HAL Id: dumas-01707315
https://dumas.ccsd.cnrs.fr/dumas-01707315
Submitted on 12 Feb 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Manifestations orales de la maladie cœliaque
Samuel Cohen
To cite this version:
Samuel Cohen. Manifestations orales de la maladie cœliaque. Sciences du Vivant [q-bio]. 2017.
�dumas-01707315�
UNIVERSITÉ PARIS DIDEROT - PARIS 7
FACULTÉ DE CHIRURGIE DENTAIRE
5, Rue Garancière 75006 PARIS
Année 2017
Thèse N° :
N° attribué par la bibliothèque :
THÈSE pour le DIPLOME D'ÉTAT DE DOCTEUR
en CHIRURGIE DENTAIRE
présentée et soutenue publiquement le 22 mars 2017
par COHEN Samuel
MANIFESTATIONS ORALES DE LA MALADIE CŒLIAQUE
Directeur de thèse : Docteur Bruno Courrier
JURY
Mme le Professeur Laurence JORDAN
Président
Mme le Docteur Vanessa BAAROUN
Assesseur
M. le Docteur Stéphane BAREK
Assesseur
M. le Professeur Yves BOUCHER
Assesseur
U N I V E R S I T É P A R I S D I D E R O T – P A R I S 7
Présidente de l’Université :
Mme la Professeure Christine CLERICI
Doyen de l’U.F.R. d’Odontologie :
M. le Professeur Robert GARCIA
Directrice Générale des Services :
Madame Pascale SAINT-CYR
______________
J U R Y
Mme le Professeur Laurence JORDAN
Président
Mme le Docteur Vanessa BAAROUN
Assesseur
M. le Docteur Stéphane BAREK
Assesseur
M. le Docteur Yves BOUCHER
Assesseur
M. le Docteur Rufino FELIZARDO
Assesseur
M. le Docteur Bruno COURRIER
Membre invité
Mme le Professeur Laurence JORDAN
Docteur en Chirurgie Dentaire
Diplôme en doctorat
Professeur des Universités – Praticien
Hospitalier
Vous me faites l’honneur de présider le jury de cette thèse. Veuillez trouver ici l’expression de mon profond respect. Pour l’intérêt de vos enseignements et votre amabilité, soyez assurée de ma profonde reconnaissance.
Mme le Docteur Vanessa BAAROUN
Docteur en Chirurgie Dentaire
Maître de Conférences des Universités –
Praticien Hospitalier
Pour l’honneur que vous me faites de participer au jury de cette thèse, veuillez trouver ici l’expression de mes sincères remerciements.
M. le Docteur Stéphane BAREK
Docteur en Chirurgie Dentaire
Diplôme de Doctorat
Maître de Conférences des Universités –
Praticien Hospitalier
Pour l’honneur que vous me faites de participer au jury de cette thèse, veuillez trouver ici l’expression de mes sincères remerciements.
M. le Professeur Yves BOUCHER
Docteur en Chirurgie Dentaire
Diplôme en doctorat
Professeur des Universités – Praticien
Hospitalier
Vous me faites l’honneur de siéger au sein du jury de cette thèse, veuillez trouver ici l’expression de mes sincères remerciements. Pour les connaissances que vous m’avez transmises, pour votre patience et votre disponibilité́, veuillez trouver ici l’expression de mes remerciements et de toute mon estime.
M. le Docteur Rufino FELIZARDO
Docteur en Chirurgie Dentaire
Diplôme en doctorat
Maître de Conférences des Universités –
Praticien Hospitalier
Pour l’honneur que vous me faites de participer au jury de cette thèse, veuillez trouver ici l’expression de mes sincères remerciements. Pour l’intérêt de vos enseignements et votre amabilité, soyez assuré de ma profonde reconnaissance.
M. le Docteur Bruno COURRIER
Docteur en Chirurgie Dentaire
Docteur en Sciences Odontologiques
Diplôme de Doctorat
Maître de Conférences des Universités –
Praticien Hospitalier
Pour l’honneur que vous m’avez fait en acceptant la direction de ce travail, veuillez trouver ici l’expression de mes sincères remerciements. Pour votre soutien, votre disponibilité et pour la confiance que vous
TABLE DES MATIÈRES
INTRODUCTION ... 3
1. Présentation de la maladie cœliaque ... 4
1.1.
Définition ... 4
1.2.
Épidémiologie ... 4
1.3.
Étiopathogénie ... 5
1.3.1. Le gluten ... 5
1.3.2. Réponse immunitaire ... 6
1.3.3. Facteurs génétiques ... 9
1.3.4. Facteurs environnementaux ... 9
1.4.
Signes cliniques généraux ... 10
1.5.
Diagnostic ... 13
1.5.1. Tests sérologiques ... 13
1.5.2. Biopsie ... 15
1.5.3. Typage HLA ... 17
1.6.
Traitement ... 18
1.6.1. Régime sans gluten ... 18
1.6.2. Nouveaux espoirs thérapeutiques ... 19
1.7.
Complications ... 21
1.8.
Diagnostic différentiel ... 24
1.8.1. Allergie au blé ... 24
1.8.2. SGNC (Sensibilité au gluten non cœliaque) ... 25
2. Manifestations orales de la maladie cœliaque ... 26
2.1.
Défauts/Altérations de l’émail ... 26
2.1.1. Prévalence ... 26
2.1.2. Effet du régime sans gluten ... 27
2.1.3. Localisation ... 28
2.1.4. Types et sévérité : Classification d’Aine ... 29
2.1.5. Étiologie ... 31
2.2.
Stomatite aphteuse récidivante ... 37
2.2.1. Aspects cliniques ... 37
2.2.2. Prévalence ... 38
2.2.3. Étiologie ... 39
2.2.4. Effet du régime sans gluten ... 40
2.2.5. Diagnostic différentiel ... 40
2.3.
Glossites ... 41
2.3.1. Aspect clinique ... 41
2.3.2. Prévalence ... 41
2.3.3. Étiologie ... 42
2.3.4. Effet du régime sans gluten ... 42
2.4.
Lichen plan buccal ... 42
2.4.1. Aspects cliniques ... 42
2.4.2. Lien avec la maladie cœliaque ... 42
2.5.
Retard d’éruption dentaire et d’âge dentaire ... 43
2.5.1. Retard d’éruption ... 43
2.5.2. Retard d’âge dentaire ... 44
2.6.
Modification salivaire et caries ... 45
2.6.1. Modification du pH ... 45
2.6.2. Modification du pouvoir tampon ... 45
2.6.3. Diminution du flux salivaire ... 46
2.6.4. Effet du régime sans gluten ... 46
2.6.5. Modification de la flore buccale et caries ... 47
CONCLUSION ... 48
BIBLIOGRAPHIE ... 50
TABLE DES ILLUSTRATIONS ... 55
INTRODUCTION
La maladie cœliaque (ou intolérance au gluten) est une entéropathie dont l’incidence n’a cessé d’augmenter durant les dernières décennies. Le nombre d’études médicales récentes témoigne de l’intérêt grandissant pour cette pathologie aux multiples aspects cliniques et aux complications pouvant engager le pronostic vital.
Malgré des outils diagnostiques de plus en plus performants, la maladie cœliaque reste encore sous-diagnostiquée dans ses formes atypiques et silencieuses.
Cependant, un examen bucco-dentaire à la recherche des manifestations buccales par un chirurgien-dentiste informé peut constituer l’une des premières étapes dans le diagnostic et donc la prise en charge de cette maladie.
Cette thèse rappellera dans une première partie les caractéristiques générales de la maladie cœliaque, sa prévalence dans la population et les diagnostics différentiels. Puis, dans une deuxième partie, les manifestations buccales y seront détaillées afin de permettre un diagnostic précoce et la mise en place d’un traitement adapté.
1. Présentation de la maladie cœliaque
1.1. Définition
La maladie cœliaque est une entéropathie auto-immune provoquée par l’ingestion du gluten, chez les individus génétiquement prédisposés.
1.2. Épidémiologie
La fréquence de la maladie cœliaque a longtemps été sous-estimée, en raison des formes silencieuses, paucisymptomatiques ou atypiques qui sont actuellement majoritaires (Green et Cellier 2007). L’importante augmentation de l’incidence de la maladie ces 30 dernières années reflète une meilleure connaissance des formes atypiques et silencieuses grâce notamment aux tests sérologiques (Graphique 1).
Graphique 1 - Evolution de l'incidence de la maladie cœliaque aux Etats-Unis depuis 30 ans.
(Murray et al. 2003)
Des études séroépidémiologiques récentes font état d’une prévalence d’environ 1 % dans la population générale en Europe et aux Etats-Unis. Mais ces études révèlent également que pour chaque cas diagnostiqué, il existerait trois à sept cas non diagnostiqués, et que 1 à 3 % de la population occidentale peut être touchée à un moment donné. Des chiffres proches de ceux observés en Europe ont été rapportés en Afrique du Nord, au Moyen-Orient, en Inde, en Nouvelle-Zélande, en Australie et en Argentine (Rewers 2005). En revanche, la maladie reste exceptionnelle en Afrique et en Asie (Farrell et Kelly 2002).
