HAL Id: hal-02824380
https://hal.inrae.fr/hal-02824380
Submitted on 6 Jun 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Etude de la diversité allélique du gène S-RNase chez
l’amandier
Ludovic Alaux
To cite this version:
Ludovic Alaux. Etude de la diversité allélique du gène S-RNase chez l’amandier. [Stage] Université d’Avignon et des Pays de Vaucluse (UAPV), Avignon, FRA. 2009, 19 p. + 9 p. annexes. �hal-02824380�
INRA AVIGNON – UNITE GENETIQUE ET AMELIORATION
DES FRUITS ET LEGUMES
GAFL – UGAFL
Domaine Saint Maurice BP 94
84143 MONTFAVET CEDEX 04 32 72 27 10
Etude de la diversité allélique du gène S-RNase
chez l’amandier
ALAUX Ludovic
Master 1 Gestion de la Qualité des Productions Végétales Université d’Avignon et des Pays de Vaucluse
Avril - Juillet 2009
Remerciements
Tout d’abord, je tiens à remercier Henri Duval pour son expérience qui m’a beaucoup apporté, sa disponibilité, son soutien et pour la confiance qu’il m’a accordée tout au long de ce stage.
J’adresse également mes remerciements à Emeline Baptiste, qui a participé à ce travail dans le cadre de son stage pour l’obtention du DUT, pour avoir effectué autant d’expériences malgré le temps accordé et pour sa bonne humeur.
Je manifeste ma reconnaissance à Jean-Paul Bouchet pour m’avoir initié à la bioinformatique, pour sa grande compétence et pour tous les conseils qu’il m’a apportés.
Pour avoir permis le bon déroulement des expériences, merci à toute l’équipe du laboratoire de biologie moléculaire et plus particulièrement à Patrick Lambert.
J’exprime ma sympathie Chantal Olivier pour son aide précieuse et Martin Masse pour sa bienveillance.
Etude de la diversité allélique du gène S-RNase chez l’amandier
Mots-clés : amandier, auto-incompatibilité, S-RNase, PCR, polymorphisme de taille, séquençage
Résumé
L’amandier (Prunus dulcis) possède un système d’auto-incompatibilité pollinique gamétophytique. Ce système est contrôlé par le locus S, multiallélique. Ce locus contient le gène S-RNase et le gène SFB.
Le but de cette étude est de déterminer les allèles de la S-RNase présents dans la collection de ressources génétiques de l’amandier de l’INRA d’Avignon. Cette étude se déroule dans le cadre du programme européen SAFENUT.
Nous avons utilisé des techniques de PCR pour détecter le polymorphisme de taille des introns du gène S-RNase et déterminer les allèles S. Ces génotypes ont été confirmés à l’aide d’amorces spécifiques d’allèles. Nous avons également séquencé les génotypes donnant des résultats différents des allèles déjà connus.
Au cours de cette étude, nous avons génotypé 224 cultivars et trouvé plusieurs nouveaux allèles. Nous avons constaté que le gène S-RNase présente une diversité génétique importante. En effet, beaucoup d’allèles différents de ce gène existent et ceux-ci sont associés à des séquences ADN très variables.
Allelic diversity study of the S-RNase gene in almond
Keywords: almond, self-incompatibility, S-RNase, PCR, size polymorphism, sequencing
Abstract
Almond (Prunus dulcis) has a gametophytic pollen self-incompatibility. This locus contains the S-RNase gene and the SFB gene.
The aim of this study is to determine the alleles of the S-RNase gene in the Avignon INRA almond genetic resource collection. This study is in the SAFENUT project.
We used PCR techniques to detect the size polymorphism of the S-RNase gene introns and determine S alleles. These genotypes are confirmed with allele-specific primers. We also sequenced the genotypes which results differed from known alleles.
During this study, we genotyped 224 cultivars and found several new alleles. We noticed that the S-RNase gene has an important diversity. Indeed, a lot of different alleles of this gene exist, and these ones are associated with sequences very variable.
