U n i v e r s i t é P A U L S A B A T I E R
THESE
Présentée par
FETTAH AJAMAA
Pour l’obtention du titre de
DOCTEUR DE L'UNIVERSITE PAUL SABATIER DE TOULOUSE
Domaine : Chimie Macromoléculaire et Supramoléculaire
Etudes conformationnelles de
phényl-fulleropyrrolidines et préparation d’édifices
supramoléculaires photoactifs incorporant du C
60Soutenue le 26 Septembre 2007 devant la commission d'examen :
Dr. J.-F. Nierengarten, LCC-CNRS, Toulouse Directeur de thèse
Prof. R. Deschenaux, Université de Neuchâtel, Suisse Rapporteur externe Prof. F. Langa, Université de Castilla-La Mancha, Espagne Rapporteur externe Prof. R. Martino, Université Paul Sabatier, Toulouse Examinateur
REMERCIEMENTS
Le travail de thèse présenté dans ce manuscrit a été réalisé en grande partie au laboratoire de chimie des fullerènes et des systèmes conjugués à l’Ecole Européenne de Chimie Polymères et Matériaux de Strasbourg (ECPM), puis au Laboratoire de Chimie de Coordination (LCC-CNRS) à Toulouse, sous la direction du Dr. Jean-François Nierengarten, que je tiens à remercier de façon très particulière et chaleureuse de m’avoir accepter au sein de son groupe, pour tous ses conseils et pour le soutien dont j’ai pû bénéficier tout au long des mes études à compter de mon année de maîtrise, ce qui m’a permis d’acquérir et de perfectionner mes capacités théoriques et expérimentales dans le domaine de la synthèse chimique.
Je tiens également à exprimer mes remerciements au Dr. Michel Holler pour le temps qu’il a consacré pour le bon déroulement de cette thèse que ce soit au début à Strasbourg, ou à la fin pour la rédaction et la correction , malgré les longues distances qui nous séparent « Merci beaucoup michmich, sachant que je ne pourrai jamais te remercier assez !? ».
Je tiens également à remercier Dr. Béatrice Delavaux-Nicot pour ses conseils, son acceuil à notre arrivée, ainsi que Messieurs Jean-Jacques Bonnet, Bruno Chaudret et Denis Neibecker pour leurs encouragements.
J’adresse toutes mes sincère remerciements aux membres du jury qui ont accepté de juger ce travail : Prof. R. Deschenaux, Université de Neuchâtel (Suisse), Prof. F. Langa, Université de Castilla-La Mancha (Espagne), Prof. R. Chauvin, Université Paul Sabatier (Toulouse), Prof. R. Martino, Université Paul Sabatier, Toulouse et le Dr. B. Delavaux-Nicot, LCC-CNRS (Toulouse).
Je remercie vivement l’Union Européenne et en particulier le réseau Organic LED’s for lighting and ICT applications (OLLA), de m’avoir octroyé un financement pour une partie de ma thèse ce qui m’a permis de terminer ma thèse dans de bonnes conditions.
Merci enfin à toutes les personnes que j’ai rencontrées au laboratoire certains avec qui j’ai eu des collaborations au sein du groupe : Teresa, Juan-luis et Maria-Teresa (c’était un plaisir de travailler avec vous), et aussi à tous les membres de notre groupe avec qui j’ai appris beaucoup de chose : Aline, François, Omar, Adrien, nos post-doc : Uwe et notre brésilienne josé, Julien, Hind, Kemal, Jean, Maxence, Laure, Régis, Mathieu, Souhaila, Khalid…et d’autres que j’aurais sans doute oublié, mais ils se reconnaîtront sans doute, et à toutes les personnes extérieures du laboratoire qui m’ont aidé même avec des paroles surtout dans les moments les plus difficiles de ma vie (des fois des mots simple ont une grande porté sur les rapports entre les gens, et ça ne coûte pas chère) .
Je suis très reconnaissant à tous mes amis de Strasbourg qui m’ont accompagné tout au long de mes études et pour leur soutien moral : N et Y. Bouazza, A. Fatmaoui, S. bouabid, B. Ramil, H. Oubssis, B. Edafaa , M. Elâachari, M. Lahrach et toutes leurs épouses respectifs, Mohamed (CTS), Nabil, Abdelwahed, Hassan (L), Mokhtar (J), Hamouda, Ahmed (W), Rachid (S), Oukssis (L), Jawad (ECPM), Ali (CTS), Chenkit (M), Ahmed (H), Khalid (H), Omar (C.T), Mohammed (R), Zaid, Hassan (O), Nabil, Tayeb (S), Elouakili (A et M), Kamal (E), Ahmed (Egypt), Karim (T), Abd Arrahim (G), …et Farid Aziat à qui je souhaite du courage et une bonne continuation pour la fin de sa thèses.
Enfin, je dois souligner le fait que cette thèse n’aurait pas vu le jour sans le soutien moral et financier de mes parents pendant toutes ces années d’études. Je ne pourrai jamais les remercier assez mais j’espère les rendre éternellement fiers de moi.
Citation :
« Ce qui se fait pour nous, et sans nous est contre nous »
CHAPITRE I
Introduction Générale
Les Fullerènes
1. Production des fullerènes
2. Structure des fullerènes
3. Propriétés Physicochimiques du Buckminsterfullerène
3.1. Solubilités
3.2. Propriétés photophysiques 3.3. Propriétés électrochimiques
4. Propriétés Chimiques du Buckminsterfullerène
4.1. Cyclopropanation : la réaction de Bingel. 4.2. Réaction de rétro-Bingel
4.3. Cycloadditions [4+2]: les réactions de Diels-Alder 4.4. Réactions de cycloaddition [3+2]
4.4.1. Les ylures d'azométhine 4.4.2. Les nitriles imines 4.5. Réactions de multi-additions
5. Objectifs
6. Références
2 3 6 6 7 9 11 11 15 19 25 25 32 38 46 47CHAPITRE II.
Synthèse et étude conformationnelle de phényl-fulleropyrrolidines
1. Introduction
2. Objectifs
3. Réalisation
3.1. Préparation d’un dérivé de la glycine 3.2. Préparation des phényl-fulleropyrrolidines
3.3. Caractérisation des phényl-fulleropyrrolidines 7a-f 3.4. Etudes conformationelles 3.4.1. Les fulleropyrrolidines 8a et 8b 3.4.2. La phényl-fulleropyrrolidine 8e : 3.4.3. Les phényl-fulleropyrrolidines 8f-h et 8j: 3.4.4. Les phényl-fulleropyrrolidines 8c-d et 8i :
4. Conclusion
5. Partie expérimentale
56 58 60 60 61 65 67 67 71 72 76 80 811. Introduction
2. Objectifs
3. Réalisation
3.1. Préparation du récepteur bis-Zn(II)-porphyrinique (L2Zn) 3.2. Préparation du ligand C60-pyridine de référence 12
3.3. Préparation du ligand C60-bispyridine 22
4. Etudes des systèmes supramoléculaires.
5. Conclusion
6. Partie expérimentale
103 112 114 114 114 117 125C
CHHAAPPIITTRREE II 11
Des observations de l’espace interstellaire par des radioastronomes suggéraient l’existence de chaînes d’atomes de carbone au sein de certaines étoiles, les géantes rouges. En cherchant à produire des conditions proches de celles existantes dans ces étoiles pour obtenir ces molécules en laboratoire, H. W. Kroto, R. F. Curl et R. E. Smalley ne se doutaient pas qu’ils étaient à l’aube d’une découverte qui révolutionna nos certitudes sur le carbone.1 En effet, l’analyse des agrégats de carbone formés au sein d’un plasma très chaud obtenu en vaporisant du graphite à l’aide d’un laser révéla l’existence de molécules en forme de cage exclusivement constituées de carbone : les fullerènes.2 Après le graphite et le diamant, c’est une troisième forme de carbone pur qui fut ainsi découverte. Alors que les deux autres variétés sont des solides avec des réseaux infinis d’atomes, les fullerènes sont des molécules bien définies. L’étude de ces composés ne sera néanmoins possible que quelques années après leur mise en évidence grâce au développement d’une méthode de synthèse en quantités macroscopiques. Nous présenterons ici cette nouvelle famille de molécules, en particulier le buckminsterfullerène (du nom de l’architecte Buckminster Fuller) ou C60 qui en est le représentant le plus abondant et le plus étudié.
1. Production des fullerènes
La technique permettant la production de fullerènes fut mise au point en 1990 par Krätschmer et Huffman.3 Cette méthode est fondée sur la vaporisation de carbone sous une atmosphère d’hélium. Le dispositif expérimental se compose de deux tiges de graphite connectées à des électrodes de cuivre (Figure 1). L’une des tiges est taillée en pointe et maintenue au contact de l’autre à l’aide d’un ressort. Le tout est enfermé au sein d’une cloche de verre munie d’un système de pompage permettant d’évacuer l’air et d’une entrée d’hélium. Lorsqu’un courant électrique passe au travers des tiges de graphite, la chaleur d’origine ohmique se dissipe principalement au niveau du petit point de contact entre les deux tiges. La
C
CHHAAPPIITTRREE II 33
température atteint alors 2500-3000°C et le graphite se vaporise en un plasma qui se refroidit au contact de l’atmosphère d’hélium pour former une suie. Cette matière première est constituée d’un mélange de fullerènes solubles (Cn, n<100), de fullerènes dits « géants » (Cn,
n>100), de nanotubes et de carbone amorphe. Par des techniques d’extraction, il est alors possible d’isoler les fullerènes solubles de la suie.4 Des méthodes chromatographiques permettent ensuite de séparer les différents fullerènes.
Figure 1. Dispositif expérimental pour la production de fullerènes (d’après : A. Hirsch, The Chemistry of the Fullerenes, Thieme, Stuttgart, 1994).
