Function and regulation of 17β-hydroxysteroid
dehydrogenase type7 (17β-HSD7) in sex hormone
biosynthesis and breast cancer: in vitro, in vivo, proteomic
and three dimensional co-culture studies
Thèse
XIAO QIANG WANG
Doctorat en biologie cellulaire et moléculaire
Philosophiae Doctor (Ph.D.)
Québec, Canada
iii RÉSUMÉ COURT
La 17β-hydroxystéroïde déshydrogénase humaine de type 7 (17β-HSD7) a une double fonction dans la cholestérologénèse et la stéroïdogénèse, et qui est impliquée à la fois dans la formation d’estradiol (E2) à partir de l’estrone (E1), et dans la dégradation de la dihydrotestostérone (DHT) en un œstrogène faible (3β-diol). Cependant, sa fonction dans le cancer du sein dépendant des œstrogènes (positif aux récepteurs oestrogéniques (RE+) n’a pas toujours été claire. L’E2 stimule la croissance des cellules cancéreuses du sein (CCS; cellules MCF-7) via les RE tandis que la DHT a un effet antiprolifératif via le récepteur des androgènes. Mes études in vitro, in vivo et de protéomique, ont apporté les résultats suivants : (1) L’inhibition de la 17β-HSD7 par un inhibiteur spécifique (INH7) dans les CCS a entrainé une baisse de l’E2, une augmentation de la DHT, une interruption du cycle cellulaire et une régulation négative de cette enzyme. De plus, l’INH7 a permis de réduire des tumeurs xénogreffes qui a été accompagnées d’une diminution de l’E2 et une augmentation de la DHT sériques. (2) L’INH7 a modulé des protéines impliquant différents processus biologiques. L’INH7 a supprimé l’expression de la protéine 78 régulée par le glucose (Grp78) et de fait a augmenté l’apoptose des CCS envers le Letrozole, un inhibiteur de l’aromatase. (3) Les interactions entre les CCS et les fibroblastes tumoraux montrent que la 17β-HSD7 était l’enzyme la plus régulée dans les CCS tandis que l’aromatase était l’enzyme les plus régulées dans les fibroblastes. De telles régulations ont mené à une augmentation de la conversion de l’E2 à partir de ses précurseurs, et a ainsi encouragé la prolifération cellulaire des CCS. Si l’augmentation de la prolifération cellulaire est bloquée par le Letrozole des résultats plus significatifs ont été observés par l’INH7 qui bloque la dégradation de la DHT. (4) L’analyse des données intégratives basée sur The Cancer
Genome Atlas (TCGA) confirme l’amplification significative du gène HSD17B7 dans
les divers cancers du sein comparé à des tissus mammaires sains. Ainsi, nous pensons que la 17β-HSD7 devrait être une nouvelle cible thérapeutique des cancers RE+ du sein.
v ABSTRACT
Human 17β-hydroxysteroid dehydrogenase type 7 (17β-HSD7) displays a dual function in cholesterogenesis and steroidogenesis. In steroidogenesis, it is both involved in the formation of the estradiol (E2) from estrone (E1) and in the degradation of dihydroterstosterone (DHT) into weak estrogen 5α-androstane-3β, 17β-diol (3β-diol). However, its function in estrogen dependent breast cancer (estrogen receptor positive, ER+) has been unclear for many years. E2 stimulates breast cancer cells (BCCs, MCF-7 cells) growth via estrogen receptor (ER) whereas DHT displays anti-proliferative effects via androgen receptor (AR). In the present thesis, the function of 17β-HSD7 in ER+ breast cancer was studied with in vitro, in vivo, proteomics and three dimensional (3D) co-culture model and results were described: (1) Inhibition of 17β-HSD7 by its selective inhibitor (INH7) in BCCs induced significant lower E2, higher DHT, cell cycle arresting and negative regulating of the same enzyme. Such inhibition induced significant shrinkage of xenograft tumors accompanied by decreased E2 and elevated DHT in plasma. (2) Inhibition of 17β-HSD7modulated 104 proteins involved in different biological processes. INH7 especially suppresses the expression of glucose regulated protein 78 (GRP78) and consequently enhanced apoptosis of MCF-7 towards aromatase inhibitor. (3) The interactions between BCCs and tumor fibroblast modulate steroidogenic enzymes. 17β-HSD7 was the most modulated enzyme in MCF-7 cells whereas aromatase was the most regulated enzyme in fibroblast (Hs578Bst). Such regulations led to an increasing of E2 conversion from precursors and promoted MCF-7 cells’ proliferation. The increased cell proliferation was blocked by aromatase inhibitor in 3D co-culture system, but more significant results were observed with INH7 which blocked DHT degradation. (4) Integrative data analysis with The Cancer Genome Atlas (TCGA) confirmed the significant amplification of 17β-HSD7 in various breast cancers compared to normal breast tissue. Thus, in the present thesis, 17β-HSD7 was characterized as a novel therapeutic target for estrogen dependent breast cancer in postmenopausal women
vii RÉSUMÉ
L’œstrogène le plus puissant, qui est formé par la réduction de l’estrone (E1) et/ou par l’aromatisation de la testostérone (T), a un double rôle dans la carcinogénèse du sein en tant qu’hormone qui stimule la prolifération cellulaire en se liant aux récepteurs d’œstrogènes (RE) ainsi qu’en tant que pro-carcinogène induisant des dommages et mutations génétiques. Donc, le blocage de la production de l’estradiol (E2) avec des inhibiteurs spécifiques contre les enzymes stéroïdogéniques telles que les inhibiteurs de l’aromatase (IA) est une de deux thérapies standards actuellement disponibles, tandis que l’autre approche implique le blocage des RE avec des modulateurs sélectifs des récepteurs des œstrogènes. Contrairement à l’E2, la dihydrotestostérone (DHT), qui est un androgène non-aromatique, a un effet anti-prolifératif envers les cellules cancéreuses du sein à RE positifs via le récepteur d’androgènes (RA) en activant p21waf1/cip1 et/ou en inhibant la Cycline D1. La co-expression du RA dans les tumeurs
de seins chez les femmes est associée à de meilleurs pronostics et résultat. De plus, les métabolites de la DHT, tels que 5α-androstane-3β, 17β-diol (3β-diol), ont une faible puissance œstrogénique vis-à-vis les cellules MCF-7 en se liant aux RE, ce qu’on croyait être un mécanisme possible d’induction de résistance à l’inhibiteur de l’aromatase. Ainsi, il est apparu un nouveau concept dans le traitement du cancer du sein ciblant de façon conjointe l’E2 (sa diminution) et la DHT (sa restauration). La 17β-hydroxystéroïde déshydrogénase humaine de type 7 (17β-HSD7) est une enzyme de la stéroïdogénèse impliquée dans la formation de l’E2, un œstrogène actif, à partir de l’E1, tout en inactivant l’androgène DHT en un œstrogène faible, soit le 3β-diol. De plus, la 17β-HSD7 est une enzyme de la stéroïdogénèse unique à rétroaction positive régulée par l’E2. Les caractéristiques biologiques et chimiques de la 17β-HSD7 suggèrent ses rôles importants dans la formation de l’hormone sexuelle et ses rôles pivots dans le cancer du sein. De fait, dans cette thèse, je démontre les rôles et la régulation de la 17β-HSD7 dans la biosynthèse des hormones sexuelles et du développement du cancer du sein dépendant à l’œstrogène avec des études in vitro, in
viii
vivo et protéomique ainsi qu’avec un modèle de co-culture en trois dimensions (3D)
basé sur le collagène.
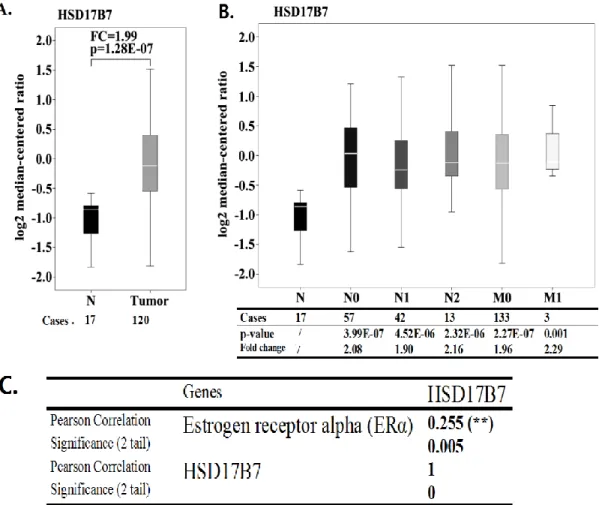
Dans un premier temps, afin de clarifier le statut d’expression de la 17β-HSD7 dans le tissu mammaire tumoral humain, une analyse intégrative a été statistiquement faite avec les données du The Cancer Genome Atlas (TCGA). L’analyse a confirmé l’amplification significative de la 17β-HSD7 dans divers carcinomes du sein (surtout dans les carcinomes canalaires infiltrants) comparée au tissu mammaire normal. Une telle amplification était corrélée avec le statut d’expression des récepteurs d’œstrogène dans les tissus mammaires tumoraux. La surexpression de la 17β-HSD7 était aussi associée à la progression de la tumeur par stades (nodules lymphatiques et autres métastases positionnelles). L’expression de la 17β-HSD7 était augmentée de façon significative dans les stades N2 et M1.
