Université de Montréal
Conception, synthèse et applications biologiques d’inhibiteurs de
biofilms à base d’imidazole et de benzimidazole
Par Jérémie Tessier
Département de chimie Faculté des arts et des sciences
Thèse présentée à la Faculté des études supérieures et postdoctorale en vue de l’obtention du grade de
Philosophiæ Doctor (Ph. D) en Chimie
Septembre 2020
Université de Montréal
Département de chimie, Faculté des arts et des sciences
Cette thèse intitulée :
Conception, synthèse et applications biologiques d’inhibiteurs de biofilms à base
d’imidazole et de benzimidazole
Présenté par : Jérémie Tessier
A été évalué(e) par un jury composé des personnes suivantes Pr. William D. Lubell Président-rapporteur Pr. Andreea R. Schmitzer Directeur de recherche Pr. Alexis Vallée-Bélisle Membre du jury Pr. Sylvie Garneau-Tsodikova Examinateur externe
Résumé
La résistance aux antibiotiques est l'une des menaces les plus graves pour la santé mondiale de nos jours. L'émergence de bactéries multirésistantes encourage les chercheurs à développer de nouveaux antibiotiques et stratégies pour compenser leurs différents mécanismes de résistance. L'un de ces mécanismes de défense est la formation de biofilms. Sous cette forme, les bactéries développent une matrice extracellulaire protectrice les rendant plus résistantes à divers traitements antimicrobiens. Nous avons conçu et synthétisé des composés de déstabilisation des membranes avec des caractéristiques clés : un cation benzimidazolium ou imidazolium, une chaîne apolaire hydrophobe et/ou un site de reconnaissance des anions lipophiles. Ces caractéristiques leur confèrent une activité antimicrobienne accrue et une grande capacité à perturber les membranes cellulaires. Ces composés perturbateurs de la membrane agissent via un mécanisme rapide et efficace et ont montré de bons résultats contre les souches de SARM (staphylococcus aureus résistant à la méthiciline) en tant que candidats antibiofilms prometteurs. Ces nouveaux agents ont le potentiel de se disperser et d'inhiber la formation de biofilms et pourraient avoir un impact positif sur la médecine humaine à l'avenir.
Mots-clés : benzimidazolium, imidazolium, membrane cellulaire, biofilms, bactéries, antibiotique, perturbateur membranaire, ammonium quaternaire.
Abstract
Antibiotic resistance is one of the most serious threats to global health nowadays. Emergence of resistant bacteria encourages researchers to develop new antibiotics and strategies to mitigate their different resistance mechanisms. One of these defense mechanisms is the formation of biofilms. In this form, bacteria develop a protective extracellular matrix making them more resistant to various antimicrobial treatments. We have designed and synthesized membrane destabilizing compounds with three key features: a benzimidazolium or an imidazolium cation, a hydrophobic apolar chain, and a lipophilic anion recognition site. These characteristics give these compounds increased antimicrobial activity and greater ability to disrupt cell membranes. These membrane-disrupting compounds act via a fast and efficient mechanism and showed good results against MRSA (methicillin-resistant staphylococcus aureus) strains as promising antibiofilms candidates. These new agents have the potential to disperse and inhibit the formation of biofilms and could have a positive impact on human medicine in the future.
Keywords : benzimidazolium, imidazolium, cellular membrane, biofilms, bacteria, antibiotic, membrane perturbator, quaternary ammonium.
Table des matières
Résumé ... v
Abstract ... vii
Table des matières ... ix
Liste des tableaux ... xvii
Liste des figures et des schémas ... xix
Liste des sigles et abréviations ... xxvii
Remerciements ... xxxiii
Chapitre 1 : Introduction générale ... 1
1.1 Découverte des pénicillines et leurs utilisations ... 1
1.2 Facteurs influençant le développement de la résistance bactérienne ... 3
1.3 Mécanisme d’action des antibiotiques ... 4
1.3.1 Inhibition de la synthèse d’ADN ... 5
1.3.2 Inhibition de la synthèse d’ARN ... 6
1.3.3 Inhibition de la synthèse de la paroi bactérienne ... 8
1.3.4 Inhibition bactérienne via la perméabilisation membranaire ... 10
1.4 La membrane cellulaire : une cible de choix ... 12
1.4.1 La bicouche phospholipidique ... 12
1.4.2 La membrane bactérienne ... 13
1.4.3 La membrane bactérienne des S. aureus ... 14
1.5 Les levures, un organisme cellulaire semblable au nôtre... 15
1.5.1 La membrane des levures ... 16
1.6.1 La concentration minimale inhibitrice ... 17
1.7 Les biofilms ... 19
1.7.1 Le cycle de développement des biofilms ... 20
1.7.2 Les biofilms mixtes ... 23
1.7.3 La détection des biofilms ... 25
1.7.4 Étude de l’activité sur les biofilms ... 27
1.8 Les composés cationiques antibactériens et antifongiques ... 28
1.8.1 Les composés de type ammonium quaternaire ... 29
1.8.2 Les composés azolés ... 32
1.8.3 Les sels d’imidazolium et de benzimidazolium ... 34
1.9 Description du projet de recherche ... 35
1.9.1 Propriétés antibactériennes d’analogues du miconazole... 35
1.9.2 Propriétés antibactériennes et antibiofilms des sels d’imidazolium et de benzimidazolium ... 37
1.10 Références ... 39
Chapitre 2 : Synthèse d’agents anti-biofilms dérivés de la molécule antifongique Miconazole .. 48
2.0 Préface ... 48
2.0.1 Les liquides ioniques organiques ... 48
2.0.2 Références ... 51
Article 1 : Anti-staphylococcal biofilm activity of micona-zoctylium bromide... 53
2.1 Abstract ... 54
2.2 Introduction ... 54
2.3 Results and discussion ... 56
2.4.1 Materials and methods ... 63
2.4.2 Synthesis ... 63
2.4.3 Bacterial strains, culture conditions and viability ... 63
2.4.4 Biofilm inhibition ... 64
2.4.5 Biofilm disruption ... 64
2.4.6 Biofilm staining and confocal laser scanning microscopy (LSM) analysis ... 64
2.4.7 Minimal bactericidal concentration (MBC) ... 65
2.4.8 Hemolysis ... 65
2.5 Conclusions ... 65
2.6 Conflicts of interest ... 66
2.7 Acknowledgements ... 66
2.8 Notes and references... 66
Chapitre 3 : Propriétés antibactériennes de plusieurs sels de lutidine-bis-benzimidazolium et l’étude de leur effet sur les biofilms bactériens ... 69
3.1 Préface ... 69
Article 2 : Antimicrobial and Antibiofilm Activity of Disubstituted Bis-benzimidazolium Salts .... 70
3.2 Abstract ... 71
3.3 Introduction ... 71
3.4 Results and Discussion ... 73
3.4.1 Design and synthesis of benzimidazolium salts ... 73
3.4.2 Antibacterial and antifungal properties ... 74
3.4.3 Inhibition of biofilm formation ... 75
3.4.4 Destruction of mature biofilms ... 76
3.4.6 Toxicity evaluation ... 82
3.5 Conclusions ... 82
3.6 Experimental Section ... 82
3.7 Conflict of interest ... 82
3.8 References ... 82
Chapitre 4 : Propriétés antibactériennes de deux nouvelles générations de sel de benzimidazolium et l’étude de leur effet sur les biofilms bactériens ... 85
4.1 Préface ... 85
Article 3 : Benzimidazolium salts prevent and disrupt methicillin-resistant Staphylococcus aureus biofilms ... 86
4.2 Abstract ... 87
4.3 Introduction ... 87
4.4 Results and discussion ... 89
4.5 Conclusions ... 96
4.6 Acknowledgements ... 96
4.7 Experimental ... 96
4.7.1 Synthesis and material ... 96
4.7.2 Bacterial strains, culture conditions, and viability assays. ... 97
4.7.3 Hemolytic assay. ... 97
4.7.4 Biofilm inhibition assay. ... 97
4.7.5 Biofilm staining and confocal laser scanning microscopy (LSM). ... 97
4.7.6 Scanning electron microscopy. ... 98
4.8 References ... 98
Chapitre 5 : Produits phytochimiques et cyclodextrines - Émulsions de Pickering: potentialisateurs naturels de l'activité antibactérienne, antifongique et antibiofilm. ... 102
5.1 Préface ... 102
5.1.1 Introduction sur les émulsions de Pickering ... 102
5.1.2 Références ... 106
Article 4 : Phytochemicals and Cyclodextrins-Based Pickering Emulsions: Natural Potentiators of Antibacterial, Antifungal and Antibiofilm Activity ... 107
5.2 Abstract ... 108
5.3 Introduction ... 108
5.4 Experimental section ... 110
5.4.1 General Information ... 110
5.4.2 Synthesis of miconazoctylium bromide ... 110
5.4.3 Emulsion preparation and phase diagram ... 111
5.4.4 Microscopy ... 111
5.4.5 X-ray powder diffraction ... 111
5.4.6 Differential scanning calorimetry ... 112
5.4.7 Multiple light scattering ... 112
5.4.8 Rheological study ... 112 5.4.9 Antimicrobial activity ... 112 5.4.10 Biofilm disruption ... 113 5.5 Results ... 114 5.6 Conclusion ... 123 5.7 Associated content ... 123 5.7.1 Supporting Information... 123 5.7.2 Acknowledgment ... 123 5.8 References ... 124