2. Structure des fullerènes
Les fullerènes sont des molécules en forme de cage comportant 2(10 + n) atomes de carbone formant 12 pentagones et n hexagones. C’est en raison de sa ressemblance avec les dômes géodésiques imaginés et créés par l’architecte Buckminster Fuller que le C60 a été
nommé Buckminsterfullerène, et le terme fullerène a été par la suite appliqué à tous les représentants de la famille. Il est intéressant de souligner que le principe de construction de
ces composés est une conséquence du théorème d’Euler : pour obtenir un polyèdre à partir d’un réseau de n hexagones, il faut 12 pentagones (sauf pour n = 1). De fait, le plus petit fullerène pouvant être imaginé en théorie est le C20. A partir de C20, n’importe quel agrégat
constitué d’un nombre pair d’atomes de carbone peut former au moins une structure de type fullerène. En augmentant n, le nombre d’isomères de fullerène augmente rapidement, de 1 pour n = 0 à plus de 20000 pour n = 29.
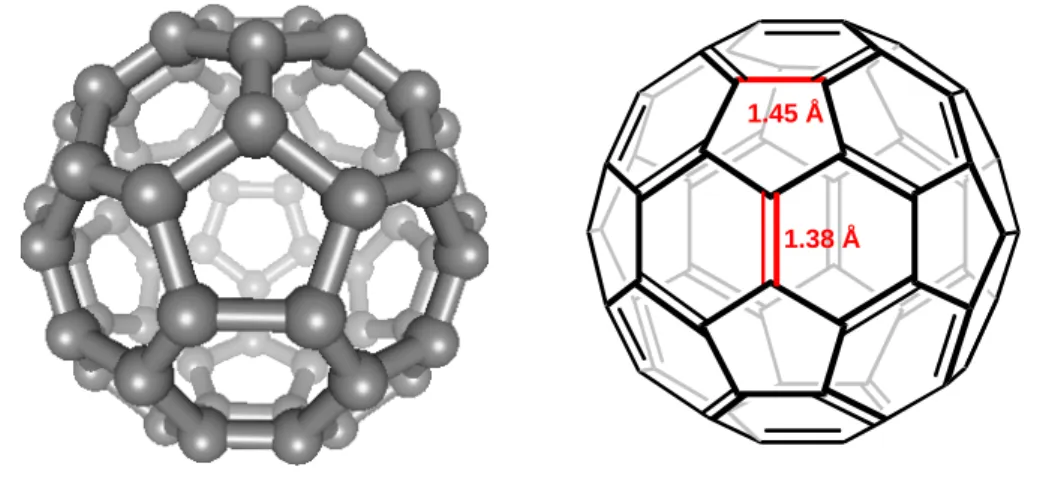
Le buckminsterfullerène (C60) est le plus petit fullerène stable. Ce composé a la forme
d’un icosaèdre tronqué, c’est en fait la réplique exacte d’un ballon de football (Figure 2). Formé de 12 pentagones et 20 hexagones, chaque pentagone étant entouré de 5 hexagones, le C60 est une molécule très symétrique dans laquelle tous les atomes de carbones sont
équivalents. Il peut aussi être noté qu’il existe deux types de liaisons carbone-carbone dans cette molécule : les liaisons situées à la jonction de deux hexagones (liaisons 6-6) et celles situées à la jonction d’un hexagone et d’un pentagone (liaisons 5-6). De fait, les liaisons 6-6 sont plus courtes que les liaisons 5-6. En d’autres termes, les liaisons 6-6 ont un caractère de liaison double alors que les liaisons 5-6 un caractère de liaison simple. Cette localisation des électrons π résulte de la pyramidalisation des atomes de carbone sp2 du fait de la structure sphérique empêchant un bon recouvrement orbitalaire. Le C60 n’est donc pas une molécule
aromatique.
1.38 Å 1.45 Å
C
CHHAAPPIITTRREE II 55
Le buckminsterfullerène est le seul isomère du C60, c’est aussi le plus petit des
fullerènes obéissant à la « règle des pentagones isolés ». Cette dernière prédit que les structures de type fullerène dont tous les pentagones sont isolés par des hexagones sont plus stables que celles possédant deux pentagones adjacents5. La déstabilisation liée à la présence de deux pentagones adjacents résulte essentiellement d’une tension de cycle importante du fait de la présence d’angles de liaison très éloignés des 120° standards.
Le second fullerène stable est le C70. Sa structure respecte la règle des pentagones isolés
et il a une forme ovale, comme un ballon de rugby. De fait, au niveau de ses deux pôles, la structure du C70 est semblable à celle du C60. En revanche, le C70 possède une ceinture
équatoriale constituée d’hexagones fusionnés (Figure 3).
Figure 3. Structure du C70.
En considérant que seuls les fullerènes Cx qui respectent la règle des pentagones isolés
sont stables, les chiffres x magiques sont : 60, 70, 72, 76, 78, 84… . Le nombre d’isomères possibles en théorie est de un pour le C60, un pour le C70, un pour le C72, un pour le C76, cinq
pour le C78, 24 pour le C84 et 46 pour le C90. Tous ces fullerènes, à l’exception du C72, sont en
fait obtenus lors de l’évaporation du graphite sous atmosphère d’hélium. A ce jour, une bonne partie de ces produits a été isolée et caractérisée.
3. Propriétés Physicochimiques du Buckminsterfullerène
Dans la suite de l’introduction, nous ne nous intéresserons plus qu’au cas du C60. C’est
en fait le plus abondant des fullerènes et certainement celui qui a été le plus étudié. Notons néanmoins que la majorité des caractéristiques physicochimiques du C60 se retrouve chez ses
homologues supérieurs.
3.1. Solubilités
Des études systématiques ont été menées afin de déterminer la solubilité du C60 dans
une large gamme de solvants organiques. Il se révéla insoluble dans les solvants polaires comme l’acétone, les alcools, ainsi que le tétrahydrofuranne, le diéthyléther ou encore le diméthylsulfoxyde, faiblement soluble dans les hydrocarbures tels que le pentane, l’hexane ou le cyclohexane. Les solvants les plus adéquats à la solubilisation du C60 demeurent les
solvants aromatiques tels le benzène (1.7 mg/mL), le toluène (2.8 mg/mL) ou encore le 1-chloronaphthalène (51 mg/mL). Il peut être noté que les solutions de C60 ont une couleur
mauve-magenta caractéristique (Figure 4).
C
CHHAAPPIITTRREE II 77
La solubilité du C60 dans les solvants organiques, bien que suffisante pour permettre sa
manipulation, a vite été jugée trop faible pour envisager de réaliser des dispositifs incorporant du C60 pur. Un autre problème réside dans la tendance des molécules de fullerène à s’agréger
les unes aux autres, ce qui modifie leurs propriétés. Pour ces raisons, la chimie du fullerène s’est ingéniée à trouver des réactions permettant de lier de manière covalente des groupes solubilisants à la sphère de carbone.
3.2. Propriétés photophysiques
Parmi les propriétés physico-chimiques tout à fait remarquables du C60, notons la non-linéarité optique et ses qualités en matière de limitation optique.6 En effet, l’absorption de lumière par le C60 dans le domaine du visible est faible si la molécule est éclairée par de
faibles intensités lumineuses. Cependant, l’absorption augmente considérablement quand l’intensité lumineuse devient plus importante : l’absorption est non-linéaire. Ce phénomène peut-être utilisé pour la protection d’un détecteur optique (une caméra, l’œil humain…) contre une agression laser sans pour autant empêcher son utilisation sous de faibles éclairements comme la lumière du jour.
La molécule de C60 présente une très grande symétrie. Grâce à cette symétrie, une
description théorique des propriétés optiques de la molécule isolée est possible, ce qui permet d’expliquer l’absorption non-linéaire observée expérimentalement : la description théorique prédit une absorption beaucoup plus importante pour la molécule dans un niveau excité que dans son niveau fondamental. Comme le nombre de molécules excitées dans un échantillon dépend directement de l’intensité lumineuse incidente, cela mène à une absorption plus forte pour une grande intensité incidente. Ce phénomène est appelé absorption saturable inverse (ASI).
solution dans du toluène. Les résultats sont interprétés par l’ASI dans le cadre d’un modèle à cinq niveaux (Figure 5).
S
0S
1S
nT
nT
1 Singulet TripletFigure 5. Modèle à 5 niveaux permettant d’expliquer les propriétés photophysiques du C60.
A chaque niveau électronique est associé un grand nombre d’état vibroniques de la molécule, ceci élargit considérablement le domaine d’absorption de la molécule. Les états vibroniques associés à des états électroniques sont proches en énergie et se recouvrent partiellement. De cette façon, pour décrire les propriétés optiques, une représentation simplifiée peut-être utilisée. Dans cette image, l’état fondamental de la molécule et ses niveaux vibroniques sont associés en un niveau S0. Tout le premier groupe d’états excités
singulets est regroupé dans un niveau S1. Notons que la transition S0→S1 est interdite par
symétrie, l’absorption associée est donc faible. Les autres états excités singulets de plus haute énergie qui sont à l’origine de transitions permises à partir de l’état fondamental sont associés à un niveau Sn (la première transition permise se situe dans le bleu-proche UV, et dans le
spectre d’absorption linéaire, une forte absorption est observée dans cette zone spectrale). Les états triplets peuvent également être répartis dans un groupe T1 et un groupe Tn. Au niveau de
C
CHHAAPPIITTRREE II 99
l’absorption non-linéaire, il s’avère que certaines transitions de l’état S1 vers des états Sn sont
permises dans le domaine du visible. Il en est de même pour les transitions entre les états constituants les niveaux T1 et Tn. De ce fait, une population dans un état excité, singulet ou
triplet, est beaucoup plus absorbante dans le visible qu’une population dans l’état fondamental. Si le C60 est éclairé par une faible intensité lumineuse, la population de
molécules dans l’état fondamental est beaucoup plus grande que celle dans des états excités, car la faible intensité engendre peu de transitions qui peuplent le niveau excité. A plus forte intensité, la population dans le premier état excité devient non négligeable et l’absorption augmente avec la population dans cet état excité, donc avec l’intensité incidente.