L’étude in vitro a démontré que l’inhibition de la 17β-HSD7 avec différentes doses d’inhibiteur sélectif (INH7) dans les cellules MCF-7 et T47D générait des effets cytostatique et cytotoxique selon la dose de l’inhibiteur. Spécifiquement, l’inhibition de la 17β-HSD7 avec une petite dose d’INH7 a supprimé l’activité de l’enzyme bloquant ainsi la formation de l’E2 et restaurant la DHT. La modulation des hormones sexuelles a arrêté en synergie les cellules dans la phase G0/G1 en activant p21waf1/cip1 et
en inhibant la Cycline D1. À l’inverse, le blocage de l’E2 a atténué l’expression de la 17β-HSD7 même. L’effet combiné peut amplifier l’effet inhibiteur de la 17β-HSD7. L’inhibition de la 17β-HSD7 avec une dose élevée d’INH7 a induit l’apoptose des cellules MCF-7 et T47D en supprimant la protéine pro-survivante Bcl-2 et en augmentant la protéine pro-apoptotique Bik.
L’étude in vivo suggère que l’inhibition spécifique de la 17β-HSD7 par l’INH7 dans une tumeur xénogreffe diminue le volume tumoral, ce qui corrèle avec la diminution du niveau de l’E2 et l’augmentation du DHT plasmatiques. De plus, l’immunohistochimie (IHC) a démontré que l’INH7 a supprimé l’expression de la 17β-HSD7 dans les tumeurs xénogreffes en accord avec l’étude in vitro. L’examen
ix histologique suggère que l’INH7 inhibe la croissance des cellules cancéreuses au sein de la tumeur.
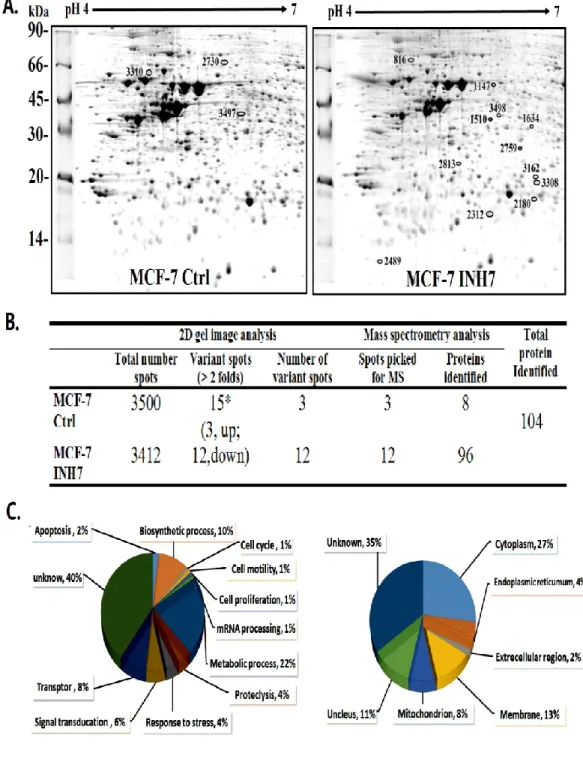
L’étude protéomique a démontré que l’inhibition de la 17β-HSD7 dans les cellules MCF-7 modulait plusieurs protéines (104 protéines) impliquées dans les processus biologiques de survie/mort cellulaire, croissance/prolifération cellulaire, traitement de l’ADN, métabolisme des glucides et traitement des protéines qui sont toutes des caractéristiques de cancer du sein. L’analyse avec le logiciel Ingenuity pathway
analysis (IPA) et les expériences subséquentes confirment que l’inhibition de la
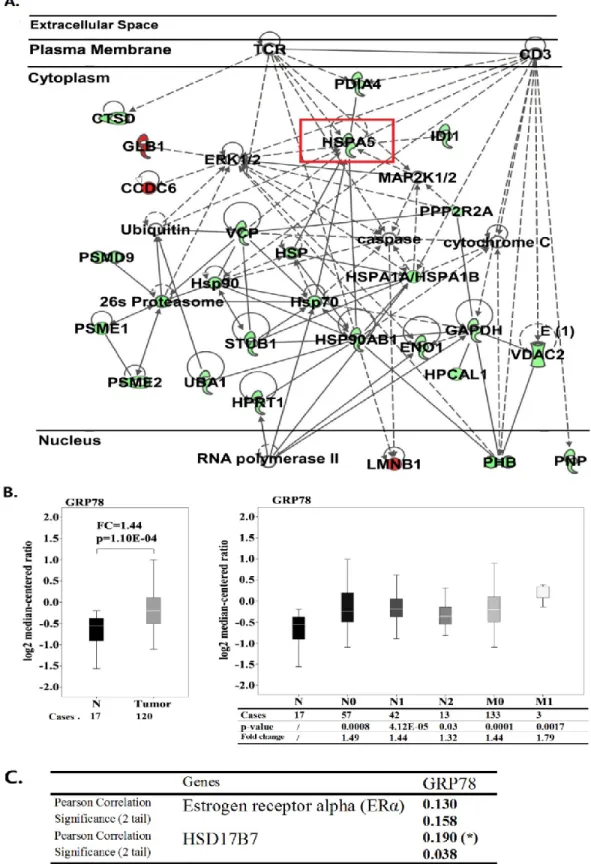
17β-HSD7 supprime l’expression de la protéine régulée par le glucose 78 (Grp78), qui joue un rôle unique dans la détermination de la réactivité et de l’apoptose anti-œstrogène/œstrogène. L’étude a démontré que l’inhibition de la 17β-HSD7 améliore l’apoptose des MCF-7 vis-à-vis l’inhibiteur de l’aromatase via la régulation négative du Grp78.
En tenant compte de l’interaction entre les tumeurs stromales fibroblastiques et les cellules épithéliales mammaires tumorales dans la modulation de la stéroïdogénèse et le développement du cancer du sein, des modèles de co-culture, soit un en deux dimensions (2D) et un en trois dimensions (3D), ont été utilisés afin d’investiguer ces interactions dans la régulation des enzymes, particulièrement la 17β-HSD7, lors de la stéroïdogénèse. Les résultats ont démontré que les enzymes de la stéroïdogénèse sont régulées à la hausse de façon complète en deux types de cellules surtout en raison d’interactions cellule-cellule. La 17β-HSD7 était l’enzyme la plus modulée au sein des cellules MCF-7 tandis que l’aromatase et la 17βHSD1 étaient les enzymes les plus régulées dans les cellules tumorales fibroblastiques (Hs578Bst). De telles régulations ont mené à l’augmentation de la conversion de l’E2 à partir de son précurseur et a ainsi encouragé la prolifération cellulaire des cellules MCF-7 avec l’activation du gène pS2 en tant que gène sensible à l’œstrogène. L’augmentation de la prolifération cellulaire a été bloquée de façon efficace par l’inhibiteur de l’aromatase dans le système de co-culture 3D, mais des résultats plus significatifs ont été observés par les inhibiteurs de la 17β-HSD7 (INH7) en raison de son puissant effet de modulation de la DHT.
x
En conclusion, dans cette thèse, basé sur les études in vitro, in vivo, protéomique et d’interactions cellule à cellule, j’ai clarifié les rôles et la régulation de la 17β-HSD7 dans la biosynthèse des hormones sexuelles et le développement du cancer du sein dépendant à l’œstrogène. La 17β-HSD7 est identifiée comme nouvelle thérapie ciblant le cancer dépendant à l’œstrogène chez les femmes post-ménopausées grâce à sa régulation en synergie de l’E2 et de la DHT. Entretemps, j’ai identifié dans cette étude un inhibiteur puissant contre la 17β-HSD7 qui détient de surcroit un potentiel thérapeutique qui pourrait se traduire cliniquement.
xi SUMMARY
The most potent estrogen, estradiol (E2), which is formed by the reduction of estrone (E1) and/or by the aromatization of 4-androstene-3,17-dione (4-dione)/testosterone (T), plays a dual role in breast carcinogenesis. E2 serves as a hormone stimulating cell proliferation through estrogen receptors (ER) as well as a pro-carcinogen inducing genetic damages and mutations. Therefore, blockage of E2 production with specific inhibitors against steroidogenic enzymes such as AIs (AIs) is the first of two currently available standard therapies, whereas the other approach involves blocking the ER with selective estrogen receptor regulators. In contrast to E2, dihydrotestosterone (DHT) exerts anti-proliferative effects towards ER+ breast cancer cells via androgen receptor (AR) by activating p21waf1/cip1 and/or inhibiting CyclinD1. Co-expression of AR in
female breast tumor is associated with better prognosis and outcome. Moreover, metabolites of DHT such as 5α-androstane-3β,17β-diol (3β-diol) displays weak estrogenic potency towards MCF-7 cells through binding to ER. Therefore, 3β-diol was thought to be a possible mechanism of inducing aromatase inhibitor resistance. Thus, a new concept for the treatment of ER+ breast cancer has emerged through the joint targeting of E2 (decrease) and DHT (increase).