Chapitre 6 : Conclusion et perspectives ... 128
6.1 Vers la modification ciblée des antibiotiques commerciaux ... 128
6.2 Vers la synthèse de nouveaux analogues antibactériens à base de sels de benzimidazolium ... 129
6.3 Vers une administration contrôlée des composés antibactériens ... 131
6.4 Références ... 132
Annexes ... 133 A1.1 Synthesis and characterization. ... I A1.2 Concepts and methods used... VII A1.2.1 Cellular reactive oxygen species (ROS) detection. ... VII A1.1.2 Ortho-nitrophenyl-β-galactoside (ONPG) hydrolysis. ... VIII A1.1.3 Biofilm staining and confocal laser scanning microscopy (LSM) Analysis. ... XI A1.3 References ... XIII A1.4 NMR spectra of newly synthesized compounds ... XIV Annexe 2 : informations supplémentaires de l’article 2 « Antimicrobial and Antibiofilm Activity of Disubstituted Bis-benzimidazolium Salts »... XXI A2.1 Materials and methods. ... XXI A2.2 Synthesis and characterization. ... XXI A2.3 Concepts and methods used... XXX A2.3.1 Bacterial strains, culture conditions, and viability assays. ... XXX A2.3.2 EYPC liposomes... XXXI A2.3.3 Lucigenin-based ion transport assays. ... XXXI A2.3.4 Biofilm inhibition assay. ... XXXI A2.3.5 Biofilm disruption assay. ... XXXII
A2.3.6 Biofilm staining and confocal laser scanning microscopy (LSM) analysis. ... XXXII A2.3.7 Biofilm formation: crystal violet assay. ... XXXIII A2.3.8 Dynamic light scattering (DLS) assay. ... XXXIV A2.3.9 Molecular modelling. ... XXXIV A2.3.9 Hemolytic assay. ... XXXV Annexe 3 : informations supplémentaires de l’article 3 « Benzimidazolium salts prevent and disrupt methicillin-resistant Staphylococcus aureus biofilms » ... XXXVIII A3.1 Synthesis and characterization. ... XXXVIII General procedure for 1st and 2nd generation benzimidazolium salt formation (... XL General procedure for benzimidazole alkylation ( ... XLVIII General procedure for 3rd generation benzimidazolium salt formation (... LIII A3.2 Concepts and methods used. ... LVIII A3.2.1 Hemolytic assay. ... LVIII A3.2.2 Biofilm inhibition assay. ... LVIII A3.2.3 Biofilm staining and confocal laser scanning microscopy (LSM) analysis. ... LIX A3.2.4. Scanning electron microscopy. ... LIX A3.3 NMR spectra and LCMS of newly synthesized compounds... LX Annexe 4 : informations supplémentaires de l’article 4 «Phytochemicals and cyclodextrins based- Pickering emulsions: natural potentiators of antibacterial, antifungal and antibiofilm activity» ... LXXXVI
Liste des tableaux
Table 2.1 Minimal concentrations (μM) required to inhibit the growth of different organisms .. 57 Table 2.2. Bacterial mortality after 24 h at different antimicrobial concentrations ... 59 Table 2.3. Bacterial mortality in preformed biofilms at different antimicrobial concentrations . 62 Table 3.1. Minimal concentrations required to inhibit microorganism growth... 74 Table 3.2. Calculated logP and proposed mechanism of action (MoA) in the membrane. ... 81 Table 4.1. Minimal inhibitory concentration (MIC) of semi-flexible benzimidazolium salts (generation II) ... 91 Table 4.2 Minimal inhibitory concentration (MIC) of flexible benzimidazolium salts (generation III) ... 94
Liste des figures et des schémas
Figure 1.1. Structures des différentes générations d’antibiotique bêta-lactames. ... 1
Figure 1.2. Structure du dichlorodiphenyltrichloroéthane (DDT). ... 2
Figure 1.3. Mécanismes d’action des antibiotiques et mécanismes de résistance bactérienne. ... 4
Schéma 1.1. Rétrosynthèse de l’acide nalixidique à partir du dérivé 6-méthylpyridin-2-amine. ... 5
Figure 1.4. Structure de l’antibiotique de deuxième génération ciprofloxacine. ... 6
Figure 1.5. Structure de l’antibiotique rifampicine. ... 7
Figure 1.6. Structure générale des céphalosporines... 8
Figure 1.7 Structure de la vancomycine et de la méthicilline. ... 9
Figure 1.8. Mécanisme d’action des agents perturbateurs de la membrane. Copyright 2011, avec la permission de Macmillan Publishers Limited. [49] ... 11
Figure 1.9. Exemple de deux phospholipides : la phosphatidylcholine et la phosphatidylsérine. 12 Figure 1.10. Représentation de l’auto-assemblage des phospholipides en solution aqueuse. .... 13
Figure 1.11. Structures des différentes membranes bactériennes. ... 14
Figure 1.12. Structure de l’acide téichoïque... 15
Figure 1.13. Structure de la membrane des levures (haut) et structure des différentes composantes de la membrane externe (bas). ... 16
Figure 1.14. Représentation d’une plaque à 96 puits utilisée lors de la détermination de la CMI. ... 18
Figure 1.15. Les différentes étapes du développement des biofilms. ... 21
Figure 1.16. Représentation de l’interaction entre deux espèces au sein d’un biofilm. ... 24
Figure 1.17. Structure du colorant cristal violet et son effet sur les biofilms. ... 25
Figure 1.18. Représentation de différents biofilms avec le test de viabilité Baclight. ... 26
Figure 1.19. Structure de l’iodure de propidium utilisé comme colorant. ... 26
Figure 1.20. Image MEB d’un biofilm S. aureus.[112] ... 27
Figure 1.21. Représentation des deux modèles utilisés pour la détermination de l’activité antibiofilm. (I) : inhibition ; (II) : perturbation/destruction. ... 27
Figure 1.23. Structure de la norspermidine et des nouveaux analogues synthétisés. ... 30
Figure 1.24. Structure des différents analogues synthétisés par J. Haldar et ses collègues... 31
Figure 1.25. Structure du noyau 1H-imidazole et de l’acide aminé essentiel L-histidine. ... 32
Schéma 1.2. Synthèse du nitrate de miconazole en trois étapes.[133] ... 33
Figure 1.26. Interaction entre le miconazole et le noyau flavohémoglobine.[136] ... 33
Figure 1.27. Structure de l’antifongique fluconazole. ... 34
Schéma 1.3. Synthèse générale des sels d’imidazolium et de benzimidazolium. ... 34
Figure 1.28. Composé NH125 et différents analogues de la librairie de Huigens.[142] ... 35
Schéma 1.4. Synthèse du dérivé amphiphile de la vancomycine.[143] ... 36
Schéma 1.5. Réaction d’alkylation afin d’obtenir divers bromures de miconazolium ... 37
Figure 1.29. Structure des antibiotiques synthétisés au sein de notre groupe de recherche. ... 37
Figure P2.1. Structures des analogues après la réaction d’alkylation du miconazole. ... 49
Figure P2.2 Structures des différents sels d’imidazolium et de pyridinium... 50
Figure P2.3 Structures des différents sels d’imidazolium et d’ammonium ... 50
Figure 2.1. Formation, maturation, and dispersion of bacterial biofilms ... 55
Figure 2.2. Synthesis of alkylmiconazolium salts ... 56
Figure 2.3. S. aureus biofilms labeled with Live/Dead stains after a 24 h incubation in growth medium (LB broth). (a) Negative control (DMSO). (b) Positive control (70% ethanol). (c) Miconazole (2.4) at 50 μM. (d) Miconazoctylium bromide (2.5c) at 50 μM. (e) Miconazoctylium bromide (2.5c) at 25 μM. (f) Miconazoctylium bromide (2.5c) at 12.5 μM. ... 58
Figure 2.4. MRSA biofilms labeled with Live/Dead stains after a 48 h incubation in growth medium (LB broth). (a) Negative control (DMSO). (b) Positive control (70% ethanol). (c) Miconazoctylium bromide (2.5c) at 25 μM. (d) Miconazoctylium bromide. (2.5c) at 12.5 μM. ... 60
Figure 2.5. Pre-formed MRSA biofilms treated with 50 μM antimicrobials in 0.9% NaCl. (a) Negative control (DMSO only, after 24 h). (b) Miconazole (2.4) (after 24 h). (c) Miconazoctylium bromide (2.5c) (after 5 min). (d) Miconazole (2.4) (after 48 h)... 60 Figure 2.6. Preformed S. aureus biofilms treated with different concentrations of miconazoctylium bromide (2.5c) over 24 h monitored in 0.9% NaCl solution. (a) Negative control
(DMSO only, after 24 h). (b) Positive control (70% ethanol). (c) 25 μM (1/2 × MIC after 5 min). (d) 12 μM (1/4 × MIC after 30 min). (e) 6μM (1/8×MIC after 6 h). (f) 3μM (1/16×MIC after 24 h). .. 61 Figure 2.7. Red blood cells hemolysis of compound (2.5c) and miconazole (2.4). ... 63 Figure 3.1. Structure of benzimidazolium salts previously reported (3.1, 3.2 and 3.3) and newly studied (3.4–3.6a–c). ... 73 Figure 3.3. Mature MRSA biofilms treated with different concentrations of 3.4a for 24 h in 0.9 % NaCl solution labeled with (A) live/dead cell staining and (B) crystal violet. (A) Live/dead cell staining of (a) negative control (DMSO only), (b) positive control (70 % ethanol), (c) 50 μg mL−1,
(d) 20 μg mL−1, (e) 12.5 μg mL−1, (f) 5 μg mL−1 (MIC), (g) 2.5 μg mL−1, and (h) 1.25 μg mL−1 of 3.4a.