Sommaire
I/ Introduction... 1
1/ Organisme d’accueil... 1
2/ Objectifs de l’étude ... 1
3/ Synthèse bibliographique ... 3
II/ Matériel et méthodes ... 4
1/ Matériel végétal utilisé... 4
2/ Obtention de l’ADN... 4
3/ Techniques basées sur des PCR ... 4
a/ Couple d’amorces EM-PC2consFD/EM-PC3consRD (figure 4.a)... 5
b/ Couple d’amorces PaConsI-F/EM-PC1consRD (figure 4.b) ... 5
c/ Amorces spécifiques d’allèles (figure 4.c) ... 6
4/ Séquençage... 6
a/ Clonage... 6
b/ Logiciels utilisés... 8
III/ Résultats ... 9
1/ Observation du polymorphisme de taille ... 9
a/ Amplification de la région du deuxième intron... 9
b/ Amplification de la région du premier intron ... 10
c/ Résultats obtenus à partir du polymorphisme de taille... 10
2/ Amorces spécifiques ... 10
3/ Séquençage... 12
a/ allèles séquencées correspondant à des allèles déjà identifiés ... 12
b/ allèles présentant une séquence peptidique identifiée et une séquence nucléotidique différente ... 13
c/ allèles séquencés ne correspondant pas à des allèles déjà identifiés : Nouveaux allèles .. 14
IV/ Discussion ... 15
1/ Cas des amorces spécifiques S10... 15
2/ Problèmes d’amplification ... 16
3/ Fiabilité des méthodes... 16
4/ Cas des allèles des clones Iran 1-4-5 (R1042) et de Narbonne (R208) ... 17
V/ Conclusion... 17
Bibliographie... 18 Glossaire
I/ Introduction
1/ Organisme d’accueil
L’Institut National de Recherche Agronomique (INRA), crée en 1946, est le premier institut de recherche agronomique en Europe et deuxième dans le monde en terme de publications en sciences agricoles et en sciences de la plante et de l’animal. L’INRA est un organisme de recherche scientifique publique finalisée, placé sous la double tutelle du ministère de l'Enseignement supérieur et de la Recherche et du ministère de l’Agriculture et de la Pêche.
Le stage a été effectué au sein de l’unité de Génétique et Amélioration des Fruits et Légumes (GAFL) situé sur le domaine St Maurice à Avignon. Cette unité a été créée en 1998 de la fusion des stations d’Amélioration des Plantes Maraîchères et de Recherches Fruitières Méditerranéennes. Cette unité est composée de plusieurs équipes de recherche dont celle de Connaissance des Génomes, Ressources et Innovation (CGRI) à laquelle j’ai été intégré.
Cette équipe a notamment pour mission de gérer et d’analyser les ressources génétiques. Elle entretient également des collections importantes de génotypes des espèces cultivées qu'elle étudie, ainsi que des espèces sauvages apparentées.
2/ Objectifs de l’étude
L’étude s’est déroulée dans le cadre du programme SAFENUT (Safeguard of hazelnut and almond genetic resources). Celui-ci est financé par subventions de la commission européenne et a pour but d’appréhender la diversité génétique de la noisette et de l’amandier afin de créer une core-collection. Cette dernière consiste en une collection d’un minimum de cultivars représentant un maximum de diversité génétique.
L’objectif de cette étude est de génotyper la collection d’amandiers de ce programme pour le locus S, responsable du système d’auto-incompatibilité pollinique. Ces informations pourront permettre d’éviter d’installer en verger deux espèces incompatibles au croisement car possédant les mêmes allèles pour le locus S et qui ne produiraient aucune amande.
Un objectif secondaire est de valider les méthodes de génotypage moléculaire de la S-RNase décrites dans la bibliographie.
Pour cela, une recherche bibliographique a été nécessaire pour comprendre le sujet. Puis, nous avons recherché un polymorphisme de taille pour deux régions de la séquence nucléotidique de la S-RNase. Enfin, nous avons effectué des séquençages d’allèles indéterminés.
Le présent rapport suit le même ordre de progression. Ce rapport est donc séparé en une synthèse bibliographique, une description des matériel et méthodes, une description des résultats, une discussion et une conclusion.