L’absorption induite par une impulsion laser peut durer des microsecondes. Pour la limitation d’impulsions longues, l’existence des états triplets est d’une importance primordiale car une limitation efficace nécessite une population excitée avec une durée de vie qui est au moins comparable à la durée de l’impulsion. Sinon un équilibre entre l’absorption S0-S1 et la relaxation S1-S0 s’installe pendant une impulsion longue. La population excitée
moyenne pendant l’impulsion sera faible et, en conséquence, la limitation aussi. Un paramètre clé dans le cas d’impulsions longues est le taux de molécules qui effectuent le transfert intersystème et s’accumulent dans le niveau T1, au lieu de se désexciter directement. Plus le
taux est élevé, plus la limitation sera efficace pour des impulsions longues qui nécessitent la présence d’une population triplet ayant une durée de vie suffisamment longue. Dans cette perspective, le C60 apparaît comme un bon candidat car en solution dans le toluène, le
croisement intersystème se fait avec un rendement proche de l’unité.
3.3. Propriétés électrochimiques
Des calculs théoriques7 avaient prédit que la molécule de C60 possédait une orbitale
moléculaire inoccupée triplement dégénérée assez basse en énergie. En termes plus simple, cette molécule devrait avoir un caractère accepteur d’électrons et, en principe, pourrait accepter jusqu’à 6 électrons pour former un hexaanion C606-. Dès l’obtention d’échantillons
purs de C60, les propriétés électrochimiques furent étudiées et les prédictions théoriques
vérifiées expérimentalement. En effet, il a été montré que le C60 pouvait accepter jusqu’à 6
électrons par six réductions monoélectroniques successives (Tableau 1).8 Il est à noter, d’une part, que toutes ces réductions demeurent des processus réversibles et d’autre part que les anions obtenus sont stables pendant plusieurs jours à basse température. Soulignons également que si le C60 est relativement facile à réduire (du moins pour la première réduction
conduisant à l’anion C60-), il est plutôt difficile de l’oxyder.
C60/C60 -C60 -/C602 -C602 -/C603 -C603 -/C604 -C604 -/C605 -C605 -/C606 -- 0.98 - 1.37 - 1.87 - 2.35 - 2.85 - 3.26
Couple monoélectronique Réduction[a]
[a] valeurs de potentiel, en V (vs. Fc+/Fc), obtenues pour une vitesse de balayage de 100 mV.s-1.
Tableau 1 : Potentiels de réduction obtenus à -10°C dans un mélange CH3CN/ toluène.
Ces résultats se révèlent particulièrement importants car ils offrent une première indication sur la réactivité chimique du C60. En effet, l’électronégativité du C60 laisse
supposer que cette molécule sera plutôt un électrophile pouvant être le siège de réactions d’addition par attaque d’un nucléophile.
C
CHHAAPPIITTRREE II 1111
4. Propriétés Chimiques du Buckminsterfullerène
Depuis la mise au point en 1990 du processus de synthèse de quantités macroscopiques du C60, sa réactivité chimique a fait l’objet de nombreuses études. Des réactions permettant de
greffer des groupements à la surface du C60 ont été découvertes et, à ce jour, de très nombreux
dérivés du C60 ont été produits. La modification chimique du C60 présente un premier intérêt,
celui de considérablement augmenter sa solubilité après greffage. Les dérivés sont ainsi nettement plus faciles à manipuler. D’autre part, de nombreux groupes possédant une fonction spécifique ont pu être greffés sur le C60 pour l’obtention de molécules à propriétés originales.
Le C60 a une réactivité chimique similaire à celle d’une oléfine déficiente en électrons.
Ainsi, le C60 est un bon électrophile et peut être le siège de réactions d’addition nucléophile.
Parmi ces réactions, les cycloaddition jouent un rôle très important dans la préparation des dérivés de C60 et une grande variété de cycloadduits a ainsi été synthétisée par cette
méthode.9,10
Nous allons présenter ici les principales réactions de cycloaddition permettant de greffer un groupement sur le C60.
4.1. Cyclopropanation : la réaction de Bingel.
Depuis la première synthèse de méthanofullerènes par l’équipe de Wudl 11, de nombreux travaux ont été effectués pour développer de nouvelles méthodes de synthèse de ce type de composés 12. La cyclopropanation est une des méthodes la plus efficace pour la préparation de dérivés de fullerènes ayant un intérêt dans les sciences des matériaux ou pour des applications biologiques13. La cyclopropanation du C60 peut être effectuée par trois
méthodes différentes :
a) addition thermique de composés diazo suivie par une thermolyse ou une photolyse b) additions de carbènes libre
Nous allons nous intéresser plus particulièrement à la réaction de Bingel car c’est une méthode qui permet de synthétiser des fullerènes cyclopropanés dans des conditions relativement douce et avec de bons rendements. La réaction de Bingel qui est en fait une réaction d’addition–élimination, peut être effectuée avec des α-halomalonates en présence d’une base 14. L’addition de l’anion α-halomalonate sur le C60 est suivie par une substitution
nucléophile intramoléculaire de l’halogénure par l’anion généré sur le fullerène pour donner le méthanofullerène correspondant (Figure 6).
O O O Br O C60 / NaH O O O O Br Base O O O O
Figure 6 : Réaction de Bingel
Ce type de cyclopropanation du C60 peuvent aussi être réalisées en générant le
α-halomalonate in situ en présence d’iode15 ou de CBr416 et une base (DBU:
1,8-diazabicyclo[5.4.0]undec-7-ene) (Figure 7). RO OR O O + I2 / DBU CBr4 / DBU RO OR O O Figure 7 : Cyclopropanation du C60
C
CHHAAPPIITTRREE II 1133
Cette méthode a permit de synthétiser de nombreuses dyades et triades C60-donneurs,
les fonctions ester permettant de connecter les donneurs à la sphère de fullerène. Les hydrocarbones polycycliques aromatiques (PAHs) possèdent les propriétés photophysiques et électrochimiques requises pour obtenir des transferts électroniques photo induits. Une réaction de cycloaddition de Bingel classique a été utilisée pour préparer la première dyade tétracène-[60]fullerène (Figure 8). 17 Cette stratégie évite d’utiliser la réaction de Diels–Alder qui conduirait à la perte de l’aromaticité du tétracène (Figure 8).
O O O O OC12H25 OC12H25 O O O O OC12H25 C12H25O O HO O O OC12H25 C12H25O OH 1. DCC, DMAP, CH2Cl2, 0ºC, r.t. 2. DDQ, benzene, reflux + toluene, r.t. C60, I2, DBU
La synthèse d’oligoporphyrines conjuguées à une ou deux sphères de fullerènes a été décrite récemment (Figure 9).18 Les auteurs ont pu montrer que ces systèmes adoptent différentes conformations en fonction des interactions fullerène-porphyrine intermoléculaires. Le dimère comportant deux porphyrines et deux C60 présente un processus de transfert
d’énergie photo-induit des fullerènes vers les porphyrines de Zinc.
N N Ar Ar N N Zn N N Ar Ar N N Zn N N Ar Ar N N Zn n n = 0, 1, 2 O O O O O O O O N N Ar Ar N N Zn N N Ar Ar N N Zn O O O O O O O O N N Ar Ar N N Zn n n =1, 2 O O O O Ar =
C
CHHAAPPIITTRREE II 1155
Une réaction de Bingel intramoléculaire entre un méthano-fullerène et un 10-éther-1,5-dinaphtho-[38]couronne a conduit à un caténane basé sur du C60 (Figure 10). Par ailleurs,
un [2]rotaxane similaire a été synthétisé par une réaction de Bingel intermoléculaire . Ces deux systèmes comportent chacun une partie aromatique électro-déficiente de type diimide.19
O O N O O N O O O O O O O O O O n n O O O O O O O O O O O O O N O N O O O O O O O O O O n n O O O O O O O O O O
Figure 10 : Rotaxane et caténane basés sur du C60
4.2. Réaction de rétro-Bingel
Les adduits méthano peuvent être retirés des fullerènes grâce à une méthode réductrice appelée réaction de rétro-Bingel20. Les bis(alkoxycarbonyl)méthanofullerènes qui sont formés
par des réactions de Bingel sont généralement assez stables. Cependant, Diederich et Echegoyen ont montré que ces adduits bis(alkoxycarbonyl)méthano peuvent être retirés électrochimiquement avec de bon rendements par réduction électrolytique à potentiel constant (CPE) (Figure 11).21 O O O O CPE, 4e-, CH2Cl2 rétro-Bingel O O O O +
Figure 11 : Réaction de rétro-Bingel
Lorsque qu’une électrolyse à potentiel contrôlé est appliquée à un isomère pur de bis-méthanofullerène, deux électrons sont déchargés par molécule et l’on observe une compétition entre une rétro-Bingel et une réaction d’isomérisation (Figure 12). Lors de cette isomérisation intramoléculaire les isomères cis ne se forment pas ; l’isomère majoritaire est le
trans bien que l’isomère-e soit obtenu avec un rendement non négligeable. Ces résultats
C
CHHAAPPIITTRREE II 1177
100% d'un seul isomère
O O O O + retro-Bingel 1. 6e- CPE 2. reoxidation 1. 2e- CPE 2. réoxydation Isomérisation O O O O O O O O O O O O O O O O + + 75% e: 11 % trans-4: 4 % trans-3: 5 % trans-2: 24 % trans-1: 5 % O O O O O O O O
Figure 12 : Réaction de rétro-Bingel versus isomérisation
La rétro-cyclopropanation peut aussi être effectuée chimiquement en traitant le bis(alkoxycarbonyl)méthane-fullerène avec un amalgame de magnésium (10% de bromure mercurique) dans le THF sous reflux pendant trois jours (Figure 13).23 Récemment, ces
réactions de rétro-cyclopropanation ont été effectuées en présence d’éther couronne pour éviter l’utilisation de HgBr2.24 O O O O Mg/Hg THF / Ar, 80 ºC C60 (73%) THF / Ar, 80 ºC Ether couronne C60 (52%) Mg
Figure 13 : Réaction de rétro-Bingel chimique
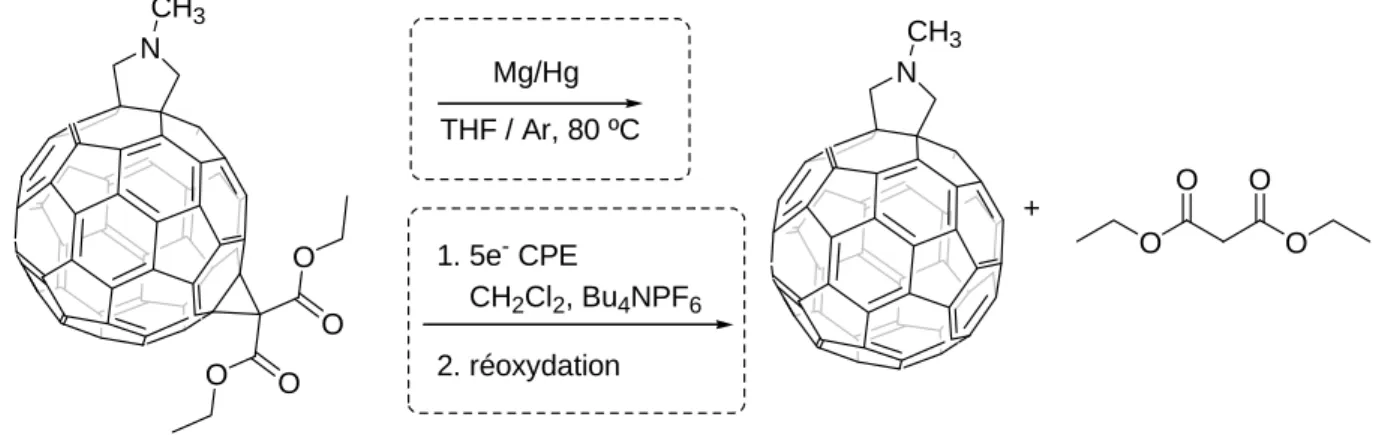
Il faut noter que ces réactions de rétro-cyclopropanation permettent de retirer des adduits de Bingel sans pour autant toucher à d’autres adduits (Figure 14) que ce soit dans des conditions électrochimiques 25 ou chimiques. 23
Mg/Hg THF / Ar, 80 ºC O O O O + 1. 5e- CPE CH2Cl2, Bu4NPF6 2. réoxydation O O O O N CH3 N CH3
C
CHHAAPPIITTRREE II 1199
4.3. Cycloadditions [4+2]: les réactions de Diels-Alder
Les doubles liaisons [6,6] des C60 sont d'excellents diénophiles. Ainsi, le fullerène peut
réagir avec différents diènes par une réaction de cycloaddition de type Diels-Alder. En fonction de la réactivité du diène, il peut être nécessaire de chauffer le milieu réactionnel sous reflux dans des solvants à haut point d'ébullition. Ceci a conduis à utiliser l'énergie des micro-ondes avec d'excellents rendements dans ce type de réactions26.
