HAL Id: dumas-01113833
https://dumas.ccsd.cnrs.fr/dumas-01113833
Submitted on 6 Feb 2015
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Particularités des essais cliniques portant sur les
biosimilaires
Maud Reynier
To cite this version:
Maud Reynier. Particularités des essais cliniques portant sur les biosimilaires. Sciences pharmaceu-tiques. 2014. �dumas-01113833�
AVERTISSEMENT
Ce document est le fruit d'un long travail approuvé par le
jury de soutenance et mis à disposition de l'ensemble de la
communauté universitaire élargie.
Il n’a pas été réévalué depuis la date de soutenance.
Il est soumis à la propriété intellectuelle de l'auteur. Ceci
implique une obligation de citation et de référencement
lors de l’utilisation de ce document.
D’autre part, toute contrefaçon, plagiat, reproduction illicite
encourt une poursuite pénale.
Contact au SICD1 de Grenoble :
[email protected]
LIENS
LIENS
Code de la Propriété Intellectuelle. articles L 122. 4
Code de la Propriété Intellectuelle. articles L 335.2- L 335.10
http://www.cfcopies.com/V2/leg/leg_droi.php
UNIVERSITEJOSEPHFOURIER
FACULTEDEPHARMACIEDEGRENOBLE
Année:2014 N°
P
ARTICULARITES DES ESSAIS CLINIQUES PORTANT
SUR LES
B
IOSIMILAIRES
THESE
PRESENTEEPOURL’OBTENTIONDUTITREDEDOCTEURENPHARMACIE
DIPLÔMED’ETAT
Melle REYNIER Maud
Née le 24 janvier 1988, à GRENOBLE (38000)
THESESOUTENUEPUBLIQUEMENTALAFACULTEDEPHARMACIEDEGRENOBLE*
Le 31 Octobre 2014
DEVANT LE JURY COMPOSE DE : Président du jury :
M. le Professeur Jean-Luc LENORMAND
Membres :
Mr le Docteur Yves DONAZZOLO Mme le Docteur Camille DUCKI Mme le Docteur Edith SCHIR
La Faculté de Pharmacie de Grenoble n’entend donner aucune approbation ni improbation aux opinions émises dans les thèses ; ces opinions sont considérées comme propres à leurs auteurs.
Page 2
Doyen de la Faculté : M. le Pr. Christophe RIBUOT Vice-doyen et Directeur des Etudes : Mme Delphine ALDEBERT
Année 2013-2014
ENSEIGNANTS A L’UFR DE PHARMACIE PROFESSEURS DES UNIVERSITES (n=12)
BAKRI Aziz Pharmacie Galénique et Industrielle, Formulation et Procédés Pharmaceutiques (TIMC-IMAG)
BOUMENDJEL Ahcène Chimie Organique (D.P.M.)
BURMEISTER Wim Biophysique (U.V.H.C.I)
DECOUT Jean-Luc Chimie Inorganique (D.P.M.)
DROUET Christian Immunologie Médicale (TIMC-IMAG)
DROUET Emmanuel Microbiologie (U.V.H.C.I)
-GODIN-RIBUOT Diane Physiologie-Pharmacologie (HP2)
LENORMAND Jean Luc Ingénierie Cellulaire, Biothérapies (THEREX, TIMC, IMAG)
MARTIN Donald Laboratoire TIMC-IMAG (UMR 5525 UJF-CNRS)
PEYRIN Eric Chimie Analytique (D.P.M.)
RIBUOT Christophe Physiologie – Pharmacologie (HP2)
WOUESSIDJEWE Denis Pharmacotechnie (D.P.M.)
PROFESSEURS DES UNIVERSITES-PRATICIEN HOSPITALIER (n=6)
ALLENET Benoit Pharmacie Clinique (THEMAS
TIMC-IMAG/MCU-PH)
CORNET Murielle Parasitologie – Mycologie Médicale (LAPM, PU-PH)
DANEL Vincent Toxicologie (SMUR SAMU / PU-PH)
FAURE Patrice Biochimie (HP2/PU-PH)
MOSSUZ Pascal Hématologie (PU-PH-THEREX-TIMC)
SEVE Michel Biochimie – Biotechnologie (IAB, PU-PH)
PROFESSEURS EMERITES (n=3)
CALOP Jean Pharmacie Clinique (TIMC-IMAG, PU-PH)
GRILLOT Renée Parasitologie – Mycologie Médicale (L.A.P.M)
ROUSSEL Anne-Marie Biochimie Nutrition (L.B.F.A)
Page 3
MAITRES DE CONFERENCES DES UNIVERSITES (n=32)
ALDEBERT Delphine Parasitologie-Mycologie (L.A.P.M)
BATANDIER Cécile Nutrition et Physiologie (L.B.F.A)
BELAIDI-CORSAT Elise Pharmacologie Physiologie –(HP2)
BOURGOIN Sandrine Biochimie – Biotechnologie (IAB)
BRETON Jean Biologie Moléculaire / Biochimie (L.C.I.B – LAN)
BRIANCON-MARJOLLET Anne Physiologie Pharmacologie (HP2)
BUDAYOVA SPANO Monika Biophysique (I.B.S)
CAVAILLES Pierre Biologie Cellulaire et génétique (L.A.P.M)
CHOISNARD Luc Pharmacotechnie (D.P.M)
DELETRAZ-DELPORTE Martine Droit Pharmaceutique
(Equipe SIS « Santé, Individu, Société »-EAM 4128)
DEMEILLIERS Christine Biochimie (L.B.F.A)
DURMORT-MEUNIER Claire Biotechnologies (I.B.S)
GEZE Annabelle Pharmacotechnie (D.P.M)
GILLY Catherine Chimie Thérapeutique (D.P.M)
GROSSET Catherine Chimie Analytique (D.P.M)
GUIEU Valérie Chimie Analytique (D.P.M)
HININGER-FAVIER Isabelle Biochimie (L.B.F.A)
JOYEUX-FAURE Marie Physiologie - Pharmacologie (HP2)
KHALEF Nawel Pharmacie Galénique (TIMC-IMAG)
KRIVOBOK Serge Biologie Végétale et Botanique (L.C.B.M)
MELO DE LIMA Christelle Biostatistiques (L.E.C.A)
MOUHAMADOU Bello Cryptogamie, Mycologie Générale (L.E.C.A)
NICOLLE Edwige Chimie Thérapeutique (D.P.M)
OUKACINE Farid Chimie Thérapeutique (D.P.M)
PERES Basile Pharmacognosie (D.P.M)
PEUCHMAUR Marine Chimie Organique (D.P.M.)
RACHIDI Walid Biochimie (L.C.I.B)
RAVEL Anne Chimie Analytique (D.P.M)
RAVELET Corinne Chimie Analytique (D.P.M)
SOUARD Florence Pharmacognosie (D.P.M)
TARBOURIECH Nicolas Biophysique (U.V.H.C.I.)
Page 4
MAITRE DE CONFERENCE DES UNIVERSITES-PRATICIEN HOSPITALIER (n=3)
BEDOUCH Pierrick Pharmacie Clinique (THEMAS TIMC-IMAG/MCU-PH)
BUSSER Benoit Pharmacie (MCU-PH-IAB-INSERM)
GERMI Raphaëlle Microbiologie (U.V.H.C.I/MCU-PH)
PROFESSEUR CERTIFIE (PRCE) (n=2)
FITE Andrée P.R.C.E
GOUBIER Laurence P.R.C.E
PROFESSEURS ASSOCIES (PAST) (n=4)
BELLET Béatrice Pharmacie Clinique
RIEU Isabelle Qualitologie (Praticien Attaché – CHU) TROUILLER Patrice Santé Publique (Praticien Hospitalier – CHU)
PROFESSEUR AGREGE (PRAG) (n=1)
GAUCHARD Pierre-Alexis (D.P.M)
ASSISTANTS HOSPITALO-UNIVERSITAIRES (AHU) (n=3)
CHANOINE Sébastien Pharmacie Clinique (UF-CHU)
GARNAUD Cécile Parasitologie-Mycologie
VAN NOLLEN Laetitia Biochimie Toxicologie (HP2-DNTP-BGM)
MEDAILLE D’OR D’ANNE D’INTERNAT SUPPLEMENTAIRE (n=2)
BERNARD Delphine période de 6 mois – novembre 2013 à avril 2014 GAUTIER Elodie période de 6 mois – mai 2014 à novembre 2014
Page 5 ATER (n= 3)
BRAULT Julie ATER Pharmacologie - Laboratoire HP2 (JR)
GRAS Emmanuelle ATER Physiologie-Pharmacologie - Laboratoire HP2 (JR)
LEHMANN Sylvia ATER Biochimie Biotechnologie (JR)
MONITEUR ET DOCTORANTS CONTRACTUELS
BEL Coraline (01-10-2012 au 30-09-2014) BERTHOIN Lionel (01-10-2012 au 30-09-2014) Laboratoire (TIMC-IMAG-THEREX) BOSSON Anthony (01-10-2013 au 30-09-2015) Laboratoire GIN CAVAREC Fanny (01-10-2011 au 30-09-2014) Laboratoire HP2 (JR) CHRISTEN Aude (01-10-2013 au 30-09-2015) DCM CRESPO Xenia (01-10-2013 au 30-09-2015) LBGE LECERF-SHMIDT Florine (01-10-2012 au 30-09-2014) Pharmacochimie (DPM) LESART Anne-Cécile (01-10-2009 au 30-09-2013) Laboratoire (TIMC-IMAG) MELAINE Feriel (01-11-2011 au 31/10.2014) Laboratoire HP2(JR) MORAND Jessica (01-10-2012 au 30-09-2014) Laboratoire HP2 (JR) NASRALLAH Chady (01-10-2011 au 30-09.2013) Laboratoire HP2(JR) OUIDIR Marion (01-10-2011 au 30-09-2014) THOMAS Amandine (01-10-2011 au 30-09-2014) Laboratoire HP2 (JR) Professeur Invité NURISSO Alessandra (01/11/13 au 31/12/2013))
CHU : Centre Hospitalier Universitaire CIB : Centre d’Innovation en Biologie
DPM : Département de Pharmacochimie Moléculaire
HP2 : Hypoxie Physiopathologie Respiratoire et Cardiovasculaire
IAB : Institut Albert Bonniot, Centre de Recherche « Oncogenèse et Ontogenèse » IBS : Institut de Biologie Structurale
LAPM : Laboratoire Adaptation et Pathogenèse des Microorganismes LBFA : Laboratoire Bioénergétique Fondamentale et Appliquée LCBM : Laboratoire Chimie et Biologie des Métaux
LCIB : Laboratoire de Chimie Inorganique et Biologie LECA : Laboratoire d’Ecologie Alpine
LR : Laboratoire des Radio pharmaceutiques
TIMC-IMAG : Laboratoire Technique de l’Imagerie, de la Modélisation et de Cognition UVHCI : Unit of Virus Host Cell Interactions
Page 6
P
ARTICULARITES DES ESSAIS CLINIQUES
PORTANT SUR LES
B
IOSIMILAIRES
Page 7
REMERCIEMENTS
A Monsieur Jean-Luc Lenormand,
Pour me faire l’honneur de présider ce jury. Merci pour l’intérêt porté à ce travail.
