml de bromure d' éthidi um. La bande qui comigre avec la forme surenroulée du recombinant est électroéluée.
4. Transformation de 1a souche E. co1i JMl.09 avec 11hétérodup1ex:
L'ADN bicaténaire du phage M13 recombinant, qui est hétéroduplex pour la mutation, doit être introduit dans des souches d'E. coli permettant une oonne infection par M13. La réplication du phage à l'intérieur des bactéries semble se faire de façon à favoriser les molécules homoduplexes pour la mutation (Zoller et Smith, 1983).
i. Préparation des bactéries compétentes
La transfonnation des bactéries compétentes se fait selon les méthodes décrites par Messing et collaborateurs (Messing et al., 1983). Un volume de 100 ml de milieu 2XTY est ensemencé avec 1 ml d'une culture de E. coli JM 109. Cette culture est ensuite incubée à 37°c avec agitation jusqu'à une densité optique à 660 nm de 0.5. Un ml de cette culture est ajoutée à 7 ml de milieu 2xTY pour produire un tapis bactérien lors de l'étalement des bactéries transformées. La culture de 100 ml est centrifugée à 4000 rpm, à 4oC, pendant 10 minutes, et le culot bactérien est resuspendu dans 20 ml d'une solution CMl (10 mM acétate de sodium, 50 :rrM MgCl2, 5 :rrM NaCl, pH 5.6). La suspension bactérienne est incubée sur glace pendant 20 minutes puis est centrifugée de nouveau à 4000 rpm pendant 10 minutes. Le culot de bactéries est resuspendu dans 2 ml d'une solution CM2 (10 :rrM acetate de sodium, 5 rrM MnC12, 70 :rrM cac12, 5% glycerol, pH 5.6) et les cellules sont maintenues sur glace pendant 30 minutes.
ii. Transfonnation des bactéries compétentes
Cent ng d'ADN mutagenisé sont ajoutés à 100 µl de bactéries compétentes puis le tout est maintenu sur glace pendant 40 minutes. Les bactéries sont soumises à un choc thermique à 42°c pendant 2 minutes puis sont incubées sur la glace. 260 µl du mélange de tapis bactérien (200 µl de tapis bacterien, 10 µl !Pm 100 :rrM, 50 µl d'XGAL 2% dissous dans du DMF) et 100 µl de cellules transformées sont ajoutées à 3 ml
d'agar mou de surface maintenu à 50
°
c.
L'agar mou est étalé immediatement sur un pétri d'agar. Le pétri est laissé à la température de la pièce jusqu'à ce que l'agar mou fige puis est incubé à 37°cpendant la nuit.
V. Analyse des nutants par "dot b1ot"
Les mutants produits par mutagenèse dirigée sont analysés tout d'abord par la méthode de "dot blot". Cette méthode consiste à utiliser l'oligonucléotide de mutagenèse, marqué au 32.p, corrane sonde pour cribler l'ADN monocaténaire phagique d'un certain nombre de clones. L'ADN phagique des clones est fixé sur un filtre de nylon et celui-ci est incubé avec la sonde. Le filtre est ensuite lavé à des températures de plus en plus élevées ce qui favorise la dissociation de la sonde des clones non-mutants. La température théorique à laquelle la sonde est censée se dissocier de l'ADN monocaténaire est estimée par la formule de Wallace (Td=4X(G+C) + 2X(A+T)) (SUggs et al.). Quelques-uns des clones mutants sont séquencés par la méthode de Sanger pour vérifier si la mutation se retrouve à l'endroit voulu.
