t
.'
1
APPROCH:C A LA SYNTHESE DE PHOSPHATES CYCLIQUES;
ETUDE D'UN NOUVEAU GROUPEMENT POUR LA PROTECTION DES AMINES
pa li .,
.
;JOAN BOIVIN
Thèse présentée à la Faculté des études avancées et a
de la recherche de l'université McGill comme exigence partielle pour l'obtention'du grade de .
Maître ès SClences
Département de chimie Université McGill
Mo~all
Québec, 'Canada ,r©
décembre 1984SYNTHESE DE PHOSPHATES CYCDIQUES; PROTECTION DES .i\MINES
c
/
1
/
f..
J
\
!
Chimie M. S'e.
APPROCHE A LA SYNTHESE DE PHOSPHATES CYCLIQUES;
ETUDE D'UN NOUVEAU GH:OUPEMENT POUR LA PROTECTION DES AMINES
par
Joan Boivin
RESUME
/
Une s8rie de composés modêles a ét6 ~tudiée en vue de-la synthèse des phosphates cycliq0es de la s8rine Il et de
• 0 "
la cystéine 12-. L'acide salicy Iphosphonque ~~,. son sel de potassium et ses esters propylique, bcnzylj.gue et allylique ont ainsi été préparés. Des essais,ont également été faits . avec des dérivés de l'acide sallcyllque portant un amide en
position 4 du noyau aromatique ainsi qu<ayec les acides anthranllique'et thiosaJicyllque.
L'emplol Ides groupements azido-4 butanoyle, ~-nitro-phényl,acétyle et o-ni'trophénoxyacétyle pour la protection d'une
,
amine a été étudié. Ces de'ux derniers subs ti tuants ont pu être enlevés par la réduction sélectlve du groupe nitro en amine et cyclisation. Cette réductlon a pu être faite sans affecter les doubles liaisons carbone-cArbone pr6sentes dans
la molécule.
~~----~-~-~--:-.
.,
1
,...
J, , , 1 Chernistryl\1.
Sc . .APPROJ...CH TO THÊ~ SYNTHESIS OF CYCLIC PHOSPHATES i
III'
STUDY OF A NEW GROUP FOR
THe
PROTeCTIoN OF AMINES\\
by
Joan Boivin
ABs'rRACT
As an approa~h to the synthesis 0 [ . the cyclic
phosphates of serine Il and cystewe l~, a series of model compounds was stud.lcd. Silllcylphosphûric dCl.d 15, ils
1
potassium s~lt and its propyl, benzyl and allyl esters were prepùred. Sorne further attempts were made with derivatives of salicylic aC1J having an amide on the 4-positlon of the
" \
\
aromdtlc ring as weIL as wilh anthranilic acid ëlnd thios"alyc1-lie acid.
The use of 4-azidobutanoyl, o-nitrophenylaeetyl and 0
,
o-nitrophenoxyaeetyl substituents as amine protecting groups vas investigated. Tt vas possible to remove the two latter by selective reduction of the nitro group into an amine and cyclisation. This rcduction could be done without affecting cirbon-carbon double bonds present in the molecule.
-"
REMERCIEME~TS
Je tiens tout d'abord à exprimer mes remerciements à
mon directeur de recherche, le professeur George Just, qui
m' a accueill ie dans son labora taire et d,-'n t 1 es suc]gestions
/ o~t conduit ~ 1'01aboration ~e cette thÔS0.
,
Mes sincÈ't'es' remerclcments vont 'C'j,1 Icnlcnt. à mes
comJi>atJnons de t.L"Llv;;all pC'ur leur anll.tl.é el leurs encoulclgements, particul ièremen t au docteur 1\. ;>admapr iyêt dont l'aide et tes
'conseils m'ont (tÉ' précieux tout au long a' cc trav'élil..
.f
Je remercie enfln le département de chimie de l'Université HcClll pour son aide fl.nancipll~, le docteur
A.O. Mam~r et ses cryllègues pour la spectroqraphie de masse, monsieur D. Stossel pour la révision d~ manuscrit, madame
4
A. eerrone-Mancinl. pour le service de daclylographie et tous ceux qui, de près ou de loin, ont collaboré à la ri'ùlisation
de ce projet.
1
Hi
-\
TABLE DES MATIERES
RESUME . ABSTRl\CT .
RÈMERCIEMENTS
. .
....
...
TABLE DES MATIERESAVERTISSEMLNT
.
.
..
....
. .
NOMENCLATURE . INTRODUCT-ION • • .ie:;;;. ...
.
.
1. PARTIE T!lEOR1QUl CHAPITRE PREMIER . . . . ...
.
....
1.1.1 Tent~live de synthèse des phosphates cycliques de la L-sêrine 11 et de la L-cystéine 12
1.1.2 Synthèse du chlorure de l'acide salicyl-phosphorique ~ . . . • . . ,
1.1.3 _Réact~on de l'acide'sa1icy11que avec le pentachlorure de phosphore. Synthèse de l ' acide salicylphospho,r~que 15 . ' . . . . PAGE i ii i i i vi vii 1 7 8 10 I l
1.1.4 Réaction du chlorure de l'acide salicyl-phosphorique 14 avec un alcool. Synthèse des esters propylique l8a, benzylique )8b
et allylique lSc . . . -~-. . . . -.-. 13 1.1.5 Synthèse du salicyrphosph~te de potassium 19 14
1.1.6 Tentative de synthèse d'acides amino-4-salicylphosphoriques ~
1.1.7 Tentative de préparation des dérivés de l'acide anthranilique 22a et de l'acide thiosalicylique 22b --- . • . . • .
- iv
-1
15
PAGE CHAPITRE 2 .
.
.
..
18 1. 2.1 Groupement azido-4-butanoy1e.
19, 1.2.2 Groupement o-nitrophénylacétyle 23 , 1. 2.3 Groupement o-nltrophénoxyacétyle.
261. 2.4 "R{>arrangement Je Cldlsen du
N-to-nltro-phénoxyacétylel .:1 yCl'1élte d'allyle -36
.
30 1.2.5 AddItIon lntramolécJlalre d'Jne amine sur ~ncarbonyle: formation de cycles à cinq et
S 1>: membres . . . . 34 CONCLUS ION . . 37 2. PARTIE EXPERIMENTALE 2.0 Génorali tés 38 CHAPITRE PREMIER CHAPITRE 2
45
REFERENCES.
. .
59.
.
v-<,
AVERTISSE.'-1ENT ,,-'
Les chiffre-s arabes seuls ont été utilisés dans cet expqsé. Soulignés, ils correspondent à des numéros de
composés chimiques. Entre parenthêses et non soulignés, ils renvoient
a
des référence~ blbllogr3phlqu~S.Les .lbbr6\'1:J.tlons usùelles t8ile:- LH, UV, RMN, etC'.,
sont employées.
-NOMENCLATURE
Les abbréviations et conventions suivantes sont employées:
Ar aromatique
CCM chromatographie sur couche mince doublet· i large m multlplet Pf point de fusion Ph phényle q quadruplet qt quintuplet 5 singulet t triplet THF tétrahydrofurane TMSCI t-BOC chlorotriméthylsilane t-butoxycarbonyle ,/ " vii -:: ,
.
\
\
\ o t. '
,"
INTRODUCTION
En .1929, un bactériologiste-anglais, Sir Alexander Fleming (1), faisait l'Ç>bservation qu'un'e mo'isissu,re verte,
Penieitti'/,fm n~tatum, avait 'une action inhib'itrice sur une
cul ture de staphylocoques. LI isolà tion :de la substànce
.1
secrétée par cette moisissure, la péniciJJfne, al'lait'marg~e:r le début d'une nouvèlle époque dans" le dOlllaine méd "Leal .
Quelques dix ans après Ja découverte faite par
, .
Fleming, l'équipe de HDward Florey et Er.ns"t Chain il .Oxford
,
réussissait à isoler la pénicIlJ i n 8 ' CJ~ qUél)J,t..ité stlff L!iant,é et
sous.. -une formE: rel a t j vement pure. Ceci a] L,) i t lç'U1 permettre de démontrer l'efficaclté
.,
dû
nouvel ,antibiotique et sa faible toxicité pour l'Qrgariisme. La pénicillin~ est util~s6e pour la première fQ,is en +941 pour traiter une infection chez un1
être huÎ'na'in.
Depuis ce temps, des proqrès fulgurants ont été faits en chimie médicinale et de nombreux antibiotiques 'ont été
p~éparés
(2)
~ On retrouve entre autres,- outre les pénicillines-!'
les céphalo~porines ~, l'acide clavulanique l,-l~'th.ienamycine
!,
l,es nocardicines 5 et les monbbactames 6'~,
n
l
..:. -1 -,/
;.
" \1 "'.
2OH
"~l=Q-S/-VNHz
.;=1yOH
N . ,o
.
.
' \
èo
2H
C02H 4 3 H ' RCON H li- RCON ' 'OD'SO-lI.1ODXCOHqOH
H "CO _ 2 H5
~. ~ \Tous ces composés ont une caractéris tique ço'mnune; la présence d'une p-laetame. Il s'agit jà d'un trait particulier qu'on ne retrouve p~ normalement dans les composés naturels (3). PoUr cette raison, les chimïstes ont été portés à croire que l'activité biologique de ces produits était dû~ à cette
structure.
Les
pénicill{n~s, l~s céphalo~rines
et, de façongénérale, tous les antibiotiques contenant une B-lactame ~gissent
sur les piJrois cellulaires en émpêchant la formation de chaînes peptidigues latérdlcs (4). L'enveloppe anormale de la cellule est ainsi incapable de retenir les composiJnts ~nternes, ce qui conduit éventuellewent à la destruction de l'organisme:
.'