On observe deux pics de fréquence, avec une révélation soit dans l’enfance ou à l’âge adulte, le plus souvent entre 20 et 40 ans. La majorité des diagnostics se fait actuellement à l’âge adulte et les révélations tardives sont en constante augmentation (Farrell et Kelly 2002; Rampertab et al. 2006; Green et Cellier 2007). La prévalence dépasse 2 % après 50 ans (Vilppula et al. 2008).
0 3 6 9 12 1950-59 1960-69 1970-79 1980-89 1990-99 2000-01 In c id e n c e c a s / 1 0 0 0 0 0 / a n Années
1.3. Étiopathogénie
La maladie cœliaque résulte de l’interaction entre le gluten, la réponse immunitaire, les facteurs génétiques et d’autres facteurs environnementaux (Green et Cellier 2007). Nous les présenterons successivement :
1.3.1. Le gluten
Figure 1 - Coupe d’un grain de blé
(http://www.ikonet.com/fr/ledictionnairevisuel/regne-vegetal/cereales/ble.php)
L'albumen du grain de blé (Figure 1) est un tissu de réserve essentiellement constitué de granules d'amidon (70 %) enchâssés dans une matrice protéique (10-15 %) (Debiton, 2010).
Après avoir pétri de la farine de froment avec 50 % de son poids d'eau, on obtient une pâte élastique. Si ensuite, on lave celle-ci sous un mince filet d'eau, peu à peu l'amidon et les protéines solubles (albumine, globuline) se dissolvent et on obtient une substance verdâtre gélatineuse, nommée « gluten » (Boudreau et Ménard 2000). Une farine riche en gluten donne une pâte qui se travaille plus facilement. Il est également responsable de la texture aérée et moelleuse des pâtes levées (Feillet 2000).
Le gluten est composé de 80 % de protéines, de 10 % d’amidon, de sucres réducteurs, de lipides, de pentosanes et de matières minérales.
Parmi les protéines du gluten, on trouve la gliadine et la gluténine qui sont des prolamines, c’est-à-dire qu’elles sont caractérisées par des séquences répétées riches en glutamine et en proline. Et du fait de leur richesse en proline, les protéines composant le gluten, et notamment la gliadine, sont difficilement dégradées par les enzymes gastro-duodéno-pancréatiques (dépourvues d’activité endoprolylpeptidase efficace), persistant dans la lumière intestinale.
La fraction a2-gliadine de la gliadine est très efficacement présentée aux LT CD4+, alors que la
région N-terminale des aß-gliadines semble exercer des effets toxiques sur l’épithélium des patients
par des mécanismes mal compris (Clément et al. 2015).
D’autres céréales ont des équivalents à la gliadine du blé : la sécaline pour le seigle, l’hordéine pour l’orge, l’avénine pour l’avoine. Ces protéines induisent les mêmes effets néfastes que la gliadine chez les patients cœliaques.
1.3.2. Réponse immunitaire
Chez les patients cœliaques, la réponse immunitaire due à la gliadine engendre une réaction inflammatoire caractérisée par l’infiltration de cellules inflammatoires chroniques dans l’épithélium et la lamina propria (Figure 3).
1.3.2.1. Immunité innée au niveau de l’épithélium
La gliadine endommage les cellules épithéliales entrainant l’augmentation de l’expression de l’interleukine-15 qui active les lymphocytes intraépithéliaux possédant le récepteur NK-G2D (marqueur des cellules tueuses). Ces lymphocytes vont alors devenir cytotoxiques et détruire les entérocytes possédant un antigène de surface induit par un stress (par exemple une infection) : MIC-A (Green et Cellier 2007).
1.3.2.2. Immunité acquise dans la lamina propria
Tandis que 90 % des protéines du gluten sont dégradées lors de leur transport à travers une muqueuse saine, seulement 50-60 % le sont à travers la muqueuse d’un patient cœliaque non traité. Ces données ont conduit à étudier si ce défaut de dégradation intraluminale pouvait favoriser le passage de peptides non digérés à travers l’épithélium intestinal.
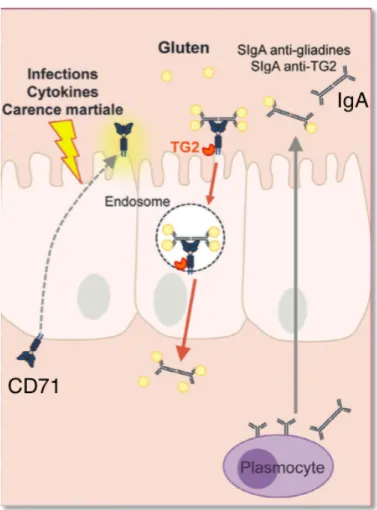
Bien qu’augmentée lors de la maladie cœliaque, la perméabilité paracellulaire semble peu compatible avec le transport de large peptide. Les travaux récents impliquent plutôt la voie transcellulaire, et notamment le transport apico-basale des immunoglobulines A (IgA) sécrétoires par le récepteur de la transferrine CD71. Il s’agit d’un récepteur ubiquitaire connu pour son rôle dans l’endocytose et le recyclage de la transferrine, indispensable à l’absorption du fer sérique par l’érythroblaste. CD71 est aussi un récepteur de faible affinité pour les IgA.
Dans un intestin normal, les IgA produites par les plasmocytes dans le chorion sont transportées et déversées dans la lumière où elles se lient aux antigènes, formant des complexes qui sont piégés dans le mucus et éliminés.
Dans la maladie cœliaque, l’expression anormale de CD71 à la surface de l’épithélium, transforme l’effet barrière des IgA produites contre le gluten en un « cheval de Troie » autorisant et pérennisant l’entrée de peptides intacts dans la muqueuse (Figure 2). Cependant le rôle de ce mécanisme dans le déclenchement de la maladie reste moins clair. L’expression de CD71 peut être induite dans l’épithélium lors d’une accélération de renouvellement épithélial au décours d’une infection, par des cytokines inflammatoires ou en cas de carence martiale (Clément et al. 2015).
Figure 2 - Transport transcellulaire du gluten dans la maladie cœliaque.
(Clément et al. 2015)
Les peptides ayant franchi la barrière intestinale vont alors interagir avec la transglutaminase, une enzyme ubiquitaire intracellulaire. On en distingue différentes isoformes, dont la transglutaminase tissulaire de « type 2 » exprimée par les cellules de l’intestin, le foie, le rein, le poumon et les capsules articulaires. Dans l’intestin, on la retrouve au niveau du tissu conjonctif qui entourent les muscles lisses de l'intestin grêle, que l’on appelle « endomysium ».
On a démontré une surexpression duodénale et une activité accrue des transglutaminases chez les patients cœliaques. Les anticorps développés dans la maladie cœliaque ne bloquent donc pas
l’activité de la transglutaminase mais au contraire la stimule, ce qui sans doute, augmente la perméabilité épithéliale et vasculaire (Király et al. 2006).
La transglutaminase tissulaire est responsable de la désamination des peptides de la gliadine et joue un rôle clef dans la présentation de l’antigène. En effet, cette modification post-traductionnelle de la gliadine favorise l’ancrage des peptides de la gliadine désaminée au sein des molécules HLA de classe II DQ2/8 présentes à la surface des cellules présentatrices de l’antigène.
L’immunité acquise est alors médiée par les lymphocytes T CD4+, spécifiques de la gliadine, à l’origine de l’augmentation de la production des cytokines pro-inflammatoires et en particulier de l’interféron-g. Il en résulte une cascade inflammatoire entrainant la synthèse de métalloprotéinases et autres médiateurs de l’inflammation. Cela provoque la formation de lésions intestinales, avec sur le plan histologique : infiltration de lymphocytes intraépithéliaux, atrophie villositaire, et hyperplasie des cryptes selon la classification de Marsh (cf. Chapitre 1.5.2). D’autre part, les lymphocytes T vont générer la multiplication des plasmocytes responsable de la production des anticorps anti-gliadine et anti-transglutaminase (Green et Cellier 2007).
1.3.3. Facteurs génétiques
La récurrence intrafamiliale et l’association avec le génotype HLA DQ2 ou DQ8 ont confirmé la responsabilité de facteurs génétiques. La prédisposition génétique est fréquente dans la population générale puisqu’environ 30 % possède le génotype HLA DQ2 ou DQ8. Or, comme nous l’avons vu, seulement 1 % des individus présenteront une des formes de la maladie cœliaque alors que tous sont exposés au gluten qui est largement présent dans l’alimentation. Cela suggère que d’autres facteurs génétiques et environnementaux interviennent dans le déclenchement du processus auto-immun. Selon les études, le statut homozygote multiplierait par 5 à 20 le risque d’évoluer vers une maladie symptomatique par rapport aux sujets hétérozygotes.