Human 17β-hydroxysteroid dehydrogenase type 7 (17β-HSD7) is dual functional steroidogenic enzyme involved in the formation of E2 from E1 and in the degradation of DHT into 3β-diol. Furthermore, 17β-HSD7 is a unique steroidogenic enzyme regulated by E2 in positive feed-back manner. The function and regulation of 17β-HSD7 suggested it as an important regulator in sex hormone formation and breast cancer. Therefore, in the present thesis, I experimentally demonstrated the function and regulation of 17β-HSD7 in sex hormone biosynthesis and estrogen-dependent breast cancer through in vitro, in vivo, proteomics and three dimensional (3D) co-culture studies.
xii
(TCGA) dataset in Oncomine to clarify the expression status of 17β-HSD7 in human breast cancer tissue. The analysis demonstrated that 17β-HSD7 is significantly amplified in various breast carcinomas (especially in invasive ductal carcinoma) compared with normal breast tissue. The overexpression of 17β-HSD7 was also associated with tumor progression by TNM staging (primary Tumor, lymph Nodes and distant Metastasis). The expression of 17β-HSD7 was significantly increased in N2 and M1 stages. Such amplification of 17β-HSD7 was correlated with the expression of estrogen receptor in breast cancer tissue.
In vitro study demonstrated that inhibition of 17β-HSD7 with different doses of a
selective inhibitor (INH7) in MCF-7 and T47D cells generated cytostatic and cytotoxic effects. Inhibition of 17β-HSD7 with a low dose of INH7 suppressed enzyme activity resulting in blocking of E2 formation and restoring of DHT. The modulation of sex hormones synergistically arrested cells in G0/G1 phase by activating p21waf1/cip1 and
inhibiting CyclinD1. The blockage of E2 conversely attenuated the expression of 17β-HSD7 itself. The combined suppression (enzyme activity and expression) magnify the inhibitory effect of 17β-HSD7 within cells. Inhibition of 17β-HSD7 with a high dose of INH7 induced apoptosis of MCF-7 and T47D cells by suppressing the pro-survival protein Bcl-2, elevating the pro-apoptotic protein Bik, and activating pro-caspase7 therefore inducing apoptosis.
In vivo study suggested that specific inhibition of 17β-HSD7in xenograft tumor
reduced the tumor volume, along with a decreased E2 level and an increased DHT level in plasma. In addition, immunohistochemistry showed that INH7 suppressed the expression of 17β-HSD7 in xenograft tumor in agreement with the in vitro study. Histological examination suggests that INH7 inhibited cancer cell growth within the tumor.
Proteomic study demonstrated that inhibition of 17β-HSD7 in MCF-7 cells with INH7 modulated 104 proteins involved in biological processes of cell death/survival, cell growth/proliferation, DNA processing, carbohydrate metabolism and protein
xiii processing. Ingenuity Pathway Analysis and subsequent experiments confirmed that the inhibition of 17β-HSD7 suppressed the expression of glucose regulated protein 78 (GRP78). GRP78 is an apoptotic protein determining the responsiveness of anti-estrogen/estrogen suppressive treatments. The study demonstrated that inhibition of 17β-HSD7 enhance apoptosis of MCF-7 towards aromatase inhibitor through down-regulation of GRP78.
In consideration of the interactions between stromal fibroblast and cancer epithelial cells in modulating steroidogenesis in breast cancer, we carried out a two dimensional (2D) and a three dimensional (3D) co-culture studies to investigate such interactions in regulating steroidogenic enzymes, particularly 17β-HSD7. The results demonstrated that steroidogenic enzymes were comprehensively up-regulated in both two cell after co-culture. 17β-HSD7 was the most modulated enzyme in MCF-7 cells whereas aromatase were the most regulated enzymes in fibroblast (Hs578Bst). Such regulations led to increasing E2 conversion from its precursor and promoted the cell proliferation of MCF-7 cells. The increasing of cell proliferation was effectively blocked by combinatory inhibitors in the 3D co-culture system. .
In conclusion, in the present thesis, based on in vitro, in vivo, proteomic and cell-cell interaction studies, the function and regulation of 17β-HSD7 in steroidogenesis and estrogen-dependent breast cancer were demonstrated. 17β-HSD7 was characterized as a novel therapeutic target toward estrogen-dependent breast cancer in postmenopausal women. Meanwhile, a druggable inhibitor toward 17β-HSD7 was identified with translational potentials.
xv
TABLE OF CONTENTS
RÉSUMÉ COURT………...iii ABSTRACT………...…...v RÉSUMÉ ………vii SUMMARY……….……….xi TABLE OF CONTENTS………...xv LIST OF TABLES………..xxiLIST OF FIGURES………..… xxiii
LIST OF ABBREVIATIONS………xxv
ACKNOWLEDGEMENTS………..xxix
FOREWORD………..xxxiii
CHAPTER 1. GENERAL INTRODUCTION………..1
1.1 BREAST CANCER...………..2
1.1.1 General Concept………...2
1.1.2 Epidemiology of breast cancer……….3
1.1.3 Histopathology and Molecular Subtype of Breast Cancer………...4
1.2, ESTROGEN DEPENDENT BREAST CANCER……...………..6
1.2.1 General definition………6
1.2.2 E2 and its roles in Estrogen receptor positive (ER+) breast cancer……6
1.2.3 Estrogen receptors (ERs) and its roles in ER+ breast cancer…………..7
1.2.4 E2/ERs and hallmarks of breast cancer………...8
1.3, E2 CONCENTRATION IN ER+ BREAST CANCER: BLOOD CIRCULATION AND INTRATUMORAL FORMATION...11
1.4, E2 FORMATION IN ER+ BREAST CANCER OF POSTMENOPAUSAL WOMEN: INTRACRINOLOGY……….11
1.5, STEROIDOGENIC ENZYMES IN E2 FORMATION AND BREAST CANCER: INTRACRINE REGULATORS………13
1.5.1 Aromatase ………..13
1.5.2 17β-HSD1………...14
1.5.3 17β-HSD7………...14
1.5.4 Steroid sulfatase (STS)………...16
1.6, ANDROGENS AND ANDROGEN RECEPTOR IN BREAST CANCER……16
1.7, MICROENVIRONMENT OF BREAST CANCER AND ITS ROLES IN ESTROGEN/ANDROGEN FORMATION……….17
1.8, MOLECULAR AND CELLULAR MECHANISM OF E2 SUPPRESSIVE AND/OR ANTI-ESTROGEN TREATMENT IN BREAST CANCER………..18
1.8.1 Anti-estrogens/estrogen suppression and cell cycle………...19
1.8.2 Anti-estrogens/estrogen suppression and apoptosis………...21
1.9, HYPOTHESES AND OBJECTIVES OF RESEARCH………...22
1.9.1 The hypotheses of research………...23
1.9.2 The objectives of research..………23
xvi
CHAPTER 2. SYNERGISTIC CONTROL OF SEX-HORMONES BY 17BETA- HSD TYPE 7: A NOVEL TARGET FOR ESTROGEN-
DEPENDENT BREAST CANCER ………....25
2.1 RÉSUMÉ EN FRANÇAIS………26
2.2 ABSTRACT………...28
2.3 INTRODUCTION……….29
2.3.1 Brief introduction of estrogen dependent breast cancer in postmenopausal women………..29
2.3.2 The roles of DHT and androgen receptor in breast cancer……….29
2.3.3 Brief introduction of intracrine E2 and DHT biosynthesis in postmenopausal women………..30
2.3.4 The function of 17β-HSD7 in breast cancer: misunderstandings And hypothes…...30
2.4 MATERIALS AND METHODS ………..32
2.4.1 Inhibitors and chemicals……….32
2.4.2 Cell culture………..32
2.4.3 Cell proliferation assay: measurement of DNA content with CyQUANT assay kit………...33
2.4.4 Cell cycle analysis by flow cytometry: Edu incorporation assay combined with propidium iodide………33
2.4.5 Determination of E2 and DHT level………...34
2.4.6 Cell viability assay using MTT ………..34
2.4.7 Analysis of cellular apoptosis by flow cytometry………...35
2.4.8 Western blotting………..35
2.4.9 MCF-7 xenograft tumor studies………..36
2.4.10 Histopathological analysis of xenograft tumor tissue ………..37
2.4.11 Immunohistological analysis of 17β-HSD7 expression level within xenograft tumor tissue………...37
2.4.12 Measurement of plasma E2/DHT level………38
2.4.13 Integrative analysis of clinical datasets: the overexpression of HSD17B7 in breast cancer………38
