© Vincent Pons, 2021
Rôle du CSF1R dans les maladies neurodégénératives
Thèse
Vincent Pons
Doctorat en médecine moléculaire
Philosophiæ doctor (Ph. D.)
Rôle du CSF1R dans les maladies
neurodégénératives
Thèse de Doctorat
Vincent Pons
Sous la direction de :
II
Résumé
La maladie d’Alzheimer (MA) est la maladie neurodégénérative la plus fréquente dans le monde, son incidence augmente au cours des années dû à un vieillissement de la population et à manque de thérapies efficaces. L’étiologie de la maladie est associée à une altération de la mémoire et du comportement ainsi qu’à l’accumulation de la bêta-amyloïde (Aβ) dans le parenchyme et les vaisseaux sanguin du cerveau. Ces dépôts aberrants sont la conséquence d’une altération de l’élimination du peptide. Depuis quelques années des évidences venant des expériences menées en laboratoire montrent que l’injection de certaines molécules ont des effets bénéfiques sur la MA, tant au niveau cognitif que sur la présence d’Aβ. Une de ces molécules est le macrophage-colony stimulating factor (m-CSF) et son récepteur (CSF1R). Cette voie de signalisation a fait l’objet de plusieurs études dans le contexte de la MA or l’implication de son récepteur dans le processus pathologique est peu connu. Les études incluses dans cette thèse de doctorat avaient pour but de mieux comprendre le rôle du CSF1R sur la prolifération et la survie microgliale dans un contexte non-pathologique ainsi que dans un modèle animal de la MA. Plusieurs études indiquent que ce récepteur est primordial pour la survie, l’activation et la prolifération microgliale. Nous avons utilisé une approche du style « perte de fonction » pour étudier le CSF1R. Grâce à un modèle de souris knock-out (KO) inductible, nous avons spécifiquement aboli la transcription du récepteur dans les microglies. Dans un premier temps nous avons comparé les fonctions du CSF1R dans deux modèles. Le premier étant purement prolifératif avec pas ou peu d’inflammation. Le second un modèle inflammatoire. Nous avons observé que les microglies dans le premier modèle étaient capables de proliférer et donc survivaient au KO. Dans le second modèle, les cellules microgliale perdaient leur capacité proliférative mais survivaient. Nous avons pu déduire que le CSF1R au stade adulte n’a vraisemblablement qu’un effet accessoire sur la prolifération et il ne semble pas être impliquer dans la survie microgliale. En utilisant la même approche, nous avons supprimé comme précédemment le CSF1R dans un modèle de souris Alzheimer APPswe/PS1. Dans cette étude nous montrons là aussi que non seulement le CSF1R n’a pas de rôle sur la survie et la prolifération des microglies, mais en plus on observe une diminution des symptômes associés à la MA. À savoir une diminution du déclin cognitif ainsi que de la charge amyloïde. Ensembles ces données nous montre de nouvelles informations sur CSF1R. Son rôle n’est pas aussi primordial dans certaines fonctions microgliale à l’âge adulte.
III
Abstract
Alzheimer’s disease (AD) is the most frequent neurodegenerative disease in the world, its incidence increases every year due to the aging of the population and a lack of therapies. AD etiology is associated with an alteration of cognitive functions and aberrant accumulation of amyloid-beta (Aβ) in the parenchyma and blood vessels in the brain. Aβ deposits are due to an impairment of phagocytosis. For many years, experimental evidence shown that injections of different molecules could have beneficial effects on AD course, either cognition or amyloid load. One of these molecules is macrophage-colony stimulating factor (m-CSF) and its receptor (CSF1R). This signaling pathway was extensively studied in AD context, but it remains largely unknown. Studies which are included in thesis, aimed to better understand the role of CSF1R on microglia proliferation and survival in both healthy and AD context. Many studies mentioned that CSF1R has a crucial impact on these functions. We used a “loss of function” approach to study this receptor. We used an inducible knock-out (KO) mice model, where microglia are specifically deleted of CSF1R. In the first time we compared CSF1R function in two models. The first model is a pure proliferative model without or barely without inflammation, the second is a model with a robust inflammatory response. We observed in the first model that microglia were still able to proliferate and survived to the KO, whereas in the second model, microglia were unable to proliferate but they also survived. We deducted that CSF1R at adult stage, has an accessory role on proliferation and has no major impact on microglia survival. Using the same approach, we have deleted the
receptor in a mouse model of AD, namely APPswe/PS1. Likewise, in this study we have
shown that CSF1R was not required for proliferation and survival, moreover we observed an improvement of cognition and a reducing level of amyloid. Altogether, these data provide a new insight on CSF1R functions, its role is not as primordial for microglia as we though.
IV
Table des Matières
RESUME ... II ABSTRACT ... III LISTE DES FIGURES, TABLEAUX, ILLUSTRATIONS ... VII LISTE DES ABRÉVIATIONS ... VIII REMERCIEMENTS ... XII AVANT-PROPOS ... XIV
INTRODUCTION GÉNÉRALE ... 1
LA MALADIE D’ALZHEIMER ... 2
L’histoire de la Maladie d’Alzheimer ... 2
Les signes cliniques ... 3
La pathologie d’Alzheimer ... 6
Les causes étiologiques ... 7
L’hypothèse amyloïde ... 7
L’hypothèse tau ... 8
L’hypothèse cholinergique ... 9
Les facteurs de risques ... 11
Facteurs génétiques ... 11
Risques non-génétiques ... 13
LE SYSTEME IMMUNITAIRE ... 14
L’immunité innée ... 14
Les monocytes ... 14
Différents sous-types de monocytes ... 15
Les monocytes classiques ... 17
Les monocytes non-classiques ... 17
Les macrophages ... 19
LES MICROGLIES ... 20
Les fonctions des microglies dans le développement ... 20
Les microglies dans le cerveau ... 21
Les microglies dans la maladie d’Alzheimer ... 23
L’AXE M-CSF/CSF1R ... 26
HYPOTHÈSE ET OBJECTIFS ... 31
CHAPITRE 1 : L’AXE M-CSF/CSF1R UNE THERAPEUTIQUE D’AVENIR ... 34
1.1 RESUME ... 35
1.2 ABSTRACT ... 35
1.3 NEW THERAPEUTIC AVENUES OF MCSF FOR BRAIN DISEASES AND INJURIES ... 36
1.3.1 Introduction ... 37
1.3.2 M-CSF in pathological conditions ... 38
1.3.3 Alzheimer’s disease (AD) ... 39
1.3.4 Multiple sclerosis (MS) ... 40 1.3.5 Glioma ... 42 1.3.6 Brain injury ... 43 1.3.7 Clinical studies ... 43 1.3.8 Conclusion ... 44 1.3.9 Authors contribution ... 45
V
1.3.10 Funding ... 45
1.3.11 References ... 45
1.3.12 Figure ... 50
CHAPITRE 2 : ROLE DU MACROPHAGE COLONY-STIMULATING FACTOR RECEPTEUR SUR LA PROLIFERATION ET SURVIE MICROGLIALE ... 51
2.1 RESUME ... 52
2.2 ABSTRACT ... 53
2.3 ROLE OF MACROPHAGE COLONY-STIMULATING FACTOR RECEPTOR ON THE PROLIFERATION AND SURVIVAL OF MICROGLIA FOLLOWING SYSTEMIC NERVE AND CUPRIZONE-INDUCED INJURIES ... 54
