Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Laboratoire Signalisation et transports ioniques membranaires - STIM (Poitiers) (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Biologie-santé - Bio-santé (Limoges)
Secteur de recherche : Aspects moléculaires et cellulaires de la biologie
Présentée par :
Madryssa de Boisvilliers
Effets anti-tumoraux du VIP dans des cellules de neuroblastome
Directeur(s) de Thèse :
Jean-Marc Muller, Corinne Chadeneau Soutenue le 12 novembre 2015 devant le jury
Jury :
Président Marie-Odile Jauberteau Professeur des Universités, Université de Limoges Rapporteur José Luis Professeur des Universités, Université d'Aix Marseille Rapporteur Sandrine Wittmann Docteur, Centre Léon Bérard, Lyon
Membre Jean-Marc Muller Professeur des Universités, Université de Poitiers Membre Corinne Chadeneau Maître de conférences, Université de Poitiers Membre Franck Festy Docteur, Stemcis, Ile de la Réunion
Pour citer cette thèse :
Madryssa de Boisvilliers. Effets anti-tumoraux du VIP dans des cellules de neuroblastome [En ligne]. Thèse Aspects moléculaires et cellulaires de la biologie. Poitiers : Université de Poitiers, 2015. Disponible sur l'Intranet de
Pour l‟obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
(Faculté des Sciences Fondamentales et Appliquées)(Diplôme National - Arrêté du 7 août 2006) Ecole Doctorale : BioSanté
Secteur de Recherche : Aspects Moléculaires et Cellulaires de la Biologie Présentée par :
Madryssa de BOISVILLIERS
************************
Effets anti-tumoraux du VIP dans des cellules de
neuroblastome
************************
Directeur de thèse : Professeur Jean-Marc MULLER
Co-directeurs de thèse : Dr Corinne CHADENEAU et Dr Franck FESTY
************************
Soutenue le 12 Novembre 2015
Devant la Commission d‟Examen
************************
JURY
Dr Sandrine WITTMANN Ŕ Rapporteur Centre Léon Bérard, Lyon Pr José LUIS Ŕ Rapporteur INSERM, UMR911, Marseille
Pr Marie-Odile JAUBERTEAU Ŕ Examinatrice EA 3842, Faculté de Médecine, Limoges Dr. Corinne CHADENEAU Ŕ Examinatrice Equipe 2RCT
Université de Poitiers
Dr. Franck FESTY Ŕ Examinateur Stemcis c/o CYROI, Ile de la Réunion Pr. Jean-Marc MULLER Ŕ Examinateur Equipe 2RCT
Mes premiers remerciements vont droit à la Région de la Réunion et l’Europe pour le financement de cette thèse et au Dr Franck Festy pour avoir accepté de co-diriger cette thèse. Je tiens à remercier infiniment le Professeur Jean-Marc Muller pour m‟avoir accueillie au
sein de son équipe, pour m‟avoir acceptée en thèse, pour son aide pour la rédaction de ce
mémoire, pour son écoute, son soutien et sa convivialité.
Je remercie tout particulièrement le Dr Corinne Chadéneau pour m‟avoir encadrée pendant mon stage de Master et ma thèse. Je vous suis reconnaissante pour m‟avoir fait partager vos connaissances et votre rigueur ainsi que votre disponibilité, votre attention et votre aide à tous les niveaux.
Je remercie les personnes, qui m’ont fait l’honneur d’accepter de juger ce travail, et vous prie de croire en l‟expression de ma reconnaissance respectueuse.
Durant ces années passées au laboratoire, Mme Annie-claire Balandre et Mme Cathérine Adolphe ont été d‟une grande aide et je les remercie vraiment pour tout, que ce soit au niveau professionnel qu‟au niveau personnel. Je remercie également les personnes qui ont participé
de près ou de loin à ce travail, notamment au Dr Vincent Huguier pour les liposuccions et le Dr David Vaudry pour les analogues.
Je remercie le Dr Souheyla Bensalma pour la colocation dans notre royaume de doctorantes. Je te remercie pour ton aide, ta présence, ton soutien. Malgré la distance, tu es toujours là pour moi !
Je remercie l‟ensemble de l‟équipe βRCT (Paule Seité, Alice Barbarin et Brigitte Vannier)
pour leur bonne humeur, les pauses gourmandes et l‟agréable séjour aux Sables d‟Olonne. Je
n‟oublie pas les anciens membres de cette équipe : Mahmoud et Donald.
Je remercie les membres de l‟équipe STIM de notre étage pour la bonne ambiance, en
particulier Amandine et Abderrahmane. Je remercie le Dr Loubna Abaamrane pour sa gentillesse, sa bonne humeur malgré le peu de temps passé ensemble.
Je remercie énormément tous mes stagiaires qui ont contribué fortement à ce travail et en particulier Cindy Michel et Salima Hebache. J‟ai une pensée également pour les stagiaires Master 2 Florian Perrin et Agnès Garnier.
Je remercie de tout mon cœur ma famille et mes amis : mes parents, ma sœur, ma nièce, Arnaud, Naima et mes autres amis de la Réunion pour leur soutien inépuisable durant les moments difficiles. Merci de tout cœur maman pour tout ce que tu as dû endurer durant ces 10 ans d‟étude.
Cette thèse fut une excellente expérience et m‟a permis de m‟investir totalement et de manière
Liste des abréviations………1
Partie I : Étude bibliographique Introduction Chapitre I : Les neuroblastomes………..7
I- Généralités………...7
II- Localisation des NB………7
III- Diagnostic………8
1- Marqueurs biologiques………9
2- Techniques de détection……….9
IV- Histologie……….…...10
V- État de différenciation du NB………..10
VI- Altérations génétiques et phénotypiques……….11
1- La ploïdie ou contenu en ADN……….1β 2- Amplification de MYCN (locus 2p24)………..1β 3- Amplification d‟autres loci………...1β 4- Mutations chromosomiques………..1β A. Trisomie pour le 17q………...…1β B. Délétion du chromosome 1p………...1γ C. Délétions chromosomiques ou pertes alléliques dans d‟autres sites……...1γ D. Mutations génétiques ponctuelles………...14
E. Expression de récepteurs à activité tyrosine kinase (RTKs)………...14
1. Les récepteurs aux neurotrophines………14
2. Le récepteur ALK……….15
VII- Développement du NB………..15
VIII- La classification des NB………17
IX- Traitements………18
1- Chimiothérapie………...18
2- Chirurgie………...…19
3- Radiothérapie………...β1
4- Immunothérapie pour maintenir la rémission………...…β1 5- Thérapie de différenciation………..β1
6- Thérapies en développement………...ββ A. Inhibition des RTKs……….….βγ 1. Inhibition d‟ALK (Anaplastic Lymphoma Kinase)………βγ 2. Inhibition de TrkB………...βγ 3. IGF-1 (Insulin-like growth factor-1)………β4 B. Ciblage de la voie p53………β4 C. Ciblage de l‟angiogenèse………β4
D. Ciblage thérapeutique de MYCN………...24
X- Les NB in vitro………...25
1- Les mécanismes moléculaires et cellulaires de la différenciation neuronale…...………25
A. Les lignées de NB : des modèles d‟études……….25
B. Marqueurs moléculaires de la différenciation neuronale………β6 C. Voies de signalisation impliquées dans la différenciation……….β7 2- Les mécanismes cellulaires et moléculaires de l‟invasion/métastase………..……29
A. Processus d‟invasion/métastase……….β9 B. Les molécules impliquées dans l‟invasion/métastase………γ0 C. Les protéases permettant l‟invasion/métastase………..γβ Chapitre II : L’oncogène MYCN………...34
I- Gène, protéine MYCN et expression dans l’organisme……….34
II- Multiples fonctions de MYCN……….…34
III- MYCN : facteur transcriptionnel………35
1- MYCN : activateur transcriptionnel……….γ5 2- MYCN et régulation épigénétique………...γ5 IV- Amplification de MYCN dans les cancers………..36
1- Dans les NB……….γ6 2- Dans les autres cancers humains………..γ7 V- MYCN dans les NB………..38 1- MYCN et la pluripotence……….γ8
4- MYCN et la prolifération/ apoptose/ arrêt du cycle cellulaire………..40
5- MYCN et la surveillance immune………41
6- MYCN et la différenciation……….4β 7- MYCN et la migration/ métastases et l‟invasion………..4β 8- MYCN et l‟angiogenèse………...…4γ VI- Régulation de l’expression de MYCN………44
1- Stabilité de l‟ARNm MYCN……….…44
2- Principales voies de signalisation régulant l‟expression de MYCN…………44
A. Les voies PI3K/AKT et RAS/MAPK……….…………44
1. Généralités………44
2. Mécanisme………45
B. La voie MDM2/p53………....46
VII- MYCN et ALK dans les NB………..47
1- Généralités sur ALK………47
2- Les mutations d‟ALK et MYCN………..48
A. Les mutations ALK F1178L et R1279Q………48
B. La mutation ALK F1174L et MYCN………..……48
3- Stratégies thérapeutiques ciblant MYCN et ALK en développement………49
A. Stratégies ciblant MYCN………...49
1. Ciblage de la liaison à l‟ADN………..50
2. Ciblage de la transcription de MYCN………..50
3. Ciblage de kinases du cycle cellulaire et de la réparation de l‟ADN induits par MYCN………...………51
4. Ciblage de la stabilisation de MYCN……….51
B. Stratégies ciblant ALK………5β Chapitre III : Le système VIP-récepteurs………...……….54
I- Les neuropeptides de la famille du VIP………..54
II- Les neuropeptides VIP et PACAP………..54
1- Découverte et structure……….54
C. Précurseurs du VIP et du PACAP………..56
III- Les récepteurs des neuropeptides VIP et PACAP……….58
1- Découverte des récepteurs………58