A Madame Camille Ducki,
Pour avoir pris la direction de cette thèse. Merci pour la confiance accordée, pour m’avoir suivie tout au long de ce travail, pour m’avoir aidée et conseillée dans mes démarches.
Merci également pour sa sympathie et sa pédagogie qui rendent le travail quotidien à ses côtés si agréable.
A Monsieur Yves Donazzolo,
Pour m’avoir fait l’honneur de participer à ce travail et de prendre part au jury. Je le remercie pour le temps qu’il m’a accordé, ainsi que pour la grande justesse et la pertinence de ses corrections.
A Madame Edith Schir
Page 8
A ma famille,
Je présente ma plus grande reconnaissance et mes remerciements sincères à toute ma famille pour m’avoir soutenue tout au long de mes études.
Tout particulièrement, je remercie mes parents pour m’avoir encouragée dans cette voie professionnelle. Leurs conseils et leur soutien m’ont été précieux. Je les remercie également pour avoir rendu possible et de m’avoir suivi au cours de mes différents périples français et étrangers !
Merci à mes sœurs, sans qui, ces années d’études n’auraient pas été les mêmes ! Merci pour leur écoute, les fous rires, les soirées … Merci d’être, avant tout, de véritables amies !
Merci à mes grands parents pour leur présence, leur tendresse et leur affection depuis mes premiers pas !
A Guillaume,
Pour sa patience et son soutien sans faille. Et pour avoir rendu ces six années d’étude à ses côtés si particulières et inoubliables!
A mes amis,
Les Grenoblois, pour tous les bons moments partagés et les futurs à venir !
Avec un clin d’œil à notre petit groupe pharma formé en P2, avec lequel j’ai passé toutes ces années et vécu des milliers de choses qui resteront marquées : on s’amusera bien quand on se remémorera nos péripéties étudiantes dans quelques années!
Et tous ceux qui me font voyager et qui, malgré la distance, sont toujours là !
Tout spécialement : Charlotte pour nos rendez-vous sportifs et nos voyages et mes trois Montpelliéraines, Belges, Parisiennes (enfin on ne sait plus vraiment…) pour nos mémorables brunchs et week-end découverte…!
A mes collègues,
Merci à l’ensemble du personnel d’Eurofins-Optimed pour son accueil, son aide lors de mes débuts dans cette nouvelle équipe.
Merci à Anne et Laurène, mes voisines de bureau, pour partager leur expérience avec moi mais surtout pour leur gentillesse et leur bonne humeur !
Page 9
TABLE DES MATIERES
REMERCIEMENTS ... 7
ABREVIATIONS ... 13
LISTE DES FIGURES ... 15
LISTE DES TABLEAUX ... 17
INTRODUCTION ... 18
CHAPITRE 1 BIOTECHNOLOGIE ET MEDICAMENTS ... 20
1. HISTOIRE DE LA BIOTECHNOLOGIE APPLIQUEE AUX MEDICAMENTS ... 21
2. PRINCIPALES DIFFERENCES ENTRE LES MEDICAMENTS TRADITIONNELS ET LES BIOMEDICAMENTS. ... 23
3. CLASSIFICATION DES PRODUITS ISSUS DES BIOTECHNOLOGIES : LE POINT DE VUE REGLEMENTAIRE ... 27
3.1 LES MEDICAMENTS IMMUNOLOGIQUES ... 28
3.2 LES MEDICAMENTS DERIVES DU SANG ET DU PLASMA HUMAIN ... 29
3.3 LES MEDICAMENTS ISSUS DE PROCEDES BIOTECHNOLOGIQUES PARTICULIERS ... 29
3.3.1 Généralités ... 30
3.3.2 L’ADN recombinant ... 31
3.3.3 L’expression contrôlée de gènes ... 33
3.3.4 Les méthodes à base d’hybridome... 34
3.3.5 Complexité de la fabrication ... 37
3.4 LES MEDICAMENTS DE THERAPIE INNOVANTE... 39
3.4.1 Les médicaments de "thérapie génique" ... 39
3.4.2 Les médicaments de "thérapie cellulaire somatique" ... 41
3.4.3 Les médicaments "issus de l’ingénierie cellulaire ou tissulaire" ... 41
3.4.4 Les médicaments " de thérapie innovante combinées "... 42
4. EXEMPLE DE BIOMEDICAMENTS ... 43
CHAPITRE 2 : PROBLEMATIQUE, REGLEMENTATION ET ESSAIS CLINIQUES PORTANT SUR DES BIOMEDICAMENTS ... 45
Page 10
2. CONCEPTION D’UN ESSAI CLINIQUE PORTANT SUR LES BIOMEDICAMENT 49
2.1 L’IMMUNOGENICITE ... 49
2.1.1 Expression des capacités immunogènes des biomédicaments ... 49
2.1.2 Recommandations pour diminuer les risques liés à l’administration de biomédicaments ... 54
2.1.2.2 Détection des ADAs ... 56
2.1.2.3 Choix de la population ... 58
2.2 RELATION PHARMACOCINETIQUE/PHARMACODYNAMIQUE ... 58
2.2.1 Connaissance PK/PD des biomédicaments ... 58
2.2.2 Recommandation pour l’évaluation ADME ... 62
2.3 CHOIX DES DOSES POUR LES PREMIERES ADMINISTRATIONS A L’HOMME ... 63
2.3.1 Problématique du calcul habituel de la dose ... 63
2.3.2 Recommandations pour les biomédicaments ... 63
2.4 AUTRES RECOMMANDATIONS ... 65
2.4.1 Interactions médicamenteuses ... 65
2.4.2 Etude QT ... 65
CHAPITRE 3 : BIOSIMILAIRES ... 67
1. CONTEXTE ... 68
2. NAISSANCE DU CONCEPT DE BIOSIMILAIRES ... 69
3. DÉFINITION DES BIOSIMILAIRES ... 72
4. DIFFÉRENCES AVEC LES GÉNÉRIQUES ... 73
5. ENJEUX ... 75
5.1 MARCHE DES BIOMEDICAMENTS ... 75
5.1.1 Dans le monde ... 75
5.1.2 En France ... 77
5.2 MARCHE DES BIOSIMILAIRES ... 79
CHAPITRE 4: ESSAIS CLINIQUES, BIOSIMILAIRES ET REGLEMENTATION .. ... 83
1. EXIGENCES GENERALES DE LA REGLEMENTATION EUROPEENNE ... 84
1.1 CHOIX DU BIOMEDICAMENT REFERENT ... 87
1.2 APPROCHE PAR ETAPE ... 87
1.2.1 Comparabilité au niveau de la qualité ... 88
1.2.2 Comparabilité Pré Clinique ... 91
1.2.3 Comparabilité Clinique ... 92
1.2.3.1 Etude clinique PK/PD ... 92
1.2.3.2 Etude clinique d’efficacité ... 93
Page 11
1.4 EXTRAPOLATION D’INDICATION ... 99
1.5 NOM ET SUBSTITUTION ... 100
1.6 PHARMACOVIGILANCE ... 100
1.7 EXIGENCES SPECIFIQUES PAR CLASSE DE MOLECULE ... 101
1.8 LE POINT DE VUE DES OPPOSANTS AUX EXIGENCES EUROPEENNES SUR LES BIOSIMILAIRES ... 105
2. COMPARAISON DES REGLEMENTATIONS EUROPEENNES AUX REGLEMENTATIONS INTERNATIONALES... 107
2.1 USA ... 107
2.2 WORLD HEALTH ORGANIZATION ... 109
2.3 AUTRES PAYS ... 110
3. PLAN DE DEVELOPPEMENT ET EXPERIENCES DE BIOSIMILAIRES PRESENTES A L’EMA ... 116 3.1 ERYTHROPOÏETINE ... 116 3.1.1 HX575 ... 118 3.1.2 SB 309 ... 119 3.2 G-CSF ... 120 3.2.1 ZARZIO® ... 121 3.2.2 XM02 ... 122 3.2.3 NIVESTIM® ... 123 3.2.4 Discussions ... 123 3.3 SOMATROPINE ... 125 3.4 INSULINE ... 126