D’autres gènes ont été également identifiés dans la progression de la maladie et la survenue de complications à long terme, notamment CD28/CTL4/ICOS 2q33, 15q26, IDDM17, 5qter (Lamireau et Clouzeau 2013).
1.3.4. Facteurs environnementaux
Quatrième facteur intervenant dans l’étiologie de la maladie cœliaque : les facteurs environnementaux ont un rôle prépondérant en particulier chez le jeune enfant.
Les infections intestinales, notamment à adénovirus et rotavirus qui augmenteraient la perméabilité intestinale, l’expression d’HLA DQ et la concentration de transglutaminase tissulaire, favorisent le développement de la maladie (Farrell 2005).
L’introduction du gluten avant l’âge de 3 mois ou après 7 mois est associée à une augmentation de la prévalence de la maladie sous toutes ses formes (Ivarsson 2005). En revanche, l’allaitement est considéré comme ayant un rôle favorisant l’acquisition d’une tolérance immunitaire, par ses facteurs immunomodulateurs et/ou la présence de faibles quantités de gliadine issue de l’alimentation maternelle. Les conseils actuels sont d’introduire le gluten en faible quantité entre 4 et 6 mois pendant la poursuite de l’allaitement maternelle (Lamireau et Clouzeau 2013).
1.4. Signes cliniques généraux
Le spectre clinique de la maladie cœliaque est large. Quatre formes sont reconnues : classique, atypique, silencieuse et potentielle.
Dans la forme classique, les signes de la maladie cœliaque sont en relation avec une malabsorption de l’intestin grêle : diarrhée avec stéatorrhée, amaigrissement, dénutrition, asthénie et douleurs abdominales. Les anomalies biologiques sont une anémie par carence de fer, folates, vitamine B12, un déficit en facteurs vitamino-K dépendants (II, VII, et X), une hypoalbuminémie, une hypocalcémie, une hypomagnésémie et un déficit en zinc (Farrell et Kelly 2002).
Les tests sérologiques sont anormaux et l’atrophie villositaire est présente.
Au cours des décennies, la forme classique est devenue minoritaire (Murray et al. 2003), alors que les formes atypiques et silencieuses, représentent actuellement la majorité des cas diagnostiqués chez l’adulte (Rampertab et al. 2006).
Dans la maladie cœliaque atypique, les signes et les symptômes de malabsorption sont absents et d’autres symptômes intestinaux ou extra-intestinaux pourraient être présents.
Les tests sérologiques sont anormaux et l’atrophie villositaire est présente.
Dans la forme subclinique ou silencieuse, la maladie évolue sous le seuil de détection clinique sans que les signes et symptômes soient suffisants pour évoquer la maladie dans la pratique clinique de routine. Ces patients subissent parfois un test de dépistage parce qu’ils présentent un risque élevé de la maladie cœliaque.
Leurs tests sérologiques sont anormaux, et l’atrophie villositaire est présente.
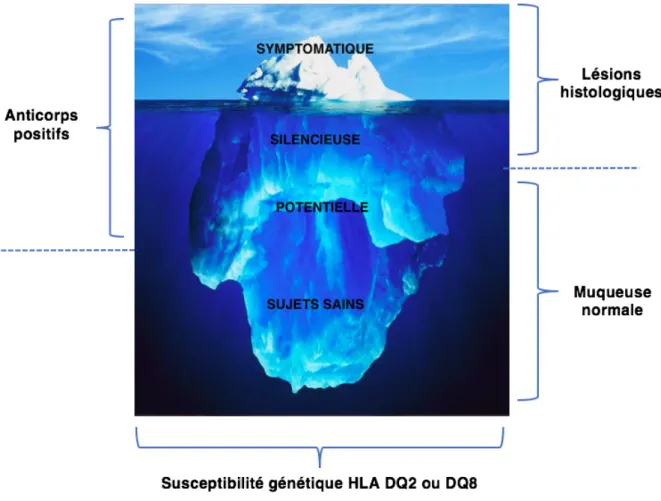
Dans la maladie cœliaque potentielle (anciennement latente), les tests sérologiques sont anormaux, mais l’histologie de la muqueuse intestinale est normale. Plusieurs de ces patients manifesteront des lésions intestinales au fil du temps, ce qui exige une surveillance et un suivi étroits.
Il existe un risque accru de maladie cœliaque chez les apparentés au premier degré de malades cœliaques (10 %), chez les patients atteints de dermatite herpétiforme ou d’autres maladies auto-immunes (diabète, thyroïdite, cirrhose biliaire primitive, alopécie, psoriasis, vitiligo, ataxie...)
(Tableau 2). Les allèles DQ2/DQ8 sont aussi des allèles de susceptibilité pour certaines maladies
auto-immunes telles que le diabète de type I et la maladie d’Addison ce qui explique probablement l’augmentation du risque de ces maladies chez les patients cœliaques (Liu et al. 2005). En théorie,
Figure 4 – Modèle de l’iceberg. D’après Fasano et Catassi (Fasano et Catassi 2001)
Le dépistage de la maladie cœliaque est indiqué dans les cas suivants : • Douleurs abdominales ou ballonnements ;
• Distension abdominale ;
• Maladie hépatique auto-immune ; • Thyroïdite auto-immune ;
• Diarrhée chronique ; • Fatigue chronique ;
• Défauts de l’émail des dents ; • Dermatite herpétiforme ; • Trisomie 21 ;
• Parents au premier degré atteints de la maladie cœliaque ; • Hausse idiopathique des transaminases ;
• Anémie ferriprive ;
• Syndrome du côlon irritable ; • Ostéopénie ou ostéoporose ;
• Neuropathie périphérique ; • Stomatite aphteuse récidivante ; • Déficit sélectif en IgA ;
• Perte pondérale inexpliquée ; • Syndrome de Turner ; • Diabète de type 1.
Les autres signes cliniques chez les enfants sont les suivants : • Anorexie ; • Constipation chronique ; • Retard de puberté ; • Retard de croissance ; • Irritabilité ; • Vomissements récurrents ; • Petite taille.
Tableau 1 - Indications cliniques pour le dépistage de la maladie cœliaque (Rashid et Lee 2016)
Maladies auto-immunes et dysimmunitaires • Dermatite herpétiforme
• Diabète insulinodépendant, dysthyroïdie, maladie d’Addison
• Myasthénie, polymyosite, polyarthrite rhumatoïde, sarcoïdose et sclérose en plaques • Anémie hémolytique et purpura thrombopénique auto-immuns
• Vascularite systémique et cutanée, lupus érythémateux diffus, syndrome de Sjögren • Cirrhose biliaire primitive, cholangite sclérosante
• Maladie de Crohn, rectocolite hémorragique • Déficit en IgA
• Néphropathie à IgA Maladies immuno-allergiques
• Atopie et asthme, maladie du poumon de fermier, maladie des éleveurs d’oiseaux Syndromes malformatifs
• Trisomie 21
• Syndrome de Turner
Tableau 2 - Principales associations morbides de la maladie cœliaque de l’adulte
1.5. Diagnostic
Sachant que la maladie cœliaque peut se manifester de manière très variée, il importe d’avoir
toujours à l’esprit cette hypothèse diagnostique.
Bien que les biopsies intestinales et l’endoscopie haute soient encore considérées à l’heure actuelle nécessaires pour confirmer le diagnostic de maladie cœliaque, les tests sérologiques sont classiquement pratiqués en première intention et identifieront les individus chez qui la procédure est indiquée. La preuve définitive sera apportée par la disparition sans équivoque des signes cliniques ou des anomalies histologiques de la muqueuse sous l’effet du retrait du gluten de l’alimentation (Heizer 2011).
1.5.1. Tests sérologiques
Le recours aux tests sérologiques, doit être facile dès lors qu’il s’agit d’étayer le diagnostic de symptômes évocateurs, de clarifier l’origine de symptômes atypiques, mais aussi de rechercher la maladie dans les groupes à risque, en particulier chez les apparentés du premier degré, et chez les patients atteints de maladies auto-immunes.
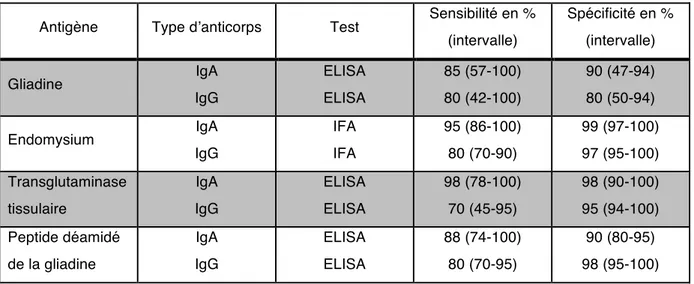
Il est important de dépister un éventuel déficit en IgA. En effet, environ 2 % des patients cœliaques sont déficients en IgA. Le dépistage de la maladie cœliaque sera donc organisé par le seul dosage des IgA anti-transglutaminase dans le cas où le taux d’IgA sériques est normal. En revanche, si ce taux est inférieur à 0,2 g/L, le dosage des IgG anti-transglutaminase ou anti-endomysium sera envisagé́ (Broutin et al. 2012). Cependant, le test des anticorps anti-endomysium possède toutefois deux inconvénients : un coût élevé (utilisation d’œsophage de singe ou de cordon ombilical) et une variabilité entre les observateurs dans l’interprétation des résultats du test (Rashid et Lee 2016).