(B) Larger pictures of crystal violet assay of (a) negative control (DMSO only), (b) positive control (70 % ethanol), (c) 50 μg mL−1, (d) 20 μg mL−1, (e) 12.5 μg mL−1, (f) 5 μg mL−1 (MIC) of 3.4a. ... 77
Figure 3.4. Mature MRSA biofilms treated with different concentrations of 3.5a for 24 h in 0.9 % NaCl solution labeled with (A) live/dead cell staining and (B) crystal violet. (A) Live/Dead cell staining of (a) negative control (DMSO only), (b) positive control (70 % ethanol), (c) 50 μg mL−1,
(d) 20 μg mL−1, (e) 12.5 μg mL−1, and (f) 5 μg mL−1 (MIC) of 3.5a. (B) Crystal violet assay of (a)
negative control (DMSO only), (b) positive control (70 % ethanol), (c) 50 μg mL−1, (d) 20 μg mL−1,
(e) 12.5 μg mL−1, and (f) 5 μg mL−1 (MIC) of 3.5a... 78
Figure 4.1. Structure of different generation benzimidazolium salts ... 88 Figure 4.2. Synthesis of second and third generation analogues ... 90 Figure 4.3. (top) MRSA biofilms labeled with Live/Dead stains after a 12 h incubation at the MIC concentration of the generation II antimicrobials. 1a) Negative control (DMSO); 1b) Compound 4.6 (PEB); 1c) Compound 4.11 (4-Bromo); 1d) Compound 4.14 (4-tert-butyl); 1e) Compound 4.21 (octyl); (bottom) SEM images of MRSA biofilms after a 12h incubation with MIC concentration of the antimicrobials. 2a) Negative control (DMSO); 2b) Compound 4.6; 2c) Compound 4.11; 2d) Compound 4.14; 2e) Compound 4.21. ... 92 Figure 4.4. (top) MRSA biofilms labeled with Live/Dead stains after a 12 h incubation with MIC concentration of the antimicrobials. 1a) Negative control (DMSO); 1b) Compound 4.34; 1c) Compound 4.39; 1d) Compound 4.42; (bottom) SEM images of MRSA biofilms after a 12h
incubation with MIC concentration of the antimicrobials. 2a) Negative control (DMSO); 2b) Compound 4.34; 2c) Compound 4.39; 2d) Compound 4.42. ... 94 Figure 4.5. Antibiofilm activity of studied compounds. Compared to commercially available BAC, all the benzimidazolium salts possess the ability to prevent the formation and disrupt mature biofilms. The dotted line represents the 25-fold selectivity towards MRSA bacteria compared to HC50. ... 95
Schéma 5.1. Structure du cation TBA utilisé comme agent de transfert de phase ... 103 Schéma 5.2. Structure de la béta-cyclodextrine et l’image d’une émulsion de Pickering à base d’eau, d’huile de parafilm et de cyclodextrine. ... 104 Schéma 5.3. Illustration de la formation du complexe huile/bCD dans un mélange eau/huile. Reproduite avec la permission (Leclercq and Nardello-Rataj, 2016[11]). Copyright 2016, Elsevier.
... 105 Figure 5.1. Structure and abbreviation of compounds used in this work. ... 109 Figure 5.2. Observations of b-CD/water/oil emulsions (10/45/45 wt.%, 3,200 rpm, 90s, 25 °C) using optical microscope (A) and WETSEM (B) and of b-CD/oil complexes using dry SEM (C). The inset shows the X-ray powder diffraction patterns of dry powders (D). ... 114 Figure 5.3. Ternary phase diagrams of the oil/water emulsion stabilized by b-CD at 25 °C. The composition is represented in weight fractions (3,200 rpm, 90s, 25 °C). ... 116 Figure 5.4. Backscattering (DBS) versus sample height and time at 25 °C (A) and shear viscosity as a function of shear rate (B) for b-CD/water/oil emulsions (10/45/45 wt.%, 3,200 rpm, 90s).... 117 Figure 5.5. Zone of inhibition obtained by the diffusion method against MRSA, E. coli and C. albicans of the b-CD/water/oil emulsions compared to the commercial cream (Monistat DermTM;
see the experimental section for more information). ... 119 Figure 5.6. Normalized biocidal factor (BF) against MRSA, E. coli and C. albicans for b-CD/water/oil emulsions (oil = carvacrol, C, or terpinen-4-ol, T) combined with [HMC][NO3] (2) or [C8MC][Br] (3).