Dans le premier exemple de réaction de Diels-Alder le C60 a été mis réagir avec le
cyclopentadiène27. L'addition du cyclopentadiène à une solution de fullerène dans le toluène à température ambiante a conduis à un produit de mono-addition avec 74% de rendement (Figure 15).
C60 + toluène
Figure 15: Premier exemple impliquant le C60 dans une réaction de Diels-Alder
Les dérives de cyclopentadiène,28 tel que le méthylcyclopentadiène et la cyclopentadiènone, conduisent aux mono-adduits correspondant, qui sont généralement plus stables que le mono-adduit de la Figure 15. Le comportement diénophilique du C60 dans les
réactions de cycloaddition [4+2] a également été vérifié dans la réaction du fullerène avec l'anthracène (Figure 16) 29, 28b et d'autres dérivés de l'anthracène.30 , 28b
C60 +
Figure 16: Réaction de Diels-Alder entre le C60 et l'anthracène
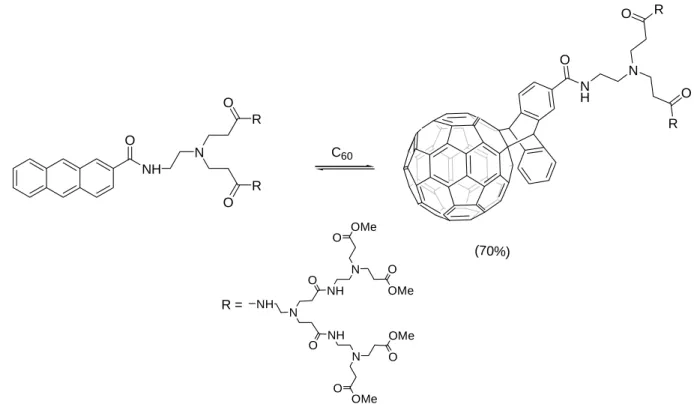
Des fullerodendrimères poly(amidoamine) hydrosoluble 31 ont pu être préparés par réaction du C60 avec un dendron anthracényle (Figure 17). Ce fullerodendrimère est soluble
dans les solvants polaire et est stable pendant plusieurs mois lorsqu'il est stocké à l'obscurité à température ambiante. O N H N R O R O NH N O NH N OMe O OMe O O NH N OMe O OMe O R = NH O N R O R O C60 (70%)
C
CHHAAPPIITTRREE II 2211
Comme nous l’avons mentionné précédemment, l'irradiation par des micro-ondes a été employée dans des réactions de cycloaddition de Diels–Alder et il a été montré que ces réactions sont accélérées et les rendements supérieurs par rapport à ceux obtenus par chauffage classique. Ceci provient du fait que les temps de réaction sont bien plus courts et l'on évite ainsi les réactions de rétro-Diels–Alder et la formation de bisadduits.32
Par ailleurs, l'efficacité des réactions de cycloaddition entre les anthracènes et le C60
peut être améliorée en effectuant les réactions dans des conditions de broyage des réactifs solides par vibration à haute vitesse.33,34
D'autres diènes ont également été utilisés dans ce type de réactions.35 Hirsch a décrit la synthèse de différentes dyades C60-accepteurs (Figure 18) par des réactions de cycloadditions
[4+2] d’anthraquinones avec le C60. 36 O O C60 + toluène reflux O O
Figure 18 : Synthèse d’une dyade C60-accepteur
L'utilisation des o-quinodiméthanes comme systèmes diéniques est l'un des outils des plus important pour fonctionnaliser le fullerène par ce type de réaction. Différentes méthodes pour générer des o-quinodiméthanes37 in situ sont disponible: 1,4-déhalogénation du
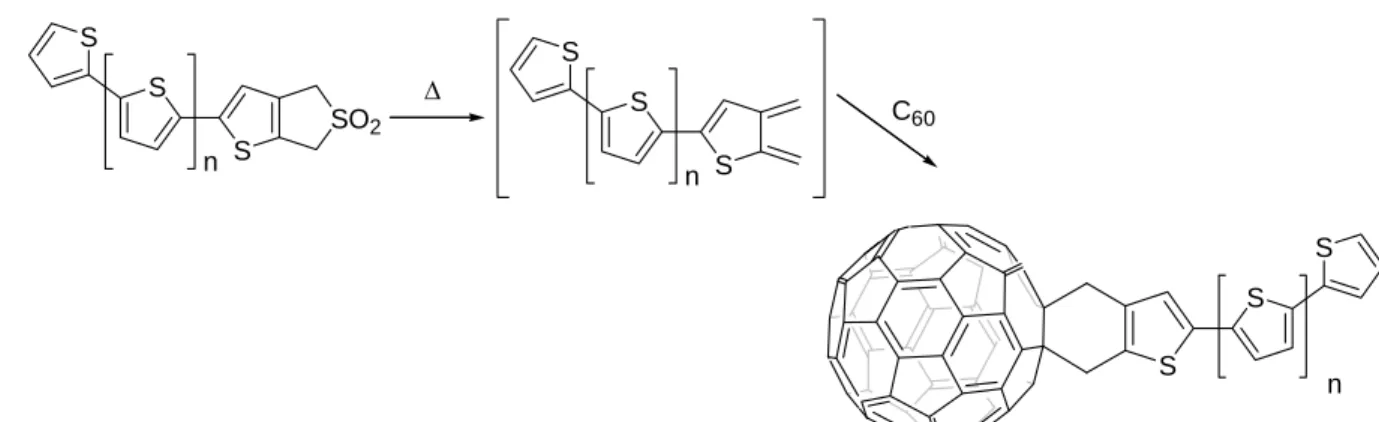
o-bis(bromométhyl) benzène grâce à l'iode,38 thermolyse de benzocyclobutènes,39 thermolyse de sulfolènes (Figure 19) 40 et thermolyse de sultines.41
SO2 S S S n S S S Δ S S S n C60 n
Figure 19 : Réaction de Diels-Alder entre le C60 et des o-quinodiméthanes générés par
thermolyse de sulfolènes.
La 1,4-déhalogénation intramoléculaire de α,α'-dibromo-o-xylènes nécessite la présence d'ions tel que les iodures pour induire les élimination-1,4 ainsi que des éthers 18-couronne-6 comme catalyseurs de transfert de phase. Des fullerènoquinoxalines ont ainsi été synthétisées en chauffant la o-bis(bromométhyl)quinoxaline en présence de NaI dans le 1,2-dichlorobenzène (Figure 20).42 N N Br Br R R' NaI, 18-crown-6 o-DCB, Δ N N R R' C60 N N R R'
Figure 20 : Synthèse de fullerènoquinoxalines.