3
La formation du réseau pep~ldique constituant une paroi cellulaire est catalysée par l'enzyme transpeptidase
(2), (4), (5)., Bien que les détails du mode d'action des
S-lactames SOlent éncore obscurs (3), l'hypothèse généralemeFlt acceptée veut que ces antiblotiques s?ient des aqents acylants qui bloquent les sltes actlfs de l'enzyme, empêchant ainsi les liens en~re les chaînes ~e ?eptlde~ d'être form~s.
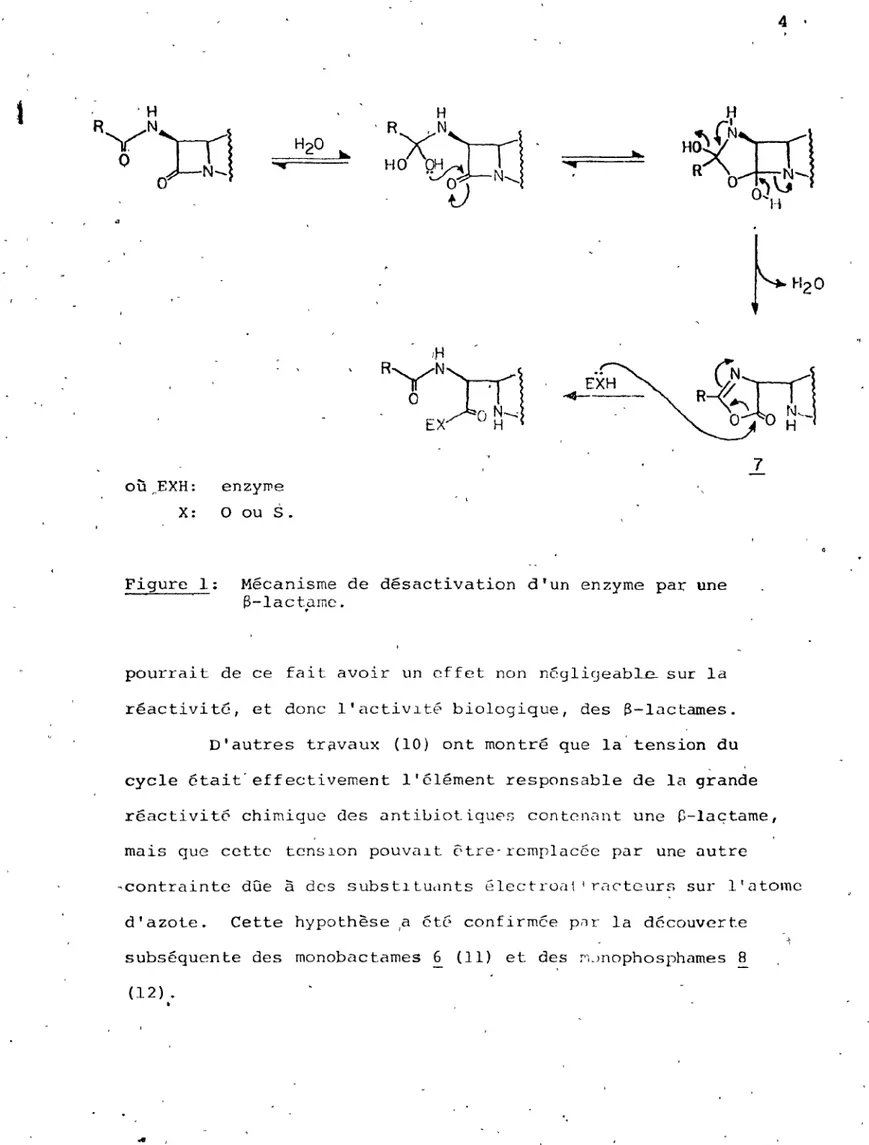
Selon lc mécanlsme proposé par KClth et ses collabora-teurs (6), la fJnction amide dl" lé; chaîr.l' 1.:lt8ri11e de la
B-l'actame seral't hydratée, ce qui condUlrait à l'ouvertlure du cycle et à la formatlon d'une oX.1zo1onc 7. Cette oxazolone est 'très réact.l\"C \"ls-à-V1S les tonct.lons .:llcool et thiol de l'enzyme. L'att-aque nucléophile de l'oxyqène qu du soufre sur la fonction carbonyle de l'oxazolone conduit B l'acylation de l'enzyme, réaction lrréverslble de désactivation des sltes. Ce mécanisme est lllustré à la figure 1.
Les am~dcs sont généralement peu réactlfs vis-à-V1S
des nucléophiles. Cependant, dans le cas des antlbiotiques à S-lactame, deux fàcteurs contribueraient à faciliter l'attaque sur la fonction carbonyle (3), (7), (8) 1 (9) la
tension du cycle à quatre membres falsant partl8 d'un
système bicyclHI1l8 et la falble résonilnce de l'i:lnllde dûe à la
..
non-planarité de ce syst_èll':e. Ces deux élE'mentt réunis pourralent expllqucr que les vitesses de réactlon soient'
au~mentées
par un facteur pouvant allerJusqu'~
1033 (3). Une contribution, mêLlc minime, d'un des deux éléments ~:écités1
4 .
'H H~~nl
RyN~DJ
'R N H20 ~HX,~)]j
....
...
....
0 -~
' - R 0 O"'H~~-~H20
IHR~~
RyN)xl
o
0 N-.; EX H OÙpEXH: enzyJTIe X: 0 ouS.
Figure 1: Mécanisme de désactivation d'un enzyme par une S-lactp.rnc.
pourrait de ce fait avoir un effet non négligeabLe sur la réactivité, et donc l'activlté biologique, des S-lactames.
n'autres trpvaux (10) ont montré que la tension du
7
cycle était'effectivement l'élément responsable de la grande réactivit6 chimique des antibiotiques contenant une O-laçtame, mais que cette tenslon pouvalt Btre-rcmplacéc par une autre
~contrainte dûe à des substl tu,mts électroai 1 racteurs sur l'atome
d'azote. Cette hypothèse ,a été confirmée pin la découverte subséquente des monobactames 6 (11) et des f'unophosphames 8
(12) .
•
(1 • < ~ H
ReON
)J
/OCH3o
...
p 07 ' 0 -8Il est de plus reconnu (13) que lracti~itG d'une
,-:.>
l3-lactame est prOt),.)rtlO)I~(ÜJ r à l ' augmentdt J on de la fréquence
dl absorption déH1S ] ' IR du groLlpement carbonyle. Il (~st donc
•
possible de supcoser n'J'un tel annde c:;cliqu'?, dont Ir: corhony-te
- 1
absorbe à une f rC!qu~'ncc de lUi'::> cm ~ et plus ( pourra.l têt_xe ' remp1 acé
p~r
une )'-lactone qUl apparaî t autour de 1780 cm-1La
SynJ1~~'C' dcc~
lact ones~
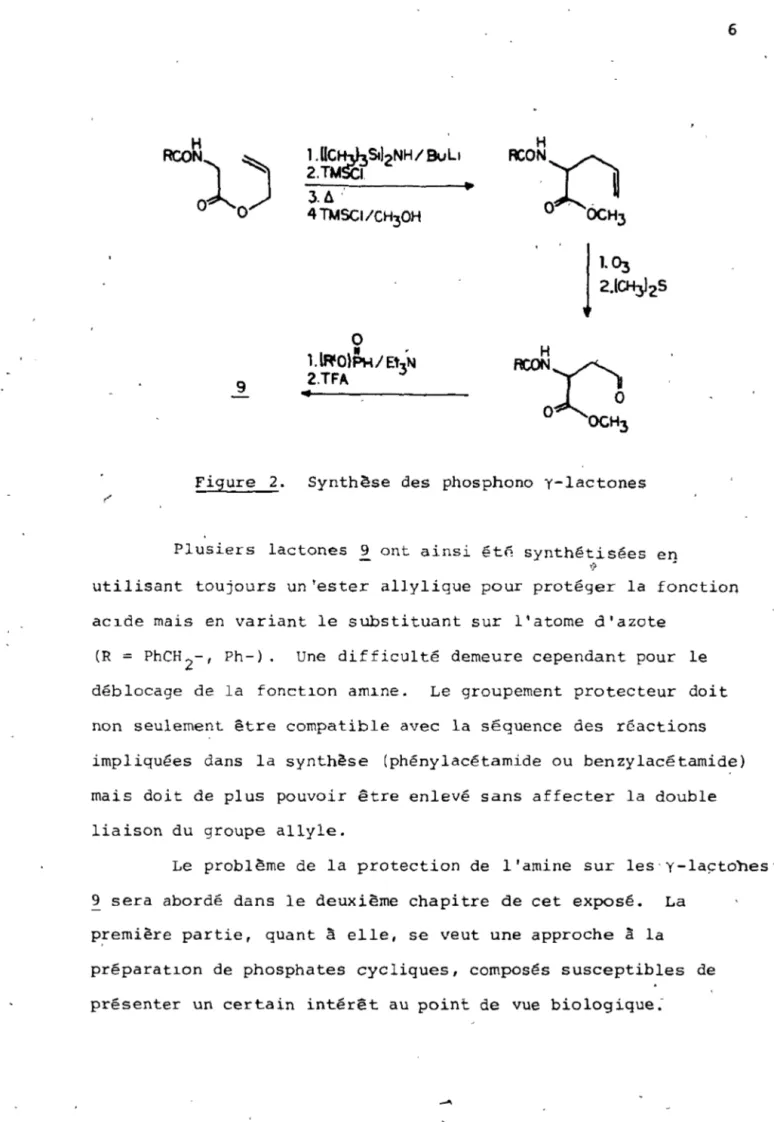
a 0té entrepr is(' dans notre lûborù Loire (14) et le d im6thyl phosphon.'1 te ~~ a é té obtenu de la mùni~re illustr~e à la flgurc 2. Tous les essais pour,
transformer cc diester en monoester 9b.ou en diacide 9c ont été infructueux. Il s'est avéré cependant gue les esters allylique~pouvLlient être ulilisés pour protéger l'acide
phosphorique, ces esters étant facilement transformés en leurs sels de potassium (15). H
/
RyN
n
a) R:PhCH2 R' : CJI 3 /ORlo
0 P b) R: PhCH 2 R' ~CH3' K O~ "ORI .~ c) R:PhCH2 RI :Ko
•
• 1.lRO)PH/Et 3N 2.TFA..