A. Le récepteur VPAC1………...58
B. Le récepteur VPAC2………...58
C. Le récepteur PAC1………..59
2- Structure des récepteurs………59
3- Distribution des récepteurs et leurs ligands………..60
A. Distribution du VIP et du PACAP……….6β B. Distribution des RCPGs VPAC1, VPAC2 et PAC1………..6β 1. Localisation tissulaire………...6β a. Le récepteur VPAC1……….…6β b. Le récepteur VPAC2……….6β c. Le récepteur PAC1………6γ 2. Localisation cellulaire………....………...6γ 4- Activation des récepteurs………..6γ 5- Signalisation des récepteurs………..64
6- Les agonistes et antagonistes de ces récepteurs………66
7- Fonctions des récepteurs et de leurs ligands……….67
A. Fonctions des peptides VIP et PACAP………..……67
1. Dans le système nerveux………...67
2. Dans le système gastro-intestinal………..68
3. Dans les autres systèmes………...69
B. Fonctions du récepteur VPAC1………..69
C. Fonctions du récepteur VPAC2………..69
D. Fonctions du récepteur PAC1……….70
E. VIP, PACAP et leurs récepteurs dans les pathologies humaines…………70
IV- Le système VIP-récepteurs dans les NB………..71
1- Découverte du système VIP-récepteurs dans les cellules de NB………..71 2- Fonctions du système VIP-récepteurs dans les cellules tumorales…………...7β A. Fonctions du système VIP-récepteurs dans les NB………7β
NB……….7γ 4- Signalisation intracellulaire liée au système VIP-récepteurs dans les cellules de
NB………74
Présentation du sujet de recherche et des objectifs Chapitre IV : Présentation du sujet de recherche et des objectifs……….76
Partie II : Étude expérimentale Présentation des travaux de recherche Présentation de l‟article 1……….78
Données annexes de l‟article 1………105
Présentation de l‟article 2………....110
Données annexes de l‟article β………133
Présentation des travaux de recherche avec « Stemcis »……….………...137
Discussion générale et Perspectives……….158
1- Synthèse des résultats……….158
2- La régulation de processus moléculaires et cellulaires par le peptide VIP dans des cellules de NB à haut risque………...159
2.1. La régulation de l‟expression de MYCN……….159
2.2. La régulation de la différenciation des cellules de NB.………..161
2.3. La régulation de l‟invasion………..161
2.4. Synergie entre le VIP et l‟acide rétinoïque dans les cellules de NB à haut risque………...162
3- Les analogues et l‟approche in vivo………16β 4- Les analogues et les autres types de cancer………....163
5- Les récepteurs nucléaires du VIP dans les NB………...164
5.1. La localisation nucléaire des récepteurs VPACβ dans les NB………164
5.2. La délocalisation du récepteur VPAC2 par son ligand……….……...167
6- La différenciation des NB induite par les ATSC………....168
Présentation de l‟article 4………...………...…172
Présentation de l‟article 5………...…………...…174
– 1 –
Abréviations
Les termes suivis d‟un astérisque (*) sont en anglais AC: Adénylate cyclase (Adenylyl cyclase*)
ALK: Anaplastic lymphoma kinase*
AMPc : Adénosine monophosphate cyclique
AP-1 : Activator Protein 1*
ARE: Elément riches en AU
ARF: ADP ribosylation factor
ATP: Adénosine triphosphate
ATSC: Adipose tissue-derived stem cell*
AT-MSC: Adipose Tissue Mesenchymal Stem Cell*
AVC: Accident vasculaire cérébral
BDNF : Facteur neurotrophique dérivé du cerveau (Brain-derived neurotrophic factor*)
BET: Bromodomain and extraterminal*
B4GALNT3: 1,4-N-acetylgalactosaminyltransferase III*
B3GNT3: 1,3-N-acetylglucosaminyltransferase-3
BMP: Bone morphogenetic protein* (protéine morphogénétique osseuse)
Ca2+: Calcium
CFTR: Cystic fibrosis transmembrane conductance regulator*
CHK1: Checkpoint kinase 1* CHO: Cellules ovariennes d‟hamster CRE: Elément de réponse à l‟AMPc
CRF1 : Récepteur 1 humain du facteur de libération des corticotropines
CyRE : Elément de réponses aux cytokines
– 2 – dbcAMP: dibutyrylAMPc
DKK1: Dickkopf-1
DMEM: Dulbecco‟s modified eagle‟s medium
Dnmt3a: ADN méthylase 3a
EC: Extracellulaire
ED/VEGF: Endocrine gland-derived vascular endothelial growth factor* ER: Endoplasmic reticulum*
ERK: Extracellular signal-regulated kinase*
FAK: Focal adhesion kinase*
FC: Facteur de croissance
FGF: fibroblast growth factor*
FISH: Hybridation in situ en fluorescence FSV: Fraction stromale vasculaire
GAP43: Growth Associated Protein 43*
GATA 2: GATA binding protein 2*
GATA 3: GATA binding protein 3*
GBM: Glioblastome multiforme
GCGR : Récepteur humain du glucagon
GFP: Green fluorescent protein
GHF-1: Growth hormone factor-1*
GHRF: Growth-hormone-releasing factor*
GIP: Glucose-dependent-Insulinotrophic-Polypeptide* GLP: Glucagon-like peptide*
GST: Génistéine
HAND2: Heart and neural crest derivatives expressed 2* hASH1: human MASH1
– 3 –
HVA: Acide homovanilique
HAT: Histones acétyltransférases
HDAC: Histones désacétylases
HES-1: Hairy/enhancer of split homologue-1*
4-HPR: N-(4-hydroxyphenyl) retinamide*
hTERT: Human telomerase reverse transcriptase* IC: Intracellulaire
IGF-1: Insulin-like growth factor*
IL: Interleukine
INPC: International Neuroblastoma Pathologic Classification*
IP3: Inositol triphosphate
INRGSS: International Neuroblastoma Risk Group Staging System* IRM: Imagerie par résonance magnétique
KO: Knock-out*
LDH: Lactodéshydrogénase
LIF: Leukemia inhibitory factor*
LMB: Leptomycine B
LSD1: Lysine-specific demethylase* MAPK: Mitogen-activated protein kinase*
MASH1: Murine achaete-scute homolog 1*
MCI : Mort cellulaire médiée par l'intégrine
MCM: Minichromosome maintenance* MEC: Matrice extracellulaire
MIBG: Métaiodobenzyl-guanidine
MMP : Métalloprotéinases
– 4 – NCAM : Neural cell adhesion molecule*
NEAA: Non essential amino acids
NGF: Facteur de croissance nerveuse (Nerve growth factor *)
NKT : Cellules tueuses naturelles T (Natural Killer T*)
NLRR1: Neuronal leucine-rich repeat protein-1*
NLRR3: Neuronal leucine-rich repeat protein-3* NLS : Nuclear localization sequence*
NRSF: Neuron-restrictive silencer factor*
NSE : Neuron Specific Enolase*
NT3: Neurotrophine 3
P: Phosphorylation
PAC1: PACAP receptor 1*
PACAP: Pituitary adenylate cyclase-activating polypeptide*
PBS : Phosphate buffered saline
PC : Prohormones convertases
PCR : Réaction de polymérisation en chaîne
PHI/PHM: Peptide having carboxyterminal isoleucine/methionine*
PHOX2A: Paired-like homeobox 2A*
PHOX2B: Paired-like homeobox 2B* PI3K: Phosphatidylinositol 3-kinase*
PINK1: PTEN-induced putative kinase 1*
PKA: Protéine kinase A
PKB: Protéine kinase B (=AKT)
PKC: Protéine kinase C
PLC: Phospholipase C
– 5 – PRP: PACAP-related peptide*
PS : Pénicilline/streptomycine
RA : Acide rétinoïque (retinoic acid*)
RCPG : Récepteur couplé aux protéines G
RE : Réticulum endoplasmique
RE-1 : Elément répresseur de type 1
RECK: Reversion-inducing cysteine-rich protein with Kazal motifs
REST: RE-1 silencing transcription factor*
RMN : Résonance magnétique nucléaire
RT : Transcription inverse
RTK : Récepteur à activité tyrosine kinase
SKP2: S-phase kinase-associated protein 2 SNC: Système nerveux central
SNP : Système nerveux périphérique
SPT : Stress post-traumatique
SVF : Sérum de veau fœtal TBE: Tris, borate, EDTA TBS: Tris buffered saline
TBS-T: Tris buffered saline-tween
TH: Tyrosine hydroxylase
TM: Transmembranaire
TP53INP1: Tumor protein p53-inductible nuclear protein 1
TRE : Elément de réponse au 12-O-tétra-décanoylphorbol 13-acétate
TrkA : Récepteur tropomyosine kinase A
TrkB : Récepteur tropomyosine kinase B
– 6 – Ub : Ubiquitinylation
uPA : Urokinase activatrice du plasminogène
uPAR : Récepteur de l‟urokinase activatrice du plasminogène VEGF: Vascular endothelial growth factor*
VIP : Peptide intestinal vasoactif (vasoactive intestinal peptide*)
VMA : Acide vanillylmendélique VPAC1 : VIP/PACAP receptor 1*
Partie I : Étude bibliographique
– 7 – I- Généralités
Le neuroblastome (NB) est une tumeur embryonnaire dérivée de cellules précurseurs du système nerveux sympathique. C‟est la tumeur solide extra-crânienne la plus mortelle chez les enfants (pour revue Brodeur, 2003). La majorité de ces tumeurs est localisée dans la médullosurrénale ou les ganglions paraspinaux. Avec une prévalence de un cas sur 7000 à 10 000 naissances, les NB représentent 8 à 10% des cancers pédiatriques et sont responsables de 15% des décès attribuables à des affections malignes chez les enfants (pour revue Janoueix-Lerosey et al., 2010). Cette tumeur est hétérogène sur le plan clinique, anatomo-pathologique, biologique, génétique et moléculaire (Brodeur et al., 1992). Une caractéristique spectaculaire du NB est que certaines tumeurs à un stade tardif, avec des métastases osseuses, peuvent régresser spontanément jusqu‟à une rémission complète, laissant la marque d'une fibrose ou une calcification (pour revue Brodeur, 2003).