1. Préparation et hybridation de 1a sonde.
L'oligonucléotide de mutagénèse est marque dans un mélange , réactionnel qui contient suffisarmnent d'eau bidistillée pour que le volurœ final totalise 30 µ 1, 3 µ 1 de tampon kinase 1 OX (lM Tris-HCl, pH
\
8.0, 100 nM MgC12), 1.5 µl de DT!' lOOrcM, 20 pmoles d'oligonucléotide, 2 rµl de [y-32P]ATP (Amersham, 10 µCi/µl) et 1 unité de T4 polynucléotide kinase. Le mélange est incubé pendant 45 minutes à 37
°
C puis la réaction est arrêtée en chauffant à 65°c pendant 10 minutes. La sonde peut être conservée à -20°c. L'hybridation de l'oligonucléotide, marqué au 32p, à l'ADN rrvnocaténaire nécessite la fixation préalable de celui-ci sur un filtre de nylon. Cecelui-ci se fait en déposant 100 µl de surnageant phagique de chacun des clones à analyser sur un filtre Hybond-N au rrvyen d'un appareil à "dot blet". Le filtre est ensuite séché puis exposé 4 minutes aux rayons ultraviolets qui fixent l'ADN defaçon covalente au filtre de nylon. Le filtre est mis dans un sac en plastique contenant 10 ml de solution de préhybridation (6X SSC (0.9 nM
NaCl, 90 rrM citrate tri sodique), Denhardt lOX ( 0. 2°~ BSA, 0. 2% Fi coll, O. Zo;b PVP), O. 2% SDS). Une fois le sac scellé, la préhybridation se poursuit pendant 1 heure à 67°c avec agitation constante. Le but de cette étape est de saturer les sites de liaison non-spécifique du filtre. Le filtre est rinçé par la suite dans 50 ml d'une solution de 6X SSC pendant 1 minute. Le filtre est placé dans un autre sac auquel est ajouté la sonde (1X106 CPM) ainsi que 4 ml d'une solution d'hybridation (6XSSC, lOX Denhardt). L'hybridation se fait à la température de la pièce pendant 1 heure lorsqu'il s'agit d'un oligonucléotide de rrvins de 20 bases. Dans le cas d'oligonucléotides qui dépassent 20 bases ou qui possèdent une structure secondaire, le filtre est hybridé à 67 ° C pendant 30 minutes puis refroidi a ' la température de la pièce pendant 30 minutes. Le filtre est lavé dans trois bains successifs de 6XSSC, 5 minutes à chaque fois, à la température de la pièce. Une fois le filtre séché, il est exposé sur
\
film Kodak XAR-5 pendant la nuit. Le lendemain, l'étape de lavage dans une solution de 6XSSC est répétée mais, cette fois, à une température qui est 5°c sous la température de dissociation estimée par la formule de Wallace. Le filtre est séché puis réexposé. Il est important de comparer les deux premiers autoradiogrammes pour voir s'il est possible de distinguer le type sauvage du mutant. Si ce premier lavage n'est pas suffisant pour favoriser la dissociation de la sonde des clones nonmutants, il est nécessaire de répéter l'étape de lavage à une température un à deux degrés plus élevée.
2. Purification sur pétri
La transformation par une molécule hétéroduplexe donne une cellule transfonnée qui, suite à quelques cycles de réplication du phage, contient un mélange de molécules mutées et non-mutées. Ceci est dû au fait que chacun des deux brins de l'hétéroduplex est utilisé lors de la réplication du phage. Par conséquent, les clones mutants positifs contiennent une certaine proportion de molécules non-mutées. Il est donc nécessaire de refaire un autre cycle d'infection avec les candidats positifs pour pouvoir ainsi isoler les phages mutants homogènes.
3. séquençage de 1 •Am des clones nutants
Le séquençage des clones se fait selon la méthode de Sanger (Sanger et al., 1977) à l'aide d'un ensemble de séquence de la compagnie Pharmacia. L'oligonucléotide de séquence est complémentaire à la région située environ 65 nt en amont de la mutation à séquencer.
Cette méthode de séquençage se base sur l'utilisation de la T7 ADN polymérase et les didéoxynucléotides. Le marquage peut être effectué
soit avec les isotopes [ 35
s]
ou [~2PJ.VI. Transfection de cellu1es prineires et essai d'iimDrtalisation
1. Préparation des fibroblastes d'embryon de rat
Des embryons de 12 à 15 jours provenant de rat Fisher sont prélevés stérilement, découpés en petits morceaux, lavés trois fois au Tris salin (140 :rrM NaCl, 5 :rrM KCl, 1 :rrM N~HP04 , 5 :rrM glucose, 25 nM Tris-HCl,
0.0015% rouge phénol, pH 7.4) puis trypsinisés avec 0.06% trypsine (75
ml Tris salin + 25 ml trypsine 0. 25%) pendant 20 minutes à 37
°
C avecagitation constante. Les cellules sont filtrées à travers de la gaze, recueillies dans 30 ml de sérum de veau foetal puis le tout est
centrifugé pendant 10 minutes à 1800 rpm. Ces mêmes cellules sont ensuite resuspendues dans du DMEM complet puis comptées au bleu de trypan. Chaque pétri de 60 mm est ensemencé avec 3Xl06 de cellules. Les cellules sont incubées à 3i°C pendant 20 à 24 heures jusqu'à ce que
la confluence atteigne une valeur de l'ordre de 30 à 50%.