... 121 Figure 5.7. Pre-formed MRSA biofilms (Live/Dead stains) treated with 1 µL of b-CD/water/oil emulsions (oil = paraffin, P, carvacrol, C, or terpinen-4-ol, T) without (1) or with [HMC][NO3] (2)
70% and aqueous solution of [C8MC][Br] 1 wt.%; see the experimental section for more
information). ... 122 Figure 6.1 Structure des différents bromures de miconazolium. ... 128 Figure 6.2. Structure des antibiotiques commerciaux le clotrimazole et le kétoconazole ... 129 Figure 6.3 Structure des différents analogues de sel de lutidine-bis-benzimidazolium ... 130 Figure 6.4. Structures des différents analogues de la nouvelle librairie d’antibiotique ... 130 Figure A1.1. Fluorescence intensity of compound (2.5c), miconazole (2.4), and BAC relative to tert-butyl hydroperoxide (100%) and DMSO (0%) in different cell lines (OD600nm = 0.1). Note: ROS production was not calculated for E. coli (MG1655) since a very low intensity was observed with positive-control. All cell lines were tested with unstained cells for autofluorescence. S. aureus ATCC 43300 and E. coli SK037. ... VIII Figure A1.2. Membrane permeabilization induced by miconazole (2.4) at different concentrations. Assay performed with a bacterial population of 2.2 × 107 CFU mL−1 (OD600nm = 0.1). ... IX Figure A1.3. Membrane permeabilization induced by miconazoctylium bromide (2.5c) at different concentrations close to the MIC value. Assay performed with a bacterial population of 2.2 × 107 CFU mL−1 (OD600nm = 0.1). ... X
Figure A1.4. Dose–response relationship between the concentration of (2.5c) and the ONPG hydrolysis rate for a bacterial population of 2.2 × 107 CFU mL−1 (OD600nm = 0.1). ... X Figure A1.5. S. aureus biofilms were labeled with Live/Dead stains after 12 h incubation in growth medium (LB broth) with a solution at 50 μM antimicrobials. (a) Negative control (LB only). (b) Positive control (70 % ethanol). (c) Miconazole (2.4). (d) Miconazoctylium bromide (2.5c) e) 50 μM BAC. ... XI Figure A1.6. Preformed S. aureus biofilms treated with different concentration of BAC over 24 h monitored in 0.9 % NaCl solution. (a) Negative control (DMSO only, after 24 h). (b) Positive control (70 % ethanol). (c) 330 μM (MIC after 5 min). (d) 165 μM (1/2 x MIC after 5 min). (e) 83 μM (1/4x MIC after 1 h). (f) 41 μM (1/8x MIC after 24 h). (g) 20 μM (1/8x MIC after 24 h)... XII Figure A2.1. Synthesis of 3.4a-c. Reagents and conditions: (i) Octylbromide, MeCN, 110 °C, MW, 82%; (ii) LiOTf, MeOH, 95%; (iii) LiNTf2, MeOH, 95%. ... XXI
Figure A2.2. Synthesis of 3.5a-c. Reagents and conditions: (i) Bromoadamantane, MeCN, 70 °C, 86% (ii) LiOTf, MeOH, 95%; (iii) LiNTf2, MeOH, 95%. ... XXII
Figure A2.3. Synthesis of (3.7). Reagents and conditions: (i) Bromoacetyl bromide, K2CO3, CH2Cl2,
88%. ... XXII Figure A2.4. Synthesis of 3.6a-c. Reagents and conditions: (i) (7), pyridine, acetone, reflux, 53%. (ii) LiOTf, MeOH, 95%; (iii) LiNTf2, MeOH, 95%. ... XXIII
Figure A2.5. MRSA biofilms labeled with Live/Dead stains after a 24 h incubation in growth medium with 50 μg mL-1 antimicrobial. (a) Negative control (DMSO 10%). (b) Positive control (70
% ethanol). (c) 3.3a-c, 50 μg mL-1 = 20xMIC. ... XXXII
Figure A2.6. Preformed MRSA biofilms treated with different concentrations of 3.4a after 24 h monitored in 0.9 % NaCl solution. Crystal violet assay (right) (a) Negative control (DMSO only). (b) Positive control (70 % ethanol). (c) 50 μg/mL. (d) 20 μg/mL. (e) 10 μg/mL. (f) 5 μg/mL (MIC). (g) 2.5 μg/mL. (h) 1.25 μg/mL. ... XXXIII Figure A2.7. Size (radius) of the aggregates formed by 3.4a at different concentrations. ... XXXIV Figure A2.8. Red blood cells hemolysis of compound 3.3a, 3.3b, and 3.3c. ... XXXV Figure A2.9. Red blood cells hemolysis of compound 3.4a, 3.4b, and 3.4c. ... XXXVI Figure A2.10. Red blood cells hemolysis of compound 3.5a, 3.5b, and 3.5c. ... XXXVII Figure A4.1. Microphotographs of oil/water emulsion (weight ratio: 1/1) stabilized with b-CD (10 wt. %) observed under: transmitted light illumination (A), reflected oblique light illumination (B) and 3D surface plot interpretation (C). ... LXXXVI Figure A4.2. Microphotographs of terpinen-4-ol/water emulsion (weight ratio: 1/1) stabilized with b-CD (10 wt. %) observed under: transmitted light illumination (A) reflected oblique light illumination (B) and 3D surface plot interpretation (C). ... LXXXVII Figure A4.3. DSC of the dry powders compared to the free b-CD... LXXXVII
Liste des sigles et abréviations
bCD : béta-cyclodextrine °C : degré Celsius
ADN : acide désoxyribonucléique AMPs : peptides antimicrobiens ARN : acide ribonucléique aq. : aqueux
Ar : aromatique atm : atmosphere
BAC : chlorure de benzalkonium Bn : benzyle
Boc : tert-butyloxycarbonyle Bu : butyle
c : concentration en g/mL cat. : catalytique
CCM : chromatographie sur couche mince CI50 : concentration inhibitrice à 50% CMI : concentration minimale inhibitrice DCE : 1,2-dichloroéthane
DCM : dichloromethane
DMAP : 4-diméthylaminopyridine DMF : dimethylformamide DMP : périodinane de Dess-Martin DMSO : diméthylsulfoxyde DO : densité optique DTT : dichlorodiphenyltrichloroéthane ESI : ionisation par électronébuliseur Et : éthyle
g : gramme
GRH : globules rouges humains h : heure
HPLC : chromatographie en phase liquide à haute performance HRMS : spectre de masse haute resolution
Hz : Hertz Im : imidazole IR : infrarouge J : constante de couplage L : litre LG : groupe partant
LRMS : spectre de masse basse resolution M : molaire
MED : microscopie à balayage électronique mg : milligramme MHz : megahertz min. : minute mL : millilitre mmol : millimole
MRSA : methicilin resistant staphylococcus aureus Ms : mésyle
N : normalité
NAG : N-acétyl-glucosamine NAM : acide N-acétyl-muramique
OMS : organisation monidale de la santé O/W : huile/eau
PEB : phényléthynylbenzyl PG : groupe protecteur Ph : phényle
Ppm : partie par million PQ : paraquat
py : pyridine
QAC : “quaternary ammonium coumpound“ quant. : quantitative
R : alkyle
rdt : rendement
RMN : résonance magnétique nucléaire ROS : espèces réactives de l’oxygène sat. : saturée
SEP : substance extracellulaire polymérique T : temperature
t.p. : temperature pièce TBA : tetra-n-butylammonium Tf : triflyle
THF tétrahydrofurane
TLC : "thin layer chromatography" TMS : tétraméthylsilane
Ts : tosyle UV : ultraviolet
δ : déplacement chimique μL : microlitre
Remerciements
Je tiens d’abord à remercier Andreea de m’avoir accueilli à bras ouverts dans son groupe. Dès les premiers jours, j’ai senti qu’elle nous donnait toute la liberté que nous voulions autant dans les horaires de laboratoire que dans les idées de projets. Cette attitude m’a permis de devenir indépendant et de travailler de manière organisée et structurée. Notre facilité à communiquer a permis à Andreea de toujours être là pour moi dans les moments un peu plus difficiles. Elle sait comment nous remettre en confiance et nous remotiver lorsque la chimie ne va pas comme on veut. Je tiens également à la remercier pour les conférences que nous avons faites ensemble : un voyage à Toronto, une semaine à Québec et un voyage en Italie que je n’oublierai jamais. Andreea, ne change jamais et merci pour tout !
Je tiens ensuite à remercier mes parents qui m’ont supporté lors de mon retour aux études. Je sais que ce n’est pas facile pour des parents de voir leur enfant retourner à l’école à 26 ans et je tiens à les remercier pour le soutien qu’ils m’ont donné.
Je tiens également à remercier Lorena qui m’a supporté pendant 4 ans de doctorat avec mes horaires atypiques et mes sorties tardives après le labo. Nous avons passé des moments difficiles durant ce doctorat et je suis fier de dire que cela nous a rendus plus forts. Je t’aime de tout mon cœur.
Ensuite, je tiens à remercier Bianca… et Alex pour qu’il ne chiale pas… encore. Elle s’est portée volontaire pour lire ma thèse et corriger les multiples, et quand je dis multiple c’est peu dire, erreurs de français. Merci beaucoup!
J’aimerais remercier Jean-Philippe qui m’a tenu compagnie dans les labos de Zargarian durant une bonne partie de doctorat avant de me délaisser pour Sherbrooke. Tous ces petits cafés pris au Café-in en chialant sur la chimie! Inoubliable.
Ensuite, je tiens à dire un énorme merci à Julien qui fut mon seul collègue masculin durant une bonne partie du début de mon doctorat. Julien, si vous ne le connaissez pas, est assez intense lorsque l’on parle de chimie. Cette qualité nous a permis de devenir de bons amis et de discuter
longuement de nos projets. Je tiens à le remercier pour tout son savoir sur la biologie qu’il a partagé avec moi. Sans toi, mon chum, j’aurais galéré à faire tout ça !
Ensuite, une mention spéciale pour ma première stagiaire dans le groupe. Margaux est arrivée avec nous un jour de verglas, les lunettes embuées et pleine de bonne volonté. Avec son enthousiasme, nous avons complété, durant son stage de M2, un article qui se retrouve dans cette thèse. Même si à ce moment tu galérais pour analyser des RMN, je sais qu’aujourd’hui tu es une étudiante exemplaire et que notre groupe avance grâce à toi.