2.4.14 Statistical analysis……….38
2.5 RESULTS ……….39
2.5.1 INH7 at low concentration suppressed cell proliferation and arrested the cell cycle in G0/G1 phase by inhibiting cyclin D1 and activating p21……….39
2.5.2 INH7 decreased E2 biosynthesis and resorted DHT ……….40
2.5.3 INH7 inhibits the expression of 17β-HSD7 in MCF-7………...41
2.5.4 INH7 at high concentration induced apoptosis by inactivating anti- apoptosis protein Bcl-2 and activating pro-apoptosis protein Bik……..41
2.5.5 INH7 induced MCF-7 xenograft tumor shrinkage ………42
2.5.6 INH7 dispersed cancer cell organization and decreased cancer cell density within xenograft tissue ………..43
2.5.7 INH7 inhibits 17β -HSD7 expression within xenograft tumor tissue….43 2.5.8 INH7 modulated E2 and DHT levels in blood serum……….44
xvii 2.5.9 Integrative datasets analysis: overexpression of 17β-HSD7
in invasive breast carcinoma………...44
2.6 DISCUSSION………46
2.6.1 The development and difficulty in developing estrogen-suppressive inhibitors………..46
2.6.2 The double catalytic activity of 17β-HSD7 in steroidogenesis ……….47
2.6.3 The overexpression of 17β-HSD7 in various breast carcinoma ………47
2.6.4 The cytostatic effect of 17β-HSD7 inhibition………47
2.6.5 The cytotoxic effect of 17β-HSD7 inhibition………48
2.6.6 The effect of 17β-HSD7 inhibition on xenograft tumor and translational applications………49
2.6.7 Conclusions and perspectives……….50
2.7 ACKNOWLEDGEMENTS ………..51
2.8 REFERENCES ………...51
2.9 TABLES……….58
2.10 FIGURES……….59
2.11 SUPPLEMENTARY MATERIALS ………...67
CHAPTER 3. INHIBITION OF 17BETA-HYDROXYSTEROID DEHYDROGENASE TYPE7 MODULATES BREAST CANCER PROTEIN PROFILE AND ENHANCE APOPTOSIS BY DOWN-REGULATION OF GRP78………..….71
3.1 RÉSUMÉ EN FRANÇAIS………72
3.2 ABSTRACT………...74
3.3 INTRODUCTION……….75
3.3.1 Brief introduction of estrogen reporter positive breast cancer. ………..75
3.3.2 The function and expression of 17β-HSD7 in breast cancer ………….75
3.3.3 The objective and methodology of present study………...76
3.3.4 The results and conclusions of present study………..76
3.4 EXPERIMENTAL PROCEDURES………..78
3.4.1 Integrative Oncomine dataset analysis………...78
3.4.2 Chemical and biological characteristics of inhibitor………..78
3.4.3 Cell culture………...78
3.4.4 E2 and DHT conversion in MCF-7 cells………79
3.4.5 Two dimensional electrophoresis, gel image analysis and mass Spectrometry………...79
3.4.6 Ingenuity Pathway Analysis for canonical pathway analysis………….80
3.4.7 Transcriptional level of selected proteins by q RT-PCR………81
3.4.8 Western blot………81
3.4.9 siRNA synthesis and transfection………...82
3.4.10 Cell apoptosis assay………..82
3.4.11 Statistical analysis……….83
3.5 RESULTS………..84
3.5.1 Elevated expression of 17β-HSD7 in primary and progressive breast cancer………...84
xviii
3.5.3 Inhibition of 17β-HSD7 modulates protein profile of MCF-7 cells…...84
3.5.4 Inhibition of 17β-HSD7 modulates biological processes through specific proteins in MCF-7 cells………...86
3.5.5 Inhibition of 17β-HSD7 induced MCF-7 apoptosis by interrupting the interaction among GRP78, Bcl-2 and BIK……….87
3.5.6 Downregulation of GRP78 by INH7 enhances apoptosis in MCF-7 cells in response to letrozole………..88
3.6 DISCUSSION………89
3.6.1 Proteomic modifications in MCF-7 cells in response to 17β-HSD7 inhibition………..89
3.6.2 Inhibition of 17β-HSD7 enhances apoptosis by down-regulation of GRP78……….91 3.7 ACKNOWLEDGEMENTS………...93 3.8 REFERENCES………..93 3.9 FIGURES………..98 3.10 TABLES……….106 3.11 SUPPLEMENTARY MATERIALS………..109
CHAPTER 4. IN VITRO INTERACTIONS BETWEEN MAMMARY FIBROBLASTS (HS578BST) AND CANCER EPITHELIAL CELLS (MCF-7) MODULATE AROMATASE, SULFATASE AND 17Β-HYDROXYSTEROID DEHYDROGENASES……….121
4.1 RÉSUMÉ EN FRANÇAIS………..122
4.2 ABSTRACT………124
4.3 INTRODUCTION………...125
4.3.1 Intratumoral E2 concentration in breast cancer………125
4.3.2 Intratumoral E2 formations through steroidogenesis pathways……...125
4.3.3 Fibroblasts and their roles in intratumoral E2 formation ……….126
4.3.4 The objectives of present study………127
4.4 MATERIALS AND METHODS……….128
4.4.1 Chemicals and inhibitors………..128
4.4.2 Cell lines and cell culture……….128
4.4.3 Two dimensional co-culture system……….129
4.4.4 Three-dimensional co-culture system………..129
4.4.5 E2 and DHT production assays in the presence of different substrates and inhibitors………...130
4.4.6 Cell proliferation/viability assay………..130
4.4.7 Morphometric analysis……….131
4.4.8 qRT-PCR………..131
4.4.9 Western-blot……….132
4.4.10 Statistical analysis………..132
4.5 RESULTS………133
4.5.1 MCF-7 and Hs578Bst cells expressed different levels of steroidogenic enzymes………...133 4.5.2 Co-cultures modulated steroidogenic enzymes in each cell line
xix
and increased E2 conversion……….133
4.5.3 Co-culture stimulated the proliferation of MCF-7 cells which was blocked by enzyme inhibitors………...134
4.5.4 Enzyme inhibitor combination suppressed MCF-7 cell growth in a 3D co-culture……….135
4.6 DISCUSSION………...137
4.6.1 The estrogen-rich microenvironment of breast cancer……….137
4.6.2 The interactions of mammary fibroblasts and cancer cells on steroidogenic enzymes and E2 formation from different steroid precursors………137
4.6.3 The combinatory inhibitions in 3D co-cultured models………..139
4.6.4 Conclusions………..141 4.7 ACKNOWLEDGMENTS………...141 4.8 REFERENCES………141 4.9 TABLES………..148 4.10 FIGURES………...151 4.11 SUPPLEMENTARY MATERIALS……….156
CHAPTER 5. DISCUSSION AND CONCLUSION……….159
5.1 THE ENZYMATIC ACTIVITY OF 17β-HSD7 IN E2 FORMATION AND DHT DEGRADATION IN BREAST CANCER CELLS: A SPECIAL REGULATON………...161
5.2 THE EXPRESSION OF 17β-HSD7 IN BREAST CANCER TISSUE……...…164
5.3 THE INHIBITION OF 17β-HSD7 INDUCED CYTOSTATIC AND CYTOTOXIC EFFECTS IN BREAST CANCER CELLS……….165
5.4 INHIBITION OF 17Β-HSD7 MODULATES PROTEIN PROFILES IN BREAST CANCER CELLS…...169
5.5 THE REGULATION OF 17β-HSD7 IN BREAST CANCER: FROM INTRACRINE TO PARACRINE………...170
5.6 MULTI-MODELS INVESTIGATING THE FUNCTION AND REGULATION OF 17Β-HSD7 IN BREAST CANCER: FROM MONOLAYER TO 3D CELL CULTURE AND XENOGRAFT ANIMAL MODEL……...…..172
5.7 CONCLUSIONS……….174
5.8 PERSPECTIVES………...175
xxi LIST OF TABLES
CHAPTER 2.
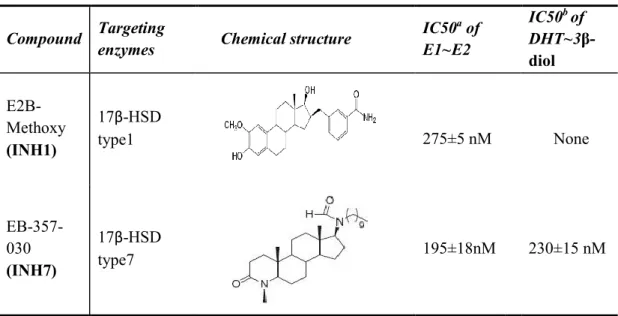
Table 1. Characteristics of inhibitors of 17β-HSD1 and 17β-HSD7 in our study…..58 Table 2. Different breast cancer cell lines………...58
CHAPTER 3.
Table 1. Mass Spectrometry identification of protein differentially expressed in INH7 treated MCF7 cells compared with non-treated MCF7 as control…106 Table 2. Top 5 biological function related to INH7 treatment……….107 Table 3. Top 5 up-stream regulators related to INH7 treatment ……….107 Table 4. mRNA quantification by qRT-PCR of network node gene involved in Network 1/2 and regulated by up-stream regulators (IPA analysis) within MCF-7 (Ctrl) and MCF-7 (INH7 treated) and
comparison with 2D gel data………..108 Supplementary Table 1. Primers for differential protein selected from 2D gels…109 Supplementary Table 2. siRNA sequence towards HSPA5/GRP78………...109 Supplementary Tables. IPA analysis of INH7 induced proteomic modulations in MCF-7 cells………..109 CHAPTER 4.
Table 1. Steroidogenic enzymes and receptors level in two cells lines………148 Table 2. E2/DHT formation and steroidogenic enzymes in MCF-7 cells
before and after co-culture with Hs578Bst cells……….148 Table 3. E2/DHT formation and steroidogenic enzymes in Hs578Bst cell
before and after co-cultured with MCF-7………...150 Supplementary Table 1. Primers for steroidogenic enzymes and
xxiii LIST OF FIGURES
CHAPTER 1.