2. Transfectian au ch1orure de calcium
La transfection des cellules se fait selon la méthode décrite par
Wigler et collaborateurs (Wigler et al., 1978). Quelques 4 heures avant la transfection, les cellules subissent un changement de milieu qui,
cette fois, ne contient pas de fongizone. Deux tubes eppendorfs sont ensui te requis. Le premier tude contient 8 µ .g d 1 ADN resuspendues dans
12 µl de TE lX ('Î mM Tris-HCl, 0.1 rrM EIYI'A, pH 8.0), 10 µg d'ADN de thymus de veau contenus dans 175 µ.1 de TE O. lx et 63 µ 1 de lM CaCl 2· Le second tube contient 250 µ 1 de tampon de HEBS 2X ( 50 mM HEPES pH 7 .12, 280 . mM NaCl, 1. 5 rrM Na2HP04) . La solution d 'ADN du premier tube est ajoutée doucement pendant 30 secondes au tampon HEBS 2X et le mélange est agité de façon constante par l'introduction lente de petites bulles d'air. Le précipité qui se forme après 30 minutes à la température de la pièce est ajouté à un pétri de 60 nnn contenant des cellules à une confluence de 30-50% dans 5 ml de DMEM avec 10% SVF. Les cellules sont incubées24 heures à 31°C puis elles sont divisées 1:2 ou 1:3 afin d'obtenir une confluence de 20-30"fe le lendemain.
3. sélection à l'antibiotique G-418 et imoortalisation
Etant donné que les plasmides transf ectés portent le gène de la résistance à la néomycine, il est possible, lors de l'essai d'irnmortalisation, de sélectionner les colonies à l'antibiotique G-418. Cette sélection, qui consiste à remplacer le milieu complet sans antibiotique par du ll1EM complet contenant du G-418 ( 400 µ g/ml) , débute 18 heures après la division des cellules. Les cellules sont maintenues dans le milieu complet contenant le G-418 et à tous les 5 jours, le milieu est changé.
VII. Transfections de lignées cellulaires établies
Les cellules FR3T3 et COS sont des lignées cellulaires établies qui proviennent respectivement de rat Fisher et de singe. Elles peuvent être utilisées pour vérifier l'expression des protéines mutantes. Ces cellules sont transfectées selon la méthode au polybrène-DMSO (Kawai et Nishizawa, 1984).
Un pétri de 10 cm2 est ensemencé avec 7xl06 cellules de la lignée FR3T3 ou COS. Le pétri est incubé 18 heures à 37°C dans un milieu complet sans fongizone (DMEM et la:'.,6 de SVF). Le lendemain, le milieu nutritif est enlevé puis les cellules sont rincées une fois avec 5 ml de DMEM contenant 10% de SVF. On ajoute ensuite aux cellules 10 µg d1ADN
dans 2 ml de DMEM avec 10% SVF et 30 tJ..l de polybrène ( 1 rrg/ml dans tampon PBSA). Les cellules sont incubées pendant 6 heures à 37°C avec agitation occasionnelle. Le milieu est enlevé puis les cellules sont soumises a un choc au DMSO (4 ml de DMEM contenant 25% de DMSO) pendant 4 minutes pour les cellules FR3T3 et 90 secondes pour les cellules COS. Les cellules sont ensuite rinçées 2 fois avec 10 ml de DMEM seul puis on ajoute 10 ml de milieu complet sans fongizone par pétri. Les cellules sont incubées entre 48 et 72 heures à 31°C.