2.3.4 Introduction ... 55
2.3.5 Materials and methods ... 56
2.3.5.1 Animals surgery ... 56
2.3.5.2 Cuprizone Diet ... 56
2.3.5.3 Conditional Csf1R KO Mice ... 57
2.3.5.4 Tamoxifen preparation and administration ... 57
2.3.5.7 Sacrifices ... 57
2.3.5.8 Immunohistochemical Staining ... 58
2.3.5.9 Immunofluorescent Staining ... 58
2.3.5.11 Quantitative Real-Time PCR ... 59
2.3.5.12 Image Acquisition and Analyses ... 61
2.3.5.13 Statistical Analyses and Figure Preparation ... 62
2.3.6 Results ... 62
2.3.6.1 Mouse model of CSF1R deletion specifically in microglia ... 62
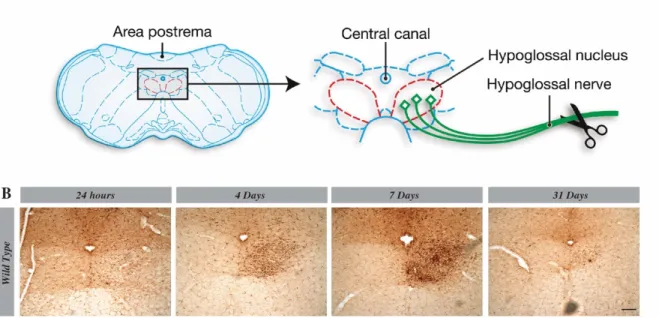
2.3.6.2 Microglial proliferation in hypoglossal nucleus is maximal 7 days after the lesion ... 63
2.3.6.3 Knocking-out CSF1R selectively in microglia does not affect cell proliferation ... 63
2.3.5.4 CSF1R depletion induces infiltration of peripheral cells ... 64
2.3.5.5 CSF1R-depleted microglia affect their proliferation in the cuprizone diet model of acute demyelination. ... 65
2.3.7 Discussion ... 65
2.3.8 Data Availability Statement ... 68
2.3.9 Ethics Statement ... 68 2.3.10 Author Contributions ... 68 2.3.11 Funding ... 68 2.3.12 Conflict of Interest ... 69 2.3.13 Supplementary Material ... 69 2.3.14 References ... 69
CHAPITRE 3 : ROLE DU MACROPHAGE COLONY-STIMULATING FACTOR RECEPTEUR DANS LA MALADIE D’ALZHEIMER. ... 80
3.1 RÉSUMÉ ... 81
3.2 ABSTRACT ... 82
3.3 CONDITIONAL GENETIC DELETION OF CSF1 RECEPTOR IN MICROGLIA AMELIORATES THE PHYSIOPATHOLOGY OF ALZHEIMER’S DISEASE ... 83
3.3.1 INTRODUCTION ... 84
3.3.2 Results ... 86
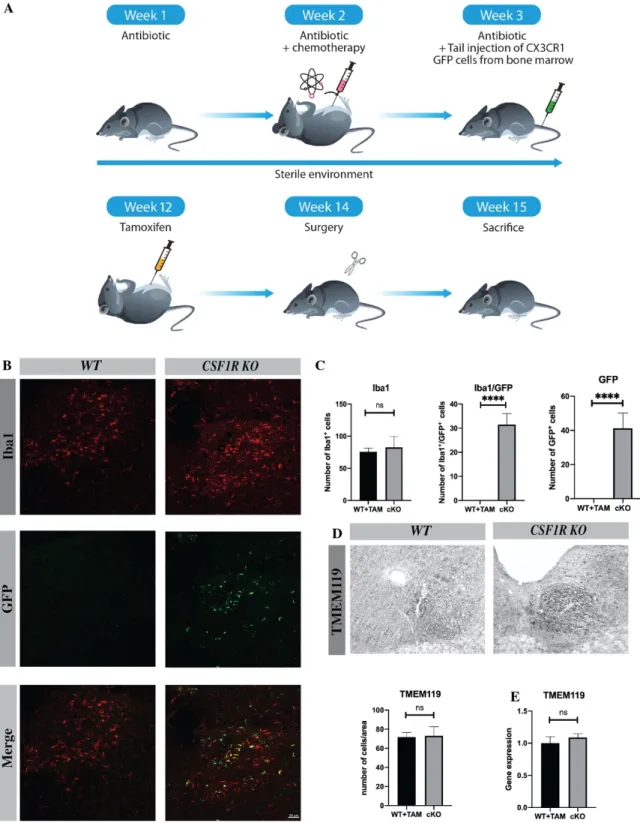
3.3.2.1 CSF1R is solely deleted in microglia ... 86
3.3.2.2 The deletion of CSF1R does not affect microglia survival and delay cognitive decline in APP cKO mice. ... 87
3.3.2.3 Long-term knock-out reduces volume and plaque number, with the onset of cerebral amyloid angiopathy (CAA). ... 88
3.3.2.4 TREM2/β-Catenin and IL-34 brain protein levels following CSF1R gene deletion. ... 90
3.3.2.5 The number of microglia following knock-out induction remains unchanged. ... 91
3.3.3 Discussion ... 91
VI 3.3.5 Data availability: ... 96 3.3.6 Ethics Statement ... 96 3.3.7 Author contributions: ... 96 3.3.8 Funding: ... 96 3.3.9 Conflict of interest: ... 96
3.3.10 Materials and methods: ... 97
3.3.10.1 Animals: ... 97
3.3.10.2 Tamoxifen preparation and administration: ... 98
3.3.10.3 Sacrifices: ... 98 3.3.10.4 Western blot: ... 98 3.3.10.5 Immunofluorescent staining: ... 99 3.3.10.6 Immunohistochemistry: ... 99 3.3.10.7 Behavioral analyses: ... 100 3.3.10.8 ELISA: ... 100
3.3.10.9 Unbiased Stereological count: ... 100
3.3.10.10 Image Acquisition: ... 101
3.3.10.11 Statistical analyses and Figure preparation: ... 101
3.3.11 References ... 102
3.3.12 Figures ... 109
CHAPITRE 4: L’IMMUNITÉ INNÉE DANS LA MALADIE D’ALZHEIMER ... 119
4.1 RÉSUMÉ ... 120
4.2 ABSTRACT ... 121
4.3 INTRODUCTION ... 123
4.3.1 Aβ, the key to understand the Disease ... 124
4.3.2 Cerebrovascular amyloid production ... 125
4.3.3 Amyloid and neuroinflammation ... 127
4.4 M-CSF/CSF1R AXIS TO MODULATE MICROGLIA AND MONOCYTES ... 128
4.5 MPL AND TLR4 LIGANDS ... 131
4.6 NOD2/MDP AND PATROLLING MONOCYTES, A PROMISING OPTION IN AD TREATMENT ... 132
4.7 EPO A NOVEL INSIGHT IN AD ... 135
4.8 CONCLUSION ... 136
4.9 ACKNOWLEDGEMENTS ... 139
4.10 REFERENCES ... 139
CHAPITRE 5 ... 153
5.1 DISCUSSION GENERALE ... 153
5.2 ROLE DE CSF1R DANS LA PROLIFERATION ET LA SURVIE MICROGLIALE ... 154
5.3 RÔLE DU CSF1R DANS LA MALADIE D’ALZHEIMER ... 156
CONCLUSION ... 158
VII
Liste des figures, tableaux, illustrations
FIGURE 1-1 DÉVELOPPEMENT ET DEVENIR DES MONOCYTES CHEZ LA SOURIS. ... 16
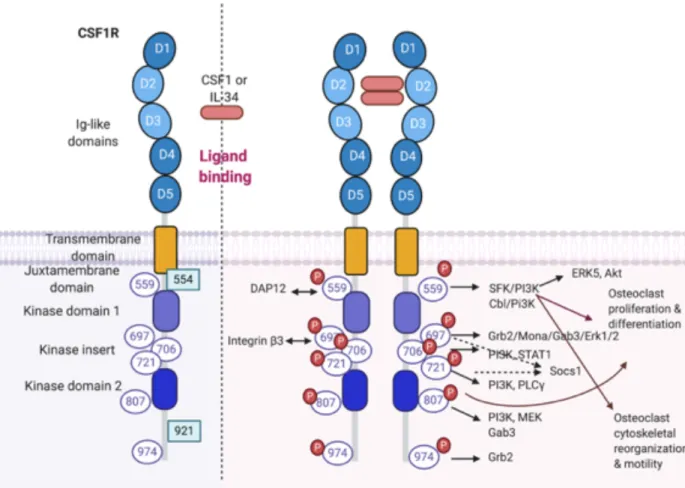
FIGURE 1-2 STRUCTURE DU CSF1R. ... 28
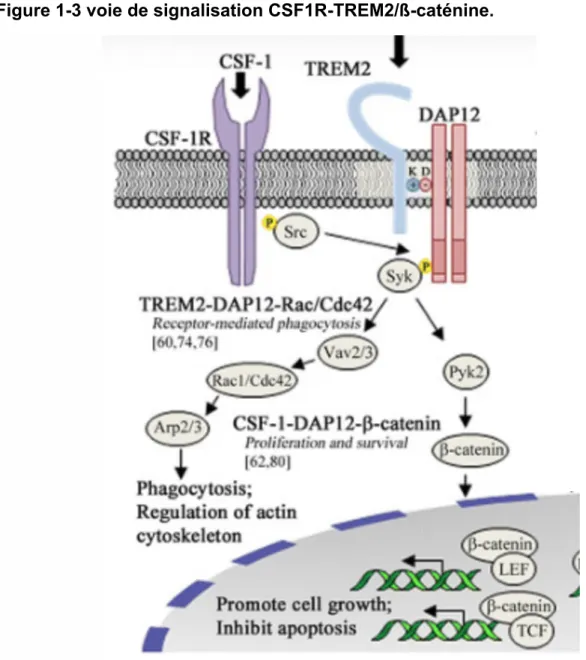
FIGURE 1-3 VOIE DE SIGNALISATION CSF1R-TREM2/ß-CATÉNINE. ... 31
FIGURE 2-1 FATE OF MICROGLIA IN BRAIN DEPENDING ON STIMULI. ... 50
FIGURE 3-1 CSF1R IS SPECIFICALLY DELETED IN MICROGLIA. ... 72
FIGURE 4-1 CSF1R DELETION IS SOLELY DELETED IN MICROGLIA ... 109
FIGURE 4-2 THE DELETION OF CSF1R DOES NOT AFFECT MICROGLIA SURVIVAL AND DELAY COGNITIVE DECLINE IN APP CKO MICE. ... 111
FIGURE 4-3 LONG-TERM KNOCK-OUT REDUCES VOLUME AND PLAQUE NUMBER, ACCOMPANIED BY CEREBRAL AMYLOID ANGIOPATHY (CAA) ONSET. ... 113
FIGURE 4-4 TREM2, Β-CATENIN AND IL-34 CAN COMPENSATE CSF1R DELETION ... 115
FIGURE 4-5 THE KNOCK-OUT HAS NO IMPACT ON MICROGLIA SURVIVAL OVER THE TIME ... 117
VIII
Liste des Abréviations
AAC angiopathie amyloïde cérébrale Aβ Bêta-Amyloïde
ACh Acétylcholine APOE Apolipoprotéine E
APP Protéine précurseur de l’amyloïde ATP Adénosine triphosphate
ARN Acide ribonucléique
AVC Accident vasculaire cérébral BACE-1 bêta-site APP cleaving enzyme 1 BDNF Brain-derived neurotrophic factor C/EBPβ CCAAT-enhancer-binding protein CCL2 Chemokine (C-C motif) ligand 2 CCR2 C-C chemokine receptor type 2 cKO conditional Knock-out
CMoP common monocytes progenitor CMP common myeloid progenitor CX3CR1 CX3C chemokine receptor 1
DAMP Danger-associated molecular patterns EPS Signes extrapyramidaux
GMP granulocyte and macrophage progenitor HSCs cellules souches hématopoïétiques
IFN-β Interféron-β IL Interleukin
IX
KO Knock-out
MA Maladie d’Alzheimer MAC-1 Macrophage-1 antigen mAChRs Récepteurs muscariniques
m-CSF Macrophage colony-stimulating factor MHC-II Complexe majeur d’histocompatibilité II MDPs macrophage and dendritic cell progenitor MDP Muramyl-dipeptide
MyD88 Myeloid differentiation primary response protein 88 nAChRs Récepteurs nicotiniques
NLRP3 NOD-like receptor family pyrin domain containing 3
NF-κB Nuclear factor kappa-light-chain-enhancer of activated B cells NOD2 Nucleotide-binding oligomerization domain-containing 2 Nur77 Nerve growth factor IB
PAMP Pathogen-associated molecular patterns PS1 Presenilin 1
PS2 Presenilin 2
ROS Espèces réactives de l’oxygène RTK Récepteur tyrosine kinase SNC Système nerveux central TAK1 TGF-β-associated kinase 1 TGF-β Transforming growth factor bêta TLR Toll-like receptor
TNF-⍺ Tumor necrosis factor-alpha
XI
« Les portes de l’avenir s’ouvrent à ceux qui savent les pousser »
XII
Remerciements
Voici maintenant quelques années que j’ai entamé cette périlleuse aventure que sont les études. Il serait prétentieux de m’attribuer tous les mérites de mon parcours. En effet sans les personnes qui m’ont entouré, m’ont tendu la main, m’ont fait confiance, je ne serai peut-être pas ici. Certaines rencontres sont plus marquantes que d’autres que ce soit d’un point de vue personnel ou professionnel. C’est grâce à ces personnes croisées parfois par hasard que j’ai pu arriver où j’en suis. C’est donc naturellement à eux que je veux dédier cette partie car sans leur soutien et leur confiance les choses auraient été je pense bien différentes.