Figure 1. Schematic illusion of multi-processes of carcinogenesis
and progression of breast cancer………2 Figure 2. Incidental and mortality of breast cancer worldwide and
in North American………...4 Figure 3. Histological and anatomical illusion of breast cancer………5 Figure 4. Different Subtype of breast cancer……….6 Figure 5. Schematic illusion of dual function of Estradiol (growth factor and pro- carcinogen) in initiation and progression of breast cancer………7 Figure 6. The contribution of estradiol in hallmark of breast cancer………9 Figure 7. Modeling of estrogen formation in pre- and post-menopausal women…….12 Figure 8. Schematic illusion of steroidogenesis (estrogen/androgen)
in intracrine tissue………13 Figure 9. Strategies of endocrine therapy for estrogen dependent breast cancer…...19
CHAPTER 2
Figure 1. Cytostatic effect of INH7/INH1 in MCF-7 cells……….59 Figure 2. Apoptotic effect INH7/INH1 in MCF-7 cells………..61 Figure 3. In vivo study of INH7-treated xenograft MCF-7 breast tumor………63 Figure 4. Integrative analysis of 17β-HSD7 overexpressed in neoplastic breast
vs. normal breast type………..65
Figure 5. Schematic mechanisms of INH7 towards ER+ breast cancer cells………..66
Supplementary Figure 1. In vitro study of INH7/INH1 with T47D cells………….67 Supplementary Figure 2. In vitro study of INH7/INH1 with BT-20 cells…………69
CHAPTER 3.
Figure 1. Integrative analysis of the expression status of 17β-HSD7
in breast cancer with TCGA dataset………98 Figure 2. Chemical and biological characteristics of the selective
xxiv
inhibitor for 17β-HSD7………...99 Figure 3. Proteomic analysis of INH7 treated MCF-7 cells vs.
untreated MCF-7 cells………..100 Figure 4. Functional analysis of proteomic data of 17β-HSD7 inhibition
in MCF-7 Cells………..102 Figure 5. Inhibition of 17β-HSD7 suppressed GRP78 expression and enhanced apoptosis in MCF-7 cells in response to letrozole………104 CHAPTER 4.
Figure 1. Activities of MCF-7 and Hs578Bst cells in Transwell
based 2D co-culture model………151 Figure 2. MCF-7 and Hs578Bst cell behavior in a collagen-based
3D co-cultured system………..153 Figure 3. Schematic presentation of the synthesis of sex steroids by specific
steroidogenic enzymes through interactions between
stromal cells and tumor cells in breast carcinoma tissue………..155 Supplementary Figure 1. E2/DHT formation and cell growth of MCF-7 and
Hs578Bst respectively in collagen gel………157
CHAPTER 5.
Figure 1. Schematic of cytostatic effect induced by 17β-HSD7 inhibition………….166 Figure 2. Schematic of cytotoxic effect induced by 17β-HSD7 inhibition………….168
xxv LIST OF ABBREVIATIONS 2D, two dimensional; 3D, three dimensional; 3α-HSD, 3α-hydroxysteroid dehydrogenase; 3β-HSD, 3β-hydroxysteroid dehydrogenase; 3β-diol, 5α-androstane-3β,17β-diol; 4-dione, 4-androstene-3,17-dione;
17β-HSD1, 17beta-hydroxysteroid dehydrogenases type 1; 17β-HSD7, 17beta-hydroxysteroid dehydrogenases type 7; AIs, aromatase inhibitors;
AKR1C3, aldo-keto reductase family 1, member C3; Akt, serine/threonine protein kinase;
AR, androgen receptor;
ATCC, American Type Culture Collection; BaA, Basal A;
Bad, Bcl-2-associated death promoter; Bax, Bcl-2-like protein 4;
Bak, Bcl-2 homologous antagonist/killer; BCCs, Breast cancer cells;
Bcl-2, B-cell lymphoma 2; Bik, Bcl-2-interacting killer;
BRCA1/2, Breast cancer type 1/2 susceptibility gene; BTG, B-cell translocation gene;
Cdc2, Cell division cycles 2; CDKs, cyclin-dependent kinases; CTLs, cytotoxic T lymphocytes; CYP19A, Cytochromes P450 A1; DBD, DNA binding domain; DCIS, ductal carcinoma in situ; DHEA, dehydroepiandrosterone;
xxvi
DMEM, Dulbecco's Modified Eagle Medium; DHT, 5α-dihydrotestosterone;
E1, estrone; E2, estradiol;
ECM, extra-cellular matrices; E1-S, estrone-sulfate;
EGF, epidermal growth factor;
ELISA, enzyme-linked immunosorbent assay; ERα, estrogen receptor α;
EREs, estrogen response elements;
FACS, Fluorescence-activated cell sorting; FBS, Fetal bovine serum;
FCM, flow cytometry;
G6PD, Glucose-6-phosphate dehydrogenase;
GRP78/HSPA5, glucose regulated protein 78/heat shock protein A5; GC/MS, Gas chromatography–mass spectrometry;
HES, hematoxylin-eosin-saffron;
HRP/DAB, horseradish peroxidase/ Diaminobenzidine; HSP1A, heat shock protein 70A;
HSP90AB1, heat shock protein 90beta;
INH1, inhibitor of 17beta-hydroxysteroid dehydrogenases type 1; INH7, inhibitor of 17beta-hydroxysteroid dehydrogenases type 7; IDC, invasive ductal carcinoma;
IHC, Immunohistological; ILC, invasive lobular carcinoma; IL-4/6/11, interleukin-4/6/11; IPA, Ingenuity pathway analysis; IPG, immobilised pH gradient strips; LBD, ligand binding domain;
Lu, luminal;
xxvii MT, Masson's Trichrome;
MTT, (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide, NADPH, nicotinamide adenine dinucleotide phosphate;
NFE2L2, Nuclear factor erythroid 2-related factor 2; NK, natural killer cells;
OVX, ovariectomized;
P21waf/cip1, cyclin-dependent kinase inhibitor 1 or CDK-interacting protein 1; P27kip1, Cyclin-dependent kinase inhibitor 1B;
PI3K,Phosphatidylinositol-4,5-bisphosphate 3-kinase; PBS, Phosphate buffered saline
PCNA, proliferating cell nuclear antigen; PDH, pyruvate dehydrogenase;
PDX, patient derived xenograft; PGE2, prostaglandin E2; PI, propidium iodide;
PKM1/2, Pyruvate kinase M1/M2; PMSF, phenylmethylsulphonyl fluoride; PRL, Prolactin; PS, phospholipid phosphatidylserine; PVDF, polyvinylidenedifluoride; qRT-PCR, Quantitative real-time PCR; rBM, reconstituted basement membrane; S.C, subcutaneous;
STS, steroid sulfatase;
TCGA, The Cancer Genome Atlas; TGFβ, transformation growth factor beta) Testo, testosterone;
TNFα, tumor-necrosis factor α; VEGF, endothelial growth factor;
VEGFR1/2, endothelial growth factor receptor ½;
xxix ACKNOWLEDGEMENTS
I would like to sincerely appreciate the following person and organisations for supporting my doctoral study to obtain the degree of Doctor of Philosophy (Ph.D.). More valuable than a degree, I appreciated the knowledge about “Intracrinology and Endocrinology” and the PhD training in the Centre de Recherche en Endocrinologie Moléculaire et Oncologique et Génomique Humaine (CREMOGH). I acknowledge for all the encouragements and trustiness in the process of study. The Ph.D. study will light up my future career in medical research and practise.
The first person I would like to acknowledge from the bottom of my heart, is the advisor of my doctoral study, Dr. Sheng-Xiang Lin. I appreciate all his supports and encouragements in the past four years and half. I sincerely acknowledge for the challenging and inspiring project offered by him with great trustiness and supports. I especially appreciate his generous mind and diligent directions in guiding young researcher to carry on the study independently and consistently. His interactive and multi-disciplinary concepts inspired me considerably, in which undoubtedly I will continuously benefit in my further career. I will try my best to be an excellent researcher as he does in his entire life. Thank you, Dr. Lin.
Especially, I would like to express my great gratitude to my co-advisor, Dr. Charles. J. Doillon, as a good advisor in research and a treasure mentor in life. I am so lucky to have him as my mentor. His diligent directions in research supported me to fulfill my study. His intelligent advices in life encouraged me to pursuit my career. I appreciate his guidance in the techniques of xenograft animal and three dimensional cell culture. I have learned many from him, not only the fundamental knowledge, but also the important of motivation and independent thinking in work. He sets me a good example to be a teacher and make me understand how important a good teacher could be to a student. I will try my best to be a good mentor in further to direct my own students. Thanks so much, Dr. Doillon.
xxx
I would like to thank all the colleagues in Dr. Lin’s Lab. I express my gratitude to Dr. Chenyan Zhang for showing me the techniques of siRNA transfection, to Mrs. Mouna Zerradi for her help in MTT assay and in courses; to Miss. Dan Xu for her advice and discussion in diagnosis and treatment of breast cancer; to Mr. Tang Li for his help in statistical analysis of Oncomine dataset; to Mr. Jian Song for his inspiriting discussion in biomedical research and experimental design. As well as other members in his lab. I would like to thank Dr. Dao wei Zhu and Dr. Ming Zhou for their help in life and study.