9Figure 2. Synthèse des phosphono y-lactones
Plusiers lactones 9 ont ainsi ét~ synthétisées eq
1')
6
utilisant toujours un~ester allylique pour protéger la fonction aC1de mais en variant le substituant sur l'atome d'azote
(R = PhCH2-, Ph-). Une difficulté demeure cependant pour le déblocage de la fonct1on am1ne. Le groupement protecteur doit non seulement être compatible avec la séquence des réactions impliquées dans la synthèse (phénylacétarnide ou benzylacétamide) mais doit de plus pouvoir être enlevé sans affecter la double liaison du groupe allyle.
Le problème de la protection de l'amine sur les'y-Iaçtones' 9 sera abordé dans le deuxième chapitre de cet exposé. La
p!emière partie, quant à elle, se veut une approche à la préparat10n de phosphates cycliques, composés susceptibles de présenter un certain intérêt au point de vue biologique:
,
1
1 PAR T l E THE 0 R l QUE
."
CHAP T'PRE PREI·1 IEf<
.
,~n observant ~a structure des antlbiotiques
!
à ~ et ~,nous~emarquo~s
qu'en plus de la lactame à quatre membres, tous sont porteurs d'un groupement acide. -Celui-ci peut être soit un acide carboxylique adlùcent à l'azote du cycle (~à ~) SOl t un <)clde sulfonJ.que .§. Ou phosphonlqUE. 1 dtLlChé directe-rrent à l'atone d'azote. Cet é1clde est un grour:e attracteur d'électrons dont la fonction est d'activer l'i1ITlide jXlur P2mettre ] 'oUverturc du cycle (figure 1) .Nous avons avancé l'hypoth~se qu'une monophosphame ~ pourrait être refermée pour donner les structures IOa, lO'a
qui s'apparen~ent de par leur torme àux pGnlcllllnes
!
et auxcéphal~porines ~. Un des groupcITents méthylène p6urralt éventuellement être remplacé p~r un atome pe soufre lOb, lO'b
ou d'oxyg~ne IOc, lO'c. Ces composés, croyons-nous, sont
susceptibles d'être biologiguement actlfs, étant donné leur ressemblance avec les antiblotigues déjà connus.
H H
RCON
hx,
~ONt:(XJ
a) X:CH 2 Y:Ol
b) X:S Y:CH 2o
N_P- OHo
N, /Y
içf
o?
P 'OH c) X:O Y:CH 2 10 101Les structures 10 et la' peuvent cependant être
.
simplifiées davantage. Tout en conservant la chaîne latérale,
-(
..
8
le groupe acide et la fondt~on carbonyle, qui sont les €l€ment~
responsables de l'action biologiqu~, II est possible d'enlever le cycle il qUùtre membres. Les structures obtenus sont alors anûlogucs <lUX compo.sés
g
etg,
dérivés de la sérine et de la'\')
cystéine.
Nous dGcrlvons dans ce premier chapitre une approche
a
Id synth~se des composés Il el 12.d'essai s a 6.tC: tent~e avec des c.1ichlorophosphates d' alkyle et les acides ~minés L-sérine et L-cystélne. Le peu de stabllit6 des produJ ts obtenus nous a cependan t condul ts
a
faire d'abord une étude de la réactlvlté de composés mode; 1 es dérivés ùes-acides sallcylique, thlosalicyllCjue et anUJranillque.-~ .,
1.1.1 Tentative de synthèse des phosphates cycliques de la L-sérine
11
et de la L-cystéine 12La synthèse du phosphate cyclique Il à partir du
dérivé t-butoxycarbonyle de la L-sérine a été tentée selon une méthode décrite par Grzéskowlak (16) pour la préparation
d'esters phosphoriques. La t-BQC L-sérine obtenue commerciale-ment est séchée toute la nuit sur chlorure de calcium sous vide pU1S mise en prGsence de dichlorophosphate de méthyle~ aussi obtenu commercialement, et de pyridlne. La rGaction se fait dans l'éther anhydre
a
aoe
et sous atmosphère d'azote.L'apparitlon d'un précipité de chlorure de pyrîdln1Uffi indique que l'attaque de l'oxygène sur le phosphore suivie de
9
présente 501.15 1<1 forme d'une huile jaune qui n ',a pas pu être
recristallisée. Les spectt-es
rou,
IR ct de masse ne permettent"f!I
pas l'identifica~ion du mélange.
H
t_HOCN:C:
- 01
o
/p-ORo
Ilo
'1
H
LBOCN /""-.... ' - / "5\
1
OR J p" O~"O./IIo
1?Un essal de synthèse du thlophosphd re cyel ique de Id cystéine 12 a éqalement ét0 fuit. ' L'acide cnr.iné est d'abord transformf en son d~riv6 t-buloxycdrbonylc. Ccci est effectué en faisant réagir la L-cyst6ine avec un éqni.valent de
di-t-.
'butyldicarbonate dans du dim6thylfotmamide'co~tenant deux équivalents de tr16thylamine.
Le prodult obtenu est ensulte traité de la façon décrite pour la t-BOC-L-sérine. On isole encore une fois une huile
jaune non identif iable à partir des spectres
Ri·m,
IR et de masse.Etant donné l ' impossibili té dans laquelle nous nous
trouVlons de ca~,ùct6rlS,er ,les projults des r{,ZlcLions précéd~ntesJ nous avons résolu d'effectuer une étude de corposés mod~les
plus stables. La préparation de dérivés de l'acide salicylique 13a ~tant déjà d6crite dans la litt:ralure ('17), (l8), (19), .
t
."
, ,
Hf
(20), (21), nous avons opté pour ce sy tème·
,
.
a) X : 0 ;' R: OH , OR' , Cl
bl X:N,S R:OH,OR' ,Cl
13
Le noyau aromatlque, avec sa ca 6c~té de r6sonance, offre un 61ément Je stablllt6 5 De pl us, nous
uelure en remplaçant
1" ac ide sallcyl ique lni Ual par l'acidè n thraniU que ou
l'acide thlosallcyllque. Un prodult 13b ontenant une llaison entre le phosphore et un autre hét6roatome pourrait ainsi
/
être obtenu.
1.1.2 Synthèse du chlorure de l'acide sallc phosphori ue 14
Le chlorure de l'acide sa1~cylphosphoriqle 14 est prépar6 selon une méthode décri te par Montgomery t ses collaborateurs (21). Un m61ange d'acide sa11cy1lq e et d'oxychlorure de phosphore est chauffé cl
heures. Le spectre RMN du prodult n'lndlque qu'un multlplet dans la région 7,2 à 8,4 ppm, correspondant au noyau aromatlque. Le spectre IR est cependant ldentique à celUl obtenu par
-
.
•
I l
Pinkus et ses collaborateurs (20). Le spectre de masse, donne un schéma de fragm~ntation confirmant la structure 14 .
14
La chloru:r;-e de l'acide salicylphosphorique peut également être préparé par réaction de l'acide ~alicylique avec le trichlorure de phosphore pUlS oxyddtlon par l'oxygène du produit obtenu (équation 1) (20)
( r
~
0UHHOH + PCJ3 NZ
°
14Cette réaction présente ,cependant le désavantage d'être faite en deux étapes. Nous avons de plus trouvé que les résultats étaient peu reproductibles, l 'oxydatj,on ne prenant place que dans 50% des cas.
1.1. 3 R~action de l'acide salicyclique avec le pentachlorure de'ph6s phore. 'Synthêse de l'acide salicylphosphorique 15
12
phosphore pour donner le composé 16 (18). Celui-ci est
hydrolysé par deux molécules d'cau pour donner l'aclde
salicyJ-phosphorique 15. L'addltion de trols 6quivnlents d'eau résulte J
•
en l'ouverture du cycle et l'obtention du phosphate salicylique
17.
n
rQÇf
OH
Il©Çr
13rOCIOHl2
o '
OH 0 0 0•
15 16 17Ce dernier est un composé stable qui peut aisement être recristallisé pour donner des ~iguliles inc01orcs_
Nous avons cependant trouvé que l ' <'le: ide sa] i c:y lphos·-phorique 15 pouvai t être pl us f acile:'1ent préparé pax;- hydrolys e du chlorure 14 avec un équivalent d'e<:1u. Le spectre RHN du produit obtenu donne deux slgnaux: un pic élargi à 12,8 ppm correspondant au groupement acide et un mu)tiplet dans la région de 7,0 à 8,3 ppm pouvant être assigné au noyau aromatique'.
Le spectre tnfrarouge prése~Le bande large de 2500
-1
à 3100 cm correspondant au groupe hydroxyl e. ~Le carbonyle \,
appara 'ît à 1730 cm -1 et le système p=o à 12·50 cm -1. Le spectre de masse donne un signdl relativement importdnt pour l'ion
mol~culaire puis un pic de base attrlbu6 ~ la perte du radical
(
13
HP0
3•• Les fragments
a
m/z 92 et 64 sont dûs' à la pertesuccessive de deux carbonyles.