Les dosages des IgG ou IgA anti-gliadine native ne sont pas suffisamment performants pour le diagnostic biologique de la maladie cœliaque et il est recommandé́ de ne plus les utiliser (Tableau 3). Ils ne sont d’ailleurs plus pris en charge par l’assurance maladie.
Les nouveaux tests dosant les IgG anti-gliadine déamidée présentent des performances comparables aux tests recherchant les anticorps anti-transglutaminase ou anti-endomysium (Tableau 3). On proposera de les doser uniquement en cas de déficit établi en IgA (Broutin et al. 2012).
Il est important de savoir qu’un test sérologique négatif n’écarte pas la maladie cœliaque. Les facteurs pouvant contribuer aux faux négatifs sont :
- Age < 2ans ;
- Erreur de laboratoire possible ;
- Réduction ou élimination du gluten dans l’alimentation ; - Déficit sélectif en IgA ;
- Emploi de corticoïdes ou d’agents immunomodulateurs.
La sensibilité des tests sérologiques étant plus faible chez les enfants de moins de 2 ans, l’ESPGHAN (European Society for Paediatric Gastroenterology Hepatology and Nutrition) recommande dans cette population, de réaliser le test des anticorps gliadine déamidée ou anti-endomysium (de types IgA et IgG) en plus du test des anticorps anti-transglutaminase de type IgA (Rashid et Lee 2016).
Antigène Type d’anticorps Test Sensibilité en %
(intervalle) Spécificité en % (intervalle) Gliadine IgA IgG ELISA ELISA 85 (57-100) 80 (42-100) 90 (47-94) 80 (50-94) Endomysium IgA IgG IFA IFA 95 (86-100) 80 (70-90) 99 (97-100) 97 (95-100) Transglutaminase tissulaire IgA IgG ELISA ELISA 98 (78-100) 70 (45-95) 98 (90-100) 95 (94-100) Peptide déamidé de la gliadine IgA IgG ELISA ELISA 88 (74-100) 80 (70-95) 90 (80-95) 98 (95-100)
ELISA : essai immunoenzymatique ; IFA : épreuve d’immunofluorescence
Tableau 3 - Tests sérologiques de dépistage de la maladie cœliaque (Leffler et Schuppan 2010)
Des tests salivaires (Ocmant et Mascart 2007) et réalisables au doigt (Raivio et al. 2006) fondés sur la détection des anti-transglutaminases sont également en cours d’évaluation.
1.5.2. Biopsie
Un test sérologique positif ne dispense pas de biopsies avant d’instaurer un régime sans gluten du fait de la possibilité rare de faux positifs, et de la nécessité d’une atrophie villositaire documentée pour obtenir le remboursement partiel de produits de substitution sans gluten (Malamut et Cellier 2013).
Il est indispensable de ne pas entreprendre un régime sans gluten avant la biopsie, car la guérison de la muqueuse intestinale qui en résulte compliquerait l’interprétation des résultats de la biopsie (Rashid et Lee 2016).
Pour minimiser le risque de manquer une anomalie de la muqueuse, au moins deux biopsies devraient être prélevées, si possible, dans chacun des segments (2e, 3e et 4e) du duodénum (Heizer 2011).
L’examen des biopsies doit être confié à un pathologiste expérimenté, qui décrira les anomalies histologiques en les classant selon Marsh-Oberhuber (Oberhuber 2000):
• Marsh 0 : muqueuse normale ;
• Marsh I : augmentation des lymphocytes intraépithéliaux (> 30 %) ;
• Marsh II : stade « infiltratif-hyperplasique » : Marsh I + hyperplasie des cryptes ; • Marsh III : stade « atrophique-hyperplasique »
ü IIIa: Marsh II + atrophie villositaire partielle, ü IIIb : Marsh II + atrophie villositaire subtotale, ü IIIc : Marsh II + atrophie villositaire totale ;
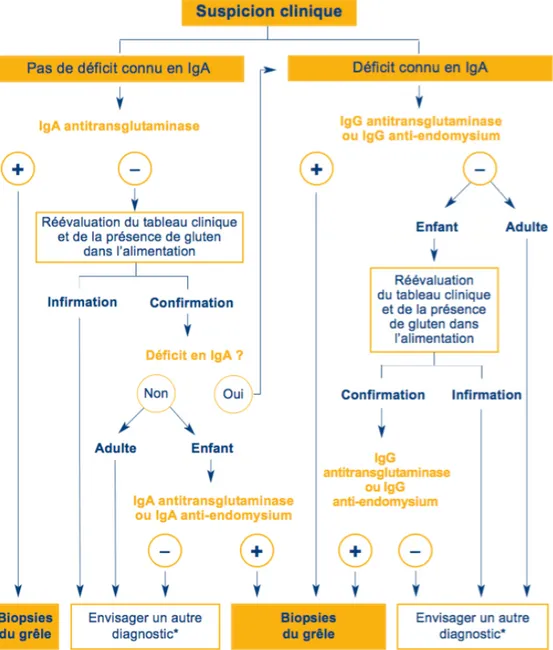
Figure 5 - Arbre décisionnel : Stratégie diagnostique d'après les recommandations
de la HAS (Haute Autorité de Santé 2008)
1.5.3. Typage HLA
On retrouve l’un ou l’autre des antigènes HLA, DQ2 (encodés par les allèles A1*05 et B1*02) et DQ8 (encodés par les allèles A1*03 et B1*0302) chez 95 % des patients atteints de la maladie cœliaque. Par conséquent, leur absence exclut le diagnostic avec une valeur prédictive négative de 99,9 %. Cependant, ces antigènes étant présents dans 30 à 40 % de la population générale, la présence de l’un ou l’autre de ces antigènes n’a une valeur prédictive positive que de 1,7 %.
Les personnes porteuses de l’HLA-DQ2 homozygotes présentent le risque le plus élevé (jusqu’à 40 %) de la maladie cœliaque.
Le typage HLA peut s’avérer utile (Heizer 2011):
ü Chez des personnes ayant une sérologie faiblement positive et des biopsies équivoques ; ü Chez celles qui ont commencé́ un régime alimentaire restrictif depuis plusieurs mois et qui se
demandent si elles ont réellement la maladie, mais ne veulent pas suspendre leur régime pendant une période prolongée ;
ü Chez les enfants des personnes atteintes de maladie cœliaque confirmée et qui espèrent éviter les essais sérologiques périodiques dans les années à venir.
Pour contenir les coûts, il faut que la requête pour le typage des HLA précise de procéder au typage des allèles HLA-DQ2 et HLA-DQ8 seulement, et non pas au typage complet des HLA.
Une fois le diagnostic de maladie posé, un certain nombre d’examens complémentaires est réalisé pour compléter l’exploration du syndrome de malabsorption et dépister d’éventuelles complications : NFS, dosage du fer sérique, des folates, de la vitamine B12, des facteurs de la coagulation (TP), calcémie et magnésémie, tests hépatiques pour rechercher une hépatopathie associée. Tout patient atteint de maladie cœliaque devrait bénéficier de la recherche d’une ostéopénie par la réalisation d’une ostéodensitométrie osseuse (Malamut et Cellier 2013).
1.6. Traitement
1.6.1. Régime sans gluten
Le seul traitement actuel de la maladie cœliaque est un régime sans gluten strict à vie. Selon Matysiak-Budnik, il doit être maintenu à vie chez l’adulte. Alors que chez l’enfant, il peut être interrompu après la puberté (Matysiak-Budnik et al. 2007). La liste de base non exhaustive des aliments autorisés/interdits chez les cœliaques est disponible auprès de l’AFDIAG (Association Française Des Intolérants Au Gluten). A ces règles diététiques il faudrait, selon les carences constatées, supplémenter le patient en fer, folates, calcium, vitamine D.