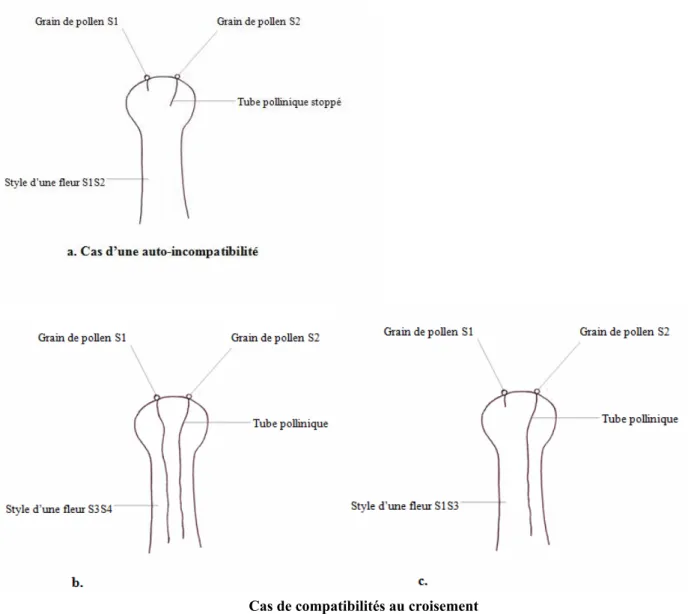
Cas de compatibilités au croisement
3/ Synthèse bibliographique
L’amandier (Prunus dulcis) est un arbre appartenant à la famille des Rosaceae et plus particulièrement au genre des Prunus. La floraison a lieu à la fin de l’hiver, début du printemps. La pollinisation est entomophile.
Beaucoup d’espèces Prunus, dont les amandiers, ont une auto-incompatibilité pollinique. Celle-ci interdit l’autofécondation et favorise le brassage génétique. Cette auto-incompatibilité pollinique est contrôlée par un locus S pour lequel il existe plusieurs formes alléliques. Ce locus est co-dominant et les deux allèles s’expriment dans le style. Cette auto-incompatibilité est de type gamétophytique. Celle-ci consiste en un blocage de la croissance du tube pollinique, issu de la germination du pollen, dans le style. Cela se produit lorsque l’allèle S porté par le génome haploïde du pollen est identique à l’un de ceux présents dans le génome du style. Ainsi, le tube pollinique ne peut atteindre l’ovaire et permettre une fécondation que si ces allèles sont différents. Pour illustrer, une variété S1S2 produit des grains de pollen S1 et des grains de pollen S2 et possède un style diploïde S1S2. Dans le cas de l’auto-incompatibilité, ceux-ci se déposent sur le stigmate d’une fleur de cette même variété ou d’une autre possédant également les allèles S1 et S2. Les tubes polliniques issus de ces deux types de pollen ne dépassent pas la partie supérieure du style (figure 1.a). Par contre, dans le cas d’une pollinisation croisée, ces mêmes grains de pollen S1 ou S2 atteignent le stigmate d’une fleur d’une variété différente S3S4, la croissance des tubes polliniques à travers le style pourra se poursuivre jusqu’à l’ovaire et aboutir à la fécondation (figure 1.b). Si ces grains de pollen S1 ou S2 arrivent sur une variété possédant les allèles S1 et S3, la croissance du tube pollinique s’arrêtera dans le cas des grains de pollen S1 mais ne sera pas interrompue dans le cas des grains de pollen S2 (figure 1.c). Dans la mesure, où même avec la moitié de pollen efficace, la récolte sera assurée, ces deux derniers cas correspondent à une compatibilité au croisement.
Une des conséquences directes de cette auto-incompatibilité pollinique est que les individus issus de la fécondation sont obligatoirement hétérozygotes pour ce locus S. En effet, un grain de pollen porteur d’un allèle donné ne peut pas féconder un ovaire d’une fleur possédant ce même allèle. Il existe un allèle auto-compatible (Sf chez l’amandier) (Grasselly et al. 1976) qui représente un intérêt agronomique. En effet, des cultivars auto-incompatibles obligent à avoir deux cultivars, une variété principale et un pollinisateur, inter compatibles et ayant des floraisons simultanées ; alors que l’on peut planter des vergers monovariétaux avec une variété auto-compatible.
Cette auto-incompatibilité peut être vérifiée de manière certaine par croisements contrôlés, et il existe aujourd’hui également des méthodes de biologie moléculaire qui permettent de génotyper les allèles du gène S.
Le locus S comprend au moins deux gènes (figure 2.a). L’un code pour une ribonucléase, la S-RNase ; l’autre pour une protéine F-box (SFB) (Ushijima et al. 1998; Tamura et al. 2000). Ces gènes sont à une distance génétique trop courte (entre 2 et 10 kpb) pour subir des recombinaisons. A une S-RNase correspond toujours la même SFB (Sutherland et al. 2008).