D’un point de vue professionnel je tiens à remercier mon directeur de recherche et en quelques sortes mon mentor en sciences Serge Rivest, sans qui rien n’aurait été possible. En 2017 il m’a fait confiance et m’a accueilli au sein de son laboratoire. Il a toujours su avec bienveillance me donner les moyens d’effectuer ma thèse. Avec son soutien et ses conseils j’ai appris à développer mes compétences et mon esprit scientifique. Sa passion pour les sciences et son parcours exceptionnel sont inspirants, il a démontré que dans la vie rien n’est impossible quand on s’en donne les moyens. J’aimerai aussi remercier les professionnels de recherches Paul Préfontaine, Marie-Michèle Plante et Nataly Laflamme qui m’ont accompagné et n’hésitons pas à le dire, supporté durant ces dernières années. Grâce à eux j’ai pu mener à bien mes projets et apprendre une énormément.
Mon doctorat à Québec a été en patie possible grâce à la confiance que m’a apporté Michel Vignes directeur de mon programme de Maîtrise en France je l’en remercie chaleureusement.
Merci aux membres de mon jury (Dr Serge Rivest, Dr David Gosselin, Dr Ayman ElAli et Dr Louis-Éric Trudeau) d’avoir pris le temps de lire ma thèse et assister à ma soutenance, ainsi que les organismes financiers IRSC et CRSNG.
D’un point de vue personnel, ma famille a énormément compté pour moi et ce depuis toujours. Même de l’autre côté de l’Atlantique ils ont sus me donner la force, l’envie d’arriver là. Je les remercie pour leur soutient et leurs encouragements malgré le fait que mon sujet de recherche sonne chinois pour eux.
Maman après toutes ces années, et les soucis que t’ont apporté mes études les révisions de partiels à la derniére minutes je ne m’en suis pas si mal sorti.
Papa, tu me demandes souvent « dé qhas trappat » au grand dam de Sylvie, voici les résultats de mes recherches en français cette fois.
XIII
Je veux aussi remercier mes grands-parents Marie et Louis, qui chaque semaine au téléphone me disaient « travaille bien cette semaine, le travail c’est la santé », en dépit des quelques cernes vestiges de ces dernières années mon travail a payé.
Simone pour qui j’ai une pensée particulière, c’est grâce à toi que je me suis orienté dans cette voie.
Aussi je ne serai pas là sans le soutien de mes Tantes et oncles, Christine, Jean, Françoise, Philippe et Marie-Josée. Le cercle familial est bien précieux car il permet aussi de se développer, de grandir et de se dépasser. À mes amis merci d’avoir été là au quotidien, pour tous ces bons moments passés au lab et en dehors Pierre-Alexandre et Romain. Un merci particulier à Sarah qui arrive à me supporter depuis pas mal de temps, merci pour tes conseils, ton soutien indéfectible et pour ta patience.
Pour finir cette partie de remerciement, le doctorat, peut être un chemin hasardeux, semé d’embuches mais ce qui est sûr c’est que, quand on est bien accompagné l’aventure n’en devient que plus intéressante.
XIV
Avant-propos
Cette thèse contient les résultats des principaux projets de mes études de doctorat divisés en 6 parties. La première est une revue de la littérature en rapport avec mes projets ainsi que la présentation des hypothèses et objectifs expérimentaux. Les chapitres 1,2,3 et 4 sont issus d’articles publiés ou en cours de publication dans des journaux scientifiques révisés par les pairs. Ces articles sont le fruit d’une collaboration étroite entre les différents acteurs de notre laboratoire. Je suis l’auteur principal des articles présentés dans cette thèse sous la tutelle du Dr Serge Rivest.
Pons V, Rivest S New Therapeutic Avenues of mCSF for Brain Diseases and Injuries. Front Cell Neurosci, 12, p. 499, 2018.
Pons V, Laflamme N, Prefontaine P, Rivest S Role of Macrophage Colony-Stimulating Factor
Receptor on proliferation and Survival of Microglia Following Systemic Nerve and Cuprizone-Induced Injuries. Front Immunol, 11, p. 47, 2020, ISSN: 1664-3224
Pons V, Levesque P, Plante MM, Rivest S Conditional genetic deletion of CSF1 receptor in
microglia ameliorates the physiopathology of Alzheimer’s disease. Soumis
Pons V, RivestS Targeting systemic innate immune cells as a therapeutic avenue for Alzheimer’s disease. En révision Pharmacological Review
Le chapitre 6 propose une discussion générale dans le domaine de la MA, ainsi qu’une discussion spécifique concernant les résultats obtenus dans les chapitres 3 et 4. La dernière partie est une conclusion générale dans laquelle on peut trouver quelques éléments spéculatifs.
1
2
La maladie d’Alzheimer
La maladie d’Alzheimer (MA) est la forme de maladie neurodégénérative la plus fréquente avec plus de 43.8 millions de personnes atteintes de la MA ou maladies apparentées en 2016 (Nichols et al., 2019). Son impact sur la société va s’aggraver avec le vieillissement de la population. Selon l’Organisation mondiale de la santé 5 à 8 % de la population atteinte par MA est agée de 60 ans ou plus. Les prévisions montrent que le nombre de personnes atteintes va doubler entre 2016 et 2030 pour atteindre près de 80 millions de cas. À l’heure actuelle aucun traitement n’existe afin de prévenir, ralentir ou guérir la maladie. Au niveau national selon le gouvernement 402 milles personnes vivent avec la MA ou maladies apparentées soit une prévalence de 7.1%. Cette maladie est lourde pour le patient qui perd ses repères et se désocialise. La famille doit s’adapter à cette situation. C’est aussi une lourde charge pour la société, car le Canada dépense selon la société Alzheimer plus de 33 milliards de dollars par année pour les démences, d’ici 2040 les prévisionnistes, prévoient une dépense dix fois supérieure par année. Malgré les avancées faites en recherche, la MA reste une maladie peu comprise. D’où le besoin urgent d’identifier des cibles thérapeutiques efficaces, afin de guérir ou du moins ralentir efficacement sa progression.
L’histoire de la Maladie d’Alzheimer
Le terme démence est utilisé vers la fin du 19ème siècle, il fait référence à tous stades
de dégradation psychologique associé à une maladie chronique du cerveau (Berrios, 1990). Le premier cas décrit de cette forme de démence a été fait par Alois Alzheimer psychiatre allemand en 1907. Il décrit une patiente, nommée Auguste D de 51 ans présentant une altération de la mémoire, une désorientation spatio-temporelle, une altération des émotions et du caractère. D’un point de vue histopathologique, il rapporte la présence de plaques, de neurofilaments, ainsi
qu’une atrophie cérébrale (Maurer et al., 1997). Ce n’est qu’en 1910 dans la 8ème
3
« Maladie d’Alzheimer ». Alzheimer classe cette maladie comme étant rare et touchant les individus d’âge moyen. Il faudra attendre les années 1960 pour qu’une étude épidémiologique montre que la MA est une maladie relativement courante chez les personnes âgées (Kay et al., 1964). La MA suscite un fort intérêt de la part des scientifiques, cependant un long chemin reste à parcourir afin de mieux caractériser et comprendre cette pathologie.