Ce régime permet chez la grande majorité des patients la guérison des symptômes digestifs (Farrell et Kelly 2002), mais également la régression de manifestations extradigestives telles que la déminéralisation osseuse (Bai et al. 1997; Mora et al. 1999) ou les cytolyses hépatiques (Kaukinen et al. 2002). En effet, l’ostéopénie régresse presque complètement chez 80 % des malades après 12 mois d'un régime sans gluten bien suivi (Cellier et al. 2000b; Vasquez et al. 2000). L'augmentation des transaminases associée à la maladie cœliaque, régresse totalement dans 90 % des cas après un an d'éviction du gluten et une biopsie hépatique n'est requise qu'en cas d'échec du régime bien suivi (Trivin et Cellier 2001). Quelques cas d'hépatopathies sévères justifiant d'une transplantation hépatique et associées à une maladie cœliaque ont même été spectaculairement améliorées par un régime sans gluten (Kaukinen et al. 2002). Les troubles neurologiques centraux, à type d'ataxie ou de migraine, ou périphériques à type de neuropathie semblent aussi bénéficier de l'éviction du gluten (Luostarinen et al. 2001). En revanche, le bénéfice du régime sans gluten en cas de troubles de la reproduction n'est pas clairement démontré mais a été rapporté (Martinelli et al. 2000).
Ce régime ne permet pas de guérir des maladies auto-immunes établies comme un diabète de type I où les lésions tissulaires au moment du diagnostic sont irréversibles, mais son efficacité pour prévenir leur apparition a récemment été démontré (Cosnes et al. 2008). Ce régime permet aussi de prévenir l’apparition d’une ostéopénie/ostéoporose (Bai et al. 1997) et les complications malignes (Holmes et al. 1989 ; Askling et al. 2002 ; Haines et al. 2008).
De façon intéressante, la disparition spontanée de l'atrophie villositaire a pu être démontrée chez un petit nombre de patients adultes diagnostiqués dans l'enfance et ayant repris un régime normal bien toléré cliniquement. Cependant, la persistance d'une augmentation des lymphocytes intraépithéliaux et souvent de sérologies positives ne permet pas chez ces patients de conclure à une restauration complète de la tolérance au gluten mais plutôt au retour vers une forme potentielle de maladie
qui restés asymptomatiques malgré le régime normal, présentaient une atrophie villositaire sévère très souvent associée à une ostéopénie, qui a conduit à restaurer un régime sans gluten. En effet bien que le risque de fractures, de complications auto-immunes ou malignes chez les patents silencieux ne soit pas établi précisément, la prescription d'un régime sans gluten est actuellement considérée comme nécessaire en cas d'atrophie villositaire pour en réduire le risque potentiel. En l'absence d'atrophie villositaire histologique (maladie cœliaque potentielle), le débat sur la prescription du régime sans gluten reste ouvert (Matysiak-Budnik et al. 2007).
1.6.2. Nouveaux espoirs thérapeutiques
Le concept d’un régime sans gluten comme un traitement valable pour la maladie cœliaque remonte aux années 1950. Depuis, cette méthode a été la seule approche disponible et efficace. Le strict respect de ce régime permet d’avoir des résultats sur la réduction des symptômes, la récupération de la muqueuse intestinale et permet également de prévenir le développement des complications liées à la maladie cœliaque.
Cependant, l’adhésion au régime peut rester insuffisant, car le régime sans gluten est coûteux et particulièrement restrictif dans la mesure où les produits de remplacement sont souvent moins agréables que les aliments contenant du gluten, et ont une disponibilité plus restreinte. Compte tenu de ces problèmes évidents avec le régime sans gluten, les patients atteints de la maladie cœliaque ressentent un besoin de thérapies alternatives pour traiter leur état. Par ailleurs, un petit sous-groupe de patients ayant la maladie cœliaque ne répond pas au régime strict sans gluten, et a donc besoin de traitements supplémentaires, notamment via l’immunothérapie (Bousquet 2015).
Aujourd’hui de nouvelles options thérapeutiques sont en cours de développement (Tableau 4 et 5). Ces traitements ne sont pas encore disponibles. Cependant, les essais pré-cliniques et cliniques semblent prometteurs pour certains d’entre eux (Bousquet 2015).
Traitement au niveau de la lumière intestinale Détoxification alimentaire
Digestion du gluten pendant production
Protéases orales : Gluténases : PEP, AVL003, STAN1 Probiotiques
Polymères séquestrant le gluten (BL-7010)
Effets bloquants au niveau de l’épithélium de l’intestin
Inhibition de la médiation des IgA-CD71 et des transports peptidiques Acétate de Larazotide : empêche la perméabilité intestinale
Blocage IL15
Traitement lamina propria et ailleurs
Inhibiteur de la TG2, découverte d’une nouvelle protéine humaine : ELAFINE Bloqueurs HLA-DQ2/Q8
Anticytokines et suppression des LT Blocage LB et production des auto-AC Antogoniste du CCR9
Vaccination peptidique
Tableau 4 - Options thérapeutiques en cours de développement (Bousquet 2015)
Traitement au niveau de la lumière intestinale Effets bloquants au niveau de l’épithélium Traitements dans la lamina propria et ailleurs Pré-clinique Polymères séquestrant le gluten (BL-7010) Inhibition de la médiation des IgA-CD71 et du transport des peptides de gliadine • Bloqueurs de HLA-DQ2 ou DQ8 • Inhibiteur de la transglutaminase 2 • Blocage des LB et des auto-anticorps Phase I Vaccin
Phase II Gluténase PEP
Gluténase AVL003 Gluténase STAN1
Probiotiques
Acétate de Larazotide Antagoniste du CCR9
1.7. Complications
L'échec du régime sans gluten impose d'abord et avant tout la réévaluation du diagnostic initial de maladie cœliaque.
Si le diagnostic initial de maladie cœliaque est confirmé, la principale cause de mauvaise réponse au régime sans gluten est une observance incorrecte de celui-ci dans plus de 50 % des cas (Vahedi et al. 2003). Une analyse réalisée par une diététicienne avertie et les dosages sériques d'anticorps anti-transglutaminase et anti-gliadine doivent être pratiqués. La positivité des anticorps cœliaques doit alors faire suspecter une mauvaise observance du régime.
Si la diarrhée persiste alors que l'atrophie villositaire a régressé, il faut rechercher une cause associée en particulier une colite collagène ou lymphocytaire (Green et Cellier 2007). Si cette colite microscopique n'est toujours pas améliorée par le régime sans gluten, elle peut néanmoins bénéficier d'un traitement spécifique tels que les aminosalicylés ou les corticoïdes (Fine et al. 1997).
Finalement après avoir exclu une mauvaise observance du régime, une résistance primitive ou secondaire au régime sans gluten doit faire suspecter une complication grave telle que la sprue réfractaire clonale, voire un lymphome T intestinal (Cellier et al. 2000a).
La sprue réfractaire correspond à une maladie cœliaque primitivement ou secondairement résistante au régime sans gluten. Il s’agit d'un type I lorsqu'il n'existe pas d'anomalie phénotypique des lymphocytes intraépithéliaux ou d'un type II, en cas de prolifération clonale de lymphocytes intraépithéliaux de phénotype anormal avec persistance d'une expression intracellulaire de CD3 mais absence d'expression en surface du récepteur T et des molécules CD3, CD8 ou CD4 (Cellier et al. 1998).
La présentation clinique de la sprue réfractaire de type II (SR II) est généralement bruyante avec une entéropathie exsudative sévère liée à la fréquente jéjunite ulcéreuse associée. Au diagnostic, les patients ont d'ailleurs une hypo-albuminémie nette plus sévère qu’en cas de sprue réfractaire de type I (SR I) (Malamut et al. 2009).
Alors que le pronostic des SR I avoisine celui des maladies cœliaques non compliquées, le pronostic de la SR Il est nettement plus sombre avec une évolution vers un lymphome T invasif dans environ 50% des cas et une survie sur cinq ans de 40 à 50 % (Al-Toma et al. 2007; Malamut et al. 2009).
Le traitement de la sprue réfractaire n’est pas codifié et repose en pratique sur la corticothérapie prolongée, l’utilisation d'immunosuppresseurs, voire sur l'autogreffe de cellules souches hématopoïétiques pour la SR II (Al-toma et al. 2007).
La SR II, caractérisée par une prolifération clonale de petits lymphocytes intraépithéliaux anormaux (Cellier et al. 2000a), est considérée comme un lymphome intraépithélial de bas grade avec une atteinte digestive diffuse mais également extradigestive fréquentes et constitue une forme de passage entre maladie cœliaque et lymphome T invasif (Cellier et al. 2000a; Malamut et al. 2009). Cependant la SR II n'est pas une étape obligatoire puisque des lymphomes T ont été décrits sur des maladies cœliaques non compliquées, souvent révélateurs de l’entéropathie sous-jacente (Gale et al. 2000; Novakovic et al. 2006; Al-Toma et al. 2007) et également au cours des SR I (Malamut et al. 2009). Après avoir été surévalué, le risque relatif de lymphome malin non hodgkinien associé à la maladie cœliaque a été ramené à 6 d’après les dernières études (Askling et al. 2002). Il s’agit dans la majorité des cas d’un lymphome de phénotype T, EATL (enteropathy associated T cell lymphoma), et exprimant en surface le marqueur CD 103, spécifiques des lymphocytes intraépithéliaux dont il est issu (Cellier et al. 2000a).