I would like to thank all colleagues in Dr. Doillon’s Lab. I especially appreciated Dr. Catherine Gerard for her help in the whole process of xenograft animal study. I also thank Mr. Louis-Jean Bordeleau for his help in 3D cell culture.
I would like to thank Dr. Donald Poirier and colleagues in his lab. I especially appreciate the knowledge from Dr. Donald Poirier in the field of medicinal chemistry. Thanks for his suggestions and advising in chemical characteristics of inhibitors. I also want to thank Dr. Jenny Roy for her help in the preparation of methylcellulose solution for animal injection, to Mrs. Diana Ayan for her help in preparation of silicone implants for xenograft animal.
I would like to thank Dr. Ezekiel Calvo, Mrs. Gina Racine and Mrs. Isabelle Kelly from Platform of Proteomics in CHU de Quebec Research Centre. I appreciated Dr. Ezekiel Calvo for his help in analysing proteomics data with Ingenuity Pathway Analysis (IPA) software and correcting the paper of proteomic study. I appreciated Mrs. Racine for her help in running 2D gel and image analysis. I appreciate for her great patience and kindness within the processes of study. Thank Mrs. Kelly for her advice in reading and analysis of GC/MS results of proteomic study.
I would like to thank Dr. Alexandre Brunet in the Laboratory of Flow-cytometry for his advice in analysing of cell cycle and apoptosis. I also appreciated Mrs. Nathalie Paquet for her advice in analysing qRT-PCR results. Thank Miss. Wafae Bouhaddioui
xxxi and Dr. Eric Boucher for their help in running qRT-PCR. I would like to thank Mr. Paul Préfontaine for his kind help with intravital microscope and image analysis in 3D cell co-culture study. I would like to thank Martin Thibault for analysing image of western-blot. I also appreciated Dr. Josée Larochelle for correcting the long and short summary in French. I also want to acknowledge Dr. Josée Lavoie and Dr. Madeleine Carreau for their help and advice in pre-doctoral exam and courses. Thank Dr. Josée Lavoie for her suggestions in heat shock proteins.
I would like to thank all administration staffs in the Research Center of CHU de Quebec (CHUL). Thanks Mme Marianne Roberge for her ordering the reagents and laboratory materials. Thanks Mme Nicole Almeras for taking care of the registration and financial documents. I would like to acknowledge staffs from animal facility. Thanks for their help with animal study.
I would like to thank all my colleagues and friends in CHU de Quebec Research Centre, who shared the great friendship and happiness with me. Dr. Mélanie Rouleau, Dr. Isabelle Laverdière, Dr. Laurent Gorsse, Miss. Sylvia Chen, without your help and encouragements, it will be hard and lonely to do my study. I do cherish the friendship with you. I also would like to appreciate all my friends in Quebec City and in Canada. Dr. Pegah Poursharifi, Dr. Paridokht Mahallati, Mr. Jin Cang, Miss. Xinxia Liang, Mr. Ruijie zhao, Dr. Yun Wang, Miss. Sirui Zhou, Mrs. Junyan Shi, Mr. Dr. Craig Milligan and Dr. Sunny Wong. Thanks for the support. I also appreciated all my friends in China, who always support me and encourage me to finish my study abroad. I always try my best to be the “international recognized professor in medicine” as they expected. I especially appreciate the Ph.D. fellowship from China Scholarship Council for four years financial support for my study in Canada as well as the scholarship from CIHR to support me to finalize my thesis.
xxxii
Finally, I am very grateful to my family, especially to my parents in China and my girlfriend Miss. Jenny Na Zhang (CPA, CGA), who always give me infinite love and encouragements to support my study and my dream. I love you, my dear family.
xxxiii FOREWORD
I fulfilled my doctoral study in the Centre de Recherche en Endocrinologie Moléculaire et Oncologique et Génomique Humaine (CREMOGH) affiliated to CHU de Quebec Research Center and Laval University. My research works involved several aspects of cellular and molecular biology using a variety of assessments exploring monolayer and three dimensional (3D) cell cultures as well as in vivo experiments with tumor xenografts. In particular, I studied the enzyme functions with newly developed chemical molecules targeting steroidogenic enzymes. The present thesis includes all results obtained during my doctoral study. This thesis contains five chapters. One paper (Chapter 2) was in press by Journal of Molecular Cell Biology (JMCB, Oxford Journals) and the other paper (Chapter 4) is under reviewed by Molecular and Cellular Endocrinology (MCE, ELSEVIER). The third paper (Chapter 3) is under manuscript preparation for Molecular & Cellular Proteomics (MCP, American Society for Biochemistry and Molecular Biology).
The Chapter 1 is the General Introduction. In this chapter, I introduced the cellular and molecular fundamentals of breast cancer from following points: 1) estrogen responsive genes and hallmarks of breast cancer; 2) estradiol (E2) concentration in estrogen-dependent breast cancer; 3) E2 formation by steroidogenic enzymes in ER+ breast cancer of postmenopausal women; 4) microenvironment of breast cancer and its roles in steroidogenesis; 5) molecular and cellular mechanisms of E2 suppressive and/or anti-estrogen treatments in breast cancer; and 6) hypotheses and objectives.
The Chapter 2: “Synergistic estradiol reduction and dihydrotestosterone restoration by inhibiting 17beta-hydroxysteroid dehydrogenase type 7 in breast cancer: tumor shrinkage and a new generation of therapy.” In this chapter, the function of 17β-HSD7 in steroidogenesis and estrogen-dependent breast cancer was experimentally demonstrated with in vitro and in vivo studies.
xxxiv
The Chapter 3: “Inhibition of 17beta-hydroxysteroid dehydrogenase type 7 modulates breast cancer protein profile and enhance apoptosis by down-regulation of GRP78”. In this chapter, the modulation of proteome profiles in MCF-7 induced by 17β-HSD7 inhibition was displayed with differential proteomic study. By integrative analysis with proteomic and clinical genome dataset, the mechanisms of cytotoxic effect induced by 17β-HSD7 inhibition through GRP78 was demonstrated.
The Chapter 4: “In vitro interactions between mammary fibroblast and cancer epithelial cells modulate aromatase, steroid sulfatase and 17β-HSDs”. In this chapter, the interactions between mammary fibroblast and cancer cells on steroidogenesis were experimentally displayed with in vitro two dimensional (2D) and three-dimensional (3D) co-culture models.
The Chapter 5: “Conclusion, Discussions and Perspectives”. In this chapter, I interactively discussed each chapter from enzyme activities to in vitro/in vivo models in breast cancer research. Based on the results in the present study, the function and regulation of 17β-HSD7 in estrogen-dependent breast cancer as well as the translational potentials and interests were discussed.
1
CHAPTER 1
2
1.1, Breast cancer
1.1.1 General concept
Breast cancer refers to a malignant tumor that has developed from cells (epithelial cells or basal cells) in mammary glands. It can grow into (invade) surrounding tissues or spread (metastasize) to distant areas of the body (1). Breast cancer emerges through multi-steps with multi-factors which involve: 1) genetic alterations comprising the amplification of oncogenes such c-Myc, Bcl-2 and erbB2 and mutation or loss of tumor suppressor genes such as p53, p27, BRCA1/2; 2) hormones (estrogens, progesterone, and prolactin) and their receptors; 3) interaction with stromal cells and microenvironments through various growth factors, cytokines (2) (Figure 1).
3 Figure 1. Schematic illusion of multi-processes of carcinogenesis and progression of breast cancer. (Beckmann, et al. 1997).
1.1.2 Epidemiology of breast cancer
Global Cancer Statistics 2011 reported that breast cancer is the most frequently diagnosed cancer and the leading cause of cancer death in females worldwide (3). Canadian Cancer Statistics 2013 (http://www.cancer.ca) reported that breast cancer is the most incidental cancer and the 2nd leading cause of cancer induced death in Canadian women. Based on 2009 estimates, about 1 in 9 Canadian women is expected to develop breast cancer during her lifetime and 1 in 30 will die from it. The breast cancer incidence rate in Canadian women rose through the early 1990s but decreased in the early 2000s. Breast cancer death rates in women have decreased in every age group since at least the mid-1980s, which reflects the impact of screening and improvements in treatment for breast cancer (4). Breast Cancer Statistics from National Cancer Institute, USA (http://seer.cancer.gov) also reported that the disease represents 14% of all new cancer cases in U.S, ranking as the most frequent cancer among US women with 6.8% of mortality of all cancer deaths. The rates of new cases remained stable over last ten years (2002-2011) whereas the death rates have been falling on average 1.9% each year over 2001-2010, with increasing of 5-years relative survival (from 72.5% in 1975 to 90.6% in 2006) (Figure 2). Female breast cancer is most common in middle and old aged women from 55~64 years old. The median age at diagnosis is 61 (5).