3.5 HEPARINES DE BAS POIDS MOLECULAIRES ... 127
3.6 HORMONE FOLLICULO-STIMULANTE ... 132
3.7 LES INTERFERONS ... 133 3.7.1 Alpha ... 133 3.7.2 Le cas de l’ALPHEON® ... 134 3.7.3 Beta ... 135 3.7.4 Gamma ... 135 3.8 LES ANTICORPS MONOCLONAUX ... 136 3.8.1 INFLECTRA®/REMSIMA®... 138 3.8.2 TRASTUZUMAB ... 139 3.8.3 Discussion ... 140
Page 12
CHAPITRE 5 : RECOMMANDATIONS POUR LA MISE EN ŒUVRE D’ESSAIS
CLINIQUES PORTANT SUR LES BIO-SIMILAIRES EN EUROPE ... 141
1. LA PLACE DU BIOSIMILAIRE DANS SON ENVIRONNEMENT CONCURRENTIEL/DE DEVELOPPEMENT ... 144
2. L’EXERCICE DE COMPARABILITE : LE DOSSIER QUALITE ET PRE-CLINIQUE COMME FONDATION ... 146
3. PLAN DE DEVELOPPEMENT, DE GESTION DES RISQUES ET PHARMACOVIGILANCE : A PREPARER EN AMONT DU DEVELOPPEMENT CLINIQUE ... 147
4. L’AVIS CONSULTATIF DES AUTORITES COMPETENTES : UN SUPPORT NECESSAIRE ... 148
5. LA PREMIERE ETUDE: DEMONTRER LA BIOEQUIVALENCE ... 149
6. LE ROLE DES ETUDES PHARMACODYNAMIQUES : TRES « CLASSE DEPENDANTES » ... 152
7. DERNIERE ETAPE DE L’EXERCICE : LA DEMONSTRATION DE L’EFFICACITE ... 154
8. L’IMMUNOGENICITE : UNE EVALUATION PARALLELE ... 157
CONCLUSION ... 159
ANNEXE ... 161
Page 13
Abréviations
Acs : Anticorps
ADAs : Anti-Drugs Antibodies
ADME : Absorption, Distribution, Métabolisation, Elimination
ADN : Acide Désoxyribonucléique
AMM : Autorisation de Mise sur le Marché
ANSM : Agence Nationale de Sécurité du Médicament
ARN : Acide Ribonucléique
ATIMP : Advanced Therapy Medicinal Product
AUC : Aire Sous la Courbe
BPF : Bonnes Pratiques de Fabrication BPL : Bonnes Pratiques de Laboratoire
CE : Commission Européenne
CHMP : Committee for Medicinal Products for Human Use
Cmax : Concentration Maximale
CMC : Chemistry Manufacturing Control
CMH : Complexe Majeur d’Histocompatibilité
CTD : Common Technical Document
DCI : Dénomination Commune Internationale
De50 : Dose Efficace Médiane
DT : Diabétique
DVP : Deep Vein Thrombosis
EMA : European Medicines Agency, Agence Européenne du Médicament
EPAR : European publie Assessment Report
EPO : Érythropoïétine
FDA : Food and Drug Administration
FIM : First-In-Man
FIV : Fécondation in Vitro
FSH : Follicle-stimulating Hormone
G-CSF : Growth Colony Stimulating Factor
GnRH : Gonado Tropin Releasing Hormon
Hb : Hémoglobine
HBPM : Héparines de Bas Poids Moléculaire
HED : Human Equivalent Dose
HER : Human Epidermal Growth Factor Receptor
HLA : Human Leukocyte Antigen
hGH : Human Groxth Hormon
IA : Interim Analysis
ICH : International Conference of Harmonization
Ig : Immunoglobulines
INF : Interférons
IM : Intramusculaire
IR : Insuffisant Rénaux
ITT : Intent To Treat
IV : Intraveineuse
LB : Lymphocyte B
Page 14
mAbs : Anticorps monoclonaux
MABEL : Minimum Anticipated Biological Effect Level
MCB : Master Cells Bank
MCID : Minimally Clinically Important Difference
MRSD : Maximum Recommended Starting Dose
MTI : Médicaments de Thérapie Innovante
NI : Non-Infériorité
NOAEL : No Observable Adverse Effect Level
OGM : Organisme Génétiquement Modifié
PAD : Pharmacologically Active Dose
PASS : Post Authorization Safety Study PAES : Post Authorization Efficacy Study PBPK : Physiologically-based pharmacokinetic
PD : Pharmacodynamique
PGR : Plan de Gestion des Risques
PK : Pharmacocinétique
PP : Per Protocol
PPV : Plan de Pharmacovigilance
PRCA : Pure Red Cell Aplasia
RCP : Résumé des Caractéristiques Produits
rEPO Recombinant Érythropoïétine
rFSH : Recombinant Follicle-stimulating Hormone
rGnRH Recombinant Gonado Tropin Releasing Hormon
rG-CSF : Recombinant Growth Colony Stimulating Factor
rhGH : Recombinant Human Groxth Hormon
rTNF : Recombinant Tumor Necrosis Factor
SC : Sous-Cutanée
TNF : Tumor Necrosis Factor
T1/2 : Temps de demi-vie d’élimination tmax : Temps nécessaire pour atteindre la Cmax
UI : Unité Internationale
Vd : Volume de distribution
Page 15
Liste des figures
Figure 1 : Différences structurales et moléculaires entre une molécule chimique
(Paracétamol) et une molécule biologique (Filgrastrim) ... 23
Figure 2 : Structure Spatiale des protéines ... 24
Figure 3 : Modification post-traductionnelle d’une protéine et conséquence fonctionnelle. ... 25
Figure 4 : Illustration de la micro hétérogénéité des biomédicaments ... 25
Figure 5: Hétérogénéité et composés présents dans le mélange d'actif ... 26
Figure 6 : Structure de l'ADN ... 30
Figure 7 : Etape de synthèse d'une protéine ... 30
Figure 8 : Technologie de l'ADN recombinant ... 31
Figure 9 : Classification des produits biologiques ... 32
Figure 10 : Illustration de la modulation de l’expression des gènes ... 33
Figure 11 : Structure d’un anticorps ... 34
Figure 12 : Procédé d’obtention d’Acs monoclonaux ... 36
Figure 13: Etape de synthèse d'un biomédicament ... 38
Figure 14 : Immunogénicité observée chez des patients traités par différentes formulations d'Interférons Alpha ... 53
Figure 15: Exemple de stratégie pour la détection et la caractérisation des ACs présentée dans les lignes directrices européennes ... 57
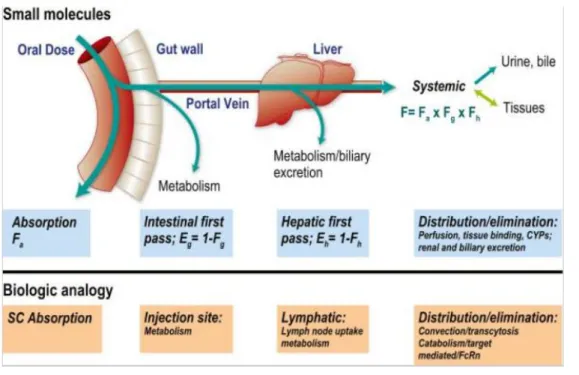
Figure 16 : Schéma représentant la PK/PD d'un médicament ... 58
Figure 17 : Analogie entre les mécanismes ADME pour les molécules chimiques et les biomédicaments ... 59
Figure 18 : Représentation de la relation Dose/Réponse ... 64
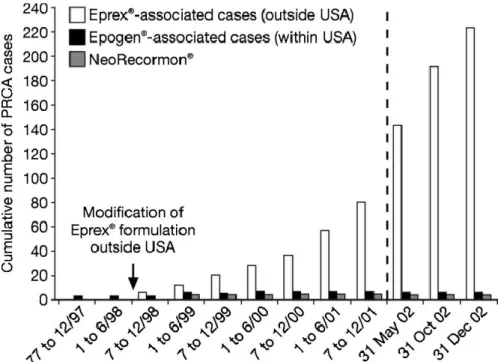
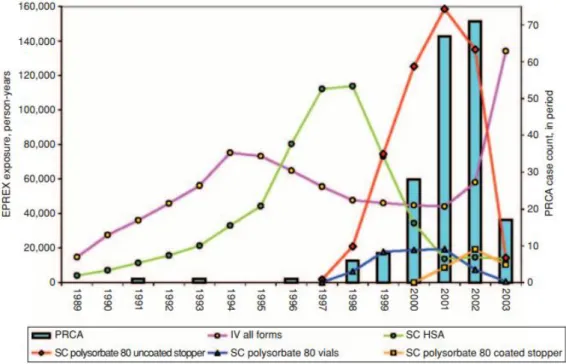
Figure 19 : Illustration du nombre de cas de PRCA de 1989 à 2007 ... 69
Figure 20 : Différence de l'incidence du nombre de cas selon les pays ... 70
Figure 21 : Corrélation entre les cas de PRCA et les différences apportées au produit ... 71
Figure 22 : Production de biomédicament : sources de variabilité entre les fabricants ... 72