Le diagnostic de lymphome invasif repose sur l’entéroscopie poussée, la capsule endoscopique, la tomodensitométrie abdominale voire le Pet-scan (Hoffmann et al. 2003). Dans certains cas, le diagnostic histologique n’est obtenu qu’après une laparotomie. Dans un cas sur deux, le lymphome est révélateur d’une maladie cœliaque silencieuse et se manifeste par une complication chirurgicale (hémorragie, perforation ou occlusion intestinale) (Egan et al. 1995).
Le pronostic clinique de l’EATL est sombre avec une survie inférieure à 20 % à cinq ans principalement en raison de l’habituelle chimiorésistance et de la dénutrition liée à l’entéropathie sous-jacente (Gale et al. 2000; Daum et al. 2003; Howdle et al. 2003).
La maladie cœliaque favorise également la survenue d’adénocarcinomes du grêle (Green et al. 2003), des cancers ORL et hépatiques (Askling et al. 2002; Howdle et al. 2003; Rampertab et al. 2003). De nombreuses études rapportent une diminution du risque de pathologies malignes, y compris lymphomateuses, chez les patients cœliaques suivant correctement leur régime sans gluten (Holmes et al. 1989; Corrao et al. 2001; Askling et al. 2002).
Principales causes de résistance du régime sans gluten : • Mauvaise observance du régime sans gluten ;
• Erreur diagnostique : exclure les autres causes d’atrophie villositaire (déficit immunitaire commun variable, entéropathie auto-immune, maladie de Whipple, sprue tropicale…) ; • Colite collagène ;
(MC : maladie cœliaque ; RSG : régime sans gluten ; AC : anticorps ; * mais présence de signes d’activation immunologiques)
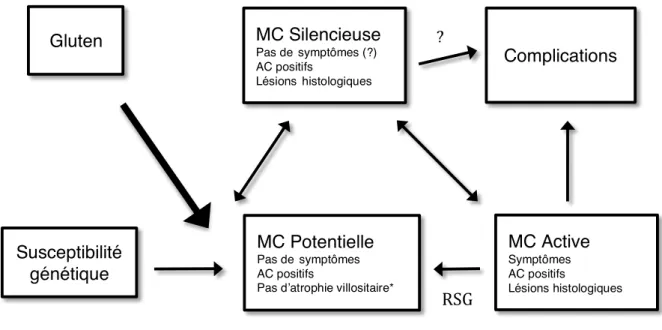
Figure 6 - Histoire naturelle de la maladie cœliaque. D’après Rewers (Rewers 2005)
Susceptibilité
génétique
Gluten
MC Silencieuse
Pas desymptômes (?) AC positifs LésionshistologiquesMC Potentielle
Pas desymptômes AC positifsPas d’atrophie villositaire*
MC Active
Symptômes AC positifs Lésions histologiquesComplications
RSG
?
1.8. Diagnostic différentiel
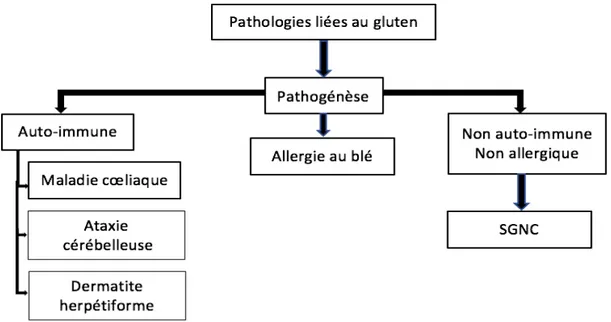
Figure 7 - Nomenclature et classification des pathologies liées au gluten (Bouteloup 2016)
1.8.1. Allergie au blé
L’allergie alimentaire est un sous-groupe de réactions d’hypersensibilité aux aliments, impliquant des mécanismes immunologiques ; elles peuvent être immunoglobulines E (IgE) médiées (allergie immédiate) et/ou non IgE médiées (allergie retardée, cellulaire type hypersensibilité), notamment chez l’enfant. Le blé est responsable d’allergies IgE et non IgE médiées et les manifestations cliniques sont liées à diverses protéines du blé et pas seulement au gluten.
Selon le mécanisme immunologique impliqué et la voie de contact avec l’allergène, quatre types d’allergie au blé sont décrits :
• L’allergie alimentaire « classique » avec atteinte digestive, cutanée et possiblement respiratoire ; • L’anaphylaxie au blé induite par l’exercice physique ;
• L’allergie respiratoire (asthme du boulanger et rhinite) ; • L’urticaire de contact, plus rare (Bouteloup 2016).
1.8.2. SGNC (Sensibilité au gluten non cœliaque)
La sensibilité au gluten non cœliaque est un phénomène décrit récemment où un patient présente des symptômes intestinaux ou extra-intestinaux qui disparaissent sous un régime sans gluten, en l’absence de maladie cœliaque. Les auto-anticorps, tels que les anticorps anti-tranglutaminase tissulaire, l’atrophie villositaire ou le risque d’autres troubles auto-immuns sont absents.
La pathogenèse de la SGNC n’est pas encore bien comprise. On ignore si elle est transitoire ou permanente, et si elle est liée à la dose de gluten ingéré. Il n’existe à l’heure actuelle aucun biomarqueur de la SGNC ; le diagnostic est donc posé après l’exclusion de la maladie cœliaque en fonction des constations sérologiques (et de la biopsie) et est fondé sur la réponse symptomatique du patient à l’élimination du gluten de l’alimentation (Rashid et Lee 2016).
2. Manifestations orales de la maladie cœliaque
Nous avons vu dans la première partie que la majorité des malades cœliaques ne présentent pas les signes cliniques classiques de la maladie (Rampertab et al. 2006). Malgré l’amélioration des tests sérologiques, les études considèrent que bon nombre de cas pourraient être diagnostiqué plus tôt. Il est donc important que les pédiatres, gastro-entérologues, internes et chirurgiens-dentistes aient une approche pluridisciplinaire. Ainsi ils pourraient découvrir des manifestations extra-digestives comme des anomalies hématologiques, dermatologiques, neurologiques, gynécologiques et orales.
Parmi les manifestations orales de la maladie cœliaque, la littérature décrit des défauts de l’émail, des stomatites aphteuses récidivantes, des glossites, des lichen plan buccaux, des retards d’éruption et d’âge dentaires et des modifications salivaires. Parfois ces manifestations orales sont les seuls signes qui mènent au diagnostic de la maladie.
2.1. Défauts/Altérations de l’émail
La maladie cœliaque peut se développer à n’importe quel âge, à partir du moment où les aliments solides sont introduits dans l’alimentation. Si celle-ci apparait pendant le développement des dents, des anomalies de structure amellaire peuvent survenir.
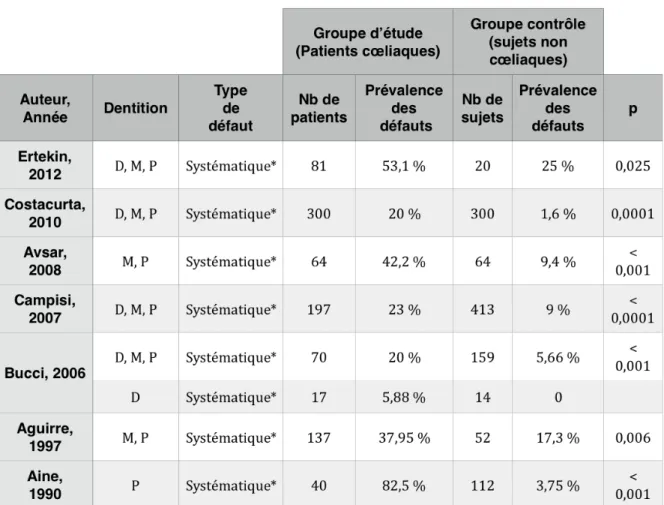
2.1.1. Prévalence
Plusieurs études ont été menées durant les dernières décennies. La prévalence des altérations amellaires systématiques chez les patients cœliaques varie entre 20 % et 53 % selon les études (Aine et al. 1990 ; Aguirre et al. 1997 ; Bucci et al. 2006 ; Campisi et al. 2007 ; Avşar et Kalayci 2008 ; Costacurta et al. 2010 ; Ertekin et al. 2012) (Tableau 6). On caractérise de « systématique », un
*Les défauts sont symétriques et touchent les mêmes dents sur 2 hémi-arcades D= Denture déciduale ; M=Denture mixte ; P=Denture permanente
Tableau 6 - Prévalence des défauts de l’émail
2.1.2. Effet du régime sans gluten
En 2005, Ciacci et al. ont mis en évidence une corrélation entre la présence de défauts de l’émail et la durée d’exposition au gluten. Aucun défaut n’a été observé chez les patients diagnostiqués précocement et suivant un régime sans gluten. 18 % des patients diagnostiqués précocement mais réexposés au gluten, présentaient des altérations de l’émail. Tandis que 26 % des adultes récemment diagnostiqués, montraient des défauts amellaires (Ciacci et al. 2005).