1.1.4 Réactlon du chlorure de l'acide salicylphosphorique 14 avec un alcool. Synthèse des esters pr_opylique l8a, benzylique 18b et allylIque lBS
1
Le chlorure de l'aclde scllicylphosphorique
!:!
réagi t avec un équlvalcnt d'alcool en présence de pyridin~ ou de tri6thylamlne pOUL donner un ester 18. lL est essentIel que les solv~nts et les r~~ctlfs s0lcnt blEG ,.
.
~ , ..(5 tan L donné que l t eau peut a tt~lCjuer le chlorure d' ac ide. Les esters propylique
l8a, benzylique !8b ct allylique 18c sont ainsi préparés.
a) R:-C3H 7 b) R:-CH2C6HS
c) R:-CI12CH=CH2
18
La purification d~ ces composés esL rendue difficile
"
par leur réactivité envers l'eau. Ce sont tous des anhydrides " et l'attaque nuc160phi le de l' cau sur un' des groupes acides résultant en l'ouverture du cycle sera relativément façile .
..
"
Les spectres IR montrent tous ~l'absorptlon du groupe
-1
P=O autour de 1250 cm et l'absorption ~u carbonyle autour de
-1
,.,)
14
-'.
dans chacun qes spectres de masse. La chaîne alkyle est . ensui te perdue et un hydrogène capté pour donner un pic à
m/z =-200 correspondant à la~ructure de l'acide'12.
1.1.5 Synthèse du sa1icylphosphate de potassium 19
Le sel de potassium de l'acide saljcylphosphorique ~ 19 est préparé à partir de l'ester allylique ISc selon une méthode déjà utilisée pour le déblocage des
phosphono-y-lactones (14), (15). Le cllvage du groupe allyle est effectué
/
-.
hexanoiiite
---..
~-~>'....
en faisant réagir l'ester avec une so]ution d'éthyl-~
de potassium dans l'acétate d'éthyle en présence de tétrakis- ) triphénylphosphine palladium.
Le sel de potassium 19 pur est obtenu avec un rendement d4 50%.
Le spectre RMN ne montre qu'un multiplet correspondant
\
l,; t \
: ! au noyau aromatique. La bande d'absor~tion de la fonction
acide dispara~t complètement dans l'infra~9uge. La fréquence
-1
du groupement p=o passe de 1270 cm pour l'acide à 1250 pour
-1
15
Ce déplacement vers des fréquences plus basses est dû à la plus grande délocalisation des électrons sur le phosphoryle. Le composé 19 ne présente aucune activité antlmicrobienne.
-~
1.1.6 Tentative de synthèse d'acides amino-4 salicylphosphori-"
Puisque les dérivés 11 et ~ que nous prévoyions
éventuellement synthétiser étaient porteurs d'une chaîne amide, nous avons voulu vérifier l'effet d'une telle chaîne placée sur le noyau aromatique de l'acide salicyl~que. Une tentative pour préparer les dérivés d'acides amino-4 salicylphosphoriques 20 à partir des acides am~no~4 salicyliq~es bloqués ~ l'amine
,co 21 a été faite.
,
'i H \ O· HRCON~I
R C O N « O H
a) R:C6HSCH'2-o
b'~
b) R:CH 3 -0 0 1 c) R:C 6HSCH2 O-20 21Les composés ~la et ~ sont préparés selon une méthode décrite par Drain et ses collaborateurs (22). L'acide amino~4 salicylique est mis en solution dans la pyridine et chauffé à lOOoe avec du chlorure de phénylacétyle ou d'acétyle. Les
produ~ts obtenus sont ensuite traités avec l'oxychlorure de
t
16
phosphore de la même façon que dans lé cas de l'acide
salicylique. Une huile brune est obtenue dans les deux cas. Les spectres IR
et
RMN ne permettent pas l'identification des composés. Nous 'croyons cependant que la fonction amide ait pu• 0
réagir- elle aussi avec l'oxychlorure de phosphore.,
Un dernier essai' est effectué avec le compos8 21e. CelUl-ci est d'abord préparé .3 partlr de l'acide amin()-4 salicylique en SUlvant une procédure employée par Gut~man et Boissonnas po,n- b t('q\lCl Id [crct 1 ~ ~ é1m~ ne Ijl ~ a s~ r: (::<' 1)
Un équivalent de chlorofbrmate de benzyle est ajout~ ~ une solution carbonatée de l'aeiae. Le produit 1solé réagit avec l'oxychlorure de phosphore pour donner aUSSI une hUI~e non identifiable.
1.1. 7 Tenta~ive de préparation des dérivés de l'acIde anthranilique 22a et de l'acide thiosalicylique 22-b
La synthèse des dérivés de l'ac1de anthranIllqll 22a et de l'acide th-iosalicylique 22b a été tentée. Le coup.lage de l'acide anthranilique avec l'oxychlorure-de phosphore ~'a donné qu'une huile brune qui ne peut être caractérisée par , les spectres. 0 ,
OC"
" 1o
['CI
" " a) X:N 0 b) X:S22
t
(\
\
17
Le chlorure de l'acide thio;;al icylphosphorique 22b
\
~ cependant pu être-obtenu. Son spectre RMN ne m9ntre que le multiplet dû au noyau aromatique. L'IR indiyue la présence du carbonyle
. /
l ;"1
à 1770 cm- et celle du-phosphoryle à 1240 -cm . La réaction de ce chlorure d'acide avec l'alcool allyllque,
selo~ la m~thode emp16yée avec le chlorurr de l'acide
sallcylphosphorlque
!l:,
n'd, cepel)da~t pas donné les réslilt'ats attendus. Les spectres montrent encOre une fois un mélange,
non identl~lablc Je produits.
SUlte aux r~sultats peu c6ncluants que nou~.avons obtenus pour les (j':;rlyés des .::lclàus anthrallllique et
thiosallcyllque alnSl Liue pour les 3.cldes ùmino-4 sallcyllquesi suite égale~ent â la dlfflculté que nous éprouvons â obtenir
-les phospha tes cyc llgues de l'acide sallcy ligue sous~ une forme sufflsaw.ment pure, nous avons décidé de ne p~'s pousser , ce ~rojet plus avant.
Les dérlv8s des acides sallcyllques devaient êtte 'les modèles stables de prodults plus réactlts. Or, i l s'est
avéré que ces. composés soient eux-mêmes trop lns tables pour en permettre une étude approfondie. Nous avons cependant' montr6 que ces produ~ts peuvent être synth6tis6s et nous en avons caractérisé quelques-uns.
"
"
CHAPITRE 2
Lors de' la synthèse des lactones ~ (14), (15), l'ester allyl~que a ~té employé pour bloquer l'acide phosphorique. Le groupement protecteur pour la fonction amine doit donc pouvoir être enlevé sans affecter la double lialson du groupe allyle et doit être compatlble ayec les,
réacti~~iquéeS
dans la synthèse (flgure 2, page 6)~'La préparation des phosphono y-lactones exige que le , groupement protecteur de l ' amln,e pUlsse rési,ster aux condi tians suivantes: - bases fortes;
- chlo~otriméthylsllane;
,
températures modérement.élevées; oxydation par l' 0
zone;-- exp::>sluon à des agents réducteurs (sulfure de à.i.rtl§thyle) ; - exposltion aux réactlfs phosphonylantsi
acide trifluoroacétlque.
Il doit de plus être ralsonablement stable- vls-à-V1S des' corditions normales d'lso1ation (aclde et base faibles én solution aqueuse), doit pouvoir être enlevé sans hydrolyser la lactone et sans rédulre la double lia~on allyllque.
Les substltuants généralement utllisés pour désactiver une fonction amlne (24) tels le t-butoxycarbonyle, le benzyl-oxycarbonyle, le groupe phtallmldo, etc., étant lncompatlbles avec les condltlons mentlonnées, nous nous sommes mlS à la recherche d'un nouveau groupement protecteur qui soit en
-19
- 1
accord avec les réactions que nous envisagions. Nous nous trouvions devant cette alternative quant au mode de clivage du groupement: la photochimie et l'hydro9~nation catalytique douce, compatible avec les llùlsons doubles.
Des travaux qui touchaient à l'emploi d'un substituant, o-nltroclnnamoyle pour protEaer une amlne ayant d6j~ ~té
effectu6s ~ans notre Laborcltolre (~5), notrL Ch01X s'est
porté sur la deuxlême possibllit6. Nous avons d'abord faltO Lme c:;érlc "j'eSsal5 5\.:1" des c:ompcc,:s IT,odülcs L'n utlll!iant des dérivés aZldo-4 butanoyle, o-nltroph§nylacGlylc et o-nitro-phénoxyacétyle d'.:rCldes amln6s, pG1S nous a\ùl1b lenlé
d'appliquer la lm'· thoc1e .3 la syn tllèse d'une phosphono y-lactone.,
1. 2.1 Groupement azido-4 butanoyle
Les travaux effectu8s par Just et Rosebery (25) ont
~ontré
que le subs tl tuant o-nl troclnnamoyle pouvai t êtreplacé sur la fonctlon affilne d'un aC:lde affiln6 et que l'hydro-gêna tion catalytique de l ' o-ni troc innamamide' 23 conduisait
à la forma~lon d:hydrocarbostyril 24 et à la libération de l'acide affilné original.
ln
20
Qne méthode semblable a ôté utilisée par Cqrey èt ses collaboré! teurs (26) pour préparer la spi rolac tone 26 à
partir de l'azido~none 25.
o
lVa
y
( 3)C5
lin
-n
25?6
croire qu'un substituant a7icJo21C)11e pourrz,il 0tre ut.ilisé
pour protéger une Clmine, la r{>duction de ] ' ,1:1.ic1e libérant
le composé () r irJ 1 nCl) pa r un 1Il6CdlH '=-inC' S 1IIll 1" 1 t l~ il CQ J II i propose>
par l'équat.ion 2. Il est à renlirqupr cepellchnt que dans les deux cas précédents, un cycle
a
six membres est formé. Comme i l est connu (27) que les cycles à cinq membres sont formés plus facilement que ceux à six membres,' nous avons penséutiliser un substrat qui 1 en se refermant après hydrogénation,
nous donnerait une y-lactame. C'est ce qui nous a amené â choisir le substituant azido-4 butahoyle pour une première sérlc d'essé1is.