Figure 23 : Illustration des données nécessaires pour un générique versus un biosimilaire lors d’une soumission aux autorités... 75
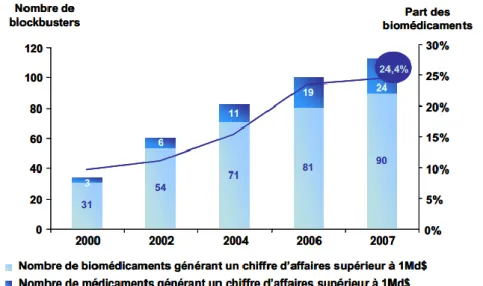
Figure 24 : Croissance du marché mondial des biomédicaments ... 76
Figure 25 : Médicaments et biomédicaments réalisant des ventes supérieures à 1 million de dollars ... 76
Figure 26: Evolution du nombre de biomédicament mis sur le marché en France au 31 Mai 2014 ... 77
Figure 27 : Classification par aires thérapeutiques des 173 biomédicaments commercialisés en France au 31 Mai 2014 ... 78
Page 16
Figure 28 : Classification pharmacologique des 173 biomédicaments commercialisés en
France (au 31 Mai 2014) ... 78
Figure 29: Prix moyen fabricant hors taxes (PFHT) et prix moyen public taxes comprise(PPUB) ... 79
Figure 30 : Coût du développement et réduction du prix de vente du médicament ... 80
Figure 31 : Volume des ventes du biosimilaire Filgastrim en Angleterre (pourcentage de changement par rapport à l'année précédente) ... 81
Figure 32 : Histogramme montrant l'évolution des quantités de biosimilaires vendus en France entre 2008 et 2012 ... 82
Figure 33 : Recommandations Européennes portant sur les biosimilaires ... 86
Figure 34 : Procédure de démonstration de la comparabilité/similarité ... 87
Figure 35 : Principales Etapes de l'exercice de comparabilité ... 88
Figure 36 : Différence entre l’exercice de comparabilité (changement d’un processus de fabrication) et l’exercice de similarité (biosimilaire) ... 90
Figure 37 : Résumé des caractéristiques d'un essai d'équivalence ... 94
Figure 38 : Résumé des caractéristiques d'un essai de non-infériorité ... 95
Figure 39:Structure du CTD ... 98
Figure 40 : Résumé des différences du dossier d’enregistrement entre un biosimilaire et un biomédicament ... 99
Figure 41 : Bénéfices et risques dans les procédures d’avis... 106
Figure 42 : Vue d’ensemble au niveau international des réglementations appliquées ... 110
Figure 43 : Etudes cliniques recommandées pour les EPOs ... 117
Figure 44 : EPOs et biosimilaires correspondants autorisés en Europe ... 118
Figure 45: Design des études de Phase I ... 121
Figure 46 : Mécanisme d'action des HBPM ... 127
Figure 47 : Liste des indications approuvées par type d'HBPM ... 129
Figure 48 : Schéma de dépolymérisation utilisé pour la préparation des HBPMs ... 131
Figure 49 : Mécanisme d'élimination des mAbs médiée par la cible ... 136
Figure 50 : Mécanisme d'élimination des mAbs ... 137
Figure 51 : Biosimilaires du TRASTUZUMAB actuellement en développement ... 139
Figure 52 : Présentation schématique des recommandations pour le développement d'un biosimilaire ... 143
Figure 53 : Place du biosimilaire ... 144
Figure 54 : Choix de la dose pour une FIM d'un biosimilaire par rapport à la courbe dose-réponse de la référence ... 150
Page 17
Liste des tableaux
Tableau 1 : Exemple de biomédicaments commercialisés selon leur classe
pharmacologique ... 44
Tableau 2 : Listes des recommandations ICH pour le développement et la fabrication des biomédicaments ... 47
Tableau 3 : Classification des biomédicaments en catégorie de risque ... 55
Tableau 4 : Stratégie d'évaluation de l'immunogénicité ... 56
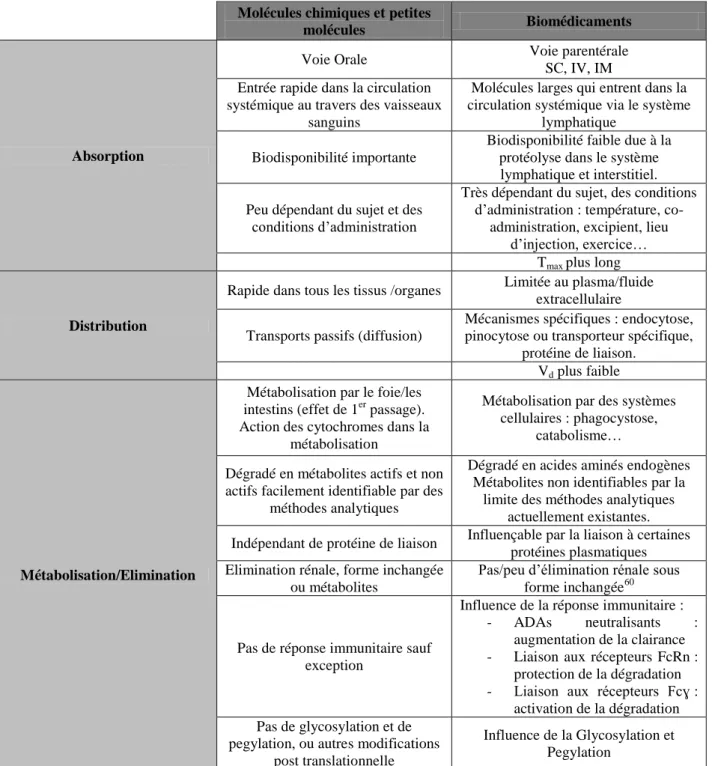
Tableau 5 : Différence des caractéristiques ADME entre molécule chimique et biomédicament ... 61
Tableau 6 : Date d'expiration des brevets de certains biomédicaments ... 68
Tableau 7 : Comparaison des coûts de développement d'un générique et d'un biosimilaire ... 74
Tableau 8: Résumé des analyses qualitatives nécessaires pour démontrer la similarité d'un biomédicament ... 89
Tableau 9 : Outils analytiques pour évaluer la structure d’une protéine. ... 90
Tableau 10 : Résumé des analyses nécessaires pour démontrer la similarité préclinique d’un biomédicament ... 91
Tableau 11 : Résumé des analyses nécessaires pour démontrer la similarité clinique d’un biomédicament ... 92
Tableau 12: Résumé des principales recommandations par classe de molécules (non exhaustif) ... 102
Tableau 13 : Principale similarités et différences entre les recommandations européennes et américaines ... 109
Tableau 14 : Résumé des réglementations et recommandations portant sur les biosimilaires ... 111
Tableau 15 : Liste des G-CSF autorisés en Juin 2014 ... 121
Tableau 16 : Résumé des études réalisée pour les biosimilaires du NEUPOGEN® ... 124
Tableau 17 : Principaux Anticorps monoclonaux autorisés en France ... 138
Tableau 18 : Type d'étude FIM en fonction de la classe pharmacologique ... 151
Tableau 19: Résumé de la nécessité des études PD par classe pharmacologique ... 152
Tableau 20: Population cible de la PD par classe pharmacologique ... 153
Tableau 21 : Suivi recommandé pour l'évaluation de l'immunogénicité des biosimilaires ... 158
Page 18
INTRODUCTION
A partir des années 1980, l’évolution des biotechnologies et l’utilisation de techniques de plus en plus innovantes ont permis d’explorer de nouvelles voies thérapeutiques par le développement de médicaments dits « biologiques ». L’évolution des biotechnologies a ouvert de nouvelles possibilités de prise en charge thérapeutique de maladies rares ou pour lesquelles il n’existait que peu de traitement. Depuis une vingtaine d’années de nouvelles molécules innovantes, issues ou produites par des organismes vivants, disposent d’AMM (Autorisation de Mise sur le Marché) en France comme dans le monde (Facteurs de croissance, Anticorps Monoclonaux, Vaccins, médicaments dérivés du sang…).
Les brevets des premières molécules mises sur le marché sont arrivés ou vont arriver à expiration dans les dix années à venir (12 médicaments de référence issus de la biotechnologie tomberont dans le domaine public d’ici à 2020). Tout comme pour les molécules chimiques, il est possible d’entreprendre le développement de médicaments dits génériques.