On retiendra donc que les anomalies de structures amellaires peuvent survenir dès l’introduction du gluten dans l’alimentation. Mais la levée de cette exposition au gluten stoppe aussitôt la progression des altérations de l’émail.
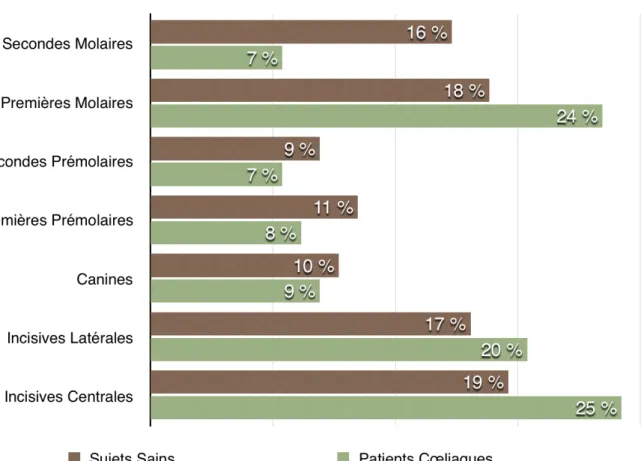
2.1.3. Localisation
Ces défauts sont observés le plus souvent sur les dents permanentes. Ils apparaissent de façon symétrique et concomitante au niveau des quatre quadrants, préférentiellement sur les incisives et les molaires maxillaires et mandibulaires sur les dents permanentes (Graphique 2). Les canines et les secondes molaires déciduales peuvent également être touchées (Graphique 3) (Costacurta et al. 2010).
L’âge de formation de l’émail des dents concernées correspond à l’âge d’introduction du gluten dans l’alimentation. Ainsi, l’implication des prémolaires, des canines et des secondes molaires permanentes indique un retard de diagnostic, puisqu’un régime sans gluten précoce aurait empêché ces altérations (Campisi et al. 2007).
Graphique 3 - Localisation des défauts d’émail sur les dents déciduales (Costacurta, 2010)
2.1.4. Types et sévérité : Classification d’Aine
On peut observer à la fois une hypoplasie et une hypominéralisation de l’émail. Le plus communément, on retrouve une bande hypoplasique amellaire avec des cuspides intactes. Une malformation de l’émail peut apparaitre en même temps que les symptômes gastro-intestinaux. Les défauts de l’émail sont courants chez les enfants ayant développés les symptômes de la maladie cœliaque avant 7 ans. Ce genre de défauts est très rarement observé chez les cœliaques adultes, soit parce qu’ils ont développé des symptômes tardivement, ou parce que leurs dents avaient déjà subi de sévères altérations (Avşar et Kalayci 2008).
Parmi les défauts de l’émail, on trouve : des trous, des rainures et parfois une perte totale de l’émail. Une classification de ces altérations a été proposée par Aine et ses collaborateurs en 1990
(Tableau 7) (Aine et al. 1990). Les différents stades des défauts sont illustrés sur les figures 8, 9 et 10. Le grade I est le plus représenté (Aine et al. 1990 ; Aguirre et al. 1997 ; Bucci et al. 2006 ; Campisi
et al. 2007 ; Avşar et Kalayci 2008 ; Costacurta et al. 2010 ; Ertekin et al. 2012), notamment dans l’étude menée par Costacurta en 2010 (Graphique 4).
Figure 8 - Grade I (Procaccini et al. 2007)
Grade I : Multiples opacités blanches et crèmes avec des marges bien définies.
Figure 9 - Grade II (Rashid et al. 2011)
Grade II : Rugosité de la surface de l'émail avec des opacités symétriques parcellaires et une décoloration.
Figure 10 - Grade III (Rashid et al. 2011)
Grade III : Rainures horizontales profondes avec de grandes fosses et une décoloration linéaire.
Graphique 4 - Distribution des grades (Costacurta et al. 2010)
2.1.5. Étiologie
L’hypocalcémie causée par la malabsorption durant la formation des dents a longtemps été considérée comme le facteur principale des défauts d’émail. Depuis, plusieurs études n’ont pas trouvé de différence de calcémie significative entre les enfants cœliaques présentant des défauts de l’émail et ceux qui n’en avaient pas (Mariani et al. 1994 ; Avşar et Kalayci 2008). De plus, des altérations de l’émail ont été observées chez des parents au premier degré sains de patients cœliaques (Mäki et al. 1991). Les pistes génétique et immunologique ont alors été envisagées. Ces pistes pourraient également expliquer l’implication de dents déciduales chez certains patients.
2.1.5.1.
Facteur génétique
Nous avons vu dans la première partie de présentation de la maladie que le typage HLA pouvait être utilisé comme critère d’exclusion avec une valeur prédictive négative de 99,9 %.
En 2010, Majorana et al. ont démontré que sur 125 enfants, âgés de 4 à 10 ans, les HLA DR52-53 et DQ7 entraînés un risque accru de défauts de l’émail (Majorana et al. 2010).
En 2011, Erriu et al. ont suivi un groupe 98 patients de Sardaigne, âgés de 7 à 77 ans, atteints de la maladie cœliaque et suivant un régime sans gluten depuis au moins un an. Parmi ces 98 patients, 28 présentaient des défauts de l’émail, 38 avaient été touchés par des aphtes au moins deux fois par
mois avant le régime sans gluten, et 60 présentaient les 2 signes. Ils ont observé que l’absence d’allèle HLA-DQB1*02 entrainait au moins une des deux manifestations orales. L’étude ne montre pas de différence significative entre les porteurs d’un ou deux allèles (Erriu et al. 2011) (Tableau 8). En 2013, Erriu a conduit une autre étude sur 44 patients plus jeunes, âgés de 6 à 16 ans. Les altérations de l’émail semblent liées à la présence ou l’absence de l’allèle HLA-DQB1*02. Cette étude confirme également les conclusions de 2011. A savoir, que l’absence de HLA-DQB1*02 se traduit cliniquement par une altération de l’émail et/ou des aphtes récidivants (Erriu et al. 2013) (Tableau 9). Ces résultats suggèrent que des prédispositions génétiques peuvent influencer l’apparition de manifestations orales. Dès lors, le typage HLA ne serait donc plus uniquement considéré comme un critère d’exclusion de la maladie cœliaque.
D’autres études avec des échantillons de population plus larges devront être menées afin de confirmer ces conclusions. L’interaction des loci HLA avec d’autres gènes reste un sujet d’étude à explorer.
DED=Dental Enamel Defects=Défauts de l’émail ;
RAS=Recurrent Aphtous Stomatisis=Stomatite aphteuse récidivante
Tableau 8 - Répartition des manifestations orales en fonction du nombre d’allèle HLA-DQB1*02
(Erriu et al. 2011)
DED=Dental Enamel Defects=Défauts de l’émail ;
2.1.5.2.
Facteur immunologique
§ Similitude dans les chaines peptidiques
Tout d’abord, au niveau de leur composition, l’amélogénine, l’améloblastine et la gliadine se caractérisent toutes par leur richesse en proline, en acides-aminés hydrophobes et en glutamine.
Puis, Munoz et al. ont recherché la présence de séquences correspondantes entre les peptides. Elles ont trouvé des taux d’identité et de similitude importants entre des chaines de gliadine et d’amélogénine (Tableau 10). La chaîne de l’amélogénine entre l’acide-aminé 44 et 180 correspond à la majorité des segments de 𝛼/𝛽 et 𝛾 Gliadine. La chaîne d’amélogénine entre l’acide-aminé 121 et 153 est particulièrement intéressante par la répétition du motif QPQP (Figure 11), qui ressemble à un motif répété de la gliadine. Concernant l’améloblastine, l’analyse révèle un faible taux d’identité (29%), mais on remarque que le motif PLVQQQ, qui fait parti de l’épitope B de 𝛼 Gliadine (QEQVPLVQQQQF), est retrouvé entre les acides-aminés 162 et 167 (Figure 11) (Muñoz et al. 2012).