II- Localisation des NB
La majorité des NB, soit 65%, est localisée dans l‟abdomen dont γ5% sont retrouvés au niveau de la surrénale. Les formes thoraciques représentent 20 % des cas et les formes pelviennes et cervicales représentent 5% des cas environ (pour revue Plantaz, 2001). Les tumeurs primaires cervicales et thoraciques sont plus fréquemment retrouvées chez les jeunes enfants. La tumeur peut progresser en envahissant le tissu adjacent, en s‟étendant au système lymphatique et aux ganglions à proximité ou à distance, et en développant des métastases. Les métastases sont présentes chez 50% environ de patients atteints de NB lors du diagnostic (pour revues Huang et Weiss, 2013; Maris et al., 2007). Les formes métastatiques de NB
envahissent typiquement l‟os et la moelle osseuse (environ 50% des cas), les ganglions
lymphatiques (28%), le foie (22%) et la peau (12%) (Figure 1). Les métastases pulmonaires ou cérébrales restent exceptionnelles (pour revues Maris et al., 2007; Plantaz, 2001). Chez les nourrissons, une expansion métastatique particulière atteignant principalement le foie ou la
Chapitre I
– 8 –
peau peut se produire et correspond au stade 4s. Malgré la présence de métastases lors du diagnostic, ces nourrissons ont généralement un bon pronostic puisque ces tumeurs peuvent
subir une régression spontanée complète (5 à 10%). En revanche, beaucoup d‟enfants de plus d‟un an avec une maladie métastatique au moment du diagnostic ont un pronostic global
médiocre (pour revue Brodeur, 2003). Environ 70% des enfants de plus d‟un an ont une forme
d‟emblée disséminée (pour revue Plantaz, 2001). A ce jour, l‟âge et le stade restent les
marqueurs pronostiques cliniques les plus validés pour les patients avec des NB (pour revue Modak et Cheung, 2010). Il existe quatre stades de NB : les stades I et II caractérisent les tumeurs localisées, les stades III et IV représentent les tumeurs métastatiques, et le stade 4s correspond aux tumeurs de stade I ou II avec des métastases limitées au foie, à la peau et à la moelle hématopoïétique (pour revue Plantaz, 2001).
III- Diagnostic
Le NB est le cancer le plus commun diagnostiqué durant l‟enfance (Gurney et al., 1997; pour revue Brodeur, 2003). L‟âge moyen lors du diagnostic est approximativement 20 mois (pour revue Øra et Eggert, 2011). Environ 40% des enfants sont diagnostiqués avant un an, 75% avant 4 ans et 98% avant 10 ans (pour revue Brodeur, 2003). Les signes cliniques les plus fréquents sont la découverte d‟une masse abdominale ou la présence de signes fonctionnels en rapport avec des phénomènes de compression digestive, urinaire, respiratoire, neurologique radiculaire ou spinale.
Figure 1 : Les sites de métastases des NB. Les NB développent des métastases de manière préférentielle dans la moelle osseuse, l‟os, le foie, la peau et les ganglions lymphatiques.
– 9 – 1- Marqueurs biologiques
Le diagnostic est établi dans plus de 90 % des cas par la présence de métabolites des catécholamines, de l‟acide homovanilique (HVA), de l‟acide vanillylmendélique (VMA) et de la dopamine dans les urines et/ou le sérum. De plus, le taux de ces molécules augmente avec
la masse de la tumeur permettant ainsi d‟observer l‟évolution tumorale. Environ 5 à 10% des
tumeurs ne produisent pas de catécholamines. Pour ces lésions, un panel de colorations immunohistochimiques démontrant la présence de neurofilaments, de synaptophysine, de Gap-4γ, d‟énolase spécifique des neurones peuvent différencier les NB des autres tumeurs « à petites cellules rondes bleues » retrouvées chez les enfants (pour revue Øra et Eggert, 2011). Un autre marqueur biologique est la lactodéshydrogénase (LDH). Son taux sanguin augmente
dans environ 75% des NB (D‟Andon et al., 2004). La mise en évidence de facteurs pronostiques génétiques au niveau tumoral, tels que l‟amplification de l‟oncogène MYCN et la ploïdie, rend indispensable une biopsie initiale permettant de définir la thérapie à mettre en place (pour revue Plantaz, 2001).
2- Techniques de détection
Le bilan d‟extension comporte des évaluations scintigraphiques et ostéo-médullaires
actuellement bien standardisées. La localisation des tumeurs situées au niveau de l‟abdomen, du thorax ou du pelvis est mise en évidence par la technique d‟échographie alors que les compressions éventuelles de la moelle épinière sont mises en évidence par la technique
d‟imagerie par résonance magnétique (IRM). Ces deux techniques permettent la révélation d‟extensions locales ou régionales des tumeurs. La visualisation des tumeurs osseuses se fait
par la technique de scintigraphie à la métaiodobenzyl-guanidine (MIBG) (Figure 2) ou au
Tc-diphosphonate. La réalisation d‟analyses histologiques et biologiques est effectuée grâce à des
biopsies tumorales permettant de caractériser la présence de certaines anomalies génétiques caractéristiques des NB (pour revue, Ishola et Chung, 2007).
– 10 –
Figure 2 : Détection des métastases par scintigraphie à la métaiodobenzyl-guanidine (MIBG). Des métastases sont détectées à divers endroits du corps, en particulier au niveau du squelette et de l‟abdomen (d‟après Maris et al., 2007).
IV- Histologie
Le NB appartient aux néoplasmes « à petites cellules rondes bleues » de l‟enfance (pour revue Triche, 1986). Les cellules ont un noyau dense hyperchromatique et un cytoplasme basophile. Le NB est composé de cellules indifférenciées de taille homogène appelées des neuroblastes qui présentent une faible différenciation neurale. Cependant, quelques tumeurs montrent une différenciation histologique partielle et sont appelées ganglioneuroblastomes (pour revue Brodeur, 2003).
V- Etat de différenciation du NB
Ces tumeurs comprennent souvent des phénotypes cellulaires multiples : il existe des NB à cellules indifférenciées, ou peu différenciées ou en différenciation (Figure 3). Des
prolongements neuritiques indiquent souvent l‟amorce de différenciation de ces neuroblastes
(pour revue Plantaz, 2001). Le faible état de différenciation du NB peut être dû à un microenvironnement hypoxique (Jögi et al., 2002).