Nous avons d'abord lent6 de pr6pClrcr l'acide azido-4 butanolque ~ partir de l'acide bromo-4 butanolquc. L'obtention de ce dernier par l'hydrolyse de y-butyroLtctone en présence d'acide bromhydrique est une réaction connue (28), (29) et
21
ne pose aucune difficulté. Selon Reeves et Bahr (30), la substitution de l'atome de brome d'un bromure d'alkyle par un groupe azido s'effectue de fàçon quantitative en utilisant un catalyseur de transfert de phase. Ces résultats ne sont cependant pas reproductibles dans le cas de l'acide bromo-4 butanoïque.
L'aclde-azido-4 butanolque est d6nc préparé en chauffrant ~ reflux une solutlon éthanolique de bro~o-4 butanoatc d'éthy1f2 en présence d'a?idure dF' sodium (31)
L'azldo-4 butûnoate d'éthyle ainsi obtenu est ensuite 'saponifié et l'acide est isolé.
Après de nombreux essalS avec différents réactifs, nous sommes arrivés à coupler l'acide avec une amine par l'intermédlaire du chlorure d'acide. Celul-ci peut être obtenu par réactlon avec le chlorure de thlonyle (32) mais
""
"de mellleurs résultats sont obtenus avec le chlorure d'oxalyle (33). En faisant réagir le chlorure d' aziclo-4 butan.oyle avec la benzylamine en présence de triéthylamine, le ~-benzyle azido-4 butanamide 27 est isolé.
(4)
1
J
Bien qu'il exis te un nombre' de façons de trans former un ~~~: en am~ne (34), notre choix s'est porté sur
l'hydrogénation catalytique en pré~ence de borure de nickel. Cette méthode est d'utilisation fac~le et est compatible avec le type de composés que nous vou~ons en ce sens qu'elle n'affecte pas les doublels ,lial.sons carbone-carbone.
22
La réduction de l'amide 27 est effectuée ~ température r
-ambiante et sous une press~on d'hydrogène légèrement supérieure à l atm. La réact~on est SUlv~e par chromatographie sur
couche mince. Au bout de quatre heures, la tache correspondant au N-benzyle aZldo-4 butanamide est complètement disparue
et une autre tache plus polaire apparaît. Un spectre IR du prodult ~rut montre la disparlt~on presque totale de la b~nde aue à 1 t azide et l"
intens~fication
de la bande â 3300 cm-llaissant supposer ,,la présence d' une c;:tm~ne. Cette hypothèse
~
est co~fiimée par le spectre de masse qu~ montre un faible pic à m/z ::: 192 dû â
r'
ion moléculalre et le plC de base à ~/z=
149 correspondant au N-benzylacétam~de, produit de latransposition de McLafferty.
L'hydrogénation est répétée,en util~sant cette fois l'oxyde de platine (35), un catalyseur plus puissant, afin d ',obtenir une réduction plus complète. Une couche mince du mélange réactionnel après une heure indique encore une fois la présence de N-benzyle am~no-4 butanamide 28 seulement.
CI
Aucun changement n'est observé même après avoir chauffé â reflux dans l'éthanol pendant 24 heures.
-23
a~
N3~N~O,
H 0 '. 29Au cours de leurs essais, Just et Rosebery (25l
avaient employé leur substituant o~nitrocinnamoyle pour bloquer la fonctlon amine des esters éthyliques de la
glycylalanlne et de la glycylphénylalanlne. Il est vraisem-blable que les aCldes aminés SOlent de meilleurs groupes partants que les amlnes slmples, telle la benzylamlne,
étant donné la capaclté qu'a la fonctlon carbonyle d'accommoder L,
)
une charge négative. Nous avons donc tenté d'effectuer la cyclisatlon avec le N-(azldo-4 butanoyle)phénylglycinate de méthyle ~. Les spectres RMN, IR et de masse du prodult obtenu après hydrogénatlon sur borure de nlckel ne nous permettent pas d'attrlbuer une structure au composé .
•
1. 2.2 Groupement o-nltrophénylacétyle
Suite aux résultats obtenus pour le groupement azido-4 butanoyle, nous avons avancé l'hypothèse que la cyclisation ne s'effectuait pas comme prévue à cause d'une trop grande souplesse de la chaîne, souplesse qUl empêche l'approche de l'amine sur la fonction carbonyle. Un système plus rlgide,
..
24
dans lequel la mobillté des substituants serait .réduite, off irait de melileures ch ... lnces de succès. Nous avon~ donc opté pour le groupement o-nitrophér.ylacétyle, le noyau
aromallque présentant la caractérlstique de rigldlté que nous recherchions.
N'ayant trouvé dans la Ilttératur0 ~ucun précédent qui lndlque que la réductlon d'J~ groupemenl nitro par le borure de nlckel salt posslble, nous avonS d'abord voulu vérifier cette SUppOSltlon. No'Js 3"ons ,,1,,-'nc effl'ctué l'hydrogénatlon catdlytlque de l'dlcool o-nltr6henzyllque en alcool o-<lmlnoLcnzyllque. L'ilbsorptlon d'hyurogène est complète al, bout dl.. Jeux heures et une coùc:he nllnCe du
mélange réactlonnel montre la disparltlon totale du produit de départ. Les spectres IR et RMN confirment ce résultat.
L'o-nitrophénylacétctte de !T'éthyle est ensuite ... préparé par estérification de l'aclde o-nitrophénylacétique.
L'hydrogénation catalytlque de l'ester suivie par chromato-graphie sur couche mlnce nous lalSsc VOlr l'apparition-de traces d'oxindole ~ en plus d'une taché correspondant vrai-senililablement à l ' o-aminophf.nylacétate de méthyle. Un reflux de 2h30 dans l'éthanol, le solvant réactionnel, résulte en la disparltlon complète de l'amlne. Les spectres IR et RMN du produit obtenu sont en tous points semblables à,ceux donn6s dans la llttérature pour l'oxindole. De plus, le spectre de massQ donne un ion mOléculalre, qui est également le pic de base, ~ m/z
=
133 et présente un schéma de25
,fragmentation en accord avec la structur~ de l'oxindole
...
30 31
Le chlorure cl' o-ni trophénylacétyle 0st préparé en fajsant
est coupll> au (;}yc1.naU' d0 lr('tl:~'l(' POùl d ' 1 J ) ' ~- le
N-(o-·nitro-phénylacétyle)qlycinatc fIé mGt~11ç 31. Cel- ~C'i est ensuite hydrogoné dans des condl tlons similaires à c'.:>] les employées pour l'o-nitroph6nylac0Late de hl~thyle.
)'
L'absorption d' hydrogène es L cornr,]
C
.j~'2
en une heure.On ob~erve sur couche mince une tache plus polaire que le
produit de départ mais aucune trùce d' oxindole. On chauffe à '
reflux d.::tos l'éthanol et de l'oxindolc apparaît l.entement. La réaction est terminée après 28 heures de reflux.
Dans l'e~poir ~e trouver une façon de catalyser la forma lion du cycle 1 le produl t de l ' hydrogénù tipn est soumis
à un certain norilire de conditlons expérimentales. Celles-ci sont données au tableau 10. En observant los résultats obtenus 1
nous ne pouvons constater aucun changement appr~ciable dans les temps de réaction qui varient entre 24 ci. 28 heures. Nous remarquons cependant la décomposition du produit dans l'acide
•
26
acétique glaclal chauffé à une température de 65 à 70°C. Cet~e décomposition est caractérisée sur couche mince par une
traînée et par le changement de couleur du mélange réactionnel qUl passe de jaunâtre à brun.
TABLEAU l
Conditlons pour la cycllsatlon du produit de l'hydroyénation de 9
1
Sol van r .: Cl Lai.~. s e ~-LT 1 t -( OC) -~h(!ures) --- - -C 2HS,OH
-
reflux 28 C 2HSOH CF 3C02H reflux 27 CH 3C02H glac la l-
65-70 J.8 (décomposition)MeOH !-ICI reflux 24
l-C
3H7OH CF3C02H reflux 28
1. 2.3 Groupement o-nltrophénoxyacétyle
Bien que les résultats obtenus pour le groupement o-nltrophénylacétyle auralenL pu nous permettre d'en~loyer ce
dernler pour protéger la fonctlon amlne du cours de la synth~se des y-lactones ~, nous avons voulu pousser plus avant nos
recherches e~ avons fait une dernlère série d'essalS avec le groupe o-nltrophŒnoxyacétyle. En effet, nous SUpposlons que notre phosphono-y-lactone résisteralt au reflux prolongé
•• 1
27
nécessaire au déblocage de l'amine. Nous aurions cependant
pr6f~r6 que l~ cycljsation sc fasse plus sponLdn€ment.
puisque, contre toutes attentes, les cycles 3 cinq
membrC'~, sernblalcn 1: ê l-ré' 1--> lus di f f j cllc·s ~ forme c que les
cycles il six Int2mbres, nous avons résolu d'employer un
substituant (JUl pldsse c1onnL'r un cycle Zl SlX memlJn!s. Prenant
exemple sur les travaux de JllsL ct Rosebcl'y (25), nous ~vons
opté pour le s ubs t l tuant o-ni trophénoxYëlc(;tyl e 32.