Cependant, ces biomédicaments « copiés » ne peuvent être réellement considérés comme des génériques : la complexité de la structure, de la production (utilisation de systèmes biologiques, protection intellectuelle des procédés de fabrication) et de leurs modes d’action rend impossible l’obtention d’un biomédicament strictement identique sur le plan chimique au produit de référence. Afin de mieux définir ces molécules semblables, la description des biosimilaires est apparue :
« Les médicaments biologiques similaires à des médicaments de référence ne remplissent habituellement pas toutes les conditions pour être considérés comme des médicaments génériques, en raison notamment des caractéristiques des procédés de fabrication, des matières premières utilisées, des caractéristiques moléculaires et des modes d’actions
thérapeutiques (...) dès lors qu’un médicament ne peut être considéré comme un médicament générique, les résultats d’essais appropriés doivent être fournis afin de satisfaire aux conditions relatives à la sécurité ou à l’efficacité.». (Directive 2004/27/CE)
Il existe donc des attentes spécifiques des autorités de santé quant à l’évaluation de la sécurité ou de l’efficacité de ces molécules. La méthodologie classique de conduite des essais de bioéquivalence, ainsi que les méthodes analytiques utilisées pour les molécules
Page 19
génériques sont donc insuffisantes pour le développement des biosimilaires : des données issues d’essais précliniques et cliniques plus complets sont attendues.
Cette thèse aura pour but d’exposer les particularités de ce type d’essai clinique ainsi que les attentes des autorités en charge de leurs mises sur le marché.
Dans les deux premières parties, les caractéristiques et l’environnement réglementaire des biomédicaments seront présentées afin de comprendre et de situer les biosimilaires. La troisième partie présentera les particularités des biosimilaires et le contexte économique qui les entoure. Enfin, au travers de la revue des différentes recommandations des autorités compétentes internationales ainsi qu’une revue bibliographique des essais cliniques réalisés pour des biosimilaires, des lignes directrices seront extraites pour aborder la réalisation d’un essai clinique portant sur un biosimilaire de façon optimale.
Page 20
CHAPITRE 1
BIOTECHNOLOGIE ET
MEDICAMENTS
Page 21
Afin d’appréhender les problématiques liés aux biosimilaires, il est nécessaire de comprendre l’origine des biomédicaments, de les différencier des médicaments traditionnels pour comprendre leurs particularités et leur contexte réglementaire.
1.
Histoire
de
la
biotechnologie
appliquée
aux
médicaments
L’utilisation des biotechnologies par l’homme remonte à l’antiquité (Brassage de la bière, Tannage). Cependant, le terme « biotechnologie » n’est apparu qu’en 1919 en Europe1 pour décrire l’utilisation de nouvelles techniques appliquées à la biologie pour la fabrication industrielle de composés biologiques ou chimiques.
Le début de l’utilisation médicinale des procédés de biotechnologie a été marqué par la découverte fortuite de la pénicilline par Alexandre Flemming en 1928. Cet antibiotique est un champignon naturellement synthétisé par la moisissure Penicillium
Notanum. Cependant, la quantité synthétisée directement par cette moisissure dans des
conditions normales était largement insuffisante pour permettre de traiter des infections. De nombreuses recherches débutèrent afin de mettre au point des techniques plus performantes pour accroitre la production de pénicilline : criblage, sélection de nouvelles souches, changement des milieux de culture et construction d'un nouveau type de fermentateur… Ces recherches sont aujourd’hui considérées comme le point de départ des biotechnologies à visée médicinale2.
En parallèle, de nombreux travaux de la fin du 19éme siècle (notamment ceux de Louis Pasteur, Robert Koch et Gregor Mendel) ont permis d’aboutir à des progrès spectaculaires dans la connaissance de la génétique et de la microbiologie.
Ces progrès ont été accompagnés de la découverte de la structure de l’ADN (par J.Watson et F.Crick en 1953) et de la synthèse réussie d’ADN recombinant3 (par S.Cohen
1 Fari M.G, Kralovanszky U.P ; The founding father of biotechnology: Károly (Karl) Ereky ; International
Journal of Horticultural Science 2006, 12 (1): 9–12 ; ISSN 1585-0404
2
AMGEN, Introduction à la biotechnologie ; 2009 ; Disponible sur http://www.amgen.be/pdfs/AMG-Biotech_brochure_04-2010_FR-06-05_FINAL.pdf consulté le 3 Mars 2014.
3 ADN recombinant : technique qui consiste à introduire un gène ciblé dans l’ADN d’un organisme hôte qui
Page 22
et F. Boyer en 1974) ; ils ont donné naissance à la biotechnologie moderne telle que définie aujourd’hui :
« Toute technique utilisant des êtres vivants (micro-organismes, animaux, végétaux),
généralement après modification de leurs caractéristiques génétiques, pour la fabrication industrielle de composés biologiques ou chimiques (médicaments, matières premières
industrielles) ou pour l'amélioration de la production agricole (plantes et animaux transgéniques ou O.G.M. [organismes génétiquement modifiés])4 ».
La première substance à avoir été entièrement synthétisée par les techniques de biotechnologie est l’insuline. Cette hormone polypeptidique, produite par le pancréas, agit sur les métabolismes protéiques, lipidiques mais surtout sur le métabolisme glucidique (régulation de la glycémie). Initialement, cette hormone était produite par extraction à partir du pancréas de porc ou de bœuf puis purifiée avant d’être administrée aux patients diabétiques. Rapidement, deux questions se sont posées : Comment accroître la production pour faire face à des besoins mondiaux en pleine expansion ? Quel sera l’effet d’un traitement à long terme par une hormone animale ? La recherche d’un procédé plus rentable et permettant de produire des protéines moins allergisantes a ainsi débuté.
En 1978, H. Boyer réussit à appliquer les techniques de biotechnologies modernes en transformant l’insuline porcine en insuline humaine et en la faisant produire par une bactérie E.coli. Ces deux insulines différant par un seul acide aminé, il a remplacé l’acide aminé en question en utilisant un processus de catalyse enzymatique. Puis, par réintroduction de cette séquence d’ADN dans le génome de la bactérie, celle-ci a commencé à produire de l’insuline humaine.
L’insuline et les antibiotiques ont ainsi été les premières molécules synthétisées à partir des nouveaux procédés biotechnologiques. C’est en partie par l’étude et les connaissances de ces premières molécules que leurs différences avec les molécules traditionnelles ont été mises en lumière.
Page 23
2.
Principales
différences
entre
les
médicaments
traditionnels et les biomédicaments
.Les médicaments traditionnels sont obtenus par extraction à partir de composés naturels (Principe actif d’origine végétal comme l’Acide Acétylsalicylique issu de la reine des prés et du saule) ou synthétisés par des procédés chimiques (comme le paracétamol). Les biomédicaments (médicaments issus de procédés de biotechnologies) différent des médicaments traditionnels par5,6,7 :
Leurs différences de structure : complexité des produits
Les médicaments traditionnels, tels que le paracétamol, sont obtenus par synthèse à partir d’ingrédients chimiques : on les qualifie de petites molécules en raison de leur faible poids moléculaire8 (une centaine de Dalton) (Figure 1). Leurs structures et leurs conformations spatiales sont caractérisables et reproductibles par réactions chimiques.
Figure 1 : Différences structurales et moléculaires entre une molécule chimique (Paracétamol) et une molécule biologique (Filgrastrim)
5 Prugnaud J.L, Trouvin J.H. ; Les Biosimilaires ; 2011 ; Edition Springer
6 EuropaBio ; Guide to Biological Medecine : A focus on Biosimilar Medicines ; Disponible sur http://www.europabio.org/sites/default/files/report/guide_to_biological_medicines_a_focus_on_biosimilar_m edicines.pdf consulté le 6 Mars2014.
7 BIOTECanada ; Démystifier les médicaments biosimilaires : Guide à l’intention des rédacteurs
scientifiques ; Disponible sur
http://www.biotech.ca/uploads/biosimilars%20guide%20french%20high%20resolution.pdf consulté le 6 Mars2014.
8 Le poids moléculaire est la somme des masses atomiques des différents atomes constituant une molécule. Il
Page 24
Les biomédicaments sont des protéines, c'est-à-dire des macromolécules constituées par l'association d'acides aminés unis entre eux par une liaison peptidique. Ces médicaments sont donc de grandes molécules, possédant une structure protéique tridimensionnelle avec un important poids moléculaire (supérieur à 10 000 Dalton). Leurs activités dans l’organisme est dépendante de leurs caractéristiques structurales.
Une protéine est d’abord définie par sa séquence d’acides aminés (structure primaire), facilement reproductible. Cet enchainement engendre des contraintes moléculaires (liaisons chimiques, ioniques, électrostatiques ou hydrogènes) qui forcent la protéine à s’organiser dans l’espace (structures secondaires, tertiaires et quaternaires) (Figure 2)9
. Ces liaisons donnent la structure spatiale de la molécule mais sont très sensibles à des changements de température, d’exposition à la lumière, à leur environnement moléculaire (ions, agents…): les structures secondaires, tertiaires et quaternaires sont ainsi très difficilement reproductibles.
