Propriétés structurales opto-électroniques élastiques et dynamiques des semi-conducteurs type II-VI
172
0
0
Texte intégral
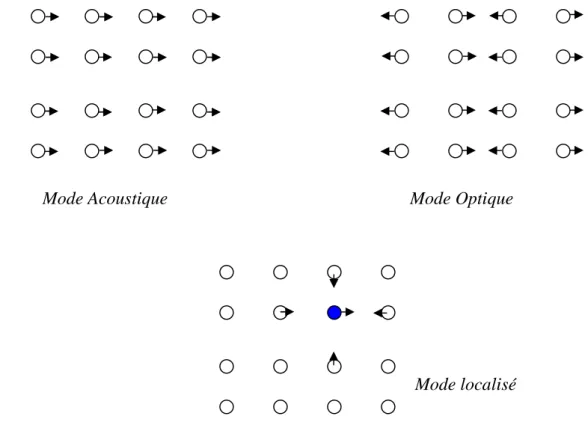
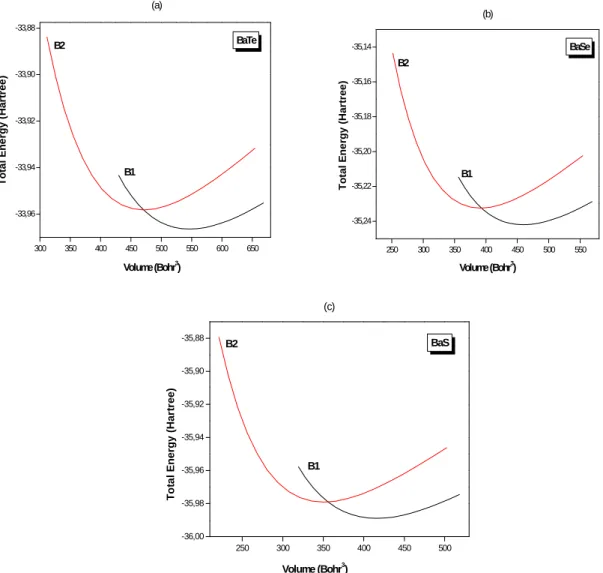
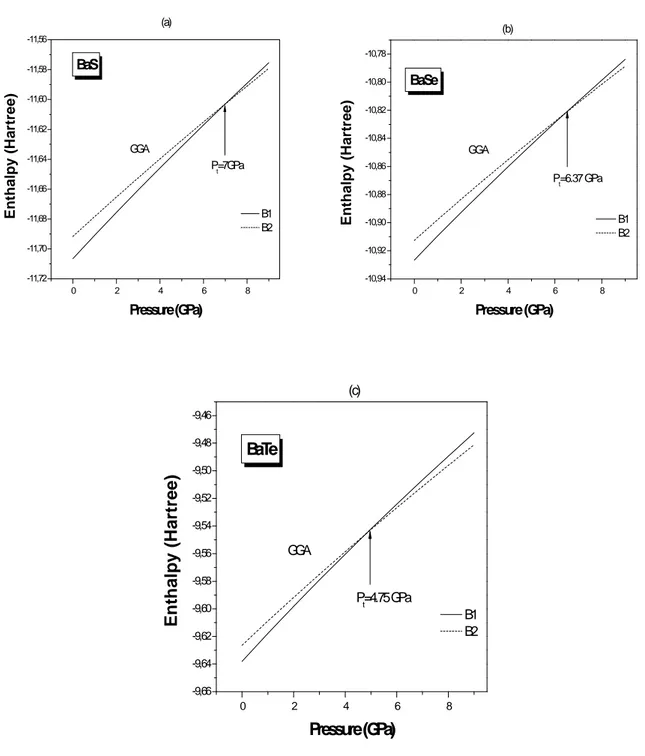
Figure
+7
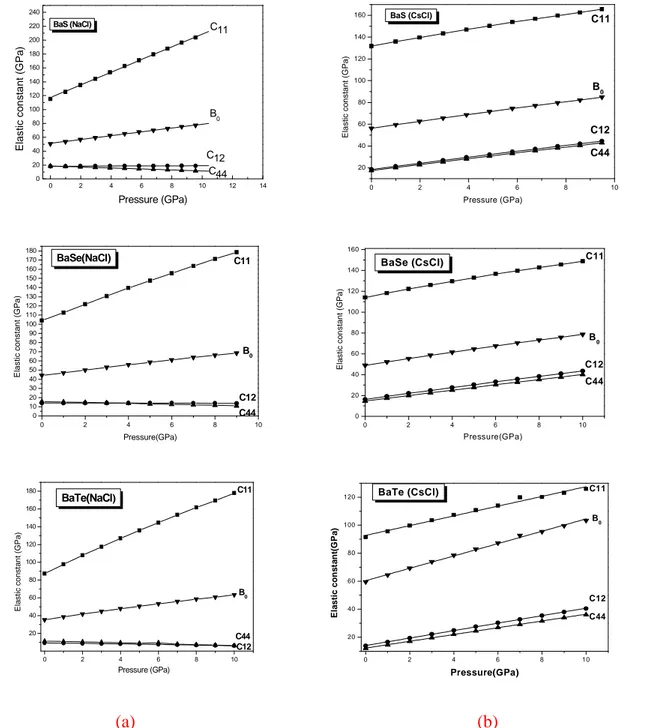
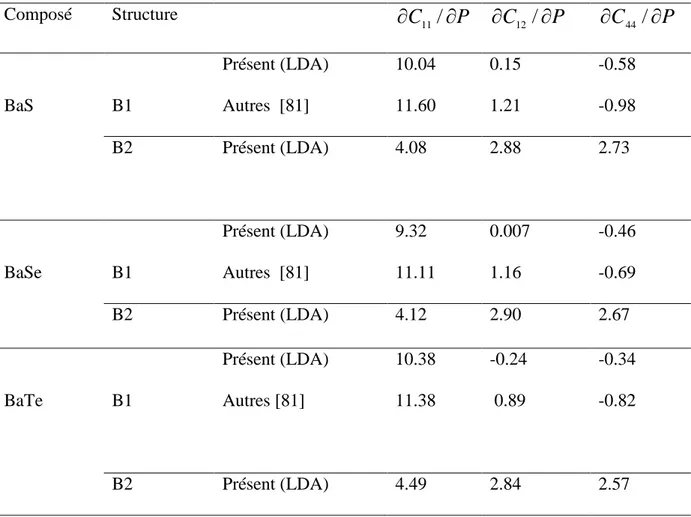

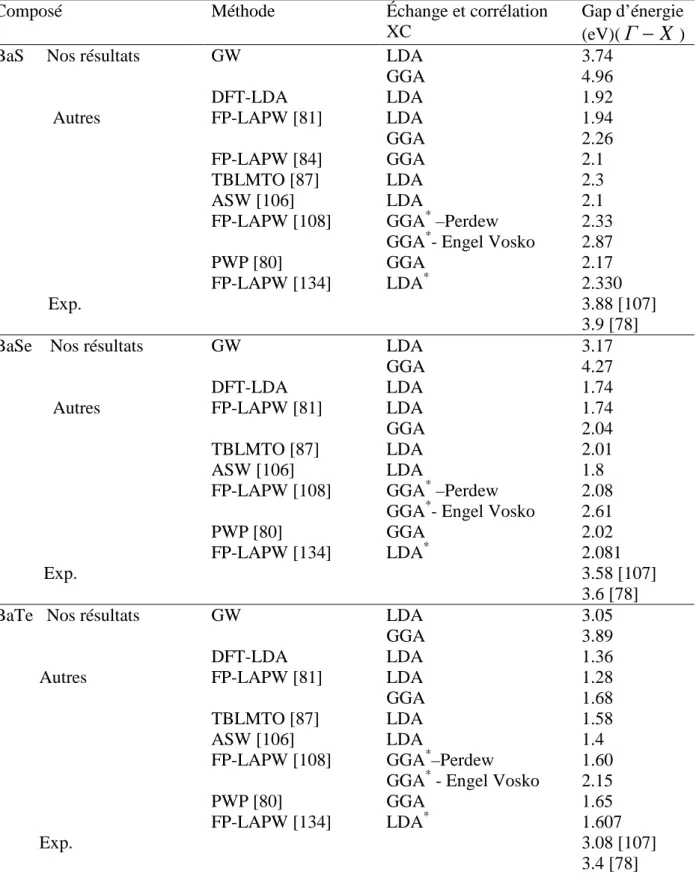
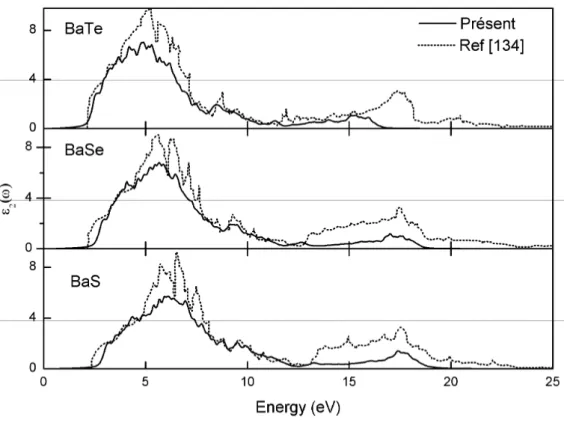

Documents relatifs