Ce type de réaction a été utilisé pour réaliser des composés photoactifs contenant une partie fullerène acceptrice d'électrons et un fragment électro-donneur comme le TTF ou ses analogues.43
La thermolyse des benzocyclobutènes nécessite des températures élevées pour obtenir une réaction d'ouverture de cycle même en présence de groupes méthoxy activateurs, qui abaissent l'énergie d'activation (Figure 21).39
C CHHAAPPIITTRREE II 2233 OMe Δ OMe C60 OCH3
Figure 21 : Réaction de Diels-Alder via thermolyse de benzocyclobutènes
Les meilleurs candidats parmi les précurseurs des o-quinodiméthanes, sont les sulfolènes et les sultines car ils sont pyrolysés à des températures relativement basses et ils sont généralement stables à température ambiante. La synthèse d'une triade porphyrin-[60]fullerène-porphyrine a ainsi été obtenue par une cycloaddition de Diels–Alder entre un sulfolène contenant une porphyrine et le C60 (Figure 22).44
S S HN O NH O R R N HN NH N R = S S HN O NH O R R S C60 hydroquinone, o-DCB reflux, 3 h O O
Les diènes peuvent être formés par élimination de dioxyde de soufre non seulement à partir de dérivés de sulfolènes mais aussi à partir de sultines. Ceux-ci sont facilement accessible à partir de la rongalite commerciale et permettent de générer de façon douce les quinodiméthanes par extrusion de dioxyde de soufre. La réaction de cycloaddition [4+2] de o-quinodiméthanes, générés in situ à partir de 4,5-benzo-3,6-dihydro-1,2-oxathiin 2-oxides (sultines), sur du C60 a été décrite (Figure 23).41a
S O O R R' R' R Δ R R' R' R C60 R R' R' R
Figure 23 : Cycloaddition [4+2] de o-quinodiméthanes sur du C60
Il a été montré que l'irradiation par des micro-ondes accélère la réaction de Diels–Alder entre le C60 et le o-quinodiméthane correspondant généré in situ à partir de dérivés sultine.45
L'irradiation par des micro-ondes dans la synthèse des dérivés de thiophène-[60]fullerène améliore les rendements et diminue le temps de réaction de façon significative (Figure 24).46
O S S R1 R2 O Δ C 60 S R1 R2 S ou micro-ondes R1 R2
Figure 24 : Utilisation de microndes dans les réactions de cycloaddition [4+2] de o-quinodiméthanes sur du C60
Bien que les réactions de Diels–Alder soient favorisées en présence de diènes riche en électrons, le C60 peut aussi réagir avec des diènes électro-défficients– comme mis en évidence
C CHHAAPPIITTRREE II 2255 C60 + N N N N Ar Ar Δ N N N N Ar Ar N N Ar Ar
Figure 25 : Synthèse de fullerènopyridazines
4.4. Réactions de cycloaddition [3+2]
4.4.1. Les ylures d'azométhine
Les réactions de cycloaddition [3+2] d'ylures azométhine avec le fullerène ont été développées par Prato et Maggini.48 C'est l'outil le plus fréquemment utilisé pour fonctionnaliser le C60 en raison de sa bonne sélectivité. En effet, la cycloaddition a toujours
lieu sur les liaisons [6,6] de la sphère de fullerène. Par ailleurs, cette réaction tolère une grande variété de groupes fonctionnels et il est possible d'introduire différents substituants dans trois positions différentes du cycle pyrrolidine en fonction des composés carbonylés (aldéhyde ou cétone) et acides aminés utilisés.
Dans cette réaction, connue sous le nom de réaction de Prato, des sels d'immonium obtenus par la condensation d'acides α-aminés avec des aldéhydes ou des cétones sont décarboxylés pour générer des ylures d'azométhine. Le premier exemple, publié en 1993, décrit la synthèse d'une N-méthylfulleropyrrolidine par réaction de la sarcosine avec du formaldéhyde et du C60 (Figure 26).48a
H N OH O + CH2O Δ -CO2 -H2O N C60 N CH3
Figure 26: Réaction de Prato
L’accessibilité à une grande variété d’aldéhydes permet d’envisager la synthèse de nombreuses dyades ou triades donneurs-accepteurs grâce à cette méthode. Il faut noter que le cycle pyrrolidine possède un centre stéréogène au niveau du carbone substitué. Lors de sa synthèse les différents stéréoisomères sont donc formés.
Les ylures d'azométhines peuvent aussi être préparés à partir d'aziridines grâce à une réaction d’ouverture de cycle thermique (Figure 27).49
N N CH2Ph CO2CH3 C60, Δ toluene CO2CH3
Figure 27 : Synthèse de fulleropyrolidines à partir d'aziridines
D'autres méthodes48b ont été utilisées pour préparer des fulleropyrrolidines : par désilylation acido-catalysée ou thermique de dérivés triméthylsilyl amine, par tautomérisation de sels d'immonium d'aminoesters et d'imines, par réaction avec des aldéhydes en présence d'ammoniaque aqueux, par réaction avec des oxazolidinones ou par des réactions photochimiques avec des dérivés aminés.
La réaction de cycloaddition d'ylures d'azométhines peut aussi être effectuée par irradiation par des micro-ondes,50 qui permettent d'utiliser des conditions plus douces.
C
CHHAAPPIITTRREE II 2277
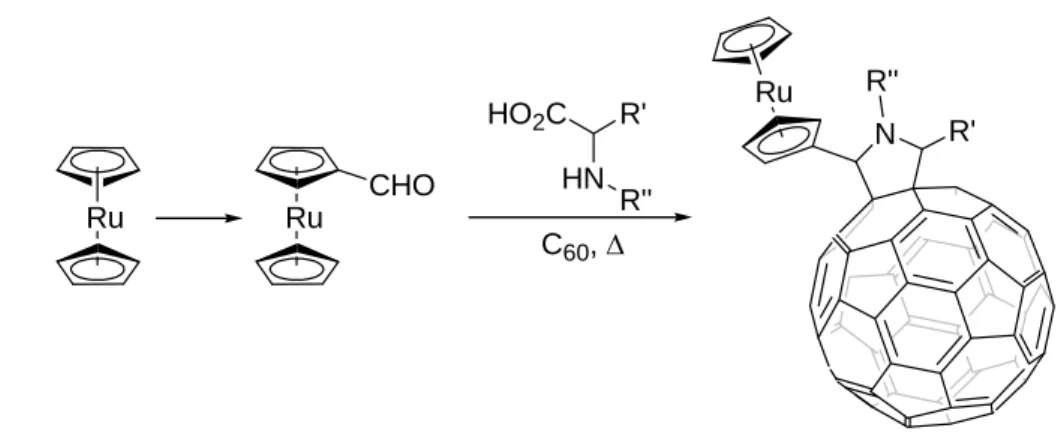
fréquemment dans des systèmes donneur-accepteur (D-A) basés sur le fullerène dans le but d'élaborer de nouveaux matériaux moléculaires de photosynthèse artificielle.51 D'autres métallocènes tel que le ruthénocène peut être incorporés dans des fragments électrodonneurs présentant des propriétés de séparations de charges phoinduites efficaces (Figure 29).52
Ru Ru CHO HO2C R' HN R'' C60, Δ N R'' R' Ru
Figure 28 : Synthèse d’un système ruthénocène-C60.
La réaction de Prato a permit de synthétiser un grand nombre de systèmes pyrrolidine[60]fullerènes sur lesquels ont été attachés différents fragments avec des propriétés électroniques importantes. Par exemple, des molécules tels que des porphyrines,53 des
subphthalocyanines,54 du TTF,55 des dendrimères56 et des oligomères conjugués57 ont été incorporés.
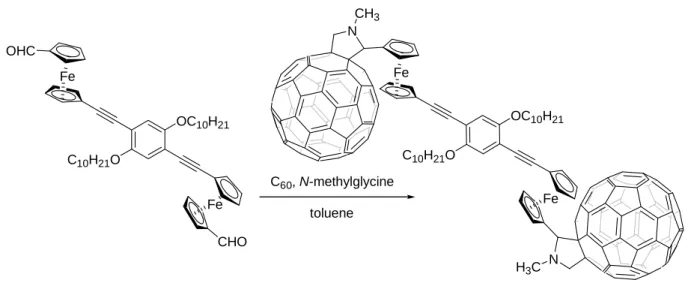
Des cycloadditions [3+2] dipolaires d’ylures d’azométhine utilisant des bis-aldéhydes permettent de préparer des molécules contenant 2 fullerènes.58 Le système bis-(ferrocènyl-fulleropyrrolidine) (Figure 29) comporte 2 fragments ferrocène electro-donneurs.
OC10H21 C10H21O Fe Fe N CH3 N H3C OC10H21 C10H21O Fe Fe OHC CHO C60, N-methylglycine toluene
Figure 29 : Synthèse d’une bis-(ferrocènyl-fulleropyrrolidine).
Bien que les N-méthylglycines soient les acides aminés les plus fréquemment utilisés pour la préparation d’ylures d’azométhines, d’autres glycines N-substitutées ont aussi été utilisées.59 Des fulleropyrrolidines N-substituées peuvent être estérifiées par des acides carboxyliques dérivés de l'anthracène pour former des systèmes C60-anthrylphénylacetylène
(Figure 30). Ces composés sont capables de former des mono-couches auto-assemblées (SAMs) grâce aux excellentes propriétés physiques et chimiques du C60 et à l’assemblage au
C CHHAAPPIITTRREE II 2299 HO NH CO2H C60, Δ toluene + HCHO N OH SAc O OH (CF3CO2)2O, benzene, 40ºC N O O SAc
Figure 30 : Synthèse de systèmes C60-anthrylphénylacetylène.
Un dérivé du C60 hydrosoluble (Figure 31), 61 préparé à partir de triéthylène glycol, est
un précurseur qui permet d’introduire de façon covalente la cage de C60 dans un heptapeptide
(H-PPGMRPP-OH) pour donner le composé A, qui a des propriétés antigéniques.62
HO N H O OH O C60, Δ toluene + HCHO N O OH PPRMGPP H2 TFA N O O A
La solubilité dans l’eau des systèmes pyrrolidino[60]fullerène peut être augmentée en
quaternarisant l’atome d’azote. Dans la Figure 32 des sels de fulleropyrrolidinium sont
préparés en méthylant les pyrrolidines correspondantes à l’aide de l’iodure de méthyle.63 Ces sels sont modérément solubles dans les solvants organiques polaires tels que le THF, le DCM, le DMSO et des mélanges DMSO/H20. Par ailleurs, ces systèmes présentent des propriétés
électro-acceptrices améliorées en raison de la présence de la partie pyrrolidinium.