Je tiens à souligner l’appui de mes autres stagiaires durant mon doctorat qui m’ont permis d’avancer grandement dans mes projets. Amélie, Thibault, Célia et Maude, je vous remercie d’avoir travaillé de manière assidue et autonome durant vos stages. Plusieurs des articles courants ou à venir sont en grande partie grâce à vous.
Je ne peux faire des remerciements sans mentionner les boys actuels du groupe. Guillaume et Pierre sont arrivés il y a près d’un an et, avec leur enthousiasme, m’ont motivé à travailler plus fort. Bonne chance à vous deux, même si je sais très bien que tout ira bien durant votre doctorat!
Ensuite, je tiens à remercier Julie V. qui m’a suivi dans nos soirées à la maisonnée et qui, avec son sourire, avait souvent le moyen de nous remonter le moral. Nous avons passé un voyage magnifique grâce à son organisation et ses suggestions de restaurants en Italie. Merci Juju.
Je tiens également à remercier Claude qui fut un collègue exceptionnel lors de mon passage à Brébeuf. Tous les cafés le matin et les soupes viet que nous avons pris ensemble sont des souvenirs chers pour moi. Même si tu as l’âge de mon père, dans ton cœur tu as 30 ans et c’est pour ça qu’on s’entend bien!
Je tiens à remercier Joëlle qui nous a toujours accueillis à bras ouverts dans ses laboratoires et qui nous a apporté beaucoup de soutien dans nos expériences de biologie. Je tiens aussi à remercier le professeur K. Wilkinson pour l’accès à ses laboratoires pour les biofilms.
Finalement, je tiens à remercier tous les membres du groupe avec qui j’ai passé des moments mémorables soit dans les sofas ou durant les fameux BBQ d’Andreea. Julie K., Solène, Marc, Philippe, Audrey, Samy, Julien D. et Thierry. Aussi, je voudrais remercier les gens des Collins qui partagent le corridor avec nous. Plus particulièrement Antoine qui, lorsque je disais bière dans les corridors, était toujours partant pour me suivre dans mes folies.
Chapitre 1 : Introduction générale
1.1 Découverte des pénicillines et leurs utilisations
Les bactéries multirésistantes ou pathogéniques sont un problème bien connu que de nombreux scientifiques considèrent comme étant la prochaine crise épidémique mondiale.[1] La
découverte de la pénicilline par Fleming en 1929, un antibiotique bêta-lactamine provenant d'une souche bactérienne de staphylocoque, qui agit comme inhibiteur de la synthèse des protéines membranaires, nous a permis de mieux gérer les infections et ainsi de sauver des millions de vies.[2]
Depuis l’introduction de cet antibiotique dans le milieu pharmaceutique, nous avons été non seulement en mesure de traiter des infections graves, mais également de diminuer de manière significative le taux de mortalité dans les pays développés et sous-développés. Dans le tiers monde, un accès minimal aux soins de santé et un environnement sanitaire déficient font en sorte que l’utilisation des antibiotiques est une solution à faible coût parfaite à ces problèmes.[3]
Figure 1.1. Structures des différentes générations d’antibiotique bêta-lactames. Cette grande découverte a mené des milliers de scientifiques vers ce domaine de recherche que nous connaissons aujourd'hui comme « l'âge d'or des antibiotiques ». En peu de temps, de nombreux antibiotiques ont été découverts.
N S H N O CO2H O N S H N O CO2H O MeO OMe N S H N O CO2H O NH2 N S H N O CO2H O CO2H Pénicilline G Méthicilline Ampicilline Carbénicilline
Les tétracyclines ont été introduites peu de temps après la découverte de la pénicilline sous forme d'antibiotiques à large spectre agissant comme inhibiteurs de la synthèse des protéines et pouvant être directement isolées de la souche bactérienne Streptomyces. De nouvelles générations d'antibiotiques (Figure 1.1) bêta-lactames ont été introduites plus tard, comme la méthicilline (2ème génération), l'ampicilline (3ème génération) et la carbénicilline (4ème génération).[4]
La plupart de ces antibiotiques nouvellement trouvés ont le même mécanisme d'action que la pénicilline, ce qui a rapidement entraîné une augmentation de la résistance bactérienne contre ces médicaments.[5] Il a fallu plus de 20 ans après la découverte de la pénicilline pour
découvrir une souche de bactéries résistantes à celle-ci, mais seulement 2 ans pour observer l’apparition de la souche résistante à la méthicilline.[6] Ces résistances nous obligent à utiliser des
doses de plus en plus élevées d’agents antibactériens et à développer de nouvelles molécules, ce qui entraîne des coûts énormes.
Ces résistances sont également observées chez les insectes et les animaux.[7] En effet, un
autre exemple est celui du dichlorodiphenyltrichloroéthane (DDT, figure 1.2), cet insecticide puissant découvert en 1939 par Paul Hermann Muller qui lui valut le prix Nobel en 1948.[8] Dix ans
après sa découverte, des résistances chez certaines espèces d’insectes étaient déjà observées et, pas moins de cinquante ans plus tard, 500 espèces d’insectes montraient une résistance à cet insecticide.[9]
Figure 1.2. Structure du dichlorodiphenyltrichloroéthane (DDT).
Cl ClCl
Cl Cl
1.2 Facteurs influençant le développement de la résistance bactérienne
De nombreux facteurs peuvent expliquer cette adaptation bactérienne rapide aux antibiotiques nouvellement trouvés. La surutilisation ou la surexposition d'un certain médicament à la fois dans le domaine de la santé et de l'agriculture, en particulier dans l'administration d'antibiotiques au bétail, est le premier facteur.[10]Ceci est particulièrement important lorsque l'on examine la résistance bactérienne, car l'approche typique pour traiter une épidémie chez l'animal était de donner à tout le troupeau la même quantité d'antibiotique pour limiter la propagation de l'infection.[11] Cette méthode qui a
été longtemps utilisée pour sauver les troupeaux est désormais interdite. En effet, les grandes quantités d'antibiotiques administrées au bétail ne sont pas entièrement métabolisées par les animaux. L’excédent d’antibiotique est excrété du système animal via l'urine ou les matières fécales. Ces excrétions, une fois en contact avec l'environnement, sont retrouvées dans les eaux de surface et/ou souterraines adjacentes à ces fermes.[12] L'exposition prolongée d’une souche
bactérienne à de faibles concentrations d’antibiotiques conduit généralement au développement d'une résistance à ceux-ci.[13]
Un autre facteur qui conduit au développement des résistances bactériennes est le manque de réglementation et la prescription inappropriée d'un médicament.[14] La structure
moléculaire et la ressemblance du mécanisme d'inhibition de plusieurs antibiotiques de différentes générations jouent également sur le développement de la résistance bactérienne.[15]
Il existe de nombreuses façons pour un microorganisme de se protéger contre un stress externe, comme l’exposition aux antibiotiques, pour éviter la mort cellulaire. Pour ce faire, les bactéries peuvent modifier les sites de liaison du médicament, perméabiliser leur membrane, induire l'excrétion de l'antibiotique via l’activation des pompes à efflux ou détruire/métaboliser simplement l'antibiotique.[16]
Ces mécanismes peuvent être utilisés par les bactéries individuellement ou en combinaison les uns avec les autres. [17] Dans le cas de la pénicilline, les souches bactériennes qui
avaient déjà été exposées aux premières générations d'antibiotiques ont très rapidement développé une résistance aux nouvelles générations d’antibiotiques.[18]
Ces analogues, plus efficaces, possèdent tous un noyau bêta-lactame au sein de leur structure. Par conséquent, ces organismes se sont protégés très rapidement via la surexpression des bêta-lactamases, enzymes permettant le métabolisme/destruction de ces antibiotiques.[18b]
Tout bien considéré, la modification structurelle d'une famille d'antibiotiques est un moyen efficace et crucial de lutter contre les infections bactériennes de plus en plus puissantes. Cependant, cette stratégie présente de graves lacunes et doit être considérée comme une solution à court terme du à l’apparition très rapide de résistance tel que décrit précédemment.[19]
1.3 Mécanisme d’action des antibiotiques
Afin de mieux comprendre les résistances bactériennes et pouvoir développer des outils pour les limiter, il faut s’attarder aux différents mécanismes d’action que peuvent utiliser les antibiotiques afin de tuer la bactérie. Ces différents mécanismes sont : l’inhibition de la synthèse D’ADN et D’ARN, l’inhibition de la synthèse protéique ou membranaire ainsi que la perméabilisation membranaire.[20]
Figure 1.3. Mécanismes d’action des antibiotiques et mécanismes de résistance bactérienne. Antibiotique
Quinolones, fluoroquinolones
Rifampicine
1.3.1 Inhibition de la synthèse d’ADN
Lorsque l’on parle de ce mode d’action antibactérien, on ne peut s’empêcher de parler de la famille des quinolones et des fluoroquinolones.[21] Ces classes d’antibactériens se différencient
des autres classes de composés découvertes pendant la même période puisque celles-ci ont été synthétisées et non extraites d’un microorganisme. C’est en 1962, dans les laboratoires de George Y. Lesher, que la recherche sur des dérivés naphthyridines comme agents antibactériens a commencé.[22] L'acide nalidixique (Schéma 1.1), obtenu par une synthèse en trois étapes à partir
du dérivé 6-méthylpyridin-2-amine, a montré une activité antibactérienne contre une variété de microorganismes causant des maladies chez l'humain et les animaux.[23] Une sélectivité contre les
bactéries Gram-négatives a également été observée lors des tests in-vivo.