Figure 2 : Structure Spatiale des protéines
D'après Abraham J.
(1) Structure primaire: ordre des acides aminés le long de la chaîne polypeptidique.
(2) Structure secondaire: repliement local des acides aminés en hélices, en feuillets, ou en d'autres formes similaires.
(3) Structure tertiaire: agencement stable dans l'espace de ces hélices et feuillets.
(4) Structure quaternaire: agencement des sous-unités entre elles, quand la protéine est constituée de plusieurs sous-unités indépendantes (comme l'est par exemple l'hémoglobine).
Ces molécules, du fait de leur structure complexe, peuvent également subir des phénomènes d’agrégation (formation d’agrégat de protéines) et des modifications post-traductionnelles ou maturations (greffe sur des acides aminés de groupements chimiques ou biologiques supplémentaires afin de leur conférer des caractéristiques supplémentaires) comme la glycosylation (Figure 3). Ces phénomènes peuvent avoir des conséquences sur la clairance, la demi-vie d’élimination et l’activité du biomédicament10,11.
9 Roger S. ; Biosimilars : How similar or dissimilar are they ?; Nephrology ;2006 ; 11, 341-346 ; 1440-1797 10 KaurChugh P. , Roy V. ; Biosimilars : Current Scientific and Regulatory Considerations ; Current Clinical
Page 25
Figure 3 : Modification post-traductionnelle d’une protéine et conséquence fonctionnelle.
D’après Kuhlmann M.et Covic A.
Leurs micro hétérogénéités et leurs variabilités reliés aux procédés de
fabrication :
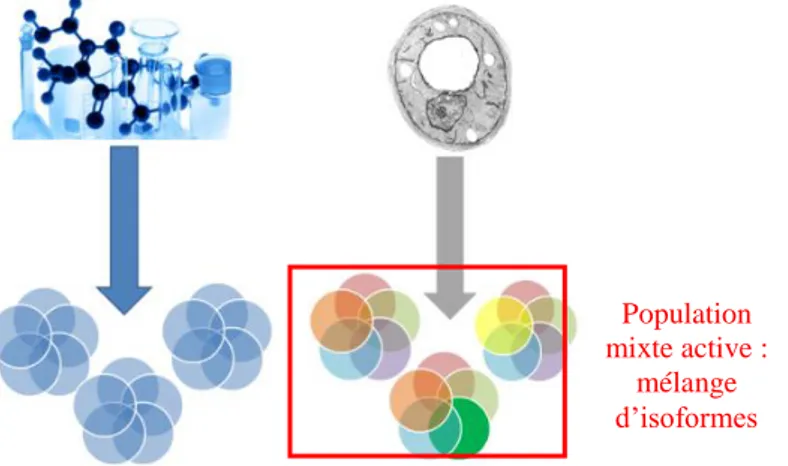
Les médicaments traditionnels sont synthétisés par des procédés chimiques contrôlés et reproductibles : une entité unique est obtenue (Figure 4). A contrario, les biomédicaments sont fabriqués à partir d’organismes vivants très dépendants de leurs conditions environnementales. La moindre variation, même mineure, peut entrainer des modifications sur le produit obtenu12. Egalement, les modifications post-translationnelles (maturations) des protéines entrainent des modifications spécifiques à une entité. Ainsi, plusieurs formes finales des biomédicaments sont obtenues formant une population mixte active (formes variantes mais structurellement proches (isoformes)) de la molécule.
On parle de « micro hétérogénéité » intrinsèque des biomédicaments.
Figure 4 : Illustration de la micro hétérogénéité des biomédicaments
11
Kuhlmann M., Covic A., The protein science of biosimilars ; Nephrology Dial Transplant ; 2006 ; 21 (5) ; 4-8
12 Ciller V. Les Biosimilaires : environnement réglementaire, particularités biologiques et spécificités de
l’évaluation de cette récente catégorie de médicament ; Th D Pharm, Marseille; 2011.
Population mixte active :
mélange d’isoformes
Page 26
A ce mélange d’isoformes s’ajoutent des molécules proches qui n’ont pu être éliminées par les étapes de purification (Figure 5) : c’est ce mélange final complexe qui va être considéré, in fine, comme le produit d’intérêt et qui justifie les difficultés de caractérisation des biomédicaments.
Figure 5: Hétérogénéité et composés présents dans le mélange d'actif
Leurs Stabilités
La conformation spatiale des biomédicaments joue un rôle important dans la structure de la molécule et dans l’action que celle-ci aura sur l’organisme.
Celle-ci est dépendante de nombreuses liaisons covalentes ou non covalentes qui peuvent rapidement être modifiées entrainant alors une modification de la molécule.
Les biomédicaments nécessitent des conditions de stockage plus strictes (réfrigérées, protégés de la lumière..) que des molécules chimiques simples. Du fait de leur instabilité, de leur sensibilité aux facteurs environnementaux et de la protéolyse par les enzymes digestives : les biomédicaments ne sont administrés que par voie parentérale.
Leurs capacités à induire une réaction immunologique : l’immunogénicité
L’immunogénicité est définie comme la capacité à induire une réponse immunitaire humorale et/ou à médiation cellulaire13.
Les molécules chimiques sont généralement trop petites pour être reconnues par le système immunitaire mais les biomédicaments, du fait de leur taille et également de
Page 27
l’existence d’éléments similaires dans le corps humain (enzyme, protéine), peuvent être reconnus par le système immunitaire humain comme indésirables et ainsi induire une réaction immunitaire.
Les conséquences de cette réaction immunitaire peuvent être une perte de l’efficacité du médicament mais également présenter un risque pour le patient.14 (Voir Chapitre 2 Partie 2.1).
Du fait de ces différences majeures, les biomédicaments ne peuvent entrer dans la définition générale des médicaments : la clarification de leur définition et de leur statut a rapidement été nécessaire.
3.
Classification des produits issus des biotechnologies : le
point de vue réglementaire
Le contexte réglementaire entourant ces substances produites à partir de cellules ou d’organismes vivants ou dérivés de ceux-ci est resté flou jusqu’aux années 2000 où la définition de biomédicament ou de médicaments biologiques a été décrite dans les directives européennes15 et reprise dans le code de la santé publique française16 :
« Médicament biologique, tout médicament dont la substance active est produite à partir d'une source biologique ou en est extraite et dont la caractérisation et la détermination de
la qualité nécessitent une combinaison d'essais physiques, chimiques et biologiques ainsi que la connaissance de son procédé de fabrication et de son contrôle » Partie I de
l’Annexe I de la Directive 2001/83/EC (amendée par la Directive 2003/63/EC).
La définition des biomédicaments englobe ainsi plusieurs catégories de médicaments : antibiotiques, insuline, facteurs de croissance, vaccins… Tous sont produits par des organismes vivants mais ils ne présentent pas tous le même degré de complexité. C’est le cas des antibiotiques qui sont connus depuis plus de 100 ans et qui n’engendrent pas d’interrogations réglementaires ou scientifiques particulières.
14 FDA ; FDA guidance for Industry : Imunogenicity assessement for therapeutic protein products ; Fev
2013 ; disponible sur http://www.fda.gov/Drugs/GuidanceCompliance Regulatory Information/Guidances/default.htm consulté le 15/02/14
15
Directive 2001/83/EC DU PARLEMENT EUROPÉEN ET DU CONSEIL du 6 novembre 2001instituant un code communautaire relatif aux médicaments à usage humain amendée par la directive 2003/63/CE du 25 Juin 2005.
Page 28
La définition des médicaments biologiques selon la directive 2001/83/EC (amendée par la Directive 2003/63/EC) est donc complétée par la phrase :
« Sont considérés comme médicaments biologiques : les médicaments immunologiques et
les médicaments dérivés du sang et du plasma humains définis respectivement à l'article
1er, paragraphes 4 et 10; les médicaments entrant dans le champ d'application de la partie A de l'annexe du règlement (CEE) n° 2309/93 (i.e médicaments issus de l’un des
procédés biotechnologiques suivants : technologie de l’acide désoxyribonucléique
recombinant ; expression contrôlée de gènes codant pour des protéines biologiquement actives dans des procaryotes et des eucaryotes, y compris des cellules transformées de
mammifères ; méthodes à base d’hybridomes et d’anticorps monoclonaux, les
médicaments de thérapie innovante définis dans la partie IV de la présente annexe » .
Quatre sous-catégories spécifiques entrent ainsi dans le cadre de cette définition : - Médicaments immunologiques
- Médicaments dérivés du sang et du plasma
- Médicaments issus de procédés biotechnologiques spécifiques - Médicaments de thérapies innovantes.
3.1
Les médicaments immunologiques
D’après le paragraphe 4 de l’article 1er
de la directive 2001/83/CE, on entend par médicament immunologique :
Tout médicament consistant en vaccins, toxines, sérums, ou allergènes: a) les vaccins, toxines ou sérums recouvrant notamment:
i) les agents utilisés en vue de provoquer une immunité active tels que le vaccin anticholérique, le BCG, le vaccin antipoliomyélitique, le vaccin antivariolique;
ii) les agents utilisés en vue de diagnostiquer l'état d'immunité, comprenant notamment la tuberculine ainsi que la tuberculine PPD, les toxines utilisées pour les tests de Schick et de Dick, la brucelline;
iii) les agents utilisés en vue de provoquer une immunité passive tels que l'antitoxine diphtérique, la globuline antivariolique, la globuline anti lymphocytique;
Page 29
b) les produits allergènes étant tout médicament destiné à identifier ou provoquer une modification spécifique et acquise de la réponse immunologique à un agent allergisant.