N R' R CH3I CHCl3, Δ N R' CH3R
Figure 32 : Synthèse de sels de fulleropyrrolidinium
L’atome d’azote des fulleropyrrolidines est environ 6 fois moins basique et 3 fois moins réactif que celui des pyrrolidines.64 Néanmoins, les pyrrolidino[60]fullerènes peuvent être substitués sur l’atome d’azote par différentes méthodes (Figure 33). Par exemple, par réaction des pyrrolidino[60]fullerènes avec des chlorures d’acides pour former des liaisons amide.65 Ou bien il est possible de faire des substitutions nucléophiles aromatiques par réaction de la fonction NH avec des composés aromatiques désactivés.66 La combinaison de la catalyse par transfert de phase en absence de solvant et la technique d’irradiation par des micro-ondes permet de faire réagir différents bromures d’alkyle avec les fulleropyrrolidines. 50a
C CHHAAPPIITTRREE II 3311 Ar Cl Alkyl Br R O Cl N H N O R N Ar N Alkyl
Figure 33 : Méthodes de substitution des pyrrolidino[60]fullerènes
On considère généralement que les pyrrolidino[60]fullerènes sont des cycloadduits très stables dans une grande variété de conditions expérimentales. Néanmoins, Martín et al.67 ont récemment décrits une réaction de rétrocycloaddition avec différents pyrrolidinofullerènes pour donner quantitativement du C60 (Figure 34). La réaction est réalisée en chauffant le
dérivé de fullerène sous reflux dans le o-dichlorobenzène en présence d’un dipolarophile très efficace et d’un métal de Lewis (CuTf2) comme catalyseur.
+ (métal) + dipolarophile o-DCB Δ N Me R2 R1
Figure 34 :Réaction de rétrocycloaddition.
4.4.2. Les nitriles imines
Le C60 est un candidat de choix pour l’élaboration de systèmes donneurs-accepteurs
(D-A) en raison de la faible valeur de son potentiel de première réduction. Ces matériaux présentent des propriétés optiques et électroniques intéressantes, c’est pour cela que différents groupes ont entrepris la synthèse de dyades C60-donneurs. Ils ont étudié les
interactions électroniques inter- ou intramoléculaires pouvant exister entre les donneurs et le fullerène à l’état fondamental ou excité.
Cependant, les potentiels de réduction des mono-adduits sont plus faibles d’environ 100 mV par rapport à ceux du C60 enraison de la saturation d’une des doubles liaisons. Il existe
très peu d’exemples dans la littérature qui montrent une meilleure affinité électronique que le C6068 et parmi eux on peut citer les sels de pyrrolidinium 63a mentionnés plus haut, qui ont une
affinité supérieure en raison de l’effet inducteur –I de l’azote quaternarisé sur la sphère de C60.
Une solution permettant de préparer des fullerènes fonctionnalisés avec une meilleure ou au moins une aussi bonne affinité électronique que le C60, consiste à lier directement des
atomes électronégatifs ou électro-défficients au fullerène.
Une des possibilités est l’addition de nitriles imines afin de préparer des systèmes pyrazolino[60]fullerène. Cette méthode n’est pas utilisée très souvent mais elle est très utile pour lier de façon covalente des groupes donneurs à la sphère de C60.
C
CHHAAPPIITTRREE II 3333
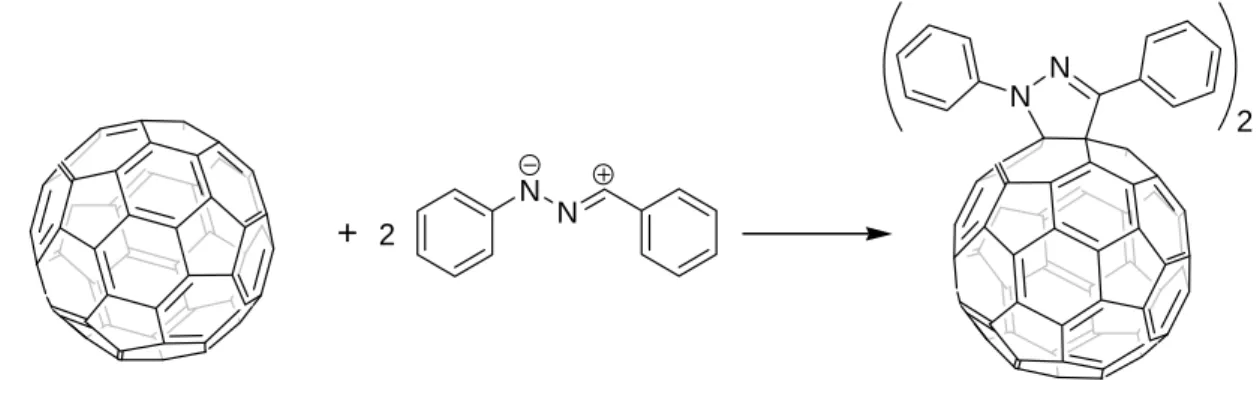
Les premièrescycloadditions 1,3-dipolaires sur le C60 ont été décrites dès les débuts de
la chimie des fullerènes mais le premier exemple décrivant la réaction entre un nitrile imine et le C60 n’a été décrit qu’en 1994, lorsque Mathews et al.69 a préparé un mélange de
bis-adduits par réaction de C60 et de 1,3-diphenylnitrile imine, généré in situ à partir de chlorure
de benzhydrazidoyl, en présence de triéthylamine. Le monoadduit par contre n’a jamais été isolé (Figure 35). 2 N N N N + 2
Figure 35 : Premier exemple d’une réaction entre un nitrile imine et le C60
La préparation de 1,3-diphényl-2-pyrazolinofullerènes a été décrite.70 Ces adduits ont été préparés par une réaction de cycloaddition entre le C60 et des nitriles imines générées in
situ à partir des dérivés de N-chlorobenzylidènes en présence de triéthylamine dans le
benzène sous reflux (Figure 36).
N N R CO2CH3 OCH3 tBu R = N R Cl HN Et3N, Benzène, reflux
Une autre voie d’accès aux pyrazolino[60]fullerènes (Figure 37) utilise des hydrazones comme composés de départ. Les dipôles 1,3 sont générés in situ en traitant des hydrazones avec du N-bromosuccinimide en présence de triéthylamine. Ces dipôles sont ensuite mis à réagir avec le C60 sous irradiation de micro-ondes.71
NBS Micro-ondes C60 1. Et3N 2. C60, MW N N Ph N H N R N N Ph N H N R Br N N R N N Ph N N R N N Ph Micro-ondes
Figure 37 : Autre voie d’accès aux pyrazolino[60]fullerènes
Ce mode opératoire a été modifié pour préparer différents pyrazolinofullerènes comportant différents groupes fonctionnels comme substituants.72 Les hydrazones de départ sont traitées avec de la N-chlorosuccinimide en présence de pyridine; les dérivés chlorés résultants sont mis à réagir avec de la triéthylamine et du C60 pour donner les cyclo-adduits
C CHHAAPPIITTRREE II 3355 R CHO NO2 HN H2N AcOH N O NO2 HN N CH R S Fe NMe2 R = 1. NCS 2. C60, Et3N R = N N R NO2
Figure 38 : Synthèse de pyrazolinofullerènes comportant différents groupes fonctionnels
Cette stratégie de synthèse a été utilisée avec une grande variété d’hydrazones pour préparer différents pyrazolino[60]fullerènes incorporant différents fragments organiques avec des propriétés électroniques intéressantes: des oligophénylènevinylènes (OPV), 73 des dendrimères de phénylènevinylène,74 des dérivésd’isoindazole75 et des ruthenocènes. 52a Des dérivés d’oligophénylenevinylènes comportant des groupes p-nitrophényl-hydrazone ont été préparés et utilisés pour la synthèse de systèmes (pyrazolino[60]fullerène)-oligophénylènevinylène76 (Figure 39).
N N O2N OC8H17 OC8H17 OC8H17 OC8H17 OC8H17 H17C8O N N NO2 n n=1, 3 Figure 39 : (Pyrazolino[60]fullerène)-oligophénylènevinylène
Cette méthode est un procédé général pour la fonctionnalisation des fullerènes en réaction one-pot à partir des hydrazones correspondantes. Les hydrazones sont des composés stables et facilement accessibles à partir des aldéhydes et les rendements obtenus sont quasi quantitatifs. 77
Un nouveau schéma de synthèse a été élaboré récemment.78 Des nitriles imines ont été générés par décomposition thermique de 2,5-diaryltétrazoles. Des cycloadduits contenant des substituants aryl- et hetaryl-, tels que des thiényl et pyridyl, dans le fragment pyrazoline ont été synthétisés grâce à cette méthode (Figure 40). L’accessibilité à des 2,5-diaryltétrazoles avec différentes structures donne accès à de nombreuses possibilités de fonctionnalisation du fullerène tandis que les autres méthodes sont limitées par la disponibilité des halogénures d’ hydrazonoyle appropriés
C CHHAAPPIITTRREE II 3377 N N R2 R1 N N R 2 PhBr, 155°C PhBr, 155°C N N N N R2 R1 R1 .
Figure 40 : Réaction de C60 avec des nitriles imines générés par décomposition thermique Des pyrazolino[60]fullerènes ont aussi été préparés par réaction de C60 avec des
diazoacétates d’alkyle à l’état solide dans des conditions de HSVM (solid-state high-speed vibration milling) ainsi qu’en solution dans le toluène. 79 La 2-pyrazoline dans la Figure 42 a été obtenue par isomérisation de la 1-pyrazoline, qui est formée par cycloaddition 1,3-dipolaire du éthyl diazoacétate, généré in situ à partir du sel de chlorure de l’ester de glycine et du nitrite de sodium avec le C60 (Figure 41).
N N H3CH2CO2C NH2CH2CO2R HCl NaNO2, HSVM N N H3CH2CO2C H
Figure 41 : Synthèse de pyrazolino[60]fullerènes par HSVM
Les 2-pyrazolino[60]fullerènes présentent des avantages par rapport aux autres dérivés de fullerènes tels que les pyrrolidino[60]fullerènes: contrairement aux pyrrolidinofullerènes, qui contiennent un centre stéréogène en position C-2 du cycle pyrrolidine, il n’y a pas de formation de stéréoisomères dans le cas des pyrazolino car l’atome de C est hybridé sp2. Par ailleurs, les systèmes présentent de meilleurs potentiels de réduction que le C60 en raison de
systèmes présentent des processus de transferts d’électrons photoinduits du cycle pyrazoline, un type de comportement qui n’a pas été observé avec d’autres dérivés tels que les fulleropyrrolidines.