Les signes cliniques
La MA est une maladie qui s’installe doucement, bien avant que les manifestations telles que nous les connaissons soient pleinement visibles. Le concept de MA
préclinique émerge à la fin du 20ème siècle (Hubbard et al., 1990). Ce qui est appelé
MA pré-symptomatique c’est le fait qu’un individu ne présente aucun symptôme visible mais exprime des marques/dispositions qui le destine à développer la MA (Dubois et al., 2016). Cela peut se caractériser par la présence dans le génome d’une mutation dominante (e.g., APP, PS1, PS2, TREM2 RH47). Le diagnostic de cette maladie, n’est pas chose facile, cependant de nombreuses recherches ont identifié des marqueurs qui sont pertinents et relativement fiables pour détecter la maladie de manière précoce. De nos jours la combinaison du dosage du liquide céphalorachidien et de l’imagerie comme la tomographie par émission de positrons (PET scan) nous aide à détecter la présence de marqueurs précoces disposants l’individu à développer les signes cliniques de la MA. Les marqueurs utilisés sont
l’amyloïde (Aβ42) et la phosphoprotéine tau. Ces molécules sont la signature de la
MA et peuvent être utilisées pour diagnostiquer la maladie des années avant. Il est montré que les modifications précoces de la protéine tau sont détectables dans le liquide céphalorachidien (“Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease,” 2012), corroboré par l’imagerie médicale qui montre une
augmentation de Aβ42 (Palmqvist et al., 2014). Ces deux phénomènes commencent
des années avant la mise en place de la MA, d’où la nécessité d’avoir à notre disposition des outils diagnostics précoces efficaces (Dubois et al., 2016).
4
Les diagnostics plus classiques sont basés sur la présence de syndromes de démences, c’est-à-dire la perte de facultés intellectuelles de gravité suffisante pour interférer avec le fonctionnement normal professionnel ou social (World Health Organization, 1993). Cependant le diagnostic de la démence seule ne constitue pas un diagnostic fiable et immuable. On détermine 4 étapes pour confirmer le diagnostic : (1) établissement du déclin des fonctions cognitives en se basant sur de précédentes données. (2) établir que le déclin des fonctions cognitives du patient interfère avec ses activités professionnelles, sociales et activités de tous les jours. (3) mesurer le déficit cognitif avec au moins 2 fonctions cognitives. (4) établir avec un bon niveau de certitude que la maladie sous-jacente correspond à l’étiologie de la démence (López and DeKosky, 2008).
Les symptômes cognitifs commencent par la perte de mémoire. C’est le principal facteur associé à la MA, dans les premiers temps cette altération est subtile (e.g., oubli de conversation, problèmes avec les noms, …). Des altérations des lobes frontal et occipital ainsi qu’un déficit dans les fonctions exécutrices sont les symptômes initiaux les plus relevant de la MA (Baddeley et al., 1997; Hof et al., 1993; Johnson et al., 1999). Le déficit du langage est la seconde manifestation la plus observée. Le patient va avoir du mal à trouver ses mots, il sera dysnomiaque (altération de l’apprentissage caractérisé par des difficultés à se souvenir de noms ou de mots) (Price et al., 1993). Il est rapporté que le déficit de langage serait annonciateur d’une avancée rapide de la MA (Boller et al., 1991). La manifestation de cette maladie n’est pas ressentie chez la plupart des patients, ils ne sont pas conscients ou peu capables de reconnaitre la présence de ces troubles (Marshall, 2004). Cette absence de prise de conscience du déficit cognitif (anosognosie) empire la maladie car cela entraine des difficultés de gestion de la personne, de sa vie au quotidien et ne facilite pas l’aide apportée par l’entourage du patient. La MA se manifeste aussi par la présence de signes neurologiques, tels que des mouvements myocloniques et des signes extrapyramidaux (EPS). Les mouvements myocloniques se produisent le plus souvent en phase de sommeil et sont très communs aux stades avancés de la maladie (Tschampa, 2001). Les signes extrapyramidaux sont communs chez les patients Alzheimer (Scarmeas et al.,
5
2004), ce sont des mouvements non-coordonnés ou désordonnés, cela inclus la dystonie (spasmes et contractions musculaires), acathésie (agitation motrice), parkinsonisme (rigidité), bradykinésie (ralentissement des mouvements), tremblements et dyskinésie tardive (mouvements irréguliers). Les plus courants dans la MA sont la bradykinésie et le parkinsonisme (rigidité). Les patients présentant des signes extrapyramidaux ont généralement une atteinte plus sévère des fonctions cognitives (López and DeKosky, 2008). La présence des EPS n’est cependant pas bien comprise, des études suggèrent plusieurs causes comme des lésions au niveau des réseaux dopaminergiques (Meguro et al., 1997; Tosto et al., 2015). D’autres rapportent que les EPS seraient en partie due à la présence d’un plus grand nombre de neurofilaments dans la substance noire de certains patients (Jang et al., 2020; Liu et al., 1997). Les EPS sont un bon indicateur la présence et de la sévérité de la MA (Azar et al., 2020; Stern et al., 1994). Dans le tableau clinique des patients Alzheimer on retrouve des altérations du comportement et des symptômes psychologiques. Parmi les symptômes psychologiques les plus fréquents on peut citer l’apparition d’hallucinations (perception incorrecte d’évènements ou d’objets) et d’illusions (fausses croyances). Quant aux changements de comportement on voit se développer de l’agressivité et une agitation. Ce sont des modifications que l’on retrouve communément chez les patients Alzheimer (Aarsland, Dag, 1996; Devanand, 1997; Segerstrom, 2018). La MA est une maladie progressive d’où la nécessité de la diagnostiquée tôt afin de mieux la gérer. Les techniques de dosage du liquide céphalorachidien et d’imagerie sont un bon point de départ corrélé avec un examen clinique neurologique. Le diagnostic permet de mettre un nom sur la souffrance du patient, l’urgence est maintenant de comprendre la maladie d’un point de vue moléculaire et cellulaire. En effet, l’espérance de vie d’une personne avec la MA est comprise entre 8 et 10 ans après l’apparition visible de la maladie. Cette statistique peut évoluer à la hausse ou au contraire à la baisse dépendamment de l’âge de la personne et de l’intensité des symptômes précédemment cités.
6
La pathologie d’Alzheimer
Depuis la première description faite par Alois Alzheimer voilà déjà plus de 100 ans, l’autopsie est le moyen le plus fiable et définitif de poser un diagnostic, dû à l’absence de biomarqueurs fiables (Perl, 2010). Lors de l’étude des sujets décédés on note que la plupart des cerveaux présentes une légère atrophie corticale frontotemporale. Lors de l’examen post-mortem, il ressort que le poids et l’épaisseur corticale du cerveau diffèrent entre une personne atteinte de la MA et un individu sain du même âge. La pathologie est associée à une perte tissulaire entrainant une dilatation des ventricules latéraux. Un examen plus approfondi des tissus peut donner un diagnostic définitif. Lors de l’étude histologique, on remarque la présence de neurofilaments dont le constituant principal est la protéine tau. Cette molécule apparait anormalement phosphorylée. Des études montre que la distribution et le nombre de filaments tau sont positivement corrélés avec le degré de démence et la durée de la maladie (Bierer et al., 1995). On note aussi la présence de plaque séniles composées d’amyloïde β. Dans l’Alzheimer cette protéine est arrangée de manière radiale. Les plaques sont entourées par des neurites anormales et des cellules microgliales activées. Cette amyloïde dérive du clivage de la protéine précurseur de l’amyloïde (APP). L’Aβ est formée suite au clivage de l’APP opéré par 2 sécrétases. S’en suit une accumulation dans le tissu cérébral. On distingue deux formes
majeures : l’Aβ42 composant principal des plaques et l’ Aβ40 que l’on retrouve plus
majoritairement autour des vaisseaux (Prelli et al., 1988). Le cœur des plaques contient en plus de l’amyloïde d’autres protéines tels que l’apolipoprotéine E (APOE) ainsi que des protéines du complément (Yin et al., 2019). Les patients Alzheimer présentent des dysfonctions vasculaires dues aux dépôts d’amyloïdes autour des vaisseaux, l’Aβ se dépose sur les parois des petits vaisseaux entrainant des angiopathies cérébrales et potentiellement une rupture vasculaire provocant des hémorragies (ElAli and Rivest, 2013). L’examen histologique révèle aussi une perte synaptique d’environ 45% chez les patients atteints de la MA (Selkoe, 2002).
7
Les causes étiologiques
Plus d’un siècle après le premier cas recensé et presque autant d’années de recherches intensives, les causes primaires entrainant le développement de la maladie d’Alzheimer restent encore inconnues. Il existe cependant de rares cas de MA familiale provoqués par une ou des mutations génétiques de manière non-exhaustive, on peut évoquer celles menant à la surproduction de Aβ (APP, presinilin 1-2 (PS1 et PS2)). La mutation touchant le Triggering receptor expressed on myeloid cells 2 (TREM2) RH47 provoquant une diminution de la phagocytose. D’autres mutations sont considérées comme des facteurs de risques tel que les mutations APOE. Ces mutations sont absentes dans la plupart des cas d’Alzheimer. Au fil des décennies plusieurs hypothèses ont vu le jour essayant d’expliquer l’étiologie de la maladie. Cependant la MA est complexe, aucune de ces hypothèses ne peut expliquer à elle seule l’ensemble de la pathologie. Cela étant dit ces hypothèses montrent la pertinence de l’étude de ces mécanismes. Les stratégies thérapeutiques à notre disposition sont peu efficaces pour prévenir l’occurrence ou la progression de la maladie.