– 11 –
Figure 3 : Représentation des phénotypes cellulaires multiples du NB. Trois types de NB sont distingués en fonction de l‟état de différenciation : les NB à cellules indifférenciées, ceux à cellules peu différenciées ou ceux à cellules en différenciation. (Adapté de « the clinical image analysis laboratory »)
VI- Altérations génétiques et phénotypiques
Beaucoup de caractéristiques génétiques des NB, comme le statut de la ploïdie,
l‟amplification d‟un oncogène ou la perte allélique, ont maintenant été identifiées (Tableau 1)
et sont en corrélation avec les résultats cliniques.
Altérations génétiques et phénotypiques
NB de bon pronostic NB de mauvais pronostic
Ploïdie Triploïde Diploïde ou Tétraploïde
Amplification - MYCN, MYCL
Mutations chromosomiques - 17q+, 1p-, 11q-, 14q-
Mutations ponctuelles - TP53, PHOX2B
Expression des RTKs TrkA TrkB, IGF-1R, ALK muté
Tableau 1 : Altérations génétiques et phénotypiques dans les NB de bon et mauvais pronostic. Ce
tableau récapitule les différentes altérations génétiques retrouvées dans les NB et détaillées dans les paragraphes qui suivent.
– 12 – 1- La ploïdie ou contenu en ADN
Un état diploïde de la cellule est associé à un mauvais pronostic alors qu‟un état triploïde est signe d‟une issue favorable. La majorité des NB primaires (55%) sont triploïdes
ou « presque triploïdes », contenant entre 58 et 80 chromosomes; les autres (45%) sont soit « presque diploïdes » (35 à 57 chromosomes) soit « presque tétraploïdes » (81 à 103 chromosomes) (Kaneko et al., 1987 ; pour revue Davidoff, 2012). Malheureusement, la ploïdie (ou contenu en ADN) perd son importance pronostique pour des patients âgés de plus
d‟un ou deux ans (pour revue Brodeur, 2003).
2- Amplification de MYCN (locus 2p24)
Schwab et ses collègues ont identifié un oncogène, apparenté à MYC, appelé MYCN
dans un large panel de lignées cellulaires de NB et une tumeur de NB (Schwab et al., 1983).
L‟amplification de MYCN apparait dans environ 25% des NB primaires chez des patients non traités et est aussi associée à une progression rapide de la tumeur et à un mauvais pronostic.
Elle est présente chez 40% des patients avec des stades avancés de la maladie alors qu‟elle n‟est que de 5 à 10% chez des patients avec une maladie de bas grade (Brodeur et al., 1984 ; pour revue Davidoff, 2012).
3- Amplification d’autres loci
D‟autres amplifications ont été retrouvées dans les lignées cellulaires de NB ou dans des tumeurs primaires. Cela inclut l‟amplification d‟ADN des régions chromosomiques βpββ
et 2p13, le gène MDM2 sur le 12q13 et le gène MYCL en position 1p32 (Brodeur et al., 1997 ; pour revue Brodeur, 2003). Ces amplifications sont toujours associées à celle de MYCN (pour revue Brodeur, 2003).
4- Mutations chromosomiques A. Trisomie pour le 17q
La trisomie 17q représente un facteur de pronostic défavorable indépendant (Bown et al., 1999). Cette trisomie est partielle dans plus de la moitié des NB et est due à la présence
– 13 –
d‟un chromosome 17 entier supplémentaire dans 40% des cas hyperdiploïdes (Caron 1995 ;
Bown et al., 1999 ; pour revue Brodeur, 2003 ; pour revue Plantaz, 2001).
B. Délétion du chromosome 1p
La délétion du bras court du chromosome 1 (1p) fut la première anomalie génétique découverte dans le NB par l‟étude du caryotype tumoral (Brodeur et al., 1993 ; Brodeur et al., 1992). Cette anomalie est communément détectée puisque sa fréquence est de 25 à 40 % (pour revue Brodeur, 2003). Les délétions du 1p sont trouvées plus fréquemment chez des patients avec des stades avancés de la maladie, et sont souvent associées à d‟autres anomalies génétiques, en particulier l‟amplification de MYCN et le caractère di ou tétraploïde (pour revue Plantaz, 2001). Cependant, une étude a démontré que les délétions 1p sont indépendamment associées à un mauvais devenir chez les patients avec des NB (Attiyeh et al., 2005).
La fréquente délétion du 1p36 dans les NB sporadiques, les délétions constitutionnelles ou les réarrangements impliquant un gène ou des gènes sur le 1p36 suggèrent un rôle de ces gènes dans la transformation maligne ou dans la prédisposition au NB. La perte allélique du 1p36 (23 à 35% des tumeurs de NB) prédit une progression de la maladie (Maris et al., 2000).
C. Délétions chromosomiques ou pertes alléliques dans d’autres sites Parmi les autres pertes de matériel chromosomique rencontrées, la délétion du bras long du chromosome 11 est observée dans β6 à 44% de patients, faisant d‟elle une autre délétion communément détectée dans les NB. Cette délétion du 11q est directement associée à la délétion 14q, mais elle est inversement corrélée à la délétion 1p et à l‟amplification de
MYCN. Néanmoins, la perte du 11q s‟avère être un prédicteur utile du devenir chez des
patients cliniquement à haut risque sans amplification de MYCN puisque sa présence est signe
– 14 – D. Mutations génétiques ponctuelles
Le gène TP53, qui code pour la protéine p5γ, est l‟un des gènes mutés le plus
communément dans les néoplasies humaines. La protéine p53 est un régulateur clé du
contrôle du cycle cellulaire, et l‟inactivation de la fonction de p53 peut contribuer à une
transformation maligne. Cependant, des mutations de TP53 sont rarement retrouvées dans les NB primaires (environ 2%) (Vogan et al., 1993 ; Hosoi et al., 1994 ; pour revue Brodeur, 2003). Elles sont observées seulement dans des NB progressifs, suggérant que les mutations de TP53 dans les NB constituent un mécanisme de résistance aux drogues cytotoxiques qui ciblent la voie p53 (Tweddle et al., 2003).
Le gène PHOX2B (paired-like homeobox 2), codant pour un facteur de transcription qui est associé à la différenciation du système nerveux sympathique et à la synthèse de catécholamines, peut être muté dans les NB. Il semble impliqué dans les cas familiaux de NB
puisqu‟il est retrouvé dans 6,4% de ces NB ; en revanche la contribution de ce gène pour le
développement de NB sporadiques est beaucoup moins évidente puisque les mutations somatiques de PHOX2B sont extrêmement rares (Van Limpt et al., 2004 ; Raabe et al., 2008).
E. Expression de récepteurs à activité tyrosine kinase (RTKs)
Des récepteurs à activité tyrosine kinase (RTKs), incluant le récepteur à l‟IGF1
(insulin-like growth factor) (IGF-1R), les récepteurs aux neurotrophines et le récepteur ALK (Anaplastic Lymphoma Kinase), peuvent réguler la stabilité de la protéine MYCN (pour revue Gustafson et Weiss, 2010).
1. Les récepteurs aux neurotrophines
Il existe trois récepteurs aux neurotrophines : TrkA, TrkB et TrkC (aussi appelés NTRK1, NTRK2 et NTRK3 respectivement) qui ont pour ligands principaux le facteur de croissance nerveuse (NGF), le facteur neurotrophique dérivé du cerveau (BDNF) et la neurotrophine 3 (NT3) respectivement.
L‟expression du récepteur de haute affinité TrkA dans les NB (25% des NB) est
corrélée à un bon pronostic, alors que l‟expression de TrkB (40% des NB) et son ligand BDNF est associée à un mauvais pronostic (pour revue Brodeur, 2003, pour revue Davidoff,
– 15 –
MYCN-amplifiées alors que TrkA et TrkC ne sont pas généralement exprimés dans des tumeurs MYCN-amplifiées (Nakagawara et al., 1994 ; Yamashiro et al., 1996 ; Rydén et al., 1996 ; pour revue Brodeur, 2003). La forte expression du récepteur TrkA induit soit
l‟apoptose soit la différenciation dans ces tumeurs. Inversement, la forte expression de TrkB
fournit une voie de survie autocrine dans les tumeurs défavorables (pour revue Brodeur, 2003).
2. Le récepteur ALK
Un autre oncogène, ALK, contribue avec MYCN à la formation des NB. ALK est un récepteur à activité tyrosine kinase (RTK) impliqué dans la genèse de plusieurs malignités incluant les lymphomes et des tumeurs myofibroblastiques infantiles par modification de la réponse de la voie MAPK (mitogen-activated protein kinase) aux facteurs de croissance. Récemment, des mutations activatrices (dans environ 50% des NB familiaux) dans le
domaine tyrosine kinase de l‟oncogène ALK sur le bras court du chromosome 2 (2p23) ont été identifiées et correspondent à des mutations germinales associées aux NB héréditaires (George et al., 2008 ; Mossé et al., 2008 ; Janoueix-Lerosey et al., 2008).