VII. Inmmoprécipitation des antigènes nntants
L'immunoprécipitation des antigènes de SV40 se fait selon les méthodes de Schaffhausen et Benjamin (1979, 1981). Les cellules à 80%
de confluence dans un pétri de 100 mm sont lavées 2 fois avec 5 ml de PBS+ (170 rrM NaCl, 3.3 rrM KCl, 10 rrM Na2HP04, 1.8 rrM KH2Po4, 5rrM MgCl ' 7 rrM CaCl2, pH 6.8) à la temperature de la pièce. Elles sont ensuite incubées pendant 60 minutes avec 3 ml de milieu Hanlcs contenant 300 µCi de [35s]-méthionine (Arnersham) avec agitation à toutes les 10 minutes. On ajoute 3 ml de DMEM avec 10";6 SVF au cellules puis l'incubation se poursuit 30 minutes avec agitation toutes les 15 minutes. Après l'étape de marquage, les cellules sont d'abord lavées une fois avec 5 ml de PBS+ froid, ensuite avec 5 ml de tampon de lavage (20 rrM Tris, 137 rrM NaCl, 0.92 rrM cac12, 0.49 rrM MgC12, pH 9.0). Une fois le tampon de lavage enlevé, on extrait les antigènes T avec 1 ml de tampon d'extraction TEB (89 ml de tampon de lavage pH 9.0, 10 ml de glycérol et 1 ml de NP-40). Les pétris sont incubés à 4°c pendant 20 minutes avec agitation toutes les 5 minutes. Les extraits sont alors transférés dans des tubes eppendorf puis centrifugés 5 minutes à 4°c. Le surnageant est transféré dans un autre tube eppendorf et on ajoute 2 µl de sérum anti-T. Le mélange est incubé 30 minutes à 4 ° C avec agitation constante. On ajoute ensuite 60 µ_1 de protéine A-sépharose 50";6 V/V
(Pharmacia) préalablement hydratée et lavée 6 fois dans de l'eau bidistillée à 4 ° C. L' incubation avec 1 'anticorps et la protéine A-sépharose se poursuit pendant 90 minutes à 4 ° C avec agitation constante. Les protéines marquées ayant formé un complexe non-spécifique avec l'anticorps sont éliminées par une série de lavages dans des tubes coniques de 15 ml (Sarstedt). On lave 2 fois avec 5 ml de PBS+, 2 fois avec 5 ml de la solution de lavage au LiCl (500 rrM LiCl, 100 rrM Tris, pH 9.0) et une fois avec 5 ml d'eau bidistillée. Chaque lavage se fait doucement en inversant le contenu du tube et est suivi
d'une centrifugation à 4°c jusqu'à ce que la vitesse atteigne 2000 rpm (Sorvall RC-3). Le culot est resuspendu dans 1 ml d'eau bidistillé puis transféré dans un tube eppendorf. On centrifuge le tube 20 secondes puis le surnageant est enlevé. Le culot est ensuite resuspendu dans 40 µl de tampon de dissociation (5% (W/V) SDS, 5% (W/V) 2-mercaptoéthanol, 20"fo
bromophénol, 125 rrM
(V/V) glycerol, 0.0075% Tris-HCl (pH 6.8).
(W/V) de bleu de Les échantillons sont immédiatement bouillis pendant 2 minutes puis déposés sur un gel de SDS-polyacrylamide à 12.5% (Laemli, 1970).
La migration se fait durant la nuit à faible voltage dans un tampon d'électrophorèse (25 rrM Tris-HCl, 192 rrM glycine, 0.1% (W/V) SDS, pH
8.3) et en présence de marqueurs protéiques précolorés (BRL). Le gel est fixé dans une solution contenant 5% (V/V) de méthanol et 7.5% (V /V) d'acide acétique pendant 30 minutes. Le gel est ensui te trempé dans la solution d'Amplify (Amersham) pendant 30 minutes avec agitation constante. Il est séché sous vide puis exposé à -70° C sur un film Kodak XAR-5.