L’hypothèse amyloïde
C’est une hypothèse très répandue, elle se base sur le dépôt anormale d’Aβ dans le cerveau des patients Alzheimer. L’Aβ est le composant principal des plaques séniles, ce phénomène serait une des causes de la pathologie et serait directement responsable de la perte cellulaire, des dommages vasculaire et de la démence (Hardy and Higgins, 1992). L’Aβ est le produit de plusieurs clivages de l’APP orchestrés par les enzymes β-sécrétase (BACE1) et le complexe enzymatique γ-sécrétase composé de PS1, nicastrin et PS2. Ce clivage va produire différentes
isoformes d’Aβ, caractérisées par le nombre d’aminoacides. L’Aβ1-40 (Aβ40) et Aβ1-42
(Aβ42) sont les isoformes les plus courantes. L’Aβ42 est la forme la plus hydrophobe,
8
s’agglomérer en différents polymères (monomère, oligomères, plaques fibrillaires) dans le cerveau, toutes ces formes ont des propriétés distinctes (Benilova et al., 2012). Des études montrent que les deux principaux marqueurs de la MA à savoir l’Aβ et l’hyperphosphorylation de tau seraient liés. Les dépôts d’Aβ seraient antérieurs au phénomène d’hyperphosphorylation et induiraient la formation des enchevêtrements de filaments tau, dérégulant par cette occasion le transport axonal et induisant possiblement la mort neuronale (Zheng et al., 2002). Il est important de noter que la sévérité du déclin cognitif est corrélée avec le taux d’oligomères de Aβ et non avec le total d’amyloïde (Lue et al., 1999). L’hypothèse amyloïde est d’autant plus crédible qu’il existe des cas de MA familiale se traduisant par une hyperproduction d’amyloïde. Cette forme de la maladie est liée à la mutation du gène APP. Le gène du précurseur de l’amyloïde est situé sur le chromosome 21, les personnes atteintes du syndrome de Down possèdent 3 copies de ce gène et développent de manière précoce l’Alzheimer (Rumble et al., 1989). Le dernier
exemple que l’on peut citer est la souris transgénique APPswe/PS1. Ces animaux
sur-expriment les protéines APP et PS1 mimant la physiopathologie induite par l’amyloïde chez l’humain. On retrouve chez ces souris accumulation aberrante d’Aβ, des déficiences synaptiques, un phénomène inflammatoire ainsi qu’une altération mnésique (Sasaguri et al., 2017). Après plus de 25 ans d’expérimentations visant à démontrer cette hypothèse, il apparaît que nous ayons encore beaucoup à apprendre sur ce sujet ainsi que sur le rôle physiologique de ces protéines et sur les phénomènes déclenchant cette accumulation d’amyloïde. Des thérapeutiques ayant pour cible l’Aβ ont été développées. Ces dernières ont toute échoué lors des essais cliniques. Cela sous-entend que l’amyloïde tient une part importante dans l’étiologie de la maladie mais n’a peut-être pas un rôle central. On peut aussi se demander si les dépôts d’amyloïde sont une réponse des cellules à un stress, ou de cellules en détresse (Nikolaev et al., 2009; Ricciarelli and Fedele, 2017).
L’hypothèse tau
La protéine tau est une protéine de structure des microtubules (élément du cytosquelette). Son interaction avec la tubuline induit l’assemblage et la stabilisation
9
des microtubules. Dans le cerveau on retrouve cet élément du cytosquelette au niveau des axones de neurones matures (Weingarten et al., 1975). Les pathologies liées à tau dont la MA résultent d’une hyperphosphorylation de la protéine (Grundke-Iqbal et al., 1986). Ce qui a pour conséquences la dissociation de tau des microtubules et son agrégation pour former des neurofilaments. Cela peut venir d’une mutation qui altère la fonction et l’expression de tau mais la plupart des patients ne présentent aucune mutation à ce niveau-là. Les avancées scientifiques n’ont pour l’instant pas pu mettre en lumière le processus d’agrégation de tau en absence de mutation. L’hyperphosphorylation résulterait de plusieurs facteurs, comme l’activité anormale de kinases ou de phosphatases, ce qui a pour effet le désassemblage des microtubules ainsi que la dérégulation du transport axonal ce qui provoque des dysfonctionnements neuronaux. On ne retrouve pas ce phénomène que dans la MA car tau joue aussi un rôle dans la maladie de Parkinson et la démence frontotemporale (Iqbal et al., 2010). Il est important de noter que l’hyperphosphorylation de tau est a été observée hors contexte pathologique lors d’un stress cellulaire. Les deux hypothèses (amyloïde et tau) sont présentées séparément mais il existe des évidences que tau n’est pas un épiphénomène de la pathologie induite par l’Aβ. En effet, des études révèlent que tau est nécessaire à la toxicité induite par l’Aβ in vivo (Roberson et al., 2007). Autrement dit l’Aβ initie les dysfonctions synaptiques tau-dépendantes. L’article de Roberson et al montre qu’une inhibition de tau diminue fortement excitotoxicité neuronale induite par Aβ. Comme l’hypothèse précédente, les facteurs déclenchants la tau pathologie restent méconnus. L’absence de mutations liées à la protéine tau semble suggérer que le dérèglement de la phosphorylation n’est pas le mécanisme premier mais une conséquence.
L’hypothèse cholinergique
Dans le système nerveux central (SNC), des neurones cholinergiques se projettent dans le bulbe olfactif, néocortex, hippocampe et amygdale (Woolf, 1991). Ces neurones synthétisent et sécrètent de l’acétylcholine (ACh). Ce neurotransmetteur agit sur deux types de récepteurs : muscariniques (mAChRs) et nicotiniques
10
(nAChRs). De par leur localisation ces neurones participent aux fonctions de mémoire, d’apprentissage et d’attention. Plusieurs groupes de recherches ont montré une perte sélective des sous-types de nAChRs α7 ou α4β2 chez les patients MA (Nordberg, 1994). Cette hypothèse a été élaborée entre-autre à partir d’études menées dans les années 70. Elles ont permis de montrer que certaines parties du cerveau des patients diagnostiqués Alzheimer présentent une diminution d’activité de l’enzyme nécessaire à la synthèse de l’ACh, la choline acétyltransférase dans l’hippocampe, le cortex et l’amygdale. (Davies, 1976; Perry et al., 1977). Des études plus poussées ont établi de la même manière que la diminution de l’activité de cette enzyme est retrouvée lors du vieillissement normal des personnes. Cependant la choline acétyltransférase est encore moins active chez les patients atteints de la MA. Le déficit de synthèse d’ACh pourrait être un marqueur de dysfonctions cognitives sous-jacentes (Perry et al., 1978). Les principaux mAChRs impliqués dans la cognition sont les récepteurs M1 postsynaptiques, ils servent à médier l’effet de l’ACh. Les seconds : M2 sont présynaptiques, ils inhibent le relargage de l'ACh. Dans la MA le couplage des récepteurs M1 avec la protéine G associée est altéré, alors que le nombre de récepteurs reste constant (Flynn et al., 1995; Tsang et al., 2006). Il y a donc un problème de diminution de ligand ainsi qu’un problème de transduction du signal. La diminution des niveaux d’ACh est commun à plusieurs pathologies tel que la maladie de Parkinson, les dommages cérébraux (Schliebs and Arendt, 2011), cependant les dépôts d’Aβ sont spécifiques de la MA. Il existe un lien entre l’Aβ et le système cholinergique, car les dépôts d’amyloïde provoque la perte de fibres des neurones cholinergiques dans le néocortex (Boncristiano et al., 2002). De plus en plus d’évidences montrent que les nAChRs interagissent avec l’Aβ, plus
particulièrement entre l’Aβ42 etnAChRs α7 (Wang et al., 2000). Cette forte affinité
peut induire l’internalisation de l’Aβ par le neurone expliquant la potentielle mort des neurones exprimant ce récepteur (Oddo et al., 2005). De plus, dans le modèle
animal APPswe/PS1, l’expression de mAChRs diminue avec l’âge (Machová et al.,
2008). Il est intéressant de mentionner que l’activité des récepteurs cholinergiques influe sur le métabolisme de l’amyloïde. Des études montrent que l’activation des mAChRs et nAChRs peut promouvoir la voie non-pathologique de l’amyloïde, l’APP
11
va être clivé par l’α-sécrétase générant un fragment soluble appelé sAPP α (Haass et al., 1993). En favorisant la voie non-amyloïde les β et γ-sécrétase vont être inhibées (Pakaski and Kalman, 2008). Le système cholinergique au vu des études est fortement impliqué dans l’évolution de la maladie d’Alzheimer. Les efforts considérables fournis pour mettre au point des thérapeutiques visant à réguler le système cholinergique ne donnent pas de résultats probants quant au ralentissement de la maladie. Cela suggère que le système cholinergique fait partie d’un ensemble de facteurs causaux entrainant la MA.
Les facteurs de risques
Comme nous avons pu le voir dans les sections précédentes, la MA est une maladie complexe multifactorielle. Les principaux facteurs de risques sont l’âge et la génétique bien que la condition médicale ainsi que les choix de vie rentrent aussi en compte. Cependant on ignore les mécanismes sous-jacents qui lient les facteurs de risques au déclenchement de la maladie, mais ils nous permettent d’avoir une vision un peu plus précise de la pathologie. On distingue 2 formes de MA. La forme familiale qui est liée aux gènes donc à l’hérédité. Elle est principalement due à la mutation des gènes APP, PS1, PS2 qui induisent la surproduction d’Aβ. Ces mutations sont en générale autosomales et dominantes. Elles entraînent de manière quasi certaine l’agrégation d’Aβ et une apparition de la maladie de manière précoce (Reitz and Mayeux, 2014). La deuxième forme de MA est la forme acquise, c’est la plus courante, elle se développe plus tardivement et a une progression plus lente. Contrairement à la forme génétique, la MA acquise peut être influencée par l’environnement, car on lui connait des facteurs de risques et de protections.