3.2
Les médicaments dérivés du sang et du plasma humain
D’après le paragraphe 4 de l’article 1er
de la directive 2001/83/CE, on entend par médicament dérivé du sang et du plasma humain un médicament à base de composants de
sang préparés industriellement par des établissements publics ou privés; ce médicament comprend notamment l'albumine, les facteurs de coagulation et les immunoglobulines d'origine humaine.
3.3
Les médicaments issus de procédés biotechnologiques
particuliers
Les biomédicaments sont caractérisés par 3 approches technologiques17 principales: o la technologie de l’ADN recombinant
o l’expression contrôlée des gènes o les méthodes à base d’hybridome.
Ces techniques sont détaillées ci-dessous car elles sont couramment utilisées pour la production de biomédicament (spécialement la technologie de l’ADN recombinant et les méthodes à base d’hybridome).
17 Steffen L., Biomédicaments en France, Etat des lieux 2013 ; Leem : les entreprises du médicament ;
Septembre2013 ; disponible sur http://www.leem.org/biomedicaments-en-france-etat-des-lieux-2013
Page 30
3.3.1 Généralités
Les éléments fondamentaux de ces techniques sont basés sur les connaissances de la structure de l’acide désoxyribonucléique (ADN) (Figure 6) et de l’expression des gènes (Figure 7).
Figure 6 : Structure de l'ADN
Une séquence d’ADN contenant de l’information génétique (information nécessaire à la synthèse d’une protéine) définit un gène.
Figure 7 : Etape de synthèse d'une protéine
La première étape d’expression de ce gène est la synthèse d’une molécule d’acide ribonucléique (ARN) par le mécanisme de transcription (processus par lequel l’information contenue dans un brin d’ADN est copiée en une molécule d’ARN simple brin possédant une séquence de base complémentaire). Les ribosomes parcourent alors cet ARN et réalisent l’étape de traduction (processus par lequel la séquence en acides aminés d’un
Page 31
polypeptide est synthétisée) donnant le polypeptide (protéine)18. Ce peptide subira alors des modifications post-translationnelles qui pourront avoir une incidence sur la fonction de la protéine dans l’organisme.
3.3.2 L’ADN recombinant
Comme le montre la Figure 9, la technique de l’ADN recombinant est la principale méthode utilisée et permet la production de la majorité des biomédicaments.
L’ADN recombinant est un ADN doté de séquences nucléotidiques issues de sources différentes :
- le fragment d’ADN d’intérêt : généralement le gène humain,
- l’ADN appelé le « vecteur » qui est une molécule d’ADN capable de se répliquer dans la cellule hôte, généralement des plasmides ou des bactériophages.
Le fragment d’intérêt (gène codant) doit être identifié, isolé, puis coupé par des enzymes de restriction (nucléases qui coupent l’ADN à chaque fois qu’il présente une séquence nucléotidique donnée). Ces fragments sont ensuite insérés dans le vecteur pour former une nouvelle combinaison (Figure 8). Cette nouvelle combinaison d’ADN est ensuite exprimée dans un micro-organisme, dans des cellules animales, végétales ou dans un organisme supérieur.
Figure 8 : Technologie de l'ADN recombinant
Page 32 Figure 9 : Classification des produits biologiques
Thérapie cellulaires &
tissulaires
Produits issus de l’ADN recombinant Produits non issus de
l’ADN recombinant
PRODUITS
BIOLOGIQUES
Médicaments dérivés du sang Enzymes, Hormones et Anticorps polyclonaux Toxines et Antibiotiques Vaccins Protéines recombinantes Acides Nucléiques Oligonucléotides& plasmides Vaccins à ADN
Protéines Thérapeutiques Thérapies Géniques Vaccins recombinants & thérapeutiques Protéines de fusion Anticorps Monoclonaux Facteurs de croissance Hormones Enzymes Cytokines Biomédicaments qui font
l’objet d’un développement de biosimilaire en 2014
Page 33
Pour assurer une production optimale et sécuritaire, une lignée cellulaire peut être créée, sélectionnée spécialement pour l’expression de l’ADN. Ces cellules sont mises en culture à grande échelle dans des bioréacteurs afin de leur faire produire la molécule d’intérêt (protéine). Elle est ensuite isolée et purifiée. La dernière étape étant la mise en forme pharmaceutique (formulation, vectorisation et conditionnement).
3.3.3 L’expression contrôlée de gènes
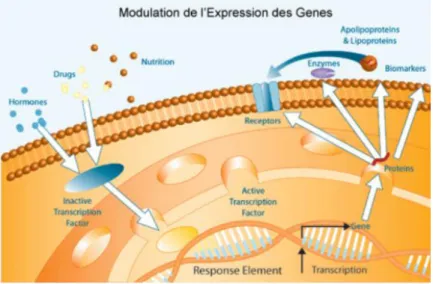
L’expression des gènes est constituée d’un ensemble de processus biologiques et biochimiques : ce phénomène est naturellement contrôlé dans l’organisme afin de n’exprimer à une période donnée que certains gènes nécessaires (par exemple lors de la différenciation cellulaire).
Cette régulation est réalisée tout au long du décodage de l’ADN : au niveau chromatinien, au niveau transcriptionnel, au niveau post-transcriptionnel, au niveau traductionnel et au niveau post-traductionnel (Figure 10).
Figure 10 : Illustration de la modulation de l’expression des gènes
D’après http://genfit.com/fr/science-discovery/capabilities-expertise/expertise-scientifique
L’expression contrôlée de gènes désigne toutes modifications de la régulation naturelle sur ces différentes étapes qui auront pour conséquence un changement de la quantité ou du moment de l'apparence du produit fonctionnel d'un gène.
Depuis de très nombreuses années des médicaments agissant sur l’expression des gènes sont connus (hormones thyroïdiennes, insuline…) mais de plus en plus de médicaments sont synthétisés dans le but d’agir notamment avec les récepteurs nucléaires afin de stimuler ou inhiber l’expression d’un gène (par exemple des anticorps bloquant
Page 34
l’interaction de la molécule entrainant la cascade cellulaire responsable de l’activation de la transcription).
La thérapie génique (voir 3.4.1), notamment, est considérée comme un mode d’expression contrôlé de gènes car la modification du génome permet un contrôle sur le gène exprimé par un remplacement direct, ou par régulation de la synthèse d’une protéine ayant elle-même un rôle particulier dans la maladie.
3.3.4 Les méthodes à base d’hybridome
Tous les organismes supérieurs ont la capacité de reconnaitre les molécules étrangères nocives et disposent comme défense d’un système immunitaire spécifique dont les anticorps constituent la composante humorale. Ces anticorps (Acs) ou immunoglobulines (Igs) reconnaissent une partie de l’agent infectieux appelé antigène (responsable de la réaction immunitaire) et sont sécrétés par les lymphocytes B (LB) (plus exactement les plasmocytes qui constituent la différenciation terminale des lymphocytes suite à l’interaction entre l’antigène et le lymphocyte B).
Chaque immunoglobuline est composée de quatre sous-unités identiques : deux chaines lourdes et deux chaines légères. Cette structure globale est identique pour tous les anticorps mais pour certaines régions des chaines lourdes ou légères la séquence en acide aminés de la chaine protéique peut varier : cette caractéristique de la partie variable confère une structure spatiale particulière et très hétérogène (Figure 11).
Page 35
Lors de l’exposition à un antigène, un clone de LB produisant des anticorps spécifiques à cet antigène prolifère : chaque LB ne peut produire qu’une immunoglobuline spécifique. Cependant, lors de l’infection des anticorps polyclonaux sont produits : c'est un mélange d’anticorps de sélectivités différentes et dirigés contre différents épitopes19
d’un même antigène.
En thérapie, les anticorps polyclonaux sont généralement utilisés pour induire une immunité chez les personnes les recevant. Ils sont obtenus à la suite de l’immunisation d’animaux. Une fois les animaux sensibilisés, ils produisent des Acs qui sont prélevés puis purifiés avant de les injecter chez l’homme. C’est sur ce principe que se base la vaccination.
Les anticorps monoclonaux quant à eux consistent en un mélange d’anticorps ayant la même sélectivité et activité. Afin d’obtenir ce type d’anticorps spécifiques en laboratoire, il faut se baser sur les techniques des hybridomes.
La première étape de cette technique consiste à immuniser un animal à l’aide d’un antigène donné (immunisation). Il faut ensuite prélever sa rate, lieu de production des lymphocytes B, afin de pouvoir les isoler (Figure 12). Ces derniers sont incapables de se multiplier et ne survivent que quelques jours en milieu de culture. Il faut donc les rendre immortels en les fusionnant avec les myélocytes (lymphocytes de cellules tumorales qui possèdent la capacité de se reproduire indéfiniment) : c’est l’étape de fusion.