RESULTATS
I. Construction de n11tants par DUtagénèse dirigée dans les régions conservées 1 et 2 de l'antigène grand T.
Les régions conservées 1 et 2 représentent deux courtes séquences d'acides aminés hautement conservés parmi les oncoprotéines virales qui lient la protéine RB. Dans la protéine ElA d'adénovirus, les deux régions forment le site de liason à RB (Whyte et al., 1989) tandis que seule la région 2 semble nécessaire à cette interaction dans le cas de l'antigène grand T de SV40 (DeCaprio et al., 1988). Le rôle de la région 1 de grand T dans cette interaction reste encore mal compris.
Afin de vérifier l'importance de l'intégrité des motifs présents dans les régions 1 et 2 de grand T quant au pouvoir inmortalisant de la protéine et de son interaction avec RB, j'ai construit une série de mutants de délétion et de substitution dans ces motifs.
La région 1 se trouve dans la partie 5' de l'exon 1 de grand T. Nous avons cloné un fragment KpnI-TagI de 798 pb (nucléotides 294 a '
4739) du génome SV40 de· type sauvage, porteur de la séquence à mutagéniser, aux sites KpnI-AccI dans la région de multiclonage du phage M13mp19 (figure 4a). L'enzyme de restriction Ace! reconnaît la même
séquence nucléotidique que TagI. La région 2 se trouve dans la partie 51 du deuxièrre exon de grand T. Un fragment KpnI-BarnHI de
A Kpnl
B
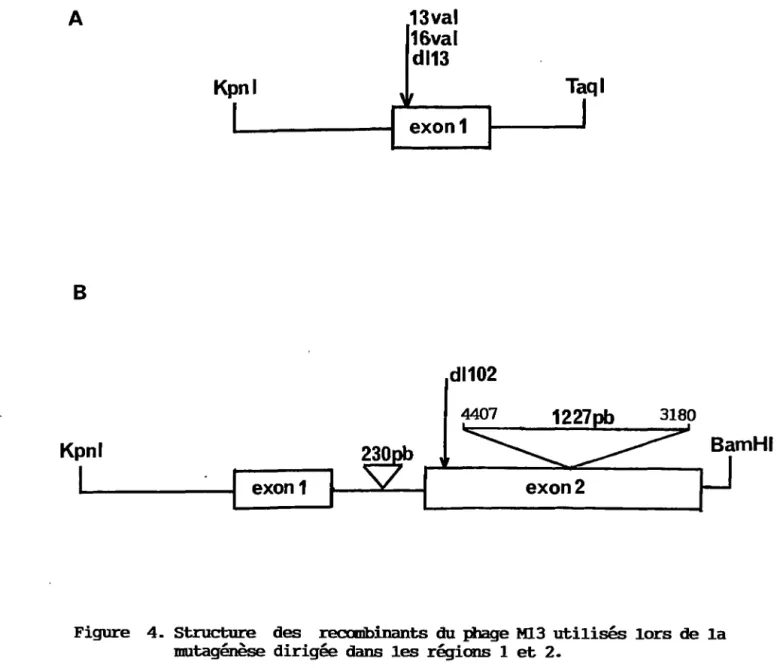
Kpnl exon1 13val 16val dl13 exon1 dl102 Taql BamHI · exon2Figure 4. Structure des recœibinants du phage Ml.3 utilisés lors de la mutagénèse dirigée dans les régions 1 et 2.
A. Représentation schématique du fragment KpnI-TagI de 798 pb (nucléotides 294 à 4739) du génome SV40 de type sauvage cloné aux sites KpnI-AccI de la région de multiclonage de la forme bicaténaire (réplicative) du phage M13rnp19. L'enzyme Ace! reconnaît la même séquence que l'enzyme TaqI. La région conservée 1 est localisée dans la partie 5' de l'exon 1. La flèche indique la position des mutants 13 val, 16 val et dll3.