Facteurs génétiques
La MA peut se classer de deux manières. La MA précoce, qui se manifeste avant l’âge de 65 ans et représente 4-6% des cas et la MA tardive qui se manifeste après
12
65 ans (Silva et al., 2019). La MA précoce est habituellement imputée à la mutation des gènes APP, PS1 et PS2 alors que la forme tardive est le plus souvent imputée au polymorphisme du gène APOE plus précisément à la présence de l’allèle 4 (Giri et al., 2016). Plus de 30 mutations ont été découvertes pour le gène APP, ces modifications génétiques sont associées à plus de 15% des cas de MA précoce. Les mutations de PS1 représentent à elles seules plus de 80% des cas alors que les mutations de PS2 ne représentent que 5% des cas. Les mutations des gènes APP
et PS1 entraînent l’augmentation du ratio Aβ42 :Aβ40, cette dérégulation favorise les
dépôts d’Aβ dans le cerveau (Bekris et al., 2010).
APOE est une protéine impliquée dans le métabolisme des lipides. Elle est encodée par le gène APOE. Il existe 3 allèles donnant trois isoformes différentes (APOE2-3 et 4). Il y a une grande variabilité de la distribution alléliques. L’allèle 4 codant pour APOE4 est l’un des principaux facteurs de risques de la MA tardive. La présence d’un allèle sur deux d’APOE4 accroit la probabilité d’avoir la maladie d’un facteur 3, cette probabilité augmente d’un facteur 12 si la personne est homozygote. De manière intéressante la présence de l’allèle APOE2 réduit les risques de développer la maladie (Corbo and Scacchi, 1999; Karch and Goate, 2015).
De larges études génomiques (GWAS) ont permis d’associer certaines variations génétiques à la MA. Certains de ces gènes associés à la maladie interviennent dans la réponse immunitaire (CD33, TREM2, CR1, EPHA1, HLA-DRB5/HLADRB1), d’autres dans le métabolisme lipidique (SORL1), la tau pathologie ( CASS4, FERMT2), ou encore la migration cellulaire (PTK2B) (Reitz and Mayeux, 2014). On peut remarquer que plusieurs de ces gènes identifiés comme facteurs de risques sont associés à l’immunité. Ce qui laisse penser que les cellules immunitaires ont un rôle important dans la MA. Il faut étudier de manière in-vivo comment ces facteurs de risques influencent la maladie afin de mieux comprendre les mécanismes pathologiques et orienter les recherches thérapeutiques.
13
Risques non-génétiques
Les maladies cérébraux-vasculaires tel que les infarctus hémorragiques, les infarctus ischémiques et les pathologies vasculaires augmentent les risques de démences. Les dommages provoqués par ces pathologies dans les régions du cerveau importantes pour la mémoire peuvent induire les démences. De plus les dommages cérébraux peuvent entrainer une augmentation du dépôt d’Aβ provoquant par la même occasion une réponse inflammatoire altérant les fonctions cognitives (Reitz and Mayeux, 2014). Des études montre que les individus ayant eu un accident vasculaire cérébral (AVC) ont 2 fois plus de risque de déclencher une démence (Vijayan and Reddy, 2016). De plus l’hypoperfusion chronique due à la diminution du débit sanguin peut entrainer la surexpression d’une sérine-thréonine kinase. L’activation de cette molécule est associée à la mort et à l’apoptose des neurones (Weishaupt, 2003) ainsi qu’à l’hyperphosphorylation de tau ce qui contribue à la formation des neurofilament tau (Wen et al., 2007).
D’autres facteurs non-génétiques sont à prendre en compte tel que le diabète de type 2, l’hypertension, le tabagisme, tous les trois associés à des pathologies de démences vasculaires. Les AVC et les démences vasculaires sont liés à la MA, ce sont des facteurs de risques comme on a pu le voir. L’âge est le facteur de risque le plus important pour la survenue d’AVC et des démences. 1/3 des patients âgés ayant fait un AVC vont déclencher une démence et des dysfonctions cognitives dans les 3 ans post-AVC (Pendlebury and Rothwell, 2009). En effet les maladies cérébraux vasculaires peuvent contribuer à la MA ce qui a pour conséquences une atrophie cérébrale et un accumulation anormale de protéines comme l’Aβ (Kalaria et al., 2012).
Il existe aussi des facteurs protecteurs sur lesquels on peut jouer, on peut citer la stimulation intellectuelle, une consommation d’acides gras polyinsaturés ainsi que de l’activité intellectuelle (Reitz and Mayeux, 2014).
14
Le système immunitaire
Le système immunitaire protège notre organisme contre les menaces extérieures et intérieures grâce aux diverses cellules qui le composent. On fait la distinction entre immunité innée et immunité adaptative. Cependant ces deux systèmes sont complémentaires et communiquent entre eux. L’immunité au sens large est fortement impliquée dans la maladie d’Alzheimer. En effet, les cellules immunitaires en présence de molécules dangereuses pour l’organisme organisent une réponse inflammatoire afin d’éliminer la menace et rétablir l’homéostasie de l’organisme. L’immunité a un rôle vital pour la survie de l’organisme.
L’immunité innée
L’immunité innée constitue la première barrière de défense face à une infection. La réponse immunitaire innée est induite par un signal de danger émis, suite à l’interaction entre les récepteurs du soi, les pattern recognition receptor (PRR) et les molécules du non-soi. Comme non-soi on distingue les pathogen-associated molecular patterns (PAMP) présentent sur les pathogènes et les damage-associated molecular patterns (DAMP), qui sont des molécules associées aux cellules endommagées ou mourantes. L’immunité innée est composée de plusieurs sous-types cellulaires, on retrouve les monocytes, les cellules natural killer, les granulocytes, les neutrophiles, macrophages et les cellules dendritiques. La famille qui nous intéresse ici est celle des monocytes. L’immunité innée est un ensemble complexe de cellules qui interagissent entre elles. Ce système est très impliqué dans les maladies neurodégénératives comme la MA, ce qui laisse penser que la modulation de ces cellules peut influencer le cours de la maladie.
15
Les monocytes et macrophages sont des cellules mononucléaires, ayant un rôles majeur dans l’homéostasie des tissus et dans l’immunité (Ginhoux and Jung, 2014). Elles représentent environ 4% des cellules du sang chez la souris et 10% chez l’humain. Les monocytes sont issus des cellules souches hématopoïétiques (HSCs) situées dans la moelle osseuse. Cette dernière est composée de plusieurs types cellulaires participant à la régulation de la différenciation des HSCs en différentes cellules progénitrices (Szade et al., 2018). La production de monocytes est continue, plus les cellules se différencies plus elles se spécialisent. Au fil des années, la communauté scientifique a établi l’arbre généalogique des monocytes. On retrouve après les HSCs, les common myeloid progenitor (CMPs), elles donnent naissances après différenciation aux granulocytes et macrophage progenitor (GMPs). On voit ensuite apparaitre les macrophages and dendritic cell progenitor (MDPs) et finalement, le début de la lignée monocytaire en tant que tel est occupé par les common monocytes progenitor (CMoP) (Hettinger et al., 2013; Schouppe et al., 2012).
Le développement et la survie des monocytes dépendent entre autres du macrophage-colony stimulating factor (m-CSF). C’est une glycoprotéine qui se lie au récepteur CSF1R aussi connu sous le nom de CD115 (Stanley and Chitu, 2014). De nombreuses études ont montré que l’absence de l’axe m-CSF/CSF1R entraine une diminution importante de la population monocytaire (Rojo et al., 2019; Sauter et al., 2014; Ushach and Zlotnik, 2016). Le m-CSF induirait aussi l’expression du facteur de transcription PU.1, important dans le devenir des HSCs. En effet ce dernier régule la différenciation des cellules myéloïdes. Lorsqu’il est assez activé il permet l’expression de gènes entrainant la différenciation en cellules myéloïdes et en inhibant les gènes responsable de la différenciation en neutrophiles (Sposi et al., 1999).
16
La plupart de nos connaissances sur les monocytes viennent de l’études des tissus murins et du sang humain. Il existe plusieurs sous-types de monocytes. On fait la distinction entre les monocytes grâce au niveau d’expression de certains marqueurs.
On retrouve les monocytes classiques, inflammatoires Ly6ChiCCR2+ chez les souris
et CD14+CD16- chez l’humain. En parallèle de ce premier groupe, on retrouve les
monocytes non-classiques, localisés au niveau de l’endothélium, ils patrouillent
dans l’organismes. Ils expriment Ly6ClowCX3CR1hi chez les souris et CD14intCD16+
chez l’humain (Mitchell et al., 2014). Il existe un troisième sous-type mais nous n’en feront pas mention afin de ne pas entrainer de confusion, car ils sont encore mal caractérisés.