Schéma 1.1. Rétrosynthèse de l’acide nalixidique à partir du dérivé 6-méthylpyridin-2-amine.
Au vu des résultats de cette étude, plusieurs scientifiques se sont penchés par la suite sur l’étude du mécanisme d’action de ce composé afin de mieux comprendre son activité antibactérienne. En 1976, un autre groupe de recherche a remarqué que l’acide nalixidique causait une accumulation de simples brins d’ADN.[23] Quelques années plus tard, les chercheurs
ont identifié une enzyme qui entaillait l'ADN chromosomique à double brin, et l'ont appelée « ADN gyrase » ou « topoisomérase II ».[24] Cette enzyme est présente autant dans les cellules
procaryotes que eucaryotes. Toutefois, les quinolones sont très spécifiques à la topoisomérase II présente dans les bactéries. Dans les années qui suivent, plusieurs nouvelles générations d’antibiotiques appartenant à cette famille ont été synthétisées afin d’en améliorer leur activité en ciblant un autre domaine de l’ADN gyrase : la topoisomérase IV.
N NH2 H3C N N Me O CO2H Et 6-méthylpyridin-2-amine Acide nalidixique
Plusieurs modifications ont été effectuées dans la structure de la molécule, mais on retient trois changements qui affectent grandement l’activité antibactérienne, comme dans le cas de la ciprofloxacine (Figure 1.4) : l’addition d’un fluor (bleu), l’addition d’un groupement pipérazine (vert) et l’ajout d’un cyclopropane (rouge). [25]
Figure 1.4. Structure de l’antibiotique de deuxième génération ciprofloxacine.
L'ajout d'un atome de fluor en position C6 augmente l'activité inhibitrice de l'ADN gyrase, facilite la pénétration dans la bactérie et fournit une activité contre les staphylocoques, une bactérie à Gram-positif. L'ajout d'un groupement pipérazine en position C7 a conduit à l’obtention d’une meilleure activité contre les bactéries à Gram-négatif et à une meilleure activité contre les bactéries à Gram-positif. Finalement, l’ajout d’un groupement cyclopropane en position N1 a conduit à une meilleure activité antibactérienne contre les pathogènes positifs et Gram-négatifs.[26]
L’utilisation excessive de ces antibiotiques à large spectre a donné lieu à une mutation au niveau de l'ADN gyrase dans les bactéries.[27] Ces mutations se produisent principalement dans
les gènes de la topoisomérase II et IV pour donner une bactérie très résistante. De plus, il existe des gènes qui influencent l'absorption du médicament dans les bactéries et la surexpression de pompes à efflux conduit à l’augmentation de l'excrétion des quinolones.[28]
1.3.2 Inhibition de la synthèse d’ARN
La rifampicine (Figure 1.5) est un antibiotique qui inhibe fortement l'ARN polymérase et bloque la synthèse d'ARN chez les bactéries à négatif Escherichia coli (E. coli) et Gram-positif Mycobactérium tuberculosis (M. tuberculosis). Celui-ci agit en se liant à l’ARN polymérase (ARNP), un complexe enzymatique responsable de la transcription de l’ADN en ARN. [29]
N C6 N N O CO2H F HN Ciprofloxacine N1 C7
Des études ont montré que cet antibiotique se lie à la poche de la sous-unité bêta de l’ARNP dans le canal ADN/ARN, mais loin du site actif. Le mécanisme d’action ainsi que les fortes interactions sont apportés par les groupements hydroxyles de la molécule (rouge) qui forment des liaisons hydrogène avec des résidus d'acides aminés de la protéine.[30]
Figure 1.5. Structure de l’antibiotique rifampicine.
Comme mentionné précédemment, les antibiotiques qui ciblent une protéine spécifique sont sujets au développement de la résistance bactérienne. En effet, les bactéries ont la capacité de muter le récepteur ciblé afin de se défendre contre ces antibiotiques. Dans le cas de la rifampicine, c’est la mutation d’un gène qui encode pour la sous-unité bêta de l’ARNP qui diminue drastiquement l’activité antibactérienne de cet antibiotique.
Cette mutation a apporté une modification du site de reconnaissance de la rifampicine, ce qui a diminué la capacité de former des ponts-hydrogènes avec la protéine et a entraîné une diminution de son activité.[31] Afin de diminuer l’apparition de cette résistance, la rifampicine est
souvent administrée en combinaison avec d’autres antibiotiques.[32] Néanmoins, le
développement de résistance a grandement affecté l’utilisation de cet antibiotique.
OH NH N N N Me OH O O O Me O OH Me Me OH HO Me Me AcO MeO MeMe Rifampicine
1.3.3 Inhibition de la synthèse de la paroi bactérienne
Une bactérie est composée d’une paroi cellulaire que l’on appelle également membrane plasmique ou cellulaire. Elle agit comme barrière entre le milieu extérieur de la cellule et le milieu intérieur de celle-ci et régit le transfert entre ces deux milieux.[33] Elle est principalement
constituée d’une bicouche lipidique qui se forme par auto-assemblage et qui contient majoritairement des phospholipides et parfois du cholestérol.[34] Elle comprend également des
lipides, des glucides et des protéines qui se modifient constamment, dépendamment des changements extérieurs que subit la cellule.
En d’autres mots, la cellule s’adapte aux différents stimuli extérieurs et permet de moduler sa fluidité.[35] Étant donné la grande importance de cette paroi, plusieurs chercheurs se
sont intéressés à des antibactériens qui peuvent inhiber ou interrompre la synthèse de la membrane cellulaire.
Comme mentionné précédemment, les pénicillines sont des antibiotiques à large spectre agissant comme inhibiteurs de la synthèse des protéines membranaires.[18b] Les pénicillines
agissent en bloquant la synthèse de la transpeptidase, une enzyme bactérienne responsable de la réticulation du peptidoglycane agissant sur la rigidité des membranes.[36] Les bactéries qui se
trouvent en phase de croissance ou de multiplication sont alors affaiblies, ce qui conduit à l’apoptose cellulaire. Nous savons désormais que les bactéries utilisent la surexpression des bêta-lactamases permettant le métabolisme/destruction de ces antibiotiques en limitant leur utilisation.[37]
Figure 1.6. Structure générale des céphalosporines.
Les céphalosporines (Figure 1.6), découvertes en 1945, sont également reconnues pour inhiber la formation de la membrane cellulaire par inhibition de la transpeptidase.
N S H N O CO2H R2 O R1 Céphalosporine
Même si ces antibiotiques ont la caractéristique d’être moins reconnus par les bêta-lactamases que les pénicillines, de nombreuses souches bactériennes résistantes à ces antibiotiques ont été découvertes. Ce phénomène s’explique par la similitude structurelle de ceux-ci (contenant le noyau bêta-lactame).
La vancomycine est une molécule qui a été isolée en 1953 des échantillons de sol. On utilisait cet antibiotique surtout pour traiter des infections au Staphylococcus aureus (S. aureus), une bactérie à Gram-positif qui est résistante à la pénicilline.[38] Toutefois, lors de sa découverte,
la vancomycine n’était jamais utilisée en premier recours puisqu’elle possède une très faible biodisponibilité orale. Des antibiotiques, comme la méthicilline (Figure 1.7), sont très adéquats contre les infections non résistantes au S. aureus, mais on a observé une toxicité accrue de ce composé au niveau des reins et de l’oreille interne, toxicité provenant d’une impureté et non de la molécule elle-même.
Cet antibiotique agit en inhibant la synthèse de la membrane cellulaire chez les bactéries Gram-positives. La vancomycine n’est pas active sur les bactéries Gram-négatives. Cette espèce de bactérie possède deux membranes cellulaires, une interne et une externe, et c’est l’incapacité de pénétrer la membrane externe qui rend cet antibiotique inactif dans ce type de bactéries.[39]
Figure 1.7 Structure de la vancomycine et de la méthicilline.