B. Représentation schématique du fragment KpnI-BamHI de 1547 pb (nucléotides 294 à 2533) provenant du mutant SV24 et cloné aux sites KpnI-BamHI de la région de multiclonage de la forme bicaténaire du phage M13mp18. La région conservée 2 se trouve dans la partie 51 de l'exon 2. La flèche indique la localisation du mutant dll02. La délétion de 230 pb se trouve dans les séquences introniques de grand T tandis que la délétion de 1227 pb élimine la majeure partie des séquences codantes de l'exon 2.
a été cloné aux sites KpnI-BamHI de la région de multiclonage du phage M13mp18. Le mutant SV24 possède une délétion de 230 pb dans les séquences introniques de grand T ainsi qu'une autre délétion de 1227 pb dans le deuxième exon (figure 4b). Le choix des vecteurs de clonage, M13mp18 ou M13mp19, est sans conséquence majeure quant à la mutation qui doit être créée. La seule différence entre les deux est l'orientation de la région de multiclonage, ce qui détermine lequel des deux brins du fragment à mutagéniser sera répliqué lorsque le phage passe de sa forme réplicative bicaténaire à sa forme rnonocaténaire encapsidée. Les oligonucléotides de mutagénèse et de séquence sont choisis afin d'être complémentaires au brin retrouvé dans la forme rnonocaténaire du phage M13.
Quatre mutants ont été produits par mutagénèse dirigée, trois dans le motif de la région 1 et un seul dans la région 2. Le mutant dll 3, créé à l'aide d'un oligonucléotide de mutagénèse de 24 bases, (5' GAA TCT TIG CAG GGT CTl' GAA AAG 3'), représente la délétion des acides aminés 13 à 17 (nucléotides 5127 a ' 5113 inclusivement) dont trois résidus leucine. Les mutants 13 val et 16 val, tous les deux créés en utilisant des oligonucléotides de 19 bases, (13 val: 5' TCT TIG CAG GTA A'IG GAC C 3' ; 16 val: 5' CTA A'IG GAC G'IT CTA GGT C 3' ) , représentent des substitutions de codons valine aux codons leucine aux positions 13 et 16 de la protéine (des transitions C à G aux nucléotides 5127 et 5118 respectivement). Le mutant dll02 représente la délétion du motif NLXCXE de la région 2 localisé entre les acides aminés 102 et 107 de la protéine (nucléotides 4496 à 4514 inclusivement). Il a été créé à l'aide d'un oligonucléotide de 29 bases, (5' CTA GAT GGC A'IT TCT TCC TCA
TI'A AAG GC 31 ). Une analyse préliminaire des mutants a été effectuée
par "dot blot". Cette technique consiste à utiliser l'oligonucléotide de mutagénèse marqué co:rmne sonde pour détecter les clones mutants. Après cette étape, quelques clones positifs de chaque mutant ont été
séquencés par la méthode de Sanger (figure 5). Suite au séquençage, toutes les mutations ont été clonées dans des constructions ou les séquences de grand T sont insérées au site BamHI du plasmide pSV2néo dans le but de vérifier la capacité des nouveaux mutants à irrnnortaliser des cellules primaires. Dans le cas de la délétion dll02, le fragment KpnI-BamHI (1547 pb) du plasmid pSV2néoA2005 a été remplacé par le même fragment mutagénisé dans M13. Le clonage a pu être vérifié par digestion enzymatique avec les enzymes KpnI et BamHI car le fragment mutagénisé dans M13, qui provient du mutant SV24, est plus court que le fragment correspondant du mutant A2005 (figure 6a). Le plasmide qui résulte de ce clonage est nommé pSV2néodll02 (figure 6b). Les mutations 13 val, 16 val et dll3 de la région 1 ont chacune été transférées aux plasmides pSV2néo A2005, pSV2néoSV24 et pSV2néodll02 d'une façon semblable en remplacant le fragment KpnI-EcoNI de 510 pb de ces constructions par le même fragment mutagénisé dans M13.
II. Essai d'i.moortalisation pour le nntant dll02 de la région conservee ,
2 du grand T.
Des quatres mutations produites par mutagénèse dirigée, seule la délétion du motif NLXCXE (mutant dll02) a été vérifiée dans un essai d'irrnnortalisation. Les autres mutations ont été faites plus tard et les essais d'irrnnortalisation sont présentement en cours. Etant donné la
Figure 5. Autoradiogranne de 1a séquence des mutants de 1a
région conservée 1 et 2 de grand T.