~I
A(~yo
W
32 24 33
Le produit 33 form0 par cyclisation ne cH f fère de
l'hydroc~rbostyri1 ~, obLenu cl partir du sùbstiLuant
o-n1troclnnélmoylc, qu'en cc qu'un ù'::os qroupè,s CH
2 est
remp] élcé par un a tome dl oxyqi'ne. Le 'j'loup'emen ta-ni trophénoxy-acétyle ne poss~cle c('pend~nt aucune 11ai50n double
carbone-carbonl~ f ce (jeU le rend compél t1ble avec les cs ters allyliques
qui seron L présen ts c1zll1s lu y-lë1ctOI1C.
L'acÏLlc ()-nJ troph(n()xYë1cét lCJUC' est préparé ~ parLir
de l'0-nitroph0nol el de l'uclde chloroQc6tlque.
\ Une première
tentative pour rl'prochnrc 1c~ rê",ult,1t:s obtl::'nus po.r van
28
tournés vers une méthode utilisée d'abord par Thate (37) puis par Jacobs et Heidelberg (38), (39). Nous avons cependant découvert qu'il était préférable d'utiliser 1,5 équivalent d'acide chloroacétique plutôt qu'un équivalent comme le font
Jacobs et Heidelberg. (
1
L'acide o-nitrophénoxyacétique étant obtenu, nous avons essayé de le transformer en chlorure l'acide en
chauffant avec du pentachlorure de phosphore selon la procédure décrite par ces auteurs (39). Celte tentativ0 s'6tant avérGe infructueuse, nous avons employé le chlorure de thionyle et avons isolé le chlorure d'o-n~trophénoxyacclyle sous [orme
"
solide.
Le 'couplage du chlorure d'aclde avec le glycinate de méthyle est effectué selon la méthode déjà employée avec le chlorure d'o-nitrophénylacétyle. Le produll ainsi obten~ 34 est ensuite hydrogéné et l'oxo-3 hydrobenzoxazine 33 est isolée. Il n' exis'te dans la littérature aucune référence à , ce composé
* .
*
Il est .... ·ressorti ct' une discussion avec le Dr. G. Just que cette c réaction aurait déjà été utilisée par le groupe Bristol. Une recherche exhaustive de la littérature n'a cependant pasQ
29·
Le spectre RMN relativement simple montre deux signaux d'intensités rélatives 2:1 correspondant respectivement au noyau aromatique, et au CH
2' adjacent au carbonyle. La bande
-1
correspondant au carbonyle apparaît dans l'IR à 1700 cm • Ceci est probablement dû à l'augmer.tation de la résonance entre le doublet libre de l'azote et les '~lectrons du cycle aromatlgue aux dépens du carbonyle. On note également dans le spectre infrarouge la présence de deux bandes fortes à 1220 et 1050 cm-1
c,lréictéristiql,~s
de l'€lher ph0ny.1igue. Le spectre de masse montre le plC dû à l'ion mol6culaire àm/z = 149 (99~) et le pic de base à m/z = 120 qUl correspond
probablement à la structure 35.
~N02 •
~o----Y~~o~
+
o
35 36
Afin de vérifier la compatibilit~ de la réaction avec les groupements allyles, l'hydrogénatlon sur borure de nickel est effectuée à nouveau en employant cette fOlS le d~rivé
du glycinate d'allyle 36. TrOlS' 2q1Jl vû.lents d' hydroyène sont
•
absorbés après Ih30. Nous nOus attendlon/ à ce que l'ester allylique de la glycine ~olt uo ~o~pos~ plutôt volatile. Pour ce_tte raison, nous n'avons p.:lS essayé de l'isoler du mélange
,
1
...
30
réactionnel. Nous nous sommes plutôt contentés d'en préparer le dérivé dinitro-2,4 phényle
12
en faisant réagir l'amine attendue avec le dinitro-2,4 fluorobenzène en présence de bicarbonate de sod~um aqueux. Un solide jaune précipite qui est en tous points identique à un témoin de N-(dinjtro-2,4 phényl)glycinate d'allyle. Un rendement global de 91% indique que l'€tape de cycllsatlon s'effectue Lrès b~en et ~ib~re, l'am~ne de façon quasi quantltdtlv,e.
H 0
~NJl...o~
02N~N02
37
1. 2.4 Réarran0èment de Cla~sen du N-(o-nitrophénoxyacétyle) glycinate d'allyle 36
Le réarrangement de Claisen du
N-(o-nitrophénoxy-acétyl~)glycinate d'allyle 36 est effectué selon une méthode
'mise au point par Bartlett et Barstow (40). Le dlanlon de
l'~stet est d'abord formé à -78°C puis silylé ~ l'alde de
ch~orotriméthylsilane et chauffé à
50-GOoe
pendant une heurg .Le composé dlsilylé ainsi formé est ensuite hydrolysé et
séparé des produits de départ par extraction. L'acidificcttion de la phase aque~se donne l'a9jde libre qui peut être estérifié à nouveau. Cette suite de réactions est illustrée à la flgure 3.
1-,
,
l
1
1
l
Fig~re'3. R~arrangoment de Clai~en d1
Gn
glycinate d'allyle.Nous avons donc tenté ce réùrJ;:"anqcm(~nt avrc , e
,
N-(o-nitrophénoxyacétyle)glycinate d'allyl~ 36. Le produit obtenu après ùcidification~'est pas caractérisé mais repris avec du méthanol et mis en présence de chlorotriméthylsilane
t
,.
" \ 39 " A'Une couchl? wince du proc1l11 t de la ,t'action montre cepend,:mt deux taches 1 la pl us po1 alre ayant un facteur de
rétention ident~icpCè à celui de 1 'o-nitroph{;nol. Le mélange
est donc repris ,:,'('C dc~ l'éthec, ) 'o-nltrop!1onoJ est extril.l.t / avec une solulio:\ ùqueusc, d'hyùro:..yd(;! de s"dium pt jdent-ifié.
La phLlSC 0l qûnique est s0chloC et 0\',lpor0c. L(~ spectr< RNN du produit olJtcnu montre trOJ S slgnaux d' inLensi tés
ré'lati ves 4: 2: 3 correspondant vriüsemblablcment à un noyau
L'Il' i r aXOUtjC
indiqul' la pr~scl.ce d' un ester et d'un groupement nitro .
.t
Ces données nous ont permis de supposer gue le produit obtenu était ] 'o-nitrop:!~noxyacétale de [T.5thyle~. Le spectre de masse confirme cette h~pothèse ùvec des pics à m/z 211 (ion moléculaire) 1 165 (perte d'un qroupe nitro) et 122 (perte
L'ester 3Y ne peut pr()v~'nil- que do lù perte du
groupcIllen t pro le c l CUl- o-ni troph0noxY(lc('ry 1('. Nous ne savons cepeudant pas ~ quel moment se prodult le clivage ami4ique et s ' i l s'agit du clivùgc du r110cJlIlt de dé'pêlrt 36 sous sa
forme dnibniqu0 ou disiJyléc>.
Ce réarrangement de Claisen avait été e~fectué à
de nombreuses reprises par d'autres membres de notre équipe de re@herche (14), (15) sur des composés qui ne différaient du nôtre que par le groupement' protecteur. Les substi tuan ts -qui avaient été
e~plOyés
incluaient les groupes acétyle,phénylacétyle, benzoyle, t-butoxycarbonyle et benzyloxy-carbony le. Il nous était dOllc cldlr que ,'5 r6sultcds
33
obtenus étaient dUs ~ une caractéristique lnhérente au groupé o-nitrophénoxyacétyle.
Nous avons refai t le réarrangement dans les mêmes .. conditions en SUL vùn~ la réaction par chroma tog raphie sur' couche mince afln de vérifler l'apparltioll cl 'o-nitrophén()l. Les premi~res traces de ce produit sont observées apr~s
l'addltlon de chlorotr1m6thylsilane et l'intensité de la tache va en augmentant avec la temp~rat~re pour ~Lteindre un
maxlmum après eWflron une heure de re flux. Le dlù.nion est
/
donc formé mais la sllylat10n subséquente entraîne une
él1m1n~tion de 1 'o-nitrophCnol plutôt gue le r6arrangement
attendu. Ce processus estAillustré à lù f1qure 4.
Quant au produit ~/ on ne peut que constater sa présence dans le mélange f1nal. Sc trouvant sous sa forme silylée ou sous forme d'ac1de Ilbre, son apparltion ne peut être SU1V1 par chromatographie sur couche mince.
En compurant ceCl aux dériv0s acétyle, phénylacétyle et benzoyle déJà étudlés, nous voyons CJu' une telle élimination est impossible. En effet, ces trOlS substltuants étant de
,
..
,
.
34
+
'
.