VII- Développement du NB
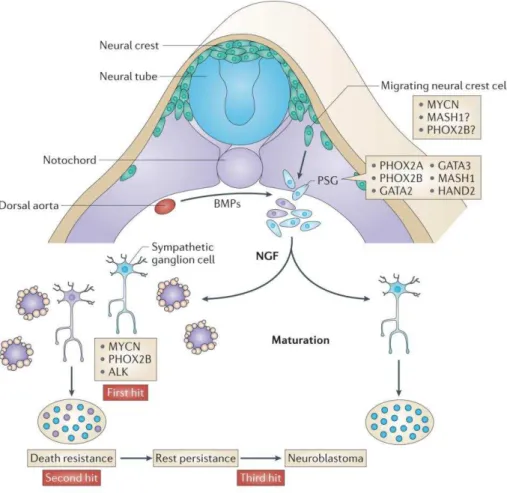
L‟accumulation de données histologiques, phénotypiques, et génétiques a conduit à l‟établissement d‟un modèle de développement du NB illustré figure 4. Les NB dérivent de cellules de la crête neurale qui migrent vers la notochorde et l‟aorte dorsale sous l‟influence de l‟oncogène MYCN, des protéines morphogénétiques osseuses (BMPs) et des facteurs de transcription tels que PHOX2B, MASH-1 et GATA-3. Sous l‟action du facteur de croissance nerveuse (NGF), les cellules ayant migré se transforment en cellules de ganglion terminal
alors qu‟en absence du NGF, les cellules subissent une mort cellulaire par apoptose. MYCN
est le premier gène touché avec l‟acquisition de mutations d‟ALK et PHOX2B permettant
ainsi une transformation maligne et conduisant à l‟émergence du NB. Certains NB ont la
– 16 –
Figure 4 : Modèle de développement du NB. Les progéniteurs neuroblastiques migrent de la crête neurale, autour du tube neural et des somites vers une région immédiatement latérale de la notocorde et de l'aorte dorsale sous l'influence de MYCN et des protéines morphogénétiques osseuses (BMPs). Sur ce site, les cellules se spécialisent en ganglions sympathiques primaires (PSG) avant de diverger en cellules neurales de ganglions sympathiques matures ou en cellules chromaffines de la médullo-surrénale. MYCN serait le premier gène touché en vertu des observations de modèles de souris transgéniques tyrosine
hydroxylase (TH)-MYCN, en concomitance avec des mutations/modifications d‟ALK (Anaplastic
Lymphoma Kinase) et PHOX2B (paired-like homeobox 2B). L'accès local au facteur de croissance nerveux (NGF) détermine si le ganglion sympathique normal mature en une cellule de ganglion terminal ou subit une mort cellulaire par apoptose. Un état pathologique relativement courant est la survie postnatale de la cellule neuroblastique au repos qui nécessite que la cellule destinée à devenir maligne soit résistante au retrait du facteur trophique avant que ces cellules au repos persistantes subissent un troisième changement pour induire la transformation, qui se présente comme une tumeur maligne clinique de la petite enfance. De plus, d‟autres gènes impliqués dans le développement embryonnaire et la neurogenèse sont présents dans les PSG. Il s‟agit des gènes MASH1 (murine achaete-scutehomolog 1), HAND2 (heart and neural crest derivatives expressed 2), GATA 2 et 3 (GATA binding protein 2 et 3), et PHOX2A (paired-like homeobox
– 17 –
Figure 5 : Mécanismes de régression spontanée. Sur ce schéma, les principales voies qui ont été proposées pour expliquer le phénomène de régression spontanée sont indiquées. Elles comprennent: la privation de neurotrophines (TrkA sans NGF) et l'activation programmée de l'apoptose (1); la mort cellulaire à médiation immunitaire par les anticorps anti-neuroblastome (la toxicité cellulaire dépend des anticorps) ou par les cellules NK (2); le raccourcissement des télomères et l'apoptose déclenchée par l‟absence ou de faibles niveaux de télomérase (3); et des modifications épigénétiques dans l'expression des gènes contrôlés par la méthylation de l'ADN, la modification d'histones, ou des altérations de remodelage de la chromatine (4). Abréviations: NGF, le facteur de croissance nerveux; NK (Natural Killer), les cellules
tueuses naturelles; TrkA, récepteur tropomyosine kinase A. (D‟après Brodeur et Bagatell β014)
VIII- La classification des NB
Différentes classifications existent telles que celle nommée INPC (International Neuroblastoma Pathologic Classification) largement inspirée de l‟ancienne classification de Shimada (Shimada et al., 1999) (Tableau β) et l‟INSS (International Neuroblastoma Staging System) prenant en compte l‟histologie, les altérations génétiques, et le grade de différenciation tumorale (Brodeur et al., 1993 ; pour revue Plantaz, 2001).
Monclair et ses collègues ont proposé un nouveau système de classification en 2009 nommé INRG (International Neuroblastoma Risk Group) (Tableau 3) prenant en comptre l‟amplification de l‟oncogène MYCN, la délétion du 11q, la différenciation et la ploïdie des cellules tumorales qui, avec l‟âge de l‟enfant, permettent de définir le traitement optimal adapté au risque (Monclair et al., 2009 ; pour revue Davidoff, 2012).
– 18 –
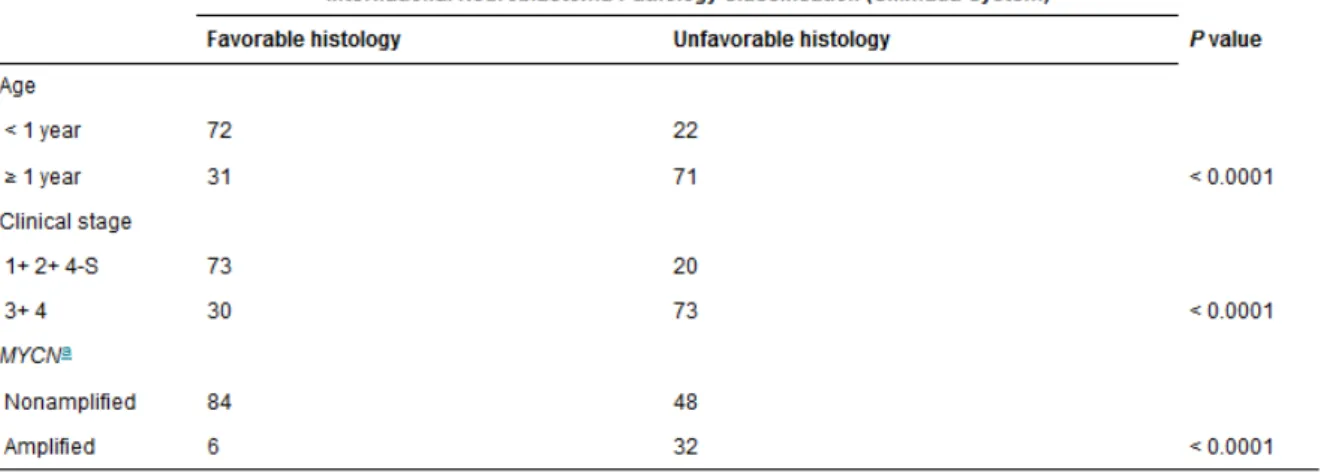
Tableau 2 : Classification INPC selon le système Shimada. Les chiffres correspondent au nombre
de cas sur 196 cas disponibles. Cette classification se base sur l‟âge, le stade clinique et le statut de MYCN. Les tumeurs histologiquement favorables se caractérisent par un âge au diagnostic inférieur à 1 an, un stade clinique non avancé (stade 1 ou 2) ou stade 4s, et aucune amplification de MYCN. Les tumeurs histologiquement défavorables se caractérisent par un âge au diagnostic supérieur à 1 an, un stade clinique avancé (stade 3 ou 4), et une amplification de MYCN. (D‟après Shimada et al., 1999)
IX- Traitements
Le traitement actuel des NB à haut risque peut être divisé en trois phases distinctes : la
chimiothérapie d‟induction, le contrôle local par chirurgie et radiothérapie, et la consolidation
de la rémission avec une prise orale du 13-cis acide rétinoïque, avec ou sans immunothérapie (pour revue Modak et Cheung, 2010).
1- Chimiothérapie
La chimiothérapie a un rôle important dans le traitement des NB puisque la majorité des patients présentent une maladie métastatique ou localement avancée au diagnostic et
requièrent un traitement systémique. La chimiothérapie d‟induction a pour but d‟induire une
réduction rapide de la tumeur entière. Elle facilite une résection complète du tissu mou malade (pour revue Modak et Cheung, 2010). Les taux de réponse tumorales sont souvent
dépendants de l‟âge : les adolescents et les adultes ont généralement des tumeurs
chimiorésistantes comparés aux NB des plus jeunes enfants (Franks et al., 1997 ; Kushner et al., 2003 ; pour revue Modak et Cheung, 2010).