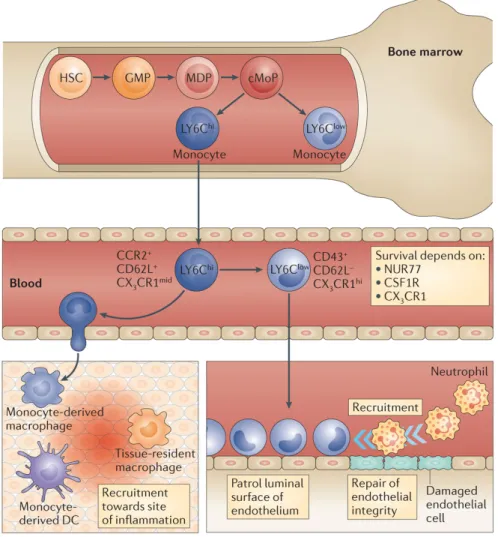
Figure 1-1 Développement et devenir des monocytes chez la souris.
Les monocytes sont produits dans la moelle osseuse. Les Ly6Chi migrent de façon
17
inflammatoire et se différencient en macrophages. Les Ly6Clow surveille leur
environnement à partir des vaisseaux sanguins. Tiré de (Ginhoux et Jung., 2014)
Les monocytes classiques
Les monocytes classiques répondent à une infection ou une injure. Ils se développent dans la moelle osseuse. On les retrouve par la suite dans la circulation sanguine. Ils sont attirés au lieu d’intérêt grâce à un gradient de chimiokine tel que CCL2 (Serbina and Pamer, 2006). Lors d’une infection par un agent pathogène, les monocytes classiques vont venir phagocyter le pathogène. Ils sécrètent en parallèle des molécules visant à recruter d’avantage de cellules immunitaires et font offices de cellules présentatrices d’antigènes via le complexe majeur d’histocompatibilité de classe 2 (CMH2) (Landsman et al., 2007). À noter aussi que les cellules immunitaires peuvent sortir des vaisseaux, ce phénomène s’appelle la diapédèse, elle met en jeu des molécules d’adhésion ainsi qu’une collaboration étroite entre les
cellules endothéliales et immunitaires. Les Ly6Chi sécrètent principalement des
espèces réactive de l’oxygène, TNF-α, oxyde nitrique et IL-1β lors d’infections bactériennes ainsi que de l’interféron en réponse aux virus. Une fois recruté via CCL2 au site d’inflammation ces monocytes se différencies en macrophages inflammatoires (Yang et al., 2014).
Les monocytes non-classiques
Les monocytes non-classiques, sont des cellules qui patrouillent dans les vaisseaux. Ce type de monocytes semblent être issus de la différenciation des monocytes classiques, dans le sang et en plus faible quantité dans la moelle osseuse sous le contrôle de Nur77 (Hanna et al., 2011; Kratofil et al., 2017). Ils se déplacent du côté luminal de l‘endothélium grâce aux intégrines. Les monocytes patrouilleurs sont impliqués dans la surveillance des vaisseaux et sont capables de recruter les neutrophiles. De même, ils ont la possibilité d’initier la réponse immunitaire. Les
18
monocytes Ly6Clow sont de potentielles cibles thérapeutiques dans la MA. Des
études ont montré que ce type de cellules était présent dans les vaisseaux à proximité des agrégats d’amyloïde et qu’ils étaient capables de les phagocyter (Jean-Philippe Michaud et al., 2013). Cette étude apporte un nouveau point de vue sur le rôle de ces monocytes dans la MA. Il existe un équilibre entre le niveau d’amyloïde dans le parenchyme et les vaisseaux. L’augmentation de la phagocytose
vasculaire par les Ly6Clow pourrait avoir un impact positif sur la maladie en raison de
cette règle de vases communicants (Marques et al., 2009). Il est intéressant de noter que les patients Alzheimer, on un plus faible nombre de monocytes patrouilleurs comparé aux sujets sains, cela peut être attribué à un plus faible taux de m-CSF chez les malades. Cette cytokine est essentielle pour la production et la survie des
Ly6Clow (Villeda et al., 2014; Yona et al., 2013). À la lumière de ces faits, cibler les
monocytes patrouilleurs serait une bonne approche thérapeutique.
Se retrouvant principalement dans les vaisseaux, ces monocytes peuvent servir à lutter contre l’angiopathie amyloïde cérébrale (AAC). Cette pathologie associée à la
MA est caractérisée par une accumulation aberrante d’Aβ40 sur la paroi des
vaisseaux (Brenowitz et al., 2015). Jellinger estime que 78 à 98% des patients Alzheimer ont aussi un AAC (Jellinger, 2002). Les conséquences de cette pathologie associée est l’accélération de la MA, donc une progression plus importante du déclin cognitif (Pimentel-Coelho and Rivest, 2012a). L’origine de l’AAC n’est pas encore bien définie (Yuan et al., 2020), ce qui est sûr c’est que les dépôts d’amyloïdes altèrent le drainage périvasculaire, ce qui entraine des hémorragies intracérébrales dues à la rupture des vaisseaux. En augmentant le nombre de monocytes patrouilleurs dans les vaisseaux, on pourrait diminuer l’impact de l’AAC.
Plusieurs molécules sont capables de recruter les monocytes. De manière non exhaustive nous pouvons citer le m-CSF qui mobilise les cellules de la moelle osseuse. Lorsque les animaux sont traités avec du m-CSF, on remarque une forte mobilisation des cellules de la périphérie accompagnée d’une infiltration dans le
19
cerveau de ces dernières. Ces cellules ont une capacité phagocytaire plus forte que les microglies.
Le Monophosphoryl lipid A, lorsqu’il est injecté chez des souris, il provoque une robuste prolifération monocytaire, associée à une potentialisation de la phagocytose de la part de ces cellules. La dernière molécule dont nous allons parler dans cette partie est peut-être la plus intéressante sinon la plus prometteuse au regard de la
conversion des monocytes Ly6Chi vers Ly6Clow, le muramyl-dipeptide (MDP). Il a été
isolé de la paroi bactérienne en 1977, il se lie au Nucleotide-binding oligomerization domain-containing 2 (NOD2) qui est un récepteur exprimé par les cellules de l’immunité innée et à CD14. Il est bien connu pour ses propriétés de conversion des
monocytes du profil Ly6Chi vers Ly6Clow (Lessard et al., 2017). Le MDP induit
l’expression de Nur77, qui va interagir avec C/EBPβ, 2 molécules requise pour la survie et la conversion des monocytes patrouilleurs (Hanna et al., 2011; Lappas, 2016; Mildner et al., 2017; Tamura et al., 2017). De plus ces cellules peuvent prévenir l’inflammation car Nur77 régule négativement TNF receptor-associated factor 6, cette molécule tient une place importante dans la réaction inflammatoire (Wu et al., 2016).
Les macrophages
Les macrophages sont des cellules mononuclées, on les retrouve dans les différents organes et leur morphologie dépend de leur état d’activation (Elhelu, 1983). Leurs fonctions principales sont la phagocytose, l’endocytose, la présentation antigénique et la sécrétion de différentes molécules immunomodulatrices (cytokines, chimiokines, facteurs de croissances) (Hume, 2006).
20
Les microglies
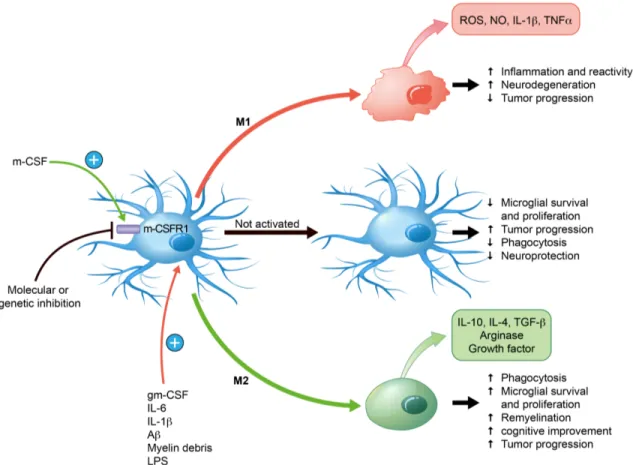
Les microglies sont les cellules immunitaires résidentes du cerveau. Elles représentent près de 10% de la totalité des cellules constituant le SNC (Tremblay et al., 2010). Elles colonisent le cerveau embryonnaire tôt dans le développement. Chez la souris c’est entre le stade embryonnaire 8.5 et 10 (Ginhoux et al., 2013). Chez l’Homme les microglies primitives apparaissent autour du tissu neural primitif vers 4.5 semaines de gestation et sont visibles dans ce même tissu aux alentours de 5.5 semaines de gestation (Alliot et al., 1999). Les microglies sont issues de la lignée monocytaire, et sont capables d’auto-renouvèlement (Thion et al., 2018). Elles surveillent constamment le SNC grâce à leurs prolongements mobiles. Ce sont les premières cellules à répondre lors d’un dommage cérébral ou d’une infection du SNC (Li and Barres, 2018; Shemer et al., 2015). Les microglies sont aussi connues pour contribuer au développement et à l’homéostasie du SNC (Li and Barres, 2018; Thion et al., 2018). Elles sont fortement impliquées dans la neurogénèse, le remodelage synaptiques et l’homéostasie neuronale (Lampron et al., 2015; Nagata, 1997; Paolicelli et al., 2011; Shemer et al., 2015). Les microglies sont des cellules très plastiques, elles peuvent se polariser, produire et sécréter une large gamme de médiateurs chimiques. Tel que des molécules inflammatoires : IL-1β, TNF-α, ROS, NO et IL-4, des facteurs trophiques qui assurent un support aux cellules du SNC, notamment le neurotrophic derived factor (BDNF), insulin growth factor 1 (IGF-1), Arginase-1 (Arg-1) and Transforming growth factor β (TGF-β) (Ueno et al., 2013). Comme on peut le voir, les microglies sont des cellules complexes ayant plusieurs rôles indispensables au bon fonctionnement du cerveau. L’étude des cellules microgliales est primordiale pour comprendre comment fonctionne le cerveau et comprendre aussi les pathologies qui y sont associées.