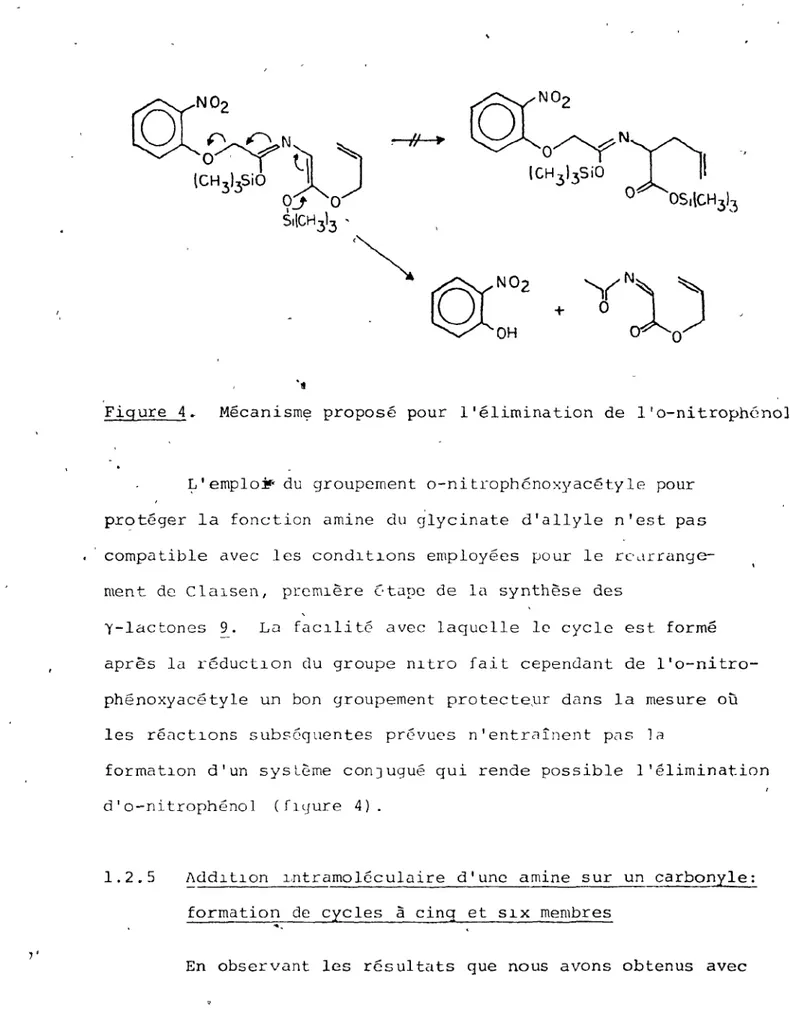
Figure 4~ Mécanisme proposé pour l'élimination de l'o-nitrophénol.
~'emploÎf' du groupement o-ni trophénoxyacéty le pour
prot6ger la fonction amine du 6lycinate d'allyle n'est pas compatible avec les condltlons employées pour le rCdrrange-ment de Clalsen, premlère 0tape de la synthèse des
y-lactones 9. La facllité avec laquelle le cycle est formé après la réductlon du groupe nltro fait cependant de l'o-nitro-phénoxyacétyle un bon groupement protectepr dans la mesure on les réactlons subséquentes prévues n'entraînent pas la
formatlon d'un sysLème conJugué qui rende possible l'élimination d'o-ni trophéno 1 (hlJure 4).
1. 2.5 l\ddl tlon l,ntramoléculùire d'une amlne s ur un carbonyle: formation de cycles
..
â cinq et SlX membres,
t
1
les trois groupements protecteurs que nous avons étudiés,
c'e5t-~-dire les groJpes azido-4 butanoyle,
o-nitrophényl-ac6tyle et o-nitrophéno~yac6tyle, nous nous apercevons que, conLrajrement- cl llOS illtc.'ntes, Je cycle à six melflbres est
formé beaucoup plus rapidement que le cy'cle à cinq membres (figure 5). ÇN3
MiR
, , (NH2 , (NHR-#--+
Qnol
.f-0 00
40
QG
0 0'~
0:l
S
0 b. ~OJ~
+-NHR NHR 24h 30 Q(NH 2 HOC
o
O"'YNHR N02o
~.-ryNHR
OC::t
+
~
0 0 33
Résultats obtenus avec les groupements azido-4
35
RNH2
RNH2 1
RNrlZ
bu tilOoyle 1 o-ni trophéllylucétyle et a-ni
trophénoxy·-acC>tyle.
D~ns le premier cas, l'arnino-4 butanamide obtenu par réduction de l'az.ido-4 butanamide ne sc referme pas pour donner la )-lactame ~O ct cc, m~Mc en chauffant. Nous savons
36
t
cependant que les y-lactones sont formés très facilement "lorsque le grpupe partant est un alcoolate. L'hypothêse qui voulait que l'amlde 40 ne soit pas formé à cause de la trop grande souplesse de la chaîne que rendrait difflcile la
rencontre des extrémités n'est pas complête. D'autres facteurs sont à considérer. Nous he sommes, au moment présent,
,
ma'lheureusement pas en mesure de fournir une expllcatlon pour ce résultat.
Le substituant o-nltrophénylacétyle, une fois réduit en o-amlnophénylacétyle, se referme pour donner l'oxlndole
30. Cette réaction ne s'effectue cependant qu'en chauffant
à, des températures d'envlron 70 C pendant 24 à 28 heures. En observant un modêle moléculalre du groupe o-nitrophényl-acétyle, nQus voyons que l'approche de l'amlne et du carbonyle est rendue dlfflclle par la rlgidlté du système aromatlque. L'angle d'attaque peu favorable pourralt ainsi être responsable de la lenteur de la réactl0n.
Ce facteur est éllmlné lorsque la chaîne latérale est allongée d'un atome, comme dans le cas du groupement
o-nltrophénoxyacétyle. La réduction du substituant nltro en amine condult à la cyclisatlon spontanée pour donner le produit 33.
t
CONCLUSIONLes' phospha,tes cycliques de la L-sérine I l et de la L-cystéine 12 sont des c'omposés plutôt instables et la
tentative qui a été faite pour les synthétjser- s'est avérée infructueuse. L'acide salicylphosphorique, son sel de
pàtasslum et ses esters propylique, berJzyl j'que et allylique ont été préparés et caractérisés. La pur 1 ;-lcatiOIl de ces composés ~st cependant difficile et resterait encore ~ faire.
Le Substltuant o-nltrophénoxyacétyle constitue un bon groupement protecteur pour la fonçtiOl, ctmine. La
réduction du groupe nl tro cr: traîne la çyc 1 LS lllon spontanée
pour donner l'oxo-3 hydrobenzoxazine l], lLbérant ainsi le substrat amlné. L'emploi de ce substltuant n'est cependant pas compatible avec les condl tlons du récn ;-clnyemen l du Cla~scn gue nous prévo:..-' ions util iser dans la synthèse des phosphono y-lactones 9.
Le substltuant o-nltrophénylacétyle pourrait aussi être employé comme groupement proteèteur pour une amine. Le déblocage exige cependant un reflux pro1ongé à des, températures modérément élevées. La réduction de l'azlde du groupe azido-4 butanoyle donne l'amine malS celle-Cl n'a pu êt~c refermée pour donner la l-lactame et ce, même en chau[ L:mt.
.,'
-t
\
2.0 Généralités
Les appareils suivants ont été utilisés pour l'enregistrement des spectres:
-- - l H-RMN: Varian T-60 ou T-60A (s tandard intern.e:
tétraméthylsilane) ;
- IR Perkin-Elmer 297;
- masse HP 5984 ou LKB 9000 (70 eV).
Les po~nts de fusion, pr~s en tube ~apillaire ouvert, na sont pas corrigés. Les abbr6viations et conventions
suivantes :,;,ont entploYL>es dûns ce tte par ~le:
38
- pf pour le pOln t de fuslon, donné' cn degrés Celsius; v pour la fréquence d'absorption en I~, dans les
un~tés
de cm-lpour les spectres RHN, l'échelle {, en ppm est
ut~lisée. J d6signe la consLJnte de couplùge et est exprlmée
en Hz. Afln d'al16ger la prEsentation, ~elletci n'est cependant, donn0e yue pour le~ couplages moins courants;
- pour les
spec~res
de masse, M+' d6signe l'lonmol~cu13lre et nVz le rapport masse/charge du fragment.
Les Chr()nlclto':Jraphl(é~S ont Eté f'::lltes sur colonne de
gel de sLlicc (0,05-0,20 mm) en applIquant une légère pression d'aIr. Les chromatographles sur couche mince (CCM) on été effectuGes SU! des [)lclqùes (j'3i.ur'llnium couvertes de gel de
sIlIce Merck fiOF
254 et révé16es par lrradiatlon UV et/ou par
irrunerslon dons èlne sol u tlon de molybdate d' dJTUT\onium et de sul fa te, c6rl<..juc ddns H
2SO 4 aqueux pUlS chauf f age, et/ou par
immersion dans une solutlon de n~nhydrine dans n-butanol puis chauffage.
CHAPITRE PREMIER
2.1.1 G:hlorure de l ' élcide sallcy Iphosphorique 14
Un mGLmge formé de 6,90 CJ (50 mmol) d'acide salJ.cylique et de 4,66 mL (50 mmol) d'oxychlorure de phosphore est chauffé doucement sur un bain d'hullc Jusqu'~ 130°C. Le mélange
réactionnel est mdlntenu J .:ette tempérdi lue Jusqu'il ce ql<e le dégagement d'acide chlornydrique SOlt terminé, puis refroidi
à température amblante. Ur. courant d':') ()tE' est ensuit'e pasRP
dans le mélange pour enlever l'excès d 'Ill:l. Un liquide Jaune-brun est obtCfl1., (lO,8 g). ~.JC prodult esl l~H1ployé sans
RNN ( 6 0 :vII [ z, C DC l -.) 15:
J 8,4-7,:: (m, Ar)
IR (pur) V: 1750 (C:::O) , 1290 (P=O)
masse (70eV) m/z: 200 (M+' - CHOH), 120 (200 - HP0 3),
92 (120 - CO), 64 (92 - CO)
1
"
2.1. 2 Acide salicylphosphorique 15
\
A une solutlon de 0,21 mL (2,6 mmo1) de pyridine sèche dans enVlron 25 mL de 'l'HF anhydre, on aJoute 0,04 mL (2,5 rrunol) d'eau. On ref~oldl â
ooe
pU1S on ajoute goutte-a-goutte0.,56 C] (2,5 mrl(1) du chlorure de l ' aClde sallcy lphosphorlque 14. On lalsse la réaction sc pourSUlvre 5 cette te~pérature pendant 0,5 heure, pU1S on lalsse revenl r à tempêtcl ture amb iùnte
•
40
et on agite pendant deux heures supplémentaires. Le mélange est maintenu sous atmosphère d'azote. Le précipité de chlor-hydrate de pyridinium est filtré et le filtrat évaporé pour donner 0,42 g d'une huile jaune.