4.5. Réactions de multi-additions
Des fullerènes bis-fonctionalisés ont été largement utilisés dans des études biologiques, appareils moléculaires, et des matériaux évolués en raison de leur structure tri-dimensionnelle définie qui peut contenir une variété de groupes fonctionnels. Après fonctionnalisation, les liaisons 6-6 restants du C60 ne sont pas identiques et, en principe, 8 différents régioisomères,
qui sont difficiles à séparer, peuvent être formés par addition d’un second adduit.
La nomenclature IUPAC80 peut être utilisée pour nommer les différents régioadduits. Cependant on utilise généralement une description différente proposée par Hirsch et al 81. Les différentes positions d’un second adduit par rapport à une première fonctionnalisation sont appelés cis-n (n = 1–3), équatorial (e) et trans-n (n = 1–4) (Figure 43).
cis-1
cis-2
cis-3
e
trans -4
trans -3
trans -2
trans -1
R
C
CHHAAPPIITTRREE II 3399
La distribution des régioadduits varie en fonction des méthodes de fonctionnalisation du C60 utilisées 48b,82 mais les positions e et trans-3 sont généralement préférées. Comme montré
dans la Figure 43, la double cyclopropanation du C60 par la réaction de Bingel avec le diéthyl
2-bromomalonate conduis à la formation de 7 isomères, qui sont isolés par chromatographie.83 Lors de la réaction avec des ylures d’azométhine symétriques, 8 isomères sont obtenus avec des sélectivités légèrement différentes que celles trouvées avec les bismalonates.84
O O O O O O O O cis-2: 0.9 % cis-3: 2.5 % e: 15.5 % trans-4: 3.7 % trans-3: 12.0 % trans-2: 5.3 % trans-1: 0.8 %
Figure 43 : Résultats de la cyclopropanation du C60 par la réaction de Bingel avec le
diethyl 2-bromomalonate
La préparation régiosélective de multiadduits des fullerènes est d’une grande importance pour la préparation de matériaux évolués fonctionnels étant donné que la géométrie joue rôle crucial dans les propriétés des dérivés de fullerènes. Un procédé élégant a été introduit par Diederich85 en 1994 afin d’obtenir un contrôle sur la régiosélectivité des bisadditions sur les fullerènes. Cette méthode est fondée sur une réaction intramoléculaire sur la sphère de carbone (Figure 44). On peut trouver de nombreuses revues sur ce sujet.86 Bien que d’autres composés bi-fonctionnels ont été utilisés dans ce type de réaction, ce sont généralement des fonctions malonates, connectées grâce à un fragment organique plus ou moins rigide, qui sont utilisés dans des cycloadditions de Bingel. La rigidité du fragment et la longueur de l’espaceur peuvent modifier la régiosélectivité de la réaction de cycloaddition.87
O O O O EtO2C CO2Et EtO2C CO2Et O O O Br O DBU Toluene O O O O CH2(CO2Et)2 DBU
Figure 44 : Synthèse régiosélective de multiadduits de fullerènes
L’utilisation de cette fonctionnalisation a permit de synthétiser des polyadduits portant différents fragments organiques. La macrocyclisation de Bingel du C60 avec un bis-malonate
contenant un fragment dibenzo[18]-couronne-6 conduit à un bis-adduit planaire-chiral trans-1 (Figure 45).88 C60, I2, DBU toluène, 20 ºC, 12h O O O O O O O O O EtO O O O O O O O O O OEt O O O O O O O O
Figure 45 : Synthèse d’un bis-adduit planaire-chiral trans-1.
Des fragments dendritiques ont été liés à la sphère de [60]fullerène par cette réaction régiosélective. Ces bis-adduits macrocycliques du C60 ont été synthétisés par une réaction de
C
CHHAAPPIITTRREE II 4411
cyclisation entre le C60 et les dérivés bis-malonate afin d’introduire des dendrimères de
différentes générations (Figure 46).89
O O O O O O O O R R O O O O O O OCH3 O O O OCH3 O O O OCH3 H3CO O O G2
Figure 46 : Systèmes dendritiques.
Le premier exemple de synthèse régio- et stéreosélective d’un bis-adduit trans-2 chiral de C60 a été récemment décrit. Elle est basée sur l’utilisation de la base de Tröger90 comme
auxiliaire chiral (Figure 47).91 Une réaction de Bingel modifiée entre un diester avec du C60 a
conduis au bisadduit trans-2 comme produit majoritaire (27%); une petite quantité (6%) de e-bisaddui a aussi été formé.
N N O O O OEt O O EtO O C60, I2, DBU toluène, 0 ºC, 1h O O O O O O O O N N
Les groupes malonate sont généralement connectés avec des systèmes organiques rigides via des espaceurs pour que les groupes réactifs soient dirigés vers une position spécifique. Néanmoins, Hirsch et collaborateurs ont étudiés la formation d’adduits bis- ou tris-fullerène en utilisant des macrocycles contenant des espaceurs alkyl entre 2 ou 3 groupes malonate réactifs (Figure 48).92 Les espaceurs rigides sont remplacés par des chaînes alkyles flexibles et les groupes malonate sont incorporés dans un macrocycle– ce type de structure permet de synthétiser des régioisomères du fullerène avec une excellente sélectivité.
C60, I2, DBU toluène O O O O (CH2)n (CH2)n O O O O n = 12 O O O O (CH2)n (CH2)n O O O O O O O O (CH2)n (CH2)n O O O O (CH2)n O O O O n = 14 C60, I2, DBU toluène O O O O (CH2)n (CH2)n O O O O (CH2)n O O O O
Figure 48 : Synthèse de régioisomères du fullerène
La formation régiosélective de tris-adduits est possible en adaptant correctement la structure. L’utilisation de systèmes tripodaux93 a permis d’obtenir un tris-adduit de [60]fullerène-symétrique avec un motif d’addition e,e,e (Figure 49).
C CHHAAPPIITTRREE II 4433 O OH HO HO O O O O O O O O O O O O O O C60, I2, DBU toluène O O O O O O O O O O + O O O O O O O O O O O O O O O O O O O O
Figure 49 : Formation de tris-adduits régiosélectifs
Une synthèse de fulleropyrrolidines utilisant la réaction de Prato via cette méthode a été décrite récemment par D’Souza et ses collaborateurs. Ils ont synthétisé des bis-adduits de Prato en utilisant un dibenzo-18-couronne-6 bisaldéhyde. Par cette approche ils ont obtenus un mélange de différents isomères qui ont été utilisés tel quel pour des études supplémentaires.94
Le premier exemple d’étude systématique de la synthèse de bis-adduits de C60
fulleropyrrolidines utilisant ce procédé a été décrit récemment. De nouveaux bis(benzaldéhydes) séparés par un espaceur rigide ont été préparés et utilisés pour diriger la seconde cyclo-addition d’un ylure d’azométhine sur le C60. Des bis-adduits équatoriaux
trans-4, trans-3, trans-2 et trans-1 ont ainsi été préparés sélectivement grâce à cette approche
(Figure 50).95 N R1 O N R2 O Espaceur
Des polyadduits ont aussi été synthétisés grâce à des doubles cycloadditions de Diels– Alder par le groupe de Rubin. Un mélange de bis(buta-1,3-diène) et de C60 a été chauffé sous
reflux pour donner un bisadduit trans-1 avec une grande quantité 30% (Figure 51).96
O O O O C60 toluène, reflux O O O O
Figure 51: Synthèse d’un bisadduit trans-1 par réactions de Diels–Alder
En utilisant un système différent, Nishimura et ses collaborateurs ont montré que la réaction du [60]fullerène avec 2 groupements α,α'-dibromo-o-xylène, connectés entre eux par une chaîne oligoméhylène (n = 2–5), conduis à 2 isomères (cis-2 et cis-3) lorsque n = 2 et 3 alors que lorsque n = 5 seul l’isomère (e) est obtenu (Figure 52).97
Br Br Br Br O O (CH2)5 C60, KI, 18-couronne-6 toluène, Δ O O (CH2)5
Figure 52: Préparation d’un isomère (e).
La régioselectivité des processus d’addition peut aussi être contrôlée en fixant temporairement des substituants sur le C60. Des adduits anthracène ont ainsi permis la
C
CHHAAPPIITTRREE II 4455
anthracène peuvent ensuite être retirés par chauffage.
En chauffant un mono-adduit de C60-anthracène en phase solide à 180° C pendant 10 minutes
un transfert régiosélectif de l’anthracène est obtenu pour former le cyclo-adduit portant 2 groupes anthracène aux pôles de la sphère du fullerène.29a La réaction de celui-ci, avec un excès de diéthyl bromomalonate et du DBU conduit à la formation d’un hexaadduit avec 95% de rendement. Le tétraadduit D2h symétrique est quant à lui obtenue avec 88% de rendement en chauffant l’hexa-adduit intermédiaire.
EtO2C CO2Et CO2Et CO2Et BrCH(CO2Et)2 DBU 2 2 Δ EtO2C EtO2C CO2Et CO2Et CO2Et EtO2C EtO2C CO 2Et
4. Objectifs
Dans le cadre de nos recherches nous nous sommes proposés d’étudier des dérivés de fullerènes. Dans une première partie de ce travail de thèse nous avons synthétisé toute une série de dérivés de (phénylpyrrolidino)fullerènes. Ensuite nous avons entrepris une analyse conformationnelle détaillée de ces composés en nous basant sur des études de RMN 1H à températures variables et des études de modélisation.
Dans la seconde partie de ce travail de thèse nous avons préparé des dérivés de C60
portant des sous unités pyridine. Ces composés ont été conçus pour qu’ils puissent s’assembler avec des récepteurs bis-Zn(II)-porphyrines pour former des complexes non-covalents macrocycliques stables.
C
CHHAAPPIITTRREE II 4477
5.
Références
1
La découverte des Fullerènes a valu à ses auteurs, H. Kroto, R. Curl et R. Smalley, l’attribution du Prix Nobel de Chimie en 1996.
2
H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl, R. E. Smalley, Nature, 1985, 318, 162.