Cette étape de fusion est réalisée par des méthodes physiques (électroporation) ou chimiques (utilisation du polyéthylene glycol) qui entrainent la fusion des deux membranes des cellules. La sélection d’une lignée cellulaire de myélome particulière est aussi importante pour rendre possible cette fusion. Cette étape conduit à la production d’une cellule appelée hybridome qui possède la capacité de synthétiser des anticorps et d’être immortelle.
19 Déterminant antigénique
Page 36
Figure 12 : Procédé d’obtention d’Acs monoclonaux
Par la suite, les myélomes non fusionnés sont éliminés et les hybridomes doivent être sélectionnés pour ne conserver que ceux produisant les anticorps ayant la spécificité recherchée20. Ils doivent ensuite être clonés afin d’obtenir une quantité suffisante d’anticorps qui permettra la réalisation de tests de type ELISA21
et ou d’immunoprécipitation22
afin de vérifier la nature des anticorps produits.
Si les Acs produits correspondent à ceux recherchés, l’hybridome est confirmé et la lignée cellulaire est cultivée afin de permettre la production de ces derniers.
20 Nelson P., Reynolds G., Waldrom E., Ward E., ; Demystified …Monoclonal Antibodies ; J Clin Pathol :
Mol Pathol ; 2000 Jun;53(3):111-7.
21 De l’anglais Enzyme-Linked Immunosorbent Assay : méthode immuno-enzymatique pour la détection et le
dosage des anticorps
22 Technique qui permet la précipitation d’un Ag en solution par un Ac
s qui agglutine spécifiquement une
Page 37
3.3.5 Complexité de la fabrication
Les trois méthodes citées précédemment ont leurs particularités pour permettre de produire la protéine d’intérêt. Toutes ont pour but la production à grande échelle de cette protéine et ont donc en commun une partie des étapes suivantes23,24 (Figure 13):
- Isolement de la séquence d’ADN humaine. - Insertion de cette séquence dans un vecteur.
- Incorporation au génome de la cellule (bactérie, levure, cellules de mammifère, cellules végétales ou d’insecte) et sélection d’une cellule, qui sera clonée pour la création de la banque de cellules (ce système est appelé le système d’expression) :
o Mise en culture d’un clone répondant aux caractéristiques désirées.
o Obtention d’une population appelée la banque cellulaire maitresse ou «Master Cells Bank » (MBC) qui est conservée.
o Pour assurer la pérennité de cette MCB : création d’une banque cellulaire de travail ou « Working Cell Bank » (WCB) à partir de cellules issues de la MCB.
- Mise en culture des WCB pour qu’elles produisent la protéine d’intérêt (fermenteur ou système de culture cellulaire).
- Récupération du surnageant qui contient la protéine. - Isolement et purification de la protéine d’intérêt. - Mise en forme pharmaceutique.
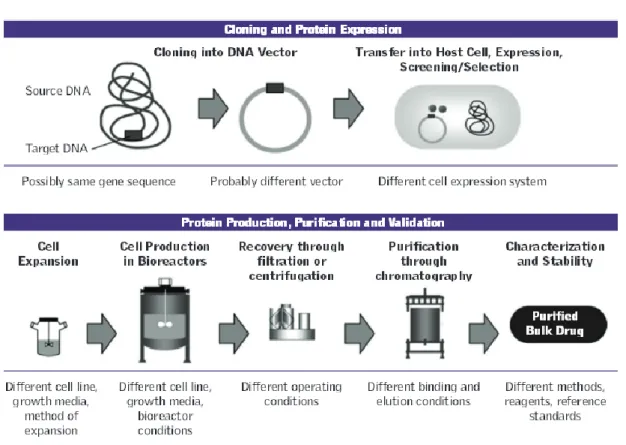
Chaque fabricant possède ses propres MCB et WCB et développe ses propres processus de fabrication (caractéristique de la fermentation ou de la purification), uniques et brevetés. Ces processus étant très sensibles, un contrôle précis est essentiel : quelques 250 essais qualité en cours de fabrication sont nécessaires, au lieu de 50 essais pour des molécules chimiques habituelles. La fabrication d’un biomédicament demande donc un très haut niveau d’expertise technique25
.
23 Roger S. ; Biosimilars : How similar or dissimilar are they ?; Nephrology ;2006 ; 11, 341-346 ; 1440-1797 24Sahoo N., Choudhury K., Manchikanti P. ; Manufacturing of Biodrugs : Need for Harmonization in
Regulatory Standards ; Biodrugs ; 2009 ; 23 (41) ; 217-218
25 Commission Européenne ; Ce qu’il faut savoir su les médicaments biosimilaires ; Document d’information
consensuel ; 2013 ; Disponible sur
Page 38
Page 39
3.4
Les médicaments de thérapie innovante
Avant 2007, il n’existait pas de cadre juridique pour les médicaments présentant un caractère complexe, novateur ainsi que des spécificités techniques. Les autorités nationales pouvaient décider du cadre réglementaire à appliquer26. L’agence européenne a pallié ces disparités en instaurant la notion de thérapie innovante définie par le règlement N° 1394/2007 du parlement européen.
Les médicaments de thérapie innovante (MTI)27 sont définis en quatre catégories : les médicaments de thérapie génique, les médicaments de thérapie cellulaire somatique, les médicaments issus de l’ingénierie tissulaire ou cellulaire, les médicaments combinés de thérapie innovante.
3.4.1 Les médicaments de "thérapie génique"
Un médicament de thérapie génique est défini par la Directive 2009/120/CE de la commission du 14 septembre 2009 modifiant la directive 2001/83/CE28 comme un médicament biologique qui dispose des caractéristiques suivantes :
a) il contient une substance active qui contient ou constitue un acide nucléique
recombinant administré à des personnes en vue de réguler, de réparer, de
remplacer, d’ajouter ou de supprimer une séquence génétique;
ET
b) son effet thérapeutique, prophylactique ou diagnostique dépend directement de la
séquence d’acide nucléique recombinant qu’il contient ou au produit de
l’expression génétique de cette séquence.
Le but de la thérapie génique est d’avoir une action par insertion de matériel génique (le « gène médicament ») au sein même de la cellule afin de pouvoir corriger une anomalie du génome ou permettre l’inhibition ou l’activation d’un protéine donnée.
26 Casado M. Les produits frontières : réflexion autour de l’attribution d’un statut réglementaire ; Th D
Pharm, Grenoble; 2012.
27RÈGLEMENT (CE) No 1394/2007 DU PARLEMENT EUROPÉEN ET DU CONSEIL du 13 novembre
2007 concernant les médicaments de thérapie innovante et modifiant la directive 2001/83/CE ainsi que le règlement (CE) no 726/2004 (J.O.C.E L 324/121du 10 Décembre 2007)
28 Directive n°2009/120/CEE de la Commission du 14 Septembre 2009 modifiant la Directive 2001/83/CE du
Parlement Européen et du Conseil instituant un code communautaire relatif aux médicaments à usage humain en ce qui concerne les médicaments de thérapie innovante (JO L 311 du 28.11.2001, p. 67)
Page 40
La thérapie génique est basée sur l’exploitation des caractéristiques d’une famille particulière de virus : les rétrovirus. Lors de l’infection d’un organisme par un rétrovirus, celui-ci utilise une transcriptase inverse qui va permettre de copier son génome ARN en une molécule d’ADN double brin. Cette copie est ensuite insérée dans le génome de la cellule infectée : celle-ci survit à l’infection mais contient la copie de l’ARN du virus dans son ADN, cet ADN est ensuite exprimé par le mécanisme cellulaire.
La thérapie génique utilise cette capacité des rétrovirus : un double brin d’ADN d’intérêt est synthétisé in vitro et, par l’utilisation de la transcriptase inverse du rétrovirus celle-ci peut être insérée dans une molécule d’ADN de la cellule.
Au sens premier, la thérapie génique est le remplacement d'un gène défectueux, c'est-à-dire une séquence d’ADN, par un gène normal et fonctionnel qui sera à l'origine de la synthèse de la protéine manquante ou défectueuse. Au sens large, la thérapie génique consiste à introduire dans la cellule un gène qui sera à l'origine de la synthèse d'une protéine, pas nécessairement présente dans l'organisme normal, mais susceptible d'avoir, dans des circonstances particulières, un effet bénéfique pour le malade. Dans un sens encore plus large, on entend par thérapie génique toute introduction dans une cellule de matériel génétique, gène, portion de gêne, DNA, RNA, oligo-nucléotide, comme moyen thérapeutique.
Il existe trois méthodes de thérapie génique29 :
La thérapie génique ex vivo consiste à prélever sur le patient les cellules cibles, à les modifier génétiquement avec le vecteur viral porteur du gène thérapeutique puis à les réintroduire chez le patient.
La thérapie génique in situ consiste à placer directement au sein du tissu cible le vecteur de transfert. Cette technique est expérimentée, notamment, dans les cas de mucoviscidose (transfert de vecteurs dans la trachée et les bronches), de cancer (injection dans la tumeur d'un vecteur portant le gène d'une toxine, par exemple), ou de dystrophie musculaire (injection dans le muscle d'un vecteur porteur du gène de la dystrophine).
29 Serusclat F. ; GÉNOMIQUE ET INFORMATIQUE : L'impact sur les thérapies et sur l'industrie