RMN (60 MHz, ÇI?C1
3) 0: 12,8 (2, OH), 7,0-8,3 (m, 4H, Ar)
IR (pur) Ornasse (70eV)
V: 3200-2500 (OH), 1730 (C=O), 1250 (P=O) m/z: 200 (M+'), 120 (200 - HP0
3), 92 (120 -CO;, 64 (92 - CO)
2.1. 3 Phosphate salicylique 17
Un mélange de 0,55 g (2,6 rnrnol) de pentach10rure de phosphore et de 0,39 g (2,7 mmol) d'acide salicylique est agité à température ambiante. Après 0,5 heure, la réaction est chauffée sur,bain-marle Jusqu'à 60-65°e pendant encore
0,5 heure puis refroidie à oDe. On aJoute alors l mL d'acétone, l mL de benzène et, goutte-à-goutte, 0,15 mL (8,3 mmo1) d'eau. On aglte pendant 0,5 heure à cette température pU1S on dilue avec 2 mL de benzène et on abandonne toute la nUlt à
température ambiante. Le précipité est flltré, séché sous vide et rearista1lisé dans un mélange d'acétone et de berizène. On obtient 0,26 9 de. cristaux incolores (45%). Pf: 156-8°C.
RMN (60 MHz, CD
30D) 0: 7,5-6,6 (m, 4II, Ar), 4,6 (s, OH)
IR (KBr) V: 3100-2800 (OH), 1700 (C=O), 1225 (P=O)
masse (70eV) m/z: 200 (M+' - H
20), 120 (200 - HP03), 92 (l20-CO)
t
2.1. 4 Esters propylique 18a, benzylique 18b et allylique ISc , Les esters sont pr6pJr6s selon la méthode dêcrite en2.1.2 pour l'acide salicylphosphorique 15 en remplaçant l'eau par 2,5 mmol de l'alcool fraîchement distillé. Une huile
j~e
est obtenue dans tous les cas. Es ter p:copylique:.0,51 g,' es-ter bcnzylique: 0,73 S, ester al'lyll.que: 0,1)5 g.
Ester propylique 18a
'
-RNN (60 MHz, CDeL:)) 0: 8,4-7 .0 (m, 4H, -,'r) 1 4.3 (t rlfdoublfr -' IR (pur) masse (70eV) OCH 2, JHP = 10 IL) 1 1,8 (m, CCH 2C) 1 l , () \ t , CE 3 ) v; 1735 , c r ) 1 ::'270 , ' , ?=O) I"[l/z: 242 (M+·),ûn
(M+· - C 3H7+2H), 200 (M+' - C 3H7+H), 120 (200 - HP03) Estèr benzylique 18b RMN (60, HHz, CDC1) 0: 8,4-7.2 (m, 4H, Ar) , 7,2 (s, 5H, Ph) , 5,1 (d, b1 2, JHP = 9 ~z ).
IR (pur) V: 1720 (C=O), 1255 (Z, ?=O) masse (70eV) m/z: 290 (M+·) , 200 (M+'
-
CH 2Ph+H) , 120 () (200-
'1?0 3) , 92 d20-
CO) 1 91 (CH 2l-]) ) Ester allylique.
RMN .( 60 MH z, C DC l 3) é: 8 , 4 - 7 , 2 (m, 4 H, Ar), 6, 8 - 6 , 2(in,
=C H) , IR (pur) masse (70eV) 6 1 2 - 5 1 5 (m, ;:::C H 2)' 5, 0 - 4 1 5 (rn , OC H 2 ) \!: 1725 (C=O), 1230 (9)/ P=O) m/z: 240 (M+:),~~~M+'
- C 3H5+2H), 20r} (M+.: - C 3HS+H), 120 (200 - HPO;) •..
#\.
/
•
"
2.1. 5 Sa~cy1phosph<ilte de potassium 19
L'éthyl-2 hexanoate de potasslum est d'abor~ préparé en aJoutant 0,8 mL (5 rnmol) d'acide éthyl-2 hexanolgue à
42
une solutlon de 0,28 g de KOH (5 mmol) dans 10 mL de méthanol. Le solvant est évaporé, le rés~du est séch~ sous vlde et
repr~s avec 10 mL d'acétate d'éthyle sec.
Une solution-de 0,25 9 de l'ester allylique 18c
(1 r~ol) dans 3 mL de d~chlorométhane anhydre est agitée avec
3 ;ni. â' ùnc seL. t ll:·n ü, >:, ': r':' (thy 1-::: he. :a:l'L(-;: d~ Pl); d~,si unI
(1,5 ~~ol) en présence de 0,025 9 de trlphény1phosphlne
( l m'TIol) et de 0,025 g de tétrakistrlphénylphosphine Pd. La réaction se fal t'-d tempéra ture amblante et S()US atmosphère
c1' azote. Au bout c1' une deIT'i -heure, 20 ~~J;., Ù 1 acétone: sont ajoutés, le prodult est t l l t r t et rlnsé avec de l'(ther. On
obtlent ainsi 0,12 9 d'une poudre blanche (50~).
R"iN (60 Ml! z, CD 3 OD) è: 8, 4 - 6 ,5 (m, Ar)
IR (KBr) V: 1720 (C=O), 1250 (P=O)
2.1.6 Acides phénylacét~mi~o-4 salicylique 21a et acéta~ido~4
sallcy11que 2Ia
L'acide amlno-4 sallcyllque (l,53 g/lO mmol) en
solution dans 8 mL de pyridlne est chauffé à
IOOee
pendant 2 à3 heures en présence de 11 rrunol de chlorure de pbénylacétyle (1,3 mL) ou de chlorure d'acétyle (0,76 mL). La sqlution est
•
•
43
refroidie et versée sur un mélange formé de 20 9 de glace et 10 mL d'HCl concentré, puis filtrée. Le solide obtenu est recristallisé dans L'acide acétlque glaclal. Acide phényl-acétdmido-4 salicyllque 21a: 1,13 9 d'une poudre jaunâtre
~
(42%), décomposition 212°C. Acide acétamido-4 salicyliq~e
0,600 9 d'une poudre blanche (30%), décomposltlon 220-3°C.
Acide phény lacétarflldo-4 salle]' llq'-1e
RMN (60 MHz, CD 3
COc6
3) 0: 7,3-6d (m, 411, Ar), 6,; (s, SH, ph), IR (KBr) , 3 1 ('" -- 3 ,C ( ' J , 1\' ~ nII. CI , ; J 1, 1, 1 (, 1 (')1 -.: .. v • '3300 (NH), 3300 - 2700 (9, OH, CO;/1) ,
1680 tC=O, arr.lje), 1640 (C=O, aC1Cle)
Acide acétamido-4 salIcylIque
"7,2-0,2 1:--, 4F, Ar), 4,J-3,4 (R.., jll,
NH, OH, C0
2H), 1,4 (s, , CH3)
IR (KBr) \) : 3300 {NH), 3300-2700 (9", OH, C0
2H), 1670 (C=O, amlde), 1640 (C=O, acide)
2.1. 7 AClde ~-(carbobenzyloxy)arnino-4 salicylique 21c
en solution da:1S 40 J11L de NaHC03 1 M. On ajoute alors en " agl tant 2 mL de chloroforcatc de benzy le (11 mmo}) et on laisse la réactIon se pours~lvre pend~nt 4 heures. On lave
avec de l'éther, on refroidIt la phdse aqueuse â ocr et on acidIfIe avec liCI concentré .. Le préclplLé est flltré, lavp avec de l'eùu et séchp sous vldc. .On obtlen t ainSI 2,35
g-44
d'une poudre blanche (89%). Pf: 180-2°C.
RHN (60 MHz, CD 3COCD3) cS: 7,4-6,4 (m, 4H, Ar) 1 6,8 (s, SH, Ph) l 4,6 (s, CH 2), 4,8-4,0 (2, 3H, NH, OH, IR (KBr) C0 2H) \): 3350 (NH), 3300-2700 (t., OH, C0 2H), 1720 (C='&, un'é'thane), 1640 (C=O 1
ilClde)
r:h-,-'.nJre
- - - -
(J( l' .. 'IUe t.ll._·salIC\·ll)i~ '~!)horiquc .. 22b~ - " - -._- --
---_.
-"rillicyligue et de 0,95 mL {ID 17·,11'<_1\ c1'ox') '!11ot-urp de phosphore!
est chauffé doucement sur un bdl.n Ù'hUl.lc Jusqu'à 130°C. Le mélange réactIonnel' est maIntenu
.
à cette tempérùtureJusqu r à ce que le déyagement ù' Hel soit terminé, puis refrOldj ~ tempCr.:lture ù.:nlnante. Lln courant d'azot~ est ensuite passé dans le mélange pour enlever
l'~xcès
d'Hel. Unliqu~de'
Jaune4brun est obtcnù (2,08 g). Le produit est emplojlé sans
*-purificatIon supplémentaIre.