3
W. Krätschmer, L. D. Lamb, K. Fostiropoulos, D. R. Huffman, Nature, 1990, 347, 354.
4
Cet extrait contient essentiellement du C60 (environ 60%) et du C70 (environ 20%) ; les
autres fullerènes (C76, C78, C82, C84, C90, C94 et C96) sont nettement moins abondants. 5
P. W. Fowler, A. Rassat, PHYSICAL CHEMISTRY CHEMICAL PHYSICS 2002, 4, 1105-1113 ; P. W. Fowler, Chem. Phys. Lett., 1986, 131, 444 ; P. W. Fowler and J. I. Steer, J. Chem. Soc., Chem. Commun., 1987, 1403 ; P. W. Fowler and C. M. Quinn, Theor. Chim. Acta, 1986, 70, 333 ; R. Taylor, J. P. Hare, A. K. Abdul-Sada and H. W. Kroto, J. Chem. Soc., Chem. Commun., 1990, 1423 ; P. W. Fowler and A. Ceulemans, J. Phys. Chem., 1995, 99, 508.
6
L. W. Tutt, A. Kost, Nature, 1992, 356, 225.
7
F. Negri, G. Orlandi, F. Zerbetto. J. Amer. Chem. Soc., 1992, 114, 8.
8
Q. Xie, E. Perez-Cordero, L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978.
9
A. Hirsch, M. Brettreich, in Fullerenes: Chemistry and Reactions, Ed. John Wiley & Sons, 2005..
10
M. A. Yurovskaya, I. V. Trushkov, Russ. Chem. Bull., Int. Ed., 2002, 51, 367.
11
T. Suzuki, Q. Li, C. Khemani, F. Wudl, Ö. Almarsson, Science, 1991, 254, 1186
12
F. Diederich, L.I saacs, D. Philp, Chem. Soc. Rev. 1994, 243.
13
J. F. Nierengarten, “Synthesis of Methanofullerenes for Materials Science and Biological Applications’’, chapter 4 in Fullerenes: From Synthesis to Optoelectronic Properties, Kluwer Academic Publishers, 2002, p. 51.
14
C. Bingel, Chem. Ber., 1993, 126, 1957.
15
. [a] J.-F. Nierengarten, V. Gramlich, F. Cardullo, and F. Diederich, Angew. Chem. Int. Ed.
Engl., 1996, 35, 2101; [b] J.-F. Nierengarten, and J.-F. Nicoud, Tetrahedron Lett., 1997, 38,
16
. X. Camps, and A. Hirsch, J. Chem. Soc., Perkin Trans 1, 1997, 11, 1595.
17
. S. Taillemite, D. Fichou, Eur. J. Org. Chem., 2004, 4981.
18
. D. Bonifazi, G. Accorsi, N. Aramaroli, F. Song, A. Palkar, L. Echegoyen, M. Scholl, P. Seiler, B. Jaun, F. Diederich, Helv. Chim. Acta, 2005, 88, 1839.
19
. Y. Nakamura, S. Minami, K. Iizuka, J. Nishimura, Angew. Chem. Int. Ed. Engl., 2003, 42, 3158.
20
. M. A. Herranz, F. Diederich, L. Echegoyen, Eur. J. Org. Chem., 2004, 2299.
21
. R. Kessinger, J. Crassous, A. Hermann, M. Rüttimann, L. Echegoyen, F. Diederich,
Angew. Chem. Int. Ed. Engl., 1998, 37, 1919.
22
. [a] R. Kessinger, M. Gómez-López, C. Boudon, J.-P. Gisselbrecht, M. Gross, L. Echegoyen, F. Diederich, J. Am. Chem. Soc., 1998, 120, 8545; [b] L. E. Echegoyen, F. D. Djojo, A. Hirsch, L. Echegoyen, J. Org. Chem., 2000, 65, 4994.
23
. N. N. P. Moonen. C. Thilgen, L. Echegoyen, F. Diederich, Chem. Commun., 2000, 335.
24
. M. A. Herranz, L. Echegoyen, M. W. J. Beulen, J. A. Rivera, N. Martín, B. Illescas, M. C. Díaz, Proc.- Electrochem. Soc., 2002, vol. 12, 307.
25
. R. Kessinger, N. S. Fender, L. E. Echegoyen, C. Thilgen, L. Echegoyen, F. Diedierich,
Chem. Eur. J., 2000, 6, 2184.
26
. F. Langa, P. de la Cruz, A. de la Hoz, A. Díaz-Ortiz, E. Diez-Barra, Contemp. Org. Synth., 1997, 373.
27
. [a] V. M. Rotello, J. B. Howard, T. Yadav, M. M. Conn, E. Viani, L. M. Giovane, A. L. Lafleur, Tetrahedron Lett., 1993, 34, 1561; [b] M. Tsuda, T. Ishida, T. Nogami, S. Kurono, M. Ohashi, J. Chem. Soc., Chem. Commun., 1993, 1296.
28
. [a] M. F. Meidine, A. G. Avent, A. D. Darwish, H. W. Kroto, O. Ohashi, R. Taylor, D. R. M. Walton, J. Chem. Soc., Perkin Trans. 2, 1994, 1189; [b] H. Takeshita, J.-F. Liu, N. Kato, A. Mori, R. Isobe, Chem. Lett., 1995, 377; [c] S. R. Wilson, M. E. Yurchenko, D. I. Schuster, A. Khong, M. Saunders, J. Org. Chem., 2000, 65, 2619.
29
. [a] J. A. Schlüter, J. M. Seaman, S. Taha, H. Cohen, K. R. Lykke, H. H. Wang, J. M. Williams, J. Chem. Soc., Chem. Commun., 1993, 972; [b] K. Komatsu, Y, Murata, N. Sugita, K. Takeuchi, T. S. M. Wan, Tetrahedron Lett., 1993, 34, 8473.
C
CHHAAPPIITTRREE II 4499
30
. [a] R. Kraütler, T. Müller, A. Duarte-Ruiz, Chem. Eur. J., 2001, 2, 3223; [b] N. Chronakis, M. Organopoulus, Tetrahedron Lett., 2001, 42, 1201; [c] I. Lamparth, C. Maichle-Mössmer, A. Hirsch, Angew. Chem. Int. Ed. Engl., 1995, 34, 1607.
31
. Y. Takaguchi, T. Tajima, K. Ohta, J. Motoyoshiya, H. Aoyama, T. Wakahara, T. Akasaka, Angew. Chem. Int. Ed. Engl., 2002, 41, 817; [b] Y. Takaguchi, Y. Sako, Y. Yanagimoto, S. Tsunoi, J. Motoyoshiya, H. Aoyama, T. Wakahara, T. Akasaka, Tetrahedron
Lett., 2003, 44, 5777.
32
. P. de la Cruz, A. de la Hoz, F. Langa, B. Illescas, N. Martín, Tetrahedron Lett., 1997, 38, 2599.
33
. Y. Murata, N. Kato, K. Fujiwara, K. Komatsu, J. Org. Chem., 1999, 64, 3483.
34
. K. Mikami, S. Mtsumoto, T. Tonoi, Y. Okubo, T. Suenobu, S. Fukuzumi, Tetrahedron
Lett., 1998, 39, 3733.
35
. [a] B. Kraütler, M. Puchberger, Helv. Chim. Acta, 1993, 76, 1626; [b] M. Ohno, Y. Shirakawa, S. Eguchi, Synthesis, 1998, 1812.
36
. M. Diekers, A. Hirsch, S. Pyo, J. Rivera, L. Echegoyen, Eur. J. Org. Chem., 1998, 1111.
37
. J. L. Segura, N. Martín, Chem. Rev., 1999, 99, 3199.
38
. [a] P. Belik, A. Gügel, J. Spickerman, K. Müllen, Angew. Chem., Int. Ed. Engl., 1993, 32, 78; [b] Y. Nakamura, T. Minowa, S. Tobita, H. Shizuka, J. Nishimura, J. Chem. Soc., Perkin
Trans. 2, 1995, 2351; [c] M. Taki, S. Sugita, Y. Nakamura, E. Kasashima, E. Yashima, Y.
Okamoto, J. Nishimura, J. Am. Chem. Soc., 1997, 119, 926.
39
. [a] A. Kraus, A. Gügel, P. Belik, M. Walter, K. Müllen, Tetrahedron, 1995, 51, 9927; [b] M. Iyoda, F. Sultana, S. Sasaki, M. Yoshida, J. Chem. Soc., Chem. Commun., 1994, 1929; [c] M. Iyoda, S. Sasaki, F. Sultana, M. Yoshida, Y. Kuwatani, S. Nagase, Tetrahedron Lett., 1996, 37, 7987; [d] H. Tomioka, K. Yamamoto, J. Chem. Soc., Chem. Commun., 1995, 1961.
40
. F. Effenberg, G. Grube, Synthesis, 1998, 1372.
41
. [a] B. M. Illescas, N. Martín, C. Seoane, E. Ortí, P. M. Viruela, R. Viruela, A. de la Hoz,
J. Org. Chem., 1997, 62, 7585; [b] J.-H. Liu, A.-T. Wu, M.-H. Huang, C.-W. Wu, W.-S.
Chung, J. Org. Chem., 2000, 65, 3395.
42
. [a] U. M. Fernandez-Paniagua. B. Illescas, N. Martín. C. Seoane, P. de la Cruz, A. de la Hoz, F. Langa, J. Org. Chem., 1997, 62, 3705; [b] M. Ohno, N. Koide, H. Sato, S. Eguchi,

![Figure 22 : Synthèse d'une triade porphyrin-[60]fullerène-porphyrine](https://thumb-eu.123doks.com/thumbv2/123doknet/2177427.10353/31.892.123.789.570.1005/figure-synthese-d-une-triade-porphyrin-fullerene-porphyrine.webp)
![Figure 24 : Utilisation de micro-ondes dans les réactions de cycloaddition [4+2] de o- o-quinodiméthanes sur du C 60](https://thumb-eu.123doks.com/thumbv2/123doknet/2177427.10353/32.892.135.705.754.880/figure-utilisation-micro-ondes-reactions-cycloaddition-quinodimethanes.webp)