NH O H N HO O O O HO HO OH O NH2 OH O H N O NH2 O N H O O N H HO HO OH HN O H N O OH Cl O O O Cl Vancomycine N S H N O CO2H O MeO OMe Méthicilline
La vancomycine est très hydrophile et est capable de former des liaisons hydrogènes (Figure 1.7, rouge) avec les fragments terminaux D-alanyl-D-alanine du peptide liant les unités N-acétyl-glucosamine (NAG) et l'acide N-acétyl-muramique (NAM). En se liant, elle empêche la synthèse du peptidoglycane, une composante essentielle des membranes cellulaires qui est principalement formée des unités NAG-NAM. Cette inhibition résulte l’affaiblissement des membranes bactériennes, ce qui mène éventuellement à la mort des cellules.
1.3.4 Inhibition bactérienne via la perméabilisation membranaire
Il est important de parler de la vancomycine, puisque même si certaines souches bactériennes ont développé des résistances à cet antibiotique, la majorité des infections se traitent encore de nos jours avec cet antibiotique. En effet, n’étant utilisée qu’en dernier recours, la vancomycine n’a pas été exposée au grand public comme c’est le cas des pénicillines. De ce fait, très peu de résistances sont observées, ce qui fait de la vancomycine une très bonne référence lorsque l’on parle d’infection bactérienne à Gram-positif.[40]
Les infections persistantes impliquent des bactéries à croissance lente ou non croissante, qui sont difficiles à traiter avec des antibiotiques qui ciblent les processus de biosynthèse présents dans les cellules en croissance. Cette problématique s’applique à tous les mécanismes d’action que nous avons présentés auparavant.[41] En effet, si on prend comme exemple l’inhibition de la
synthèse de la membrane bactérienne, ce processus d’inhibition a lieu au niveau de la croissance bactérienne. Toutefois, les bactéries ont la capacité de se mettre en dormance, c’est-à-dire un état dans lequel la croissance et le développement sont temporairement arrêtés.[42] Ces bactéries
sont en mode conservation d’énergie et sont donc très difficiles à cibler avec les processus d’inhibition classiques que nous avons décrits auparavant.[43]
Une stratégie pour cibler autant les bactéries en croissance que celle en dormance est de perturber ou de perméabiliser leurs membranes.[44] Les antimicrobiens endommageant la
membrane sont généralement de nature lipophile et interagissent directement avec la bicouche membranaire, perturbant sa fonction (ou ses fonctions) et son intégrité physique.[45] Le fait de
cibler directement la membrane fait abstraction de l’état dans lequel la bactérie se retrouve dans l’organisme.
Récemment, l'efficacité de la perturbation ou de la fragilisation de la membrane bactérienne a été démontrée pour la daptomycine, un lipopeptide antibactérien.[46] Cet
antibiotique est généralement utilisé cliniquement pour traiter les infections au S. aureus. Lorsque l’on parle d’infection bactérienne, il est important de parler des infections au S. aureus, mais plus précisément des souches résistantes à la méthicilline (MRSA). Cette bactérie possède une capacité impressionnante d’adaptation à plusieurs antibiotiques connus et est rapidement devenue un danger dans les milieux hospitaliers.[47] En plus d’avoir la capacité de
développer des résistances sous leur forme planctonique, les MRSA ont la capacité de se protéger dans une matrice pour former ce que l’on appelle des biofilms.[6] Ce mécanisme de défense sera
détaillé plus loin dans la thèse. Les infections par les biofilms et la tuberculose sont deux exemples majeurs de besoins cliniques non satisfaits dus à des bactéries à croissance lente ou dormante et donc difficiles à traiter.[48]
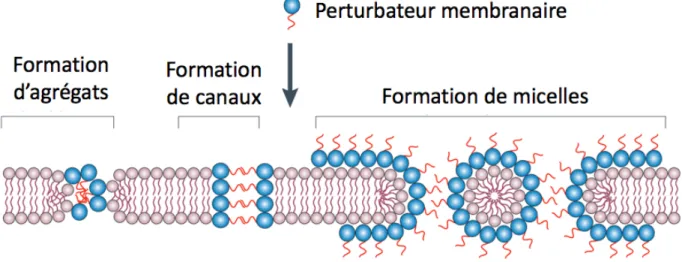
Les agents qui visent spécifiquement à perturber la membrane s’y insèrent et la déstabilisent (Figure 1.8).[44] Pour ce faire, ces composés peuvent soit former des agrégats dans
la membrane, soit ouvrir des canaux au travers de la membrane et/ou former des micelles.
Figure 1.8. Mécanisme d’action des agents perturbateurs de la membrane. Copyright 2011, avec la permission de Macmillan Publishers Limited. [49]
Les mécanismes de perméabilisation membranaire sont généralement rapides, ce qui limite le développement de résistance chez les bactéries. Par exemple, une résistance à la daptomycine, qui agit comme agent perturbateur membranaire, a été détectée peu de temps après son introduction clinique, mais les souches résistantes à ce composé sont extrêmement rares. [46]
1.4 La membrane cellulaire : une cible de choix
1.4.1 La bicouche phospholipidique
Peu importe la nature de la cellule, elle possède une membrane externe composée d’une bicouche phospholipidique.[50] Cette bicouche est imperméable aux molécules hydrophiles, tel
que les ions, ce qui permet aux bactéries de, par exemple, réguler le pH et la salinité de leur espace intracellulaire.[51] Les membranes biologiques sont principalement constituées de
phospholipides qui représentent plus de la moitié de leur composition.
Figure 1.9. Exemple de deux phospholipides : la phosphatidylcholine et la phosphatidylsérine.
Les phospholipides sont composés d’une tête hydrophile chargée (bleue) et de deux queues hydrophobes, des longues chaînes carbonés aliphatiques, non chargées (rouge) (Figure 1.9).[52] P O O O O Me3N O O R1 O O R2 P O O O O O O R1 O O R2 NH3 O O Phosphatidylcholine Phosphatidylsérine
Tête hydrophile polaire
Deux queues hydrophobes apolaires
Dans le cas de la phosphatidylcholine, une molécule isolée à partir de la lécithine de jaune d’œuf, on dénote un groupement ammonium (NMe+) et un groupement phosphate (PO4-) chargé,
formant la tête polaire, ainsi que deux longues chaînes d’acide gras (R1 et R2), formant les queues
hydrophobes. En solution aqueuse, les phospholipides s’auto-assemblent pour former des bicouches lipidiques, des liposomes ou des micelles (Figure 1.10).[53]
Figure 1.10. Représentation de l’auto-assemblage des phospholipides en solution aqueuse. Lors de l’auto-assemblage, les têtes polaires sont orientées de façon à faire le plus d’interactions possible avec le milieu aqueux extérieur, laissant ainsi les chaînes aliphatiques hydrophobes former des interactions de van der Waals entre elles et limiter leurs contacts avec les molécules d’eau, phénomène que l’on appelle effet hydrophobe.[54] Ces interactions
stabilisantes sont au cœur de la formation des bicouches lipidiques.
1.4.2 La membrane bactérienne
Les bactéries sont des entités unicellulaires dépourvues de noyau et entourées d’une membrane. Le matériel génétique, tels que l’ADN, est dispersé dans le cytoplasme.[55] Les
organismes eucaryotes unicellulaires ou multicellulaires, tel que les cellules humaines, sont généralement plus grosses et possèdent un noyau interne entouré d’une membrane qui contient tout le matériel génétique. Plusieurs mécanismes d’action antibactérienne se basent sur cette différence majeure entre les cellules procaryotes et eucaryotes pour cibler les protéines ou l’ADN plus facilement accessibles chez les bactéries que chez l’humain. Il en résulte des composés plus efficaces contre les bactéries et peu toxiques pour l’être humain.[56]
Bicouche de
Lorsqu’on s’intéresse à développer de nouveaux agents déstabilisateurs des membranes cellulaires, on doit également se pencher sur la composition des différentes membranes bactériennes, cette barrière qui leur confèrent une protection contre les éléments externes. Ces membranes diffèrent grandement d’une souche bactérienne à une autre (Figure 1.11).[55]
Par exemple, les membranes des bactéries Gram-négatives (Gram-) sont constituées d’une
couche phospholipidique externe et interne, entrelacée d’une monocouche de peptidoglycanes, alors que dans le cas des bactéries Gram-positives (Gram+), la bactérie est composée d’une seule
membrane phospholipidique, surplombée d’une couche de peptidoglycanes plus épaisse que dans le cas des Gram-négatives.[57] Il est donc, en théorie, plus facile de perturber et déstabiliser
la membrane des bactéries Gram+ telle que celle des bactéries MRSA. Malgré le fait que les
bactéries Gram- peuvent causer des infections graves, nous nous sommes concentrés, dans les
travaux présentés dans cette thèse, sur l’inhibition de la prolifération des bactéries Gram+, et en
particulier sur les MRSA.