– 19 –
Dans les NB localisés avec une amplification de MYCN, la réalisation d‟une
chimiothérapie lourde et d‟une irradiation de la tumeur semble améliorer le pronostic (Rubie
et al., 1997). Les NB à haut risque sont sensibles à une dose intensive de chimiothérapie : une
majorité de patients parvient à une rémission après chimiothérapie d‟induction, chirurgie et
radiothérapie ; mais la plupart rechute même avec un traitement de consolidation. Une étude a montré que les NB de rechute présentent des mutations de la voie Ras/MAPK (Eleveld et al., 2015). Dans les formes métastatiques, chez l‟enfant de plus d‟un an, une chimiothérapie lourde de consolidation est proposée aux patients en rémission complète ou partielle avec un bénéfice démontré par rapport à la chimiothérapie conventionnelle, et un petit bénéfice
supplémentaire apporté par un traitement adjuvant par l‟acide rétinoïque (Matthay et al., 1999).
Les molécules standards de chimiothérapie des NB sont des agents alkylants tels que le cyclophosphamide, le busulfan, l‟iphosphamide et le melphalan, des analogues du platine tels que le cisplatine et le carboplatine, des vinca-alcaloïdes tels que la vincristine, des épipodophyllotoxines telles que le VP16 et le VM26, et des anthracyclines telles que la doxorubicine. Au cours des dernières années, d‟autres agents tels que le topotecan,
l‟irinotecan, et le temozolomide ont prouvé leur efficacité clinique, et des combinaisons
incluant ces médicaments sont testées dans des études de phase II en cours (Kushner et al., 2010 ; Kushner et al., 2011 ; Bagatell et al., 2011 ; Rubie et al., 2010).
2- Chirurgie
La chirurgie reste un traitement généralement incontournable du NB (pour revue Øra et Eggert, 2011). En effet, bien que beaucoup de tumeurs répondent à la chimiothérapie, la chirurgie est cruciale pour beaucoup de patients pour parvenir à une rémission complète dans le site primaire. Le traitement des NB localisés sans amplification de MYCN repose avant tout
sur l‟exérèse complète, avec plus de 90 % de guérison (Rubie et al., 1997). Celle-ci est réalisée après chimiothérapie préopératoire dans les formes "inopérables", la moins intensive possible chez le nourrisson (Rubie et al., 1998). La présence d‟un résidu postopératoire (stade
β) ne nécessite pas de traitement complémentaire en l‟absence de critères de mauvais
P ar tie I : E tud e B ib liog ra ph iqu e C hap it re I : L es N eu robl a st o m es – 20 –
Tableau 3: Classification INRG (International Neuroblastoma Risk Group) de prétraitement.
Les analyses ont déterminé que 7 variables pronostiques (stade INRG, l'âge [moins /plus de 18 mois], l'histologie, la différenciation tumorale, le statut de MYCN, le statut 11q, et la ploïdie) pourraient définir 16 différents groupes de prétraitement à risque. Ces groupes pourraient ensuite être divisés en quatre catégories basées sur la survie à 5 ans : très faible (> 85%, 28,2% des patients), faible (>75 et ≤ 85%, 26,8% des patients), intermédiaire (≥ 50 jusqu‟à 75%, 9,0% des patients) et un risque élevé (<50%, 36,1% des patients). NA= Non amplified. (D‟après Davidoff, β01β).
– 21 – 3- Radiothérapie
Les NB sont radiosensibles et des doses tumoricides appliquées dans la gamme de 15 à
γβ Gy dépendent du site, du volume de la tumeur et de l‟âge du patient (pour revue Øra et
Eggert, 2011). La radiothérapie est un composant essentiel du contrôle local du site primaire.
Elle est appliquée dans le cadre d‟une maladie résiduelle minime après chimiothérapie d‟induction et chirurgie.
4- Immunothérapie pour maintenir la rémission
Le développement de nouvelles approches thérapeutiques, dont l‟immunothérapie, est indispensable pour améliorer encore le pronostic des formes graves, en particulier des formes
métastatiques de plus d‟un an (pour revue Plantaz, 2001). Le disialoganglioside GD2 est un
glycolipide abondant à la surface des cellules de NB et généralement des tumeurs d‟origine neuroectodermique (Modak et Cheung, 2007). En revanche, dans des tissus normaux son expression est restreinte aux neurones qui sont protégés des effets des anticorps monoclonaux thérapeutiques intraveineux par la barrière hémato-encéphalique (Hakomori, 2001). L'anticorps anti-GD2 monoclonal semble avoir une activité efficace pour l'éradication des cellules de NB résiduelles à la fin du traitement cytotoxique (pour revue Cheung et Dyer, 2013).
5- Thérapie de différenciation A. Généralités
La différenciation neuronale est un processus développemental essentiel qui détermine une connexion synaptique précise (cf paragraphe X-1 page 25). Plusieurs molécules sont connues pour induire la différenciation des cellules de NB par l‟induction de différentes voies
de signalisation. Il s‟agit notamment de l‟acide rétinoïque et de ses dérivés (Fernandes et al., 2007 ; Miloso et al., 2004 ; Akkuratov et al., 2015) tel que nous le développons ci-après.
D‟autres molécules ont également été découvertes comme induisant la différenciation des
cellules de NB, telles que le 7-(4-hydroxyphenyl)-1-phenyl-4E-hepten-3-one (Cpd 1), un diarylheptanoïde issu d‟une plante nommée Alpinia officinarum et capable d‟induire la différenciation neuronale des cellules Neuro-2A de NB par activation des voies de
– 22 –
signalisation ERK et PI3K-AKT (Tang et al., 2015), le ganglioside GM1 (Singleton et al., 2000), le FK506 (Price et al., 2003), la clozapine par activation de la PKA (Jeon et al., 2015), des facteurs protéiques comme le NGF (Emdal et al., 2015), des facteurs de croissance (Nakagawara et al., 2001), des neurotransmetteurs (Anelli et al., 2013) ainsi que le VIP et le PACAP (Chevrier et al., 2008 ; Alleaume et al., 2004 ; Héraud et al., 2004 ; Héraud et al., 2008; Monaghan et al., 2008a).
B. Thérapie de différenciation par des rétinoïdes
L‟acide rétinoïque (RA) est un métabolite biologiquement actif de la vitamine A. Il est
un puissant régulateur de la morphogenèse, de la prolifération cellulaire, et de la différenciation cellulaire (Cañón et al., 2004 ; De Luca et al., 1991 ; Konta et al., 2001 ; Love et Gudas, 1994). Il joue un rôle essentiel durant le développement normal du neurone (Durston et al., 1989 ; Hunter et al., 1991 ; Jackson et al., 1991 ; Maden, 2001), où il induit la croissance des neurites et la différenciation neuronale (Bain et al., 1995 ; Corcoran et Maden, 1999 ; López-Carballo et al., 2002 ; Pennypacker et al., 1989 ; Rebhan et al., 1994). Il régule la transcription de gènes cibles spécifiques (Boukhtouche et al., 2006) et contrôle le taux de la synthèse protéique (Carta et al., 2006). Il est un mitogène neuronal et protège les neurones
contre le stress oxydatif et l‟apoptose induite par différents stimuli (Ahlemeyer et Krieglstein,
1998 ; Dheen et al., 2005 ; Hoehner et Prabhakaran, 2003 ; Jackson et al., 1991 ; Moreno-Manzano et al., 1999 ; Ronca et al., 1999). Ce rétinoïde inhibe efficacement la prolifération cellulaire et favorise la croissance des neurites des cellules de NB in vitro (pour revue Westermark et al., 2011). Le Fenrétinide, un rétinoïde de synthèse, a été proposé pour cibler les rares cellules de NB résiduelles qui survivent après chimio-radiothérapie intensive (pour revue Maris, 2010). Actuellement, l‟isotrétinoïne ou acide 1γ-cis-rétinoïque est utilisé en thérapie dans le traitement des NB (pour revue Barone et al., 2013).
6- Thérapies en développement
Les thérapies en développement ont pour but de cibler spécifiquement les gènes, protéines et voies de signalisation responsables de la transformation maligne et de la progression dans les NB. Ces approches pourraient être plus efficaces et moins toxiques que
– 23 –
la thérapie conventionnelle (pour revue Davidoff, 2012). Certaines sont actuellement testées dans des essais cliniques.