Résultats obtenus par le programme LALIGN
𝛼/𝛽 Gliadine : 286 acides-aminés ; 𝛾 Gliadine : 302 acides-aminés ; Amélogénine : 191 acides aminés n : nombre de résidus correspondants qui se chevauchent
I : Identité (les deux résidus sont identiques)
S : Similitude (Identité + substitutions qui conservent les propriétés biochimiques)
Tableau 10 - Similarité des chaînes d’acides-aminés entre les segments de gliadine et d’amélogénine
A - Similarité entre l’amélogénine et 𝛼/𝛽 Gliadine : 35,9% d’identité et 55,4% de similitude C - Similarité entre l’améloblastine et 𝛼/𝛽 Gliadine : 29,1% d’identité et 45,5% de similitude
: = Identité . =Substitutions sp∣Q99 : Amélogénine sp∣P02 : 𝛼/𝛽 Gliadine sp∣Q9N : Améloblastine
Figure 11 - Illustration d’alignements des séquences d’acides-aminés obtenus par le programme
ALIGN (Muñoz et al. 2012)
§ Réaction croisée des anticorps anti-gliadine avec l’amélogénine
En 2016, Sóñora et al. ont utilisé des coupes de germes de premières molaires provenant de porcelets nouveaux-nés. Des tests sérologiques ont été réalisés sur 21 patients cœliaques. N’ont été retenus que les patients présentant des IgG gliadine déamidée seulement, et pas des IgG anti-actine ou anti-transglutaminase (protéines ubiquitaires chez les patients cœliaques) (Sóñora et al. 2016).
Les tests immunohistochimiques pratiqués montrent une réaction contre la matrice amellaire et les améloblastes lorsque le sérum des patients sélectionnés est mis en contact avec les germes des porcelets (Figure 12D). Les taches observées sur les coupes histologiques sont similaires à celles observées lors de l’incubation du germe dentaire avec des anticorps anti-amélogénine (Figure 12B et
12D). Le signal étant encore plus fort au niveau de l’émail immature, là où l’amélogénine est encore
(A) Test de contrôle négatif (PBS: Phosphate-Buffered Saline)
(B) Incubation avec des anticorps anti-amélogénine
(C) Test avec le sérum d’un patient sain (contrôle)
(D) Incubation avec le sérum d’un patient cœliaque sélectionné (IgG anti-gliadine déamidée positif)
Am : Améloblastes, D : Dentine, E : Email décalcifié, Em : Matrice amellaire, pB : prédentine, oD : Odontoblastes, P : Pulpe, * Artéfacts
Figure 12 - Analyse immunohistochimique d’un germe de molaire d’un porcelet (Sóñora, 2015)
Une équipe norvégienne (Petronijevic et al. 2016) a poussé les recherches sur cette réaction croisée des anticorps anti-gliadine sur l’amélogénine sur 99 enfants. Leur étude a mis en évidence :
- La réactivité croisée des anticorps anti-gliadine avec l’amélogénine ;
- Une réactivité plus spécifique aux IgA anti-amélogénine chez les patients cœliaques qu’aux IgG anti-amélogénine ;
- Une réactivité significativement augmentée aux IgG anti-amélogénine chez les patients présentant des signes plus sévères de la maladie (Marsh 3c) ;
- Une réactivité spécifique des anticorps anti-amélogénine avec l’amélogénine et pas avec la gliadine ;
- Un sous-groupe d’enfants témoins sains présentant une forte réactivité immunitaire aux IgA et IgG anti-amélogénine semblable à celle d’enfants malades. Ce qui suggère que les anticorps dirigés contre l'amélogénine peuvent interférer dans l’amélogénèse des enfants cœliaques ou sains.
2.1.6. Diagnostic différentiel
Tableau 11 - Diagnostic différentiel des défauts de l’émail (Rashid et al. 2011)
Puisque que les défauts de l’émail peuvent être observés dans plusieurs dérèglements, d’autres facteurs étiologiques doivent être envisagés avant que la maladie cœliaque ne soit reconnue comme étant la cause principale (Tableau 11).
Le chirurgien-dentiste peut facilement déterminer si l’étiologie est systémique, auquel cas toutes les dents qui se sont développées en même temps seront affectées (par exemple les incisives centrales
Le praticien doit également distinguer l’hypoplasie amellaire de l’hypocalcification. L’hypoplasie amellaire est aisément identifiable : l’émail est malformé ou absent sur une partie ou sur toute la surface (Tableau 11, ligne A et C). Une hypoplasie amellaire localisée peut prendre la forme de trous, de rainures ou de rides profondes sur la surface de l’émail, qui peut être minéralisé normalement (brillant et dur). Une surface amellaire lisse et intacte avec des taches ou des lignes opaques blanches ou jaunâtres indique une faible hypocalcification.
Parmi les atteintes amellaires, la fluorose (Tableau 11, ligne B) est un défaut d’émail commun (Vieira et al. 2005). L’amélogenèse imparfaite (Tableau 11, ligne C) est un dérèglement assez rare, le plus souvent caractérisé par un émail creusé mais parfois hypocalcifié (Kim et al. 2006). Si une partie de l’émail semble normal, on recherchera un antécédent de carence en vitamine D (Fraser et Nikiforuk 1982), de sévères infections ou une prématurité (Hall 1989). Lorsque l’enfant guérit de ses problèmes systémiques, la formation de l’émail redevient normale.
2.2. Stomatite aphteuse récidivante
2.2.1. Aspects cliniques
La stomatite aphteuse récidivante est caractérisée par une ou plusieurs ulcérations douloureuses et récurrentes de la muqueuse buccale. Ces ulcérations sont rondes ou ovoïdes. Elles se caractérisent par un halo érythémateux et un fond jaunâtre ou grisâtre.
Environ 80 % des patients atteints de stomatite aphteuse récidivante présentent des aphtes mineurs mesurant entre 2 et 8 mm de diamètre et qui disparaissent spontanément en deux semaines.
Figure 13 - Aphte mineur
Les aphtes géants sont moins communs. Les ulcérations peuvent dépasser le centimètre de diamètre et persister plusieurs semaines.
Figure 14 - Aphte géant
(Sedghizadeh et al. 2002)
La forme la moins répandue est dite « herpétiforme » et se caractérise par des grappes de petites ulcérations (pouvant aller jusqu’à 100, de 1 à 3 mm de diamètre) qui vont se regrouper et former de larges et irrégulières ulcérations (Pastore et al. 2008).
Figure 15 - Forme herpétiforme
(Sedghizadeh et al. 2002)
Les patients cœliaques peuvent présenter le plus souvent des aphtes mineurs. Mais certains cas d’aphtes majeurs et herpétiformes ont également été rapportés (Pastore et al. 2008).
2.2.2. Prévalence
Tableau 12 - Prévalence des stomatites aphteuses récidivantes
La plupart des études montrent une différence de prévalence significative entre les patients atteints de la maladie cœliaque et le groupe contrôle (Campisi et al. 2007 ; Costacurta et al. 2010 ; Acar et al. 2012 ; Ertekin et al. 2012 ; de Carvalho et al. 2015) (Tableau 12).
2.2.3. Étiologie
Ø Carence hématinique
La corrélation entre la maladie cœliaque et la stomatite aphteuse récidivante peut être expliquée par des carences hématiniques (fer, acide folique ou vitamine B12) due à la malabsorption. En effet, les taux de fer, d’acide folique et de vitamine B12 sériques sont souvent faibles chez les patients cœliaques non traités. Et l’on sait qu’environ 20 % des stomatites aphteuses récidivantes présentent une carence hématinique.
En outre, les taux d’hémoglobine et des folates sont significativement faibles chez les patients cœliaques atteints d’une stomatite aphteuse récidivante, par rapport aux non cœliaques atteints de stomatite aphteuse récidivante (Pastore et al. 2008).
Ø Facteur génétique
La prévalence du HLA DRw10 et DQw1 est significativement plus élevée chez les patients cœliaques souffrant de stomatite aphteuse récidivante par rapport aux cœliaques sans stomatite (Majorana et al. 1992 ; Meini et al. 1993).
Les études d’Eriu et al. sur l’absence de l’allèle HLA DQB1*02, ont démontré que les patients cœliaques ne portant aucun allèle de HLA DQB1*02 présentent au moins l’une des deux manifestations orales les plus fréquentes de la maladie cœliaque, à savoir une altération de l’émail et/ou une stomatite aphteuse récidivante (Erriu et al. 2011, 2013). (cf. Chapitre 2.1.4)
2.2.4. Effet du régime sans gluten
Plusieurs auteurs ont montré une amélioration significative des stomatites aphteuses récidivantes chez les patients cœliaques qui ont suivi un régime sans gluten, allant jusqu’à la rémission complète des symptômes. Une réapparition de la récidive des aphtes a également été observée lors de la réintroduction du gluten (Pastore et al. 2008).
D’autre part, Hunter et al. ont examiné les effets du régime sans gluten sur des patients non cœliaques présentant des aphtes récidivants lors d’une étude randomisée en double aveugle. Celle-ci n’a pas montré de différence significative dans l’effet du régime sans gluten entre les deux groupes (Hunter et al. 1993).
2.2.5. Diagnostic différentiel
Chez la plupart des individus, les aphtes sont bénins. Le caractère récidivant ou persistant de l’aphtose nous orientera vers une pathologie sous-jacente. Il faudra alors écarter les infections orales, les maladies auto-immunes (maladie de Crohn, maladie de Behçet) ou les états d’immunodéfience.