4
Figure 2. Incidental and mortality of breast cancer worldwide as well as in north American (Canada and United States) based on global and local cancer statistics. (Jemal A et al. 2011; http://www.cancer.ca; http://seer.cancer.gov )
1.1.3 Histopathology and Molecular Subtype of Breast Cancer
Breast cancer begins in the breast tissue that is made up of glands for lactation called lobules, which were composed of secretory epithelial cells, myoepithelial cells and basal cells. Breast cancer also begins in the ducts that connect the lobules to the nipple and transport milk, which were composed of ductal epithelial cells and myoepithelial cells. The rest of the breast is made up of adipose, connective, and lymphatic tissues named as stromal tissue (6). The most common type of breast cancer is ductal carcinoma which begins in the ducts, derived from ductal epithelial cells or basal cells. This type of breast cancer was accounted for 70%~80% cases of breast cancer. Another type of breast cancer is lobular carcinoma, which begins in the lobules, derived from lobular epithelial cells or basal cells, accounted for 5% of new cases (7, 8) (Figure 3).
5 Figure 3. Histological and anatomical illusion of breast cancer (Download from Alila Medical Image: http://www.alilamedicalmedia.com/)
Breast cancer is a heterogeneous disease with varied morphological appearances, gene expression features and responsiveness to therapy. Therefore, by using a clustered analysis of gene expression profiling, a molecular classification has been derived with prognostic value and to predictive chemotherapy response. Breast cancers were classified as 1) Luminal subtype (70% of cases) with high expression level of estrogen receptor and well responsiveness to endocrine therapy; 2) HER2 subtype (10~15 % of cases) with high expression of HER2 gene and responded to trastuzumab treatment; 3) Basal like subtype (15~20% of cases) with high expression of basal epithelial genes without responsiveness neither endocrine therapy nor trastuzumab (9, 10). (Figure 4)
6
Figure 4. Different Subtype of breast cancer (clinical subtype and molecular subtype) with biological characteristics. (Polyak, et al. 2012).
1.2, Estrogen dependent breast cancer
1.2.1 General definition
Estrogen dependent breast cancer is characterized by the expression of estrogen receptors (ERs), therefore it is also named ER positive (ER+) breast cancer. The initiation and progression of carcinoma are directly associated with estrogen, especially estradiol (E2). Estrogen dependent breast cancer refers to Luminal subtype by molecular subtype classification. Most of breast cancers, approximately 60% in premenopausal women and 75% in postmenopausal women, are estrogen dependent breast cancers (11, 12).
1.2.2 E2 and its roles in ER+ breast cancer
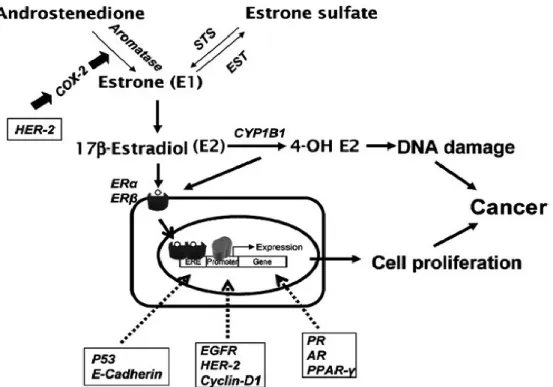
Epidemiological evidence indicates that prolonged exposure of the mammary glands to high levels of E2 represents the highest risk factors (13). E2 serves a dual role in breast carcinogenesis, as a growth factor as well as a pre-carcinogen (14) (Figure 5).
7 On the one hand, E2 is a hormone driving cancer cell proliferation by binding to ERs, and activating down-stream transcriptions (15). On the other hand, E2 is a pro-carcinogen inducing genetic damages and mutations. The metabolites of E2 mediate DNA damage, chromosomal instability and genetic mutation (16), therefore driving the initiation and progression of breast cancer.
Figure 5. Schematic illusion of dual function of Estradiol as growth factor and pro-carcinogen in initiation and progression of breast cancer. (Yang et al. 2007). 1.2.3 Estrogen receptors (ERs) and their roles in ER+ breast cancer
Human estrogen receptors (ERα and ERβ) are intercellular proteins mainly found in nucleus. They play vital roles in the malignant progression of breast cancer by comprehensively regulating down-stream estrogen response genes. The expression of ERα is closely associated with breast cancer while the roles of ERβ in breast cancer is not as clear as ERα (17, 18). The ERs are composed by three functional domains: 1) AF1/2 domain contains activation function contributing to the transcriptional activity of ER; 2) The DNA binding domain (DBD) named hinge domain contains the nucleus
8
harboring signal, binding to specific response element known as estrogen response elements (EREs) located in the promoters of estrogen responsive genes; 3) The ligand binding domain (LBD) binds to different ligands such as estrogens and anti-estrogens (19). ERs are activated by four manners. 1st is the classical genomic pathway: E2
bounds to LBD, DBD binds to ERE and recruit co-activators that will activate the transcription of target genes; 2nd, the non-classical genomic pathway involves ER
interactions with other transcription factors like AP-1 (c-myc, fos); 3rd is the
E2-independent pathway which activates ER through phosphorylation induced by growth factors; 4th is the non-genomic pathway which involves a small pool of ER located
close to the membrane. It activates signaling cascades (Akt, MAPK) through recruitment of protein kinases (Src and PI3K) (20). Through multi-pathways activation, E2-ERs play comprehensively roles in initiation and progression of breast cancer. 1.2.4 E2/ERs interactions and hallmarks of breast cancer
E2/ER interactions contribute to the hallmarks of breast cancer by comprehensively regulating downstream genes involved in cell cycle progression, apoptosis, metabolism, encoding growth factors and cytokines as well as encoding transcription factors and co-regulators (21, 22, 23). There are ten hallmarks of cancer named as: 1st,
self-sufficiency in growth signals; 2nd, evading growth suppressors; 3th, resisting cell
death and avoiding apoptosis 4rd, enabling replicative immortality; 5th, genome
instability & mutation; 6th, avoiding immune destruction; 7th, tumor-promoting
inflammation; 8th, inducing angiogenesis; 9th, activating invasion & metastasis; 10th,
9 Figure 6. The contribution of estradiol in the hallmarks of breast cancer. (Figures adapted based on Hanahan et al. 2011).
1st, E2/ER interactions induce a general up-regulation of genes encoding growth
factors, cytokines, hormones, receptors and signaling kinases such as insulin-growth factor proteins, Interleukin 4/6 (IL4/6) as well as prostaglandin E2 (PGE2), therefore maintaining self-sufficiency in growth signals (23). The latter two cytokines up-regulate E2 converting enzymes such as aromatase, STS and 17β-HSD1, consequently reinforcing the E2 formation and the E2/ERs activation. (26).
2nd, E2/ER interactions down-regulate several auto-suppressive or
environmental-suppressive signals towards cell proliferation such as p21, p27, BTG (B-cell translocation gene) families, TGFβ (transformation growth factor-beta) and inhibitin (23, 27), therefore evading growth suppressors.
10
3rd, E2/ER interactions induce down-regulation of several transcriptional repressors
such as anti-apoptosis such as Bik and pro-apoptotic gene including Bcl-2, caspases 9 (23, 28), therefore breast cancer could resist cell death and avoid apoptosis.
4th, E2/ER interactions acquire capabilities of growth signal autonomy, insensitivity to
antigrowth signals and resistance to apoptosis, all lead to the uncoupling of cell growth from autocrine and paracrine signals. Furthermore, several genes involved in cell cycle progression such as CyclinD1, CyclinA2 and Cell division cycles 2 (CDC2) were activated by E2-ERs interactions and therefore maintain the replicative immortality (23, 27, 29).
5th, E2 and its metabolites such as 2/4-hydroxyestradiol and 2/4-methoxyestradiol were
identified as epigenotoxic and genotoxic carcinogens in breast cancer which cause DNA damage, mutation and genome instability (16, 17).
6th, E2/ER interactions interfere the immune surveillance by protecting breast cancer
cells from immune cells induced cell death. E2 induces the expression of Fas-L on the surface of breast cancer cells, therefore inducing the apoptosis of Fas bearing T lymphocytes (30). Furthermore, E2 strongly induces the human granzyme B inhibitor, proteinase inhibitor 9 (PI-9), which interfering the apoptosis induced by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells (31). Such regulations consequently protect tumor cells escaping immune surveillance.
7th, E2 induces the expression of inflammatory meditators such as PGE2, IL4/6,
TGFα/β as well as chemokines, which activate the macrophages and promote their infiltrating into tumor tissue (32). The infiltrated inflammatory cells especially tumor-associated macrophage further promote the progression of breast cancer by secreting various inflammatory mediators and pro-angiogenic factors such as vascular endothelial growth factor (VEGF). Such regulations enhance the evasion of tumor cells and ultimately metastasis. (33, 34).
8th, E2 is the precise mediator for angiogenic actions in breast cancer by regulating the
mediator of angiogenesis named as VEGF and its receptors (1 and VEGFR-2). (35, 36).
9th, E2 comprehensively regulates genes involved in invasion and metastasis. On the
11 stroma cells especially tumor associated macrophage, generating an invasive microenvironment (37). On the other hand, E2 regulates specific genes in cancer cell, leading to the acquisition of the metastatic phenotype (38, 39).
10th, E2 modulates the metabolic pathways in breast cancer cells, therefore adapting
themselves to available glucose. E2 promotes MCF-7 to switch metabolic pathways from glycolysis to pyruvate dehydrogenase (PDH) pathway depending on local glucose conditions (40). Furthermore, E2 enhances MCF-7 cells to increase glucose uptake so as to meet the energy demands of the high rate of proliferation. (41).