Figure 1.11. Structures des différentes membranes bactériennes.
1.4.3 La membrane bactérienne des S. aureus
La membrane de cette souche bactérienne Gram+ est très intéressante puisqu’elle
possède des propriétés bien spécifiques.[58] De par sa structure, la S. aureus est très résistante au
lysosyme, une protéine qui est impliquée dans la défense contre les infections bactériennes.[59]
Membrane cytoplasmique Couche de peptidoglycanes Membrane cytoplasmique Membrane externe Couche de peptidoglycanes Lipoprotéine Lipopolysaccharides Gram-positive Gram-négative
Cette caractéristique est innée chez cette souche, ce qui lui confère une résistance naturelle contre notre système immunitaire. En plus de la bicouche lipidique, les membranes des bactéries Gram+ sont composées d’une couche de peptidoglycane qui contient de l’acide
téichoïque (Figure 1.12). [60]
Figure 1.12. Structure de l’acide téichoïque.
L’acide téichoïque peut s’attacher fortement à la membrane en formant des liens covalents avec l’acide N-acétylmuramique des peptidoglycanes ou tout simplement on se liant à la membrane cytoplasmique avec une ancre lipidique. Par conséquent, la couche de peptidoglycane des bactéries Gram+ est plus épaisse que celle des Gram- qui ne possèdent pas
d’acide téichoïque.[61] La fonction principale de cet acide est de fournir une rigidité accrue à la
membrane en attirant et fixant des cations, tels que le sodium et le potassium, en formant des paires d’ions avec les anions phosphate (PO4-). De ce fait, la membrane de S. aureus attirera
fortement les composés cationiques, caractéristique que nous utiliserons à notre avantage lors de la conception des antibiotiques présentés dans cette thèse, afin de cibler cette membrane bactérienne.
1.5 Les levures, un organisme cellulaire semblable au nôtre
Les levures, ou champignons, sont des organismes unicellulaires qui se rapprochent plus des cellules humaines que les bactéries, puisque celles-ci sont des cellules eucaryotes.[62] Dû à
leur forme unicellulaire, les levures et leur génome sont souvent utilisés comme modèle des cellules humaines, permettant de comprendre des aspects fondamentaux du fonctionnement de nos cellules.[63] En utilisant ce modèle simplifié, les travaux du Dr. Yoshinori Ohsumi sur le
renouvellement cellulaire par autophagie se sont vus octroyer le prix Nobel de médecine en 2016.[64] Dans un autre domaine, les travaux du Dr. Roger D. Kornberg sur le modèle moléculaire
de la transcription chez les eucaryotes lui valut le prix Nobel de chimie en 2006.[65]
O P O O O O OH P O O OH O HO NH O O P OH O O OH n
Il n’existe pas moins de 1500 espèces de levures aux usages multiples, comme la fermentation dans la bière et le vin, ou pour la fabrication du pain.[66] Toutefois, les mycoses ou
infections fongiques (aux levures) causées par le Candida albicans (C. albicans) sont les plus fréquentes dans le monde.[67] De plus, comme les souches de MRSA, les C. albicans sont reconnus
pour se protéger des composés antifongiques en formant des biofilms.[68] Une partie des travaux
présentés dans cette thèse se penchera sur l’inhibition de cette souche et la possibilité d’empêcher la formation des biofilms.
1.5.1 La membrane des levures
Bien que les levures soient des organismes qui se rapprochent énormément des cellules humaines, leur membrane se rapproche plus de celle des bactéries Gram+. En effet, la membrane
des levures est composée d’une bicouche phospholipidique surplombée d’une membrane externe.[63] Cette dernière est principalement constituée de manoprotéines, de polysaccharides,
des glucanes et de chitine (Figure 1.13). Ces composantes forment un réseau très organisé/rigide déterminant la forme et la taille de la cellule.[69]
Figure 1.13. Structure de la membrane des levures (haut) et structure des différentes composantes de la membrane externe (bas).
Membrane cytoplasmique O OH CH2OH O OH n β-glucane-1,3 O OH OH O OH m β-glucane-1,6 O O OH CH2OH NHAc n O O NHAc CH2OH OH Chitine Membrane externe
Dans le cas des levures, la membrane externe subit une pression interne que l’on appelle pression de turgescence, supérieure à celle observée dans le cas des bactéries.[70] Les
perturbateurs ou interférents membranaires sont donc des candidats idéaux pour ce genre de microorganisme, puisqu’une fragilisation de la membrane mène généralement à une lyse cellulaire amplifiée par la pression de turgescence.[71]
1.6 Détermination de l’activité antibactérienne
1.6.1 La concentration minimale inhibitrice
Afin de déterminer la capacité ou l’activité antimicrobienne des antibiotiques présentés dans cette thèse, on déterminera d’abord la concentration minimale inhibitrice (CMI).[72] Cette
concentration est définie comme étant la plus petite concentration d’un antibiotique qui inhibe la prolifération des bactéries. Afin de déterminer cette valeur, on prépare une solution mère de bactéries à partir de bactéries gardées en dormance à -80 degré Celsius que l’on met dans un bouillon stérile de nutriments. Cette solution aqueuse nutritive est généralement composée de tryptone, de levure nutritionnelle et de sel (NaCl).[73] Une fois en contact avec ces nutriments, les
bactéries se multiplient jusqu’à l’obtention de la concentration bactérienne désirée. Cette dernière est déterminée en mesurant l’absorbance de la solution à 600 nm, qu’on appelle également la densité optique (DO).[74] Cette longueur d'onde est spécifiquement choisie pour les
mesures de DO bactérienne, car contrairement aux longueurs d'onde UV, le 600 nm n'est pas nocif pour la bactérie. La concentration bactérienne est très importante puisqu’elle nous informe sur la nature des bactéries dans la solution.[75] L’absorbance désirée se situe entre 0,1 et 0,2 afin
d’obtenir des bactéries saines et en pleine croissance. En dessous de cette valeur, les bactéries sont en phase de latence (aucune multiplication ou très peu) et, au-dessus de cette valeur, les bactéries sont généralement en phase stationnaire ou bien elles sont tout simplement mortes.[76]
Figure 1.14. Représentation d’une plaque à 96 puits utilisée lors de la détermination de la CMI.
Une fois la solution mère préparée, on fait le mélange dans une plaque à 96 puits avec différentes concentrations d’antibiotique (Figure 1.14). Cette plaque est ensuite incubée à 37 °C pendant une période de 12 à 24 h. On utilise généralement trois rangées de puits par antibiotique, afin d’obtenir un triplicata sur chaque mesure, ce qui assure l’obtention d’une valeur précise et exacte de la CMI. Après 24 h, on analyse les plaques visuellement, ou par spectrométrie d’absorbance, afin de déterminer la CMI. Pour ce faire, on note simplement la concentration à laquelle les bactéries ont arrêté de proliférer. Une différence majeure d’absorbance est généralement visible entre les puits.
Pour chaque expérience de croissance bactérienne il faut vérifier sa validité. Dans l’exemple de la figure 1.14, l’antibiotique X donne une valeur de CMI reproductible, tandis que l’antibiotique Y donne des valeurs exactes, mais non précises. C’est un exemple où nous devons éliminer cette expérience et la refaire, afin d’obtenir un triplicata exact et précis pour l’antibiotique Y.
De même, il faut toujours s’assurer que les conditions d’expérimentation sont adéquates. Pour ce faire, on utilise trois tests qui nous permettent de confirmer l’obtention d’un résultat valable. Premièrement, on incube un antibiotique de référence qui possède une valeur de CMI connue pour la souche étudiée et on compare ensuite notre expérience avec cette valeur théorique (Figure 1.14, rangée H).
Gradient décroissant de 12 concentrations d’antibiotique Composé de référence Antibiotique X Antibiotique Y CMI







![Figure 1.26. Interaction entre le miconazole et le noyau flavohémoglobine. [136]](https://thumb-eu.123doks.com/thumbv2/123doknet/7563809.230028/68.918.110.809.102.429/figure-interaction-miconazole-noyau-flavohémoglobine.webp)