Les séquences des mutants 13 val, 16 val et dll3 de la région 1 et dll02 de la région 2 sont montrées ici. Les nucléotides indiquées à la gauche de chaque autoradiogrannne représentent la région modifiée par mutagénèse dirigée. Les flèches indiquent, dans le cas du mutant dll3, l'endroit de délétion d'une partie du motif de la région 1 et dans le cas de dll02 la délétion du motif NLXCXE. Les motifs des régions 1 et 2 de grand T ainsi que la séquence modifiée des mutants correspondants sont également représentés ici. Le séquençage se fait selon la méthode de Sanger. L'oligonucléotide de séquence est marqué au 35s et l'ADN monocaténaire du phage M13, qui contient la région mutagénisée, sert de matrice lors de la réaction. Les fragments produits par la réaction de polymérisation sont séparés sur un gel dénaturant de polyacrylamide de
-,
)
tl
A\
~ ~
J!
-c
ê
t- ' G T A A T G G Ac
G T T Tc
TaJ
CRl:C
~T
A'" .
-..
... -••
~-~ ..
GAA TCT Tl'G CAG CTA ATG GAC CTI' CTA GGT CTI' Glu Ser Leu Gln Leu Met Asp Leu Leu Gly Leu nvtif: E/D XXX XXX XXX L XXX E/D L XXX XXX L
13 val: GAA Ter Tl'G CAG GTA ATG GAC C'IT CTA GGT C'IT
Val
16 va1: GAA Ter Tl'G CAG CTA ATG GAC GTI' CTA GGT CTI' Val
d1 13: GAA Ter Tl'G CAG GGT CTI'
A
c
G T-
...
-
--~
-l::
..::&f--
~.
~.
-
...
A...
c-T~ .... ~
c
....
c
...
-T...
-...,. T-
-~
-
~ T T -A...
...
c
G.,,,..
-~1-
-A .,,., GA-
-..,.
-.... ~
-
..
..-:. 101 102 107 108brin sequence: ,. ,. CTC CTT TTG GAC AAA ACG AGT CTT CTT TAC CR2 : GAG GAA AAC CTG TTT TGC TCA GAA GAA ATG
Glu Glu Asn Leu Phe Cys Ser Glu Glu Met
motif: N L XXX
c
XXX E101 108 dll02 (brin codant): GAG GAA GAA ATG dll02 (brin ,. sequence : ,. ) CTC CTT CTT TAC
Figure 6. Clonage du Dlltant d.1102 dans le plasmide pSV2néo.
A. La photo ici montre la vérification de la substitution du fragment KpnI-BamHI du mutant A2005, soudé au plasmide pSV2néo, par le même fragment du mutant dll02. On peut voir deux clones de la construction pSV2néodll02 avant et après digestion avec les enzymes KpnI et BamHI (puits 2,4 et 1,3 respectivement) de même que la construction pSV2néo 2005 non-digérée (puit 6) et digérée (puit 5). Dans le puit 7 on trouve un marqueur de poids moléculaire (1 Kb). La flèche du centre point vers le fragment KpnI-BamHI de 2774 pb du mutant â2005 qui est remplacé par le fragment de 1529 pb du mutant dll02 (flèche du bas). La flèche du haut pointe vers une bande de 5723 pb qui correspond au plasmide pSV2néo. La bande juste en dessous de celle-ci (puits 1 et 3) représente une digestion partielle au site KpnI des séquences du mutant dll02. La migration des fragments se fait sur un gel d'agarose de 0.7%. Les digestions ont été effectuées pendant 30 minutes à 37°c.
B. Représentation schématique du plasmide pSV2néodll02 utilisé dans l'essai d'immortalisation. Les séquences codantes de l'antigène grand T proviennent du mutant dll02 et sont clonées au site unique BamHI du plasmide pSV2néo. Le gène de la résistance à la néomycine est représenté par une boîte noire. La délétion de 230 pb dans l'intron inactive l'expression du petit T tandis que la délétion de 1227 pb dans le deuxième exon élimine 60% des séquences codantes du grand T. La délétion du motif NLXCXE qui caractérise le mutant dll02 est également indiquée.
B
NLXCXE
1227pb
3180BamHI