Etant donné que la masse des noyaux est beaucoup plus grande que celle des électrons, le mouvement des noyaux est beaucoup plus lent [1]. A chaque instant le système
Nous venons d’établir que la densité de l’état fondamental est en principe suffisante pour obtenir toutes les propriétés intéressantes d’un système
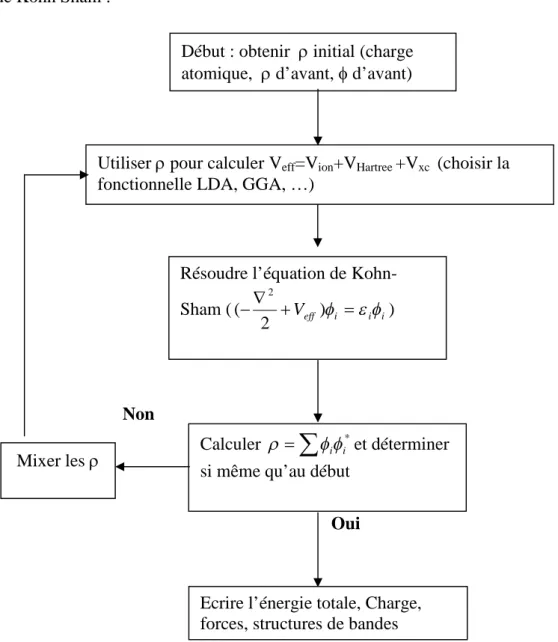
La méthode utilisée dans notre travail est l’une des plus précises, actuellement, pour le calcul de la structure électronique des solides dans le cadre de la