A. Inhibition des RTKs
1. Inhibition d’ALK (Anaplastic Lymphoma Kinase)
Le gène ALK a été identifié comme un gène de prédisposition au NB puisqu‟il est
présent dans environ 50% des cas familiaux de NB et environ 7% des cas de NB sporadiques (Mossé et al., 2008, George et al., 2008 ; Janoueix-Lerosey et al., 2008 ; pour revue Huang et Weiss, β01γ). L‟activation constitutive du RTK ALK par mutation ou translocation semble contribuer au phénotype malin de différents cancers, tels que le lymphome anaplasique à large cellule et le NB, ce qui fait de lui une cible thérapeutique potentielle pour ces cancers (pour revue Davidoff, β01β). L‟efficacité d‟antagonistes pharmacologiques du domaine kinase
d‟ALK a été mise en évidence dans des lignées de NB (George et al., 2008). Des inhibiteurs
d‟ALK sont actuellement testés pour la thérapie du lymphome anaplasique à large cellule et
pourraient potentiellement profiter à un sous-ensemble de patients atteints de NB (pour revue Modak et Cheung, 2010). La petite molécule inhibitrice d‟ALK, PF-02341066, (administrée oralement) est actuellement testée dans les tumeurs solides réfractaires de NB en essai de phase I/II (pour revue Davidoff, 2012).
2. Inhibition de TrkB
Le récepteur à la neurotrophine TrkB est préférentiellement exprimé dans les tumeurs de NB agressifs. La voie de signalisation BDNF/TrkB forme une boucle autocrine ou paracrine dans ces tumeurs, spécialement dans les tumeurs MYCN-amplifiées (Nakagawara et al., 1994 ; Scala et al., 1996 ; Schramm et al., 2005 ; pour revue Davidoff, 2012). Un blocage de la voie de signalisation TrkB/BDNF avec des inhibiteurs de tyrosines kinases spécifiques
de Trk pourraient induire l‟apoptose en bloquant les voies de survie essentielles (pour revue
Davidoff, 2012). Plusieurs composants de la voie de signalisation associée à TrkB incluant les tyrosines kinases Trk, PI3K (Phosphatidylinositol 3-kinase), Akt et des éléments en aval peuvent être ciblés par des petites molécules inhibitrices, comme le CEP-751 qui est un inhibiteur des Trk (pour revue Modak et Cheung, 2010 ; pour revue Davidoff, 2012).
– 24 – 3. IGF-1 (Insulin-like growth factor-1)
IGF-1 régule la croissance de cellules de NB par les voies AKT et MAPK (Sartelet et al., 2008). Des antagonistes du récepteur IGF-1 ont montré une activité anti-NB dans des modèles de xénogreffes (Coulter et al., 2008 ; Kolb et al., 2008).
B. Ciblage de la voie p53
Les mutations du gène TP53 sont rares dans les NB au moment du diagnostic (Tweddle et al., 2003 ; Tweddle et al., β001). L‟apoptose induite par chimiothérapie dans des
tumeurs MYCN-amplifiées peut être p53 dépendante (Chesler et al., 2008). Cependant,
l‟inactivation de p5γ par une mutation ou une activation de MDMβ est souvent observée dans
des tumeurs de rechutes, et est associée à une résistance aux drogues (Tweddle et al., 2001 ; Carr et al., β006). L‟un des mécanismes d‟activité anti-NB des inhibiteurs HDAC (Histones
désacétylases) in vitro est la restauration de la voie p53 dans les lignées cellulaires de NB (Condorelli et al., 2008). Cette activité anti-NB a aussi été démontrée dans des modèles de xénogreffes de NB (Coffey et al., 2001 ; Coffey et al., 2000).
C. Ciblage de l’angiogenèse
Les tumeurs neuroblastiques à haut risque présentent une densité de micro-vaisseaux importante (pour revue Modak et Cheung, 2010). Plusieurs drogues montrent une activité anti-angiogénique dans des modèles précliniques de NB. Elles incluent des agents chimio-thérapeutiques comme la vinblastine et le topotécan, les rétinoïdes (Ribatti et al., 2003) et le thalidomide (Kaicker et al., 2003).
D. Ciblage thérapeutique de MYCN
Plusieurs stratégies thérapeutiques sont envisagées pour cibler MYCN directement ou indirectement. Celles-ci seront développées dans le paragraphe VII-3-a. du chapitre II (page 49) portant sur MYCN. Elles comprennent le ciblage de la liaison de MYCN à l‟ADN, la régulation de sa stabilité et de son expression.
– 25 – X- Les NB in vitro
1- Les mécanismes moléculaires et cellulaires de la différenciation neuronale
A. Les lignées de NB : des modèles d'études
Les lignées de NB et dérivées de celles-ci sont de longue date utilisées comme des modèles in vitro d‟étude de la neuritogenèse et de la survie neuronales. Les NB sont des cellules indifférenciées ou peu différenciées (pour revue Maris et Matthay, 1999) en
conséquence d‟altérations des mécanismes de différenciation des cellules progénitrices
embryonnaires de la crête neurale dont elles dérivent (Tonini et al., 1993). La croissance de neurites (ou neuritogenèse) est un processus clé lors de la différenciation neuronale (Figure 6), et est une condition nécessaire pour l‟établissement d‟un réseau fonctionnel précis de neurones au cours du développement (Read et Gorman, 2009).
Figure 6 : Les étapes du processus de différenciation neuronale. La différenciation neuronale est un processus permettant de différencier une cellule souche (exprimant le marqueur nestine) en neuroblaste (exprimant différents marqueurs dont PHOX2B et MASH-1) par les processus de neurogenèse et de neuritogenèse sous l‟action de protéines morphogénétiques osseuses (BMP) aboutissant à la formation de neurites. Les neurites sont les ébauches des futurs axones et dendrites des neurones obtenus après
maturation en présence de NGF. Des contacts synaptiques vont pouvoir ensuite s‟établir après
– 26 –
Cette croissance est également cruciale pour la plasticité neuronale (Hrvoj-Mihi et al., 2013), ainsi que la régénération neuronale (Schiwy et al., 2009). La différenciation neuronale est un processus se caractérisant au niveau morphologique par la formation de pseudoganglions et une neuritogénèse pouvant aboutir à la mise en place d‟un réseau neuritique dans les cellules de NB LA-N-1, LA-N-5, IMR-32 et SH-SY5Y (Sidell, 1982 ; Pence et Shorter, 1990 ; Lovat et al., 1997). Parmi ces lignées, la différenciation neuronale des cellules de NB SH-SY5Y se traduit par un remodelage neuritique associé à l‟émergence
de varicosités le long des neurites et d‟un réseau de neurites longs et ramifiés, faisant d‟elle un modèle d‟étude très largement utilisé à cet égard (Alleaume et al., 2004, Lim et al., 2015). Les neurites constituent les ébauches des futurs axones ou dendrites des neurones.
B. Marqueurs moléculaires de la différenciation neuronale
Au niveau moléculaire, cette neuritogenèse est caractérisée par l‟augmentation de l‟expression de marqueurs neuronaux tels que la protéine GAP43 (Growth Associated Protein
43) dans les cellules Neuro-2a, les neurofilaments de haut poids moléculaire et la tyrosine
hydroxylase TH, ainsi que l‟augmentation de l‟activité de l‟énolase spécifique des neurones
(NSE pour Neuron Specific Enolase) dans les cellules IMR-32 (Morton et Buss, 1992 ; Chu et al., 2003 ; Påhlman et al., 1984). Dans les cellules de NB SH-SY5Y, l‟expression des
protéines du cytosquelette neuronal, telles que les neurofilaments, la - tubuline III et les
protéines MAP associées aux microtubules spécifiques des neurones croît avec le degré de différenciation neuronale (Héraud et al., 2004). De plus, il a également été démontré dans ces cellules que la protéine SNAP-β5 est impliquée dans l‟élongation neuritique et le maintien de la ramification (Héraud et al., 2008).
D‟autres marqueurs associés à la différenciation neuronale ont également été
identifiés. HNK-1 est un antigène exprimé sur les cellules neuroectodermiques de la crête neurale (Cooper et al., 1992). NLRR3 (Neuronal leucine-riche repeat protein-3) est une protéine membranaire préférentiellement exprimée dans les cellules de NB humains SH-SY5Y à pronostique favorable (Akter et al., 2014). Plusieurs gènes pro-neuraux ont été associés à la différenciation neuronale des cellules de NB F11 et SH-SY5Y : hASH1 (human MASH1, Ichimiya et al., 2001 ; López-Carballo et al., 2002), HES-1 (hairy/enhancer of split homologue-1, Grynfeld et al., 2000), Id (inhibiteur de la différenciation neuronale, Jögi et al.,
– 27 –
2002), NeuroD (López-Carballo et al., 2002), et neurogénine 1 (Kim et al., 2002). Une étude récente a mis en évidence que la croissance des neurites dans les cellules SH-SY5Y induit
l‟expression de la TH, des transporteurs à la dopamine, et du transporteur vésiculaire
monoamine 2 (Lim et al., 2015).