21
Les microglies sont connues pour participer activement au développement du cerveau d’une part via la sécrétion de différents facteurs trophiques et d’autres parts via la phagocytose (Lenz and Nelson, 2018). Les microglies phagocytes plusieurs éléments dans l’encéphale tel que les synapses, les axones, les débris cellulaires et les cellules mourantes via la stimulation des TLRs, des scavenger récepteurs et TREM2 (Arcuri et al., 2017; Kettenmann et al., 2013). Les cellules microgliales participent à la neurogénèse, les cellules progénitrices des neurones exprimant des marqueurs reconnus par les microglies via MerTK, vitronectine, complément récepteur 3 (CR3) et siglecs (Sierra et al., 2010) qui active la phagocytose microgliale (Cunningham et al., 2013). La genèse cellulaire ce fait entre autres par l’intermédiaire de l’insuline-like growth factor (IGF1) et des cytokines TNF-α, IL-1β, IL-6 (Wlodarczyk et al., 2017). De récentes études montrent que d’autres cellules comme les oligodendrocytes sont impliquées dans la survie des neurones via la sécrétion de IGF1 (Ueno et al., 2013; Wilkins et al., 2001). Cependant, les microglies semblent être la pierre angulaire du le développement et la neurogénèse. De plus amples recherches restent à faire pour démystifier le rôle précis des microglies dans le développement du cerveau. On peut se demander si ce sont les microglies qui régulent le développement ou les cellules progénitrices qui influencent les microglies.
Les microglies dans le cerveau
Les microglies dans le cerveau ont plusieurs fonctions, toutes plus essentielles les unes que les autres. En tant que cellules immunitaires elles vont surveiller leur microenvironnement grâce à leurs prolongements (Nimmerjahn, 2005). Cette surveillance a pour but de vérifier l’intégrité des cellules, ainsi que des synapses. Les cellules microgliales sont très dynamiques, en effet, elles peuvent s’adapter rapidement aux moindres changements d’environnement. Les microglies sont essentielles à la formation et au développement des structures cérébrales en éliminant les débris cellulaires et les synapses surnuméraires (Paolicelli et al., 2011;
22
Tremblay et al., 2010). Les cellules microgliales promeuvent la plasticité et le remodelage synaptique via la sécrétion de plusieurs facteurs tel que BDNF (Paolicelli et al., 2011). De récentes études ont montré l’implication du système du complément dans le remodelage synaptique. Les microglies vont reconnaitre la molécule C1q exprimée par les synapses et ainsi engager la phagocytose de la synapse (Tremblay and Majewska, 2011). De même CD47 est une protéine de surface qui agît comme un signal négatif de phagocytose (Rivest, 2018). Les microglies ont un rôle dans la modulation de la neurogénèse et la connectivité neuronale. Elles sont capables de phagocyter une portion des axones grâce à leurs prolongements ce qui limite la connectivité entre les neurones. Elles peuvent aussi de manière physique s’interposer entre la région pré et post-synaptique afin d’empêcher les connections (Chen et al., 2014; Filipello et al., 2018; Paolicelli et al., 2011).
L’activité phagocytaire des microglies participe à l’élimination des agents infectieux ainsi que des tissus nécrotiques et apoptotiques durant le développement ou au cours d’une pathologie. Les microglies vont participer au nettoyage des débris cellulaire lors du développement du cerveau et au cours de la vie, mais aussi participer à l’élimination des protéines toxiques tel que la β amyloïde (Magnus et al., 2001). Des études montrent que les neurones peuvent réguler et/ou maintenir l’activation microgliale via la sécrétion de neurotransmetteurs ainsi que de chimiokines (Biber et al., 2008). La communication entre les neurones et les microglies est crucial pour le maintien de l’homéostasie du cerveau. Il y a une accumulation de preuves montrant que les neurones expriment des signaux « on » et « off » qui ont pour but d’informer les microglies de leur état ; dépendamment de la santé du neurone, le signal envoyé peut influencer les microglies aux alentours. Le signal « off » est constitutivement exprimé par les neurones, contrairement au signal « on » qui lui est produit sur demande (Biber et al., 2007). Les signaux sont médiés par des purines, des neurotransmetteurs, des chimiokines ou des molécules membranaires (Hanisch and Kettenmann, 2007; Pocock and Kettenmann, 2007). La première chimiokine à être mise en évidence est CX3CL1 connue sous le nom de fractalkine. Cette molécule est exprimée par les neurones et se fixe sur son
23
récepteur CX3CR1. Elle est impliquée dans la communication entre neurones et microglies. CX3CL1 est fortement exprimé au niveau du cortex, hippocampe,
thalamus et bulbes olfactifs (Harrison et al., 1998; Nishiyori et al., 1998). Il est
intéressant de noter que cette molécules est aussi très exprimée dans les organes périphériques (Pan et al., 1997). Les souris déficientes en CX3CR1 ont une plus forte activité lors de la neuroinflammation induite par Lipopolysaccharide (Zujovic et al., 2000). CX3CL1 réduit l’inflammation et in vitro Boehme et ses collègues ont montré que CX3CL1 entraine une diminution de l’apoptose Fas-Ligand dépendent (Boehme et al., 2000). De plus, dans certains modèles de sclérose en plaques, les souris déficientes en CX3CR1 ont des microglies qui phagocytent moins la myéline mais une activité inflammatoire équivalente (Lampron et al., 2012).
CCL2/MCP-1 est une molécule chimio attractante exprimée par les cellules immunitaires mais aussi par les neurones (Banisadr et al., 2005). La production de CCL2 par les neurones peut survenir après une ischémie, un dommage axonal comme la lésion du nerf facial (Che et al., 2001; Flügel et al., 2001). Son récepteur est retrouvé sur les microglies ainsi que sur les monocytes. Mussel est collègues montrent que l’axe CCL2/CCR2 est impliqué dans l’activité neurotoxique des microglies (Muessel et al., 2002). Les chimiokines peuvent avoir autant un effet bénéfique que délétère sur la réponse microgliale. Cependant la modulation de la réponse immunitaire grâce à ce type de molécules est une voie thérapeutique intéressante dans les maladies neurodégénératives.
Les microglies dans la maladie d’Alzheimer
Comme nous l’avons vu les microglies sont impliquées dans bon nombres de pathologies, la maladie d’Alzheimer n’y faisant pas exception. Dans cette maladie, les microglies peuvent à la fois être neurotoxiques et neuroprotectives. Les microglies vont induire de la neuroinflammation, ce qui est efficace lors d’une injure ou d’une infection. Cependant lorsque la réaction inflammatoire dure trop longtemps, elle devient délétère et endommage les tissus (Ransohoff, 2016). L’effet d’une
24
inflammation chronique sur les microglies est aussi dommageable car elle provoque une diminution de leur capacité phagocytaire ce qui se traduit par une accumulation de débris cellulaires ainsi qu’une augmentation du niveau d’amyloïde dans le contexte de la MA. De plus il y aurait un remodelage synaptique aberrant (Vilalta and Brown, 2018).
L’inflammation dans la MA est une composante essentielle dans la compréhension de la maladie. Dans les stades les plus avancés, l’inflammation est chronique et donc néfaste pour le cerveau. Comprendre ce phénomène et le réguler est primordial. Une des voies menant à l’inflammation implique NOD-like receptor family pyrin domain containing 3 (NLRP3). NLRP3 va être induit par 2 types de signaux, le premier via les PAMPs et DAMPs qui en se liant aux TLRs et vont mener à la libération de NF-κB. Ce dernier va alors stimuler la production de NLRP3 et de pro-IL-1β entre autres. Le second signal est médié par l’ATP et des ARNs, qui entrainent un efflux de potassium, nécessaire à la dimérisation de NLRP3 (He et al., 2016). Dans la MA, NLRP3 est activé de manière chronique (Cribbs et al., 2012), des évidences chez la souris montrent que l’inhibition de la voie de l’inflammasome a un effet neuroprotecteur. Les animaux APP/PS1 privés de NLRP3 ont une diminution d’amyloïde bêta ainsi qu’une amélioration des performances cognitives (Dempsey et al., 2017; Heneka et al., 2013). Il se pourrait qu’en inhibant NLRP3 les microglies phagocytent plus efficacement et provoquent moins de dégâts sur leur microenvironnement grâce à la diminution d'IL-1β (Sheng et al., 1996; Zhang et al., 2020). Comme on peut le voir les microglies via l’inflammasome peuvent être délétères dans la MA. Une autre voie d’activation a une grande importance, c’est la voie du complément. Le complément peut être activer de plusieurs manières suivant la voie classique, alternative ou la voie des lectines (Morgan, 2018). Dans la MA les molécules du complément sont surexprimées (Iqbal et al., 2013). De manière intéressante la suppression des microglies qui sur-expriment la voie du complément a un effet neuroprotecteur, réduisant la perte synaptique et le déclin cognitif (Chen et al., 2014; Chung et al., 2015; Spangenberg et al., 2019). Un paterne spécifique de l’activation de la voie du complément par les microglies est observé au cours la MA. Dans les stades précoces C1q, C4d et C3d sont retrouvés autour des plaques