1.3, E2 concentration in ER+ breast cancer: blood circulation and intratumoral formation
Epidemiologic studies have consistently reported that E2 concentration in blood circulation or even more important in tumor tissue serves as the predominant risk of breast cancer in postmenopausal women (42, 43). In postmenopausal women with breast cancer, plasma E2 was higher (37pmol/L) than that in normal women (28pmol/L) (44, 45).
In neoplastic mammary glands, however, intratumoral estrogen can be as much as 10~40 folds higher than circulation levels in postmenopausal women (46, 47) and 5 times higher than that in the area considered as morphologically normal (48, 49). Furthermore, intratumoral estradiol/estrone (E2/E1) ratio was significantly higher in postmenopausal than in premenopausal breast cancer (50). The increase of intratumoral estrogens suggests that the in situ production and accumulation of estrogens plays an even more important role in the growth of ER+ breast cancer in postmenopausal women.
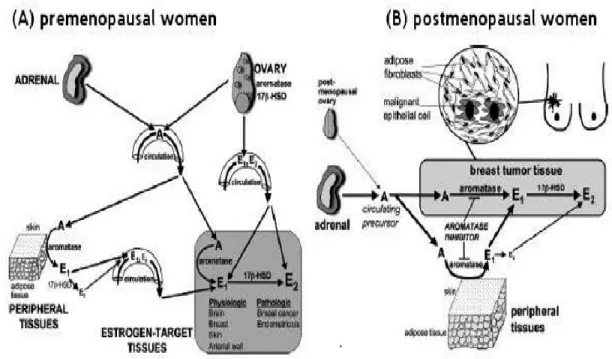
1.4, E2 formation in ER+ breast cancer of postmenopausal women: Intracrinology In premenopausal women, estrogens were predominantly produced in ovary and transported by circulation to target organs such as mammary glands. In postmenopausal women, however, ovary-derived circulatory estrogens were dramatically decreased and
12
replaced by estrogen produced in peripheral tissues such as adipose tissue, skin as well as mammary glands themselves in intracrine manner (51, 52) (Figure 7).
Figure 7. Modeling of Estrogen formation in pre- and post-menopausal women (Bulun et al. 2005).
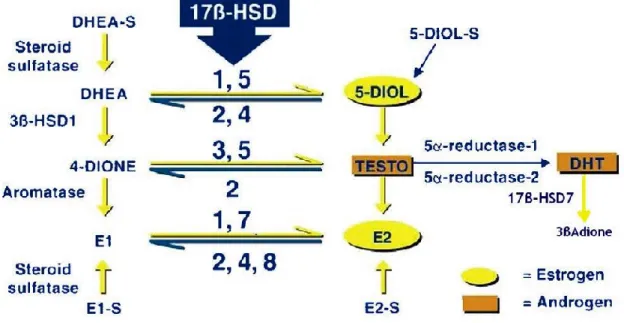
In postmenopausal women, estrogens are predominantly synthesized through converting adrenal precursor steroids such as dehydroepiandrosterone (DHEA), 4-androstene-3,17-dione (4-dione) as well as circulating estrone-sulfate (E1-S) (53) by several steroidogenic enzymes (54). DHEA and 4-dione are the two important sources of estrogen production in postmenopausal women, moreover 4-dione is the direct source of E1 by aromatase (55, 56). Another great amount of estrogen in circulation is present in sulfated form as E1-S, which is transformed into activated E1 by steroid sulfatase (STS) (57, 58). Two intracrine estrogen conversion pathways consequently have been described. First, the ‘aromatase pathway’ by aromatase, which transforms mainly 4-dione and, to a minor degree, testosterone into estrogens (mainly E1 and, to a minor degree, E2) (59);Second, the ‘sulfatase pathway’ by STS, which converts E1-S into E1(57); and the reductive 17beta-hydroxysteroid dehydrogenase type 1 and type 7 (17β-HSD1 and 17β-HSD7) that are integrated in both pathways and convert E1
13 into potent E2 (60). In breast cancer tissue of postmenopausal women, these enzymes were up-regulated (enzymes activity and/or expression level) in order to generate and maintain an estrogen-rich intratumoral microenvironment so as to sustain the tumor growth and development (61, 62) (Figure 8).
Figure 8. Schematic illusion of steroidogenesis (estrogen/androgen) in intracrine tissue. (Aka JA, et al. 2009).
1.5, Steroidogenic enzymes in E2 formation/DHT degradation in breast cancer: intracrine regulator
1.5.1 Aromatase
Aromatase enzyme is the product of CYP19 gene, which catalyzes C19 steroid into estrogens (stronger potency of 4-dione into E1 and less potent of Testo into E2) (59). Aromatase localizes in the endoplasmic reticulum of estrogen-producing cells (63). Aromatase overexpression has been shown to be critical in breast cancer since the AIs has been therapeutically used successfully (64, 65). Aromatase was characterized to mainly express in stromal fibroblast cells rather than in cancerous epithelial cells in breast carcinoma tissue (63). Cytokines such as interleukin-6 (IL-6) and prostaglandin E2 (PGE2) derived from malignant cancer cells and/or macrophage cells stimulate the expression of aromatase in stromal cells through paracrine manner, consequently
14
increase the intratumoral E2 formation (66). The success of AIs in the management of ER+ breast cancer in post-menopausal patients confers aromatase as an example of translated therapeutic target towards a micro-environmental component in breast cancer (67).
1.5.2 17β-HSD1
17β-HSD1 is the first identified enzyme in 17β-HSDs family. Its major impact in the biosynthesis of estrogens had been demonstrated since 1950 (68). In 1993, Zhu et al (74) determined its three-dimensional structure as the first human steroid enzyme structure. Gangloff et al (69) demonstrated its alternative binding affinity with DHT and therefore characterized the long-term believed estrogen formation enzyme as a dual catalytic enzyme both in E2 formation and DHT inactivation. The further functional studies by Aka et al (70) has demonstrated that 17-HSD1 promotes the proliferation of breast cancer cells by producing E2 and degrading DHT into 3α-diol (90%) and 3β-diol (10%). The expression of 17-HSD1 is mainly regulated by IL-6 and TNFα derived from fibroblast and immune cells in tumor stromal (26). Over-expression of 17β-HSD1 in breast cancer tissue plays an important role in sustaining intratumoral E2 production (71). Therefore, 17β-HSD1 (72) was considered as an additional pivotal player in intratumoral estrogen deposition and a potential target complementary to aromatase. In particular, the most significant efforts during recent years have concentrated on the design of 17β-HSD1 inhibitors. Progress has been made through structure-activity relationship studies based on small molecular structures coupled with the elimination of residual estrogenic activity, as well as rational inhibitor design based on the enzyme 3D-structure. Several inhibitors have been reported (73), but no drugs have been developed due to the unavoidable estrogenic activity of inhibitors designed as estrogen analogues (74).
1.5.3 17β-HSD7
Expression of human 17β-HSD7 has been reported in the ovary, placenta, mammary gland, and also in the liver and brain (75, 76). At present, it is generally accepted that 17β-HSD7 is primarily involved in cholesterol synthesis rather than in steroidogenesis
15 (77, 78, 79). Such acceptance has had a marked effect on the direction of studies involving this enzyme, which explains the limited number of studies addressing its function in steroid hormone biosynthesis and related diseases including breast cancer. 17β-HSD7 was first detected as prolactin receptor-associated protein in rat (80). Detection of a high expression level in the corpus luteum of pregnant mice supported the assumption of its role in E2 synthesis (81). The predominant involvement of 17β-HSD7 in cholesterol metabolism rather than in sex steroid synthesis, was further supported by the observation that although 17β-HSD7 knockout mice were fertile, they bred nonviable fetuses due to defective in situ cholesterol biosynthesis in the brain (82). In order to gain a better understanding of the role of 17β-HSD7 in this field, we established this functional study of 17β-HSD7 in estrogen-dependent BC (83, 84) with an emphasis on clarifying its contribution to sex hormone biosynthesis and breast cancer stimulation. In steroid biosynthesis, 17β-HSD7 can convert E1 into E2, and to a significant extent, it can inactivate DHT to the weak estrogen 3β-diol, thereby making it a dual intracrine regulator for the most potent estrogen and androgen (75). Immunohistochemistry studies confirmed that 17β-HSD7 mainly expressed in ductal carcinoma with extensive expression in pathological ductal (both in invasive and in situ carcinoma) as compared to adjacent normal ducts (85, 86). 17β-HSD7 is the unique enzymes characterized to be feed-forward regulated by E2. Such strong positive feedback mechanism increase local E2 synthesis causing growth of estrogen-dependent breast cancers (86). The overexpression of 17β-HSD7 in carcinoma tissue vs. normal tissue (3.5 folds in Tumor/Normal) contributes to intratumoral estrogen deposition, which was correlated with an increased ratio of E2 to E1 in breast tumors compared with normal tissue (87). Such expression and regulation pattern of 17β-HSD7 identified it as an important regulator in intraturomal estrogen deposition regulator. The inhibitors reported towards 17β-HSD7 were developed by Dr Donald Poirier (88).
1.5.3 Steroid sulfatase (STS)
STS is the key enzyme catalyzing E1-S directly into E1 as well as DHEA-S into DHEA (89, 90). STS is mainly expressed in normal and malignant breast tissues (90). The