C. Voies de signalisation impliquées dans la différenciation
Le phénotype différencié est associé à un meilleur pronostic pour les patients atteints de NB. Ceci a incité les investigateurs à étudier ce mécanisme in vitro, dans des lignées
cellulaires de NB, notamment par l‟utilisation d‟agents différenciateurs. Des mécanismes
complexes de signalisation intracellulaire sont impliqués dans le processus de neuritogenèse conduisant à l'initiation et l'allongement des neurites. En effet, plusieurs récepteurs couplés aux protéines G (notamment le système VIP-récepteurs) et des RTK (notamment TrkA) jouent un rôle important dans le contrôle de la croissance des neurites (Georganta et al., 2013, Emdal et al., 2015). L‟expression des récepteurs des neurotrophines (TrkA, TrkB, et TrkC) et de leurs ligands respectifs (NGF, BDNF et NT3) est un régulateur de la survie, de la croissance et de la différenciation neuronale in vivo (pour revue, Nakagawara, 2001). Il a été démontré que dans les cellules de NB SH-SY5Y le NGF induit la croissance des neurites par
l‟activation de TrkA (Emdal et al., 2015) permettant ainsi d‟activer la voie PI3K/AKT et des GTPases de la famille Rho telles que Rac et Cdc42 (Hirose et al., 1998 ; Alleaume et al., 2004 ; Yamauchi et al., 2006). Le système VIP-récepteurs (développé au chapitre III page 54) va permettre la croissance neuritique en activant les voies PKA, PKC et MAPK dans les cellules de NB (Tang et al., 2014) (Figure 7). D‟autres RTK ont également été identifiés comme étant impliqués dans la différenciation neuronale. En effet, le récepteur ALK est exprimé dans le développement du système nerveux avec un rôle dans la différenciation neuronale (Iwahara et al., 1997), et l‟IGF-1R est impliqué dans la régulation de la
prolifération, la survie, la différenciation et la transformation cellulaire (Bähr et Groner, 2005)
– 28 –
Figure 7 : Processus de neuritogenèse. La neuritogenèse est un processus aboutissant à la croissance neuritique par activation du système NGF/TrkA et/ou du système VIP-récepteurs. Le système NGF/TrkA va activer différentes voies de signalisation, notamment la voie PI3K/AKT et la voie des petites protéines G comprenant Rac, Ras, Rho, ROCK et Cdc42. Le système VIP-récepteurs va activer quant à lui les voies PKA, PKC et MAPK conduisant à la transcription de gènes impliqués dans cette croissance neuritique, notamment NeuroD et neurogénine. Toutes ces voies peuvent interagir entre elles, ce qui pourrait induire une amplification du phénomène.
D‟autres molécules sont connues pour leurs propriétés d‟induction de la
différenciation (cf paragraphe IX-5-A page 21). En effet, le RA augmente l‟activité du complexe transcriptionnel AP-1 (Activator Protein-1) dans les cellules de NB N1E-115 aboutissant à la neuritogenèse (de Groot et Kruijer, 1991). De plus, dans les lignées de NB humain Neuro-2a, N1E115 et SH-SY5Y, les molécules différenciatrices peuvent activer les kinases MAPK et JNK qui sont nécessaires à la croissance neuritique (Singleton et al., 2000 ; Singh et al., 2003 ; Price et al., 2003 ; Monaghan et al., 2008a ; Wang et al., 2011 a et b ; Yu
et al., 2003 ; Yamauchi et al., 2006 ; Tezuka et al., 2013), alors que la voie PI3K-Akt (Read et Gorman 2009, Chong et al., 2012 ; Park et al., 2015) est requise pour la différenciation en
diminuant l‟expression des protéines Id (inhibitor of differentiation), qui séquestrent des
– 29 –
PI3K-Akt et MAPK/ERK régulent non seulement la différenciation neuronale et la survie mais aussi plusieurs aspects de la croissance des neurites, y compris l'allongement, le calibre et le branchement des neurites (Tang et al., 2015 ;Salto et al., 2015).
D‟autres kinases de voies de signalisation ont également été mises en évidence comme
induisant la croissance des neurites dans les NB. Il s‟agit notamment des kinases de la famille Src (Formoso et al., 2015), de la PKA (Singleton et al., 2000, Dagda et al., 2014), de la PKC (Singleton et al., 2000 ; Miloso et al.,2004), de la kinase ATM (Fernandes et al., 2007), de la kinase FAK (focal adhesion kinase, Suzuki et al., 2014), de la kinase PINK1 (ou PTEN-induced putative kinase 1, Dagda et al., β014), de la voie Wnt/ -caténine (Vasileiou et al., 2015), et des GTPases de la famille Rho dont Cdc42 (Suzuki et al., 2014 ; Reddy et al., 2015). Cdc42 est un gène candidat suppresseur de tumeurs dans les NB qui contrôle
l‟organisation du cytosquelette, la formation de lamellipodes et la différenciation cellulaire du NB, et dont l‟expression est diminuée par MYCN (Valentijn et al., 2005). Une étude récente a mis en évidence un nouveau mécanisme qui couple fonctionnellement la signalisation AMPc/PKA au renouvellement protéolytique de NOGO-A (un inhibiteur majeur de la croissance des neurites dans le cerveau des mammifères) où la protéolyse de NOGO-A induite par la signalisation AMPc/PKA est liée à la croissance des neurites (Sepe et al., 2014).
2- Les mécanismes cellulaires et moléculaires de l’invasion/métastase A. Processus d’invasion/métastase
La première étape de la métastase est l‟invasion locale, qui exige que les cellules malignes perdent l‟adhérence cellule-cellule au sein de la tumeur et deviennent mobiles, un
processus qui leur permet d'envahir les tissus environnants. Au cours de la deuxième étape,
appelée l‟intravasation, les cellules tumorales pénètrent l'endothélium des vaisseaux sanguins
ou lymphatiques par dégradation protéolytique de la matrice extracellulaire (MEC) et entrent dans la circulation vasculaire ou lymphatique (Wyckoff et al., 2000). Peu de cellules tumorales circulantes sont cependant capables de survivre et former des agrégats tumoraux ou des embolies avec des leucocytes et des plaquettes circulants et de gagner des sites distants. Ces cellules tumorales vont alors acquérir un phénotype migratoire et pourront sortir des vaisseaux sanguins et lymphatiques (extravasation), et ainsi coloniser les sites smétastatiques (pour revue Ara et DeClerck, 2006) et envahir différents tissus comme l‟os (Figure 8).
– 30 –
L‟instabilité génétique dans les cellules tumorales et les modifications épigénétiques
contribuent à un phénotype invasif (Macaluso et al., 2003; Guo et Giancotti, 2004). L'interaction entre les cellules tumorales et les cellules stromales environnantes crée un micro-environnement local qui favorise les changements épigénétiques dans les cellules malignes (pour revue Ara et DeClerck, 2006).
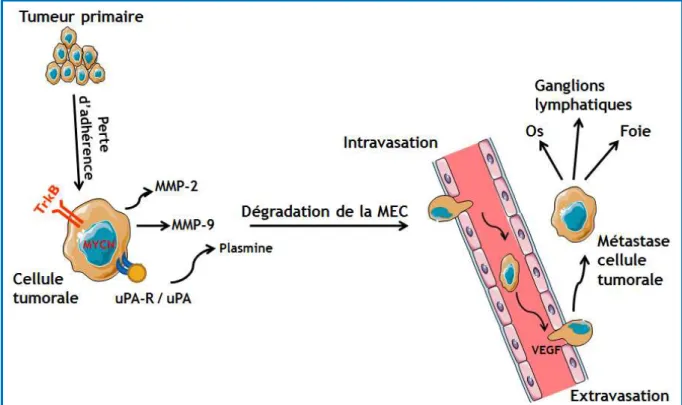
Figure 8 : Les étapes multiples du processus d’invasion/métastase. La progression tumorale dépend de l'invasion locale, l‟intravasation, la survie dans la circulation, l‟extravasation et la colonisation. Après la perte d‟adhérence à la tumeur primaire, les cellules tumorales exprimant le récepteur TrkB et une amplification de MYCN sécrètent plusieurs facteurs, y compris des protéases comme les MMPs et la plasmine qui dégradent la matrice extracellulaire (MEC) facilitant leur migration et invasion. Puis, les cellules tumorales passent à travers le revêtement endothélial des vaisseaux sanguins (intravasation) et se retrouvent dans la circulation sanguine. Après extravasation, elles envahissent les organes distants, comme les ganglions lymphatiques, les os, le foie. Les cellules cancéreuses prolifèrent dans le tissu envahi, dispersant ainsi la maladie.
B. Les molécules impliquées dans l’invasion/métastase
Un gène clé impliqué dans l‟invasion/métastase des cellules de NB est l‟oncogène
MYCN puisqu‟il est capable d‟agir et interagir avec des molécules permettant ou inhibant ce