L’arrêt de croissance du tube pollinique dans le cas d’une incompatibilité est dû à la dégradation de l’ARN du tube par les RNases du style. Pour l’allèle Sf de l’amandier, la transcription de la S-RNase est trop faible (Toshio Hanada et al. 2009). Ceci est à confirmer par le fait qu’il n’y a pas
Gène S-RNase
Gène SFB ~2-10kpb
Sens de lecture du
gène Sens de lecture du
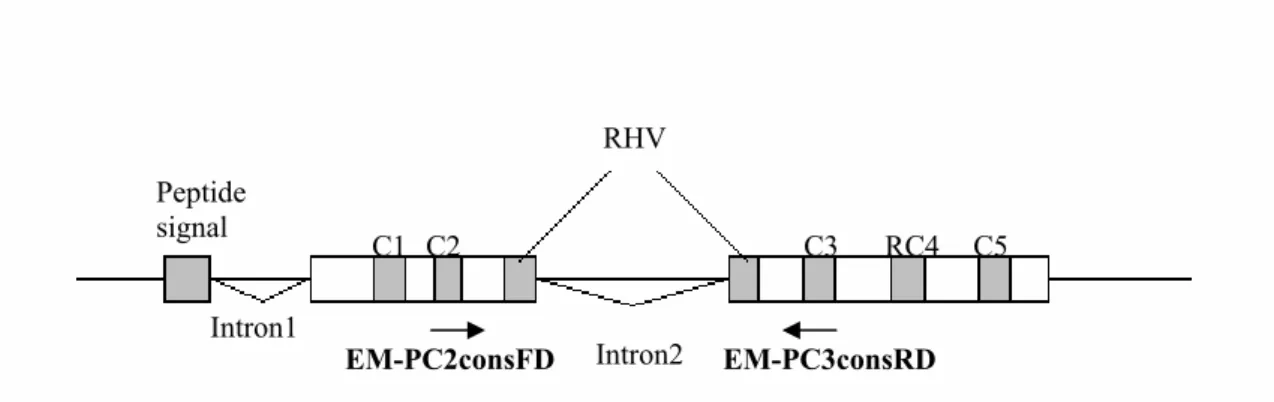
gène a. Organisation du locus S RHV Peptide signal C1 C2 C3 RC4 C5
Exon1 Exon2 Exon3
Intron1
Intron2
b. Structure du gène S-RNase
C1, C2, C3, RC4 et C5 représente les régions conservées et RHV la région hypervariable (Ushijima et al. 1998)
d’activité S-RNase (Bošković et al. 1999). Cette faible transcription ne serait pas due à la séquence nucléotidique de l’allèle Sf car il a été découvert un génotype, Ponç, avec la séquence Sf et un phénotype auto-incompatible (Kodad et al. 2009)
Le fonctionnement réel de la reconnaissance du pollen par le style et son rejet n’est pas encore élucidé, même si plusieurs modèles sont à l’étude (Hua et al. 2008).
Le locus S limite le nombre d’individus homozygotes, et privilégie les individus hétérozygotes qui présentent souvent un avantage sélectif au cours de l’évolution. Le locus S possède pour cela un degré de diversité allélique élevé par rapport à la majorité des gènes de plantes. Dans le cas du locus S, l’apparition de nouveaux allèles n’est probablement pas linéaire du fait de la co-adaptation des deux gènes du locus S (de la S-RNase et de la SFB). En effet, le bon fonctionnement du locus S dépend de l’interaction entre la S-RNase et de la SFB. L’apparition d’une nouvelle spécificité nécessite des mutations complémentaires dans les deux gènes du locus S. Une mutation dans un seul des deux gènes pourrait entraîner la perte de l’auto-incompatibilité. En effet, chez Prunus mume (abricotier du Japon), l’allèle Sf a été causé par une insertion de 6,8 kpb dans la région codant pour la SFB. De même, T. Sonneveld a trouvé, chez Prunus avium (cerisier), un mutant S3’ auto-compatible provoqué par une délétion complète de la SFB S3 et un mutant S4’ dont le cadre de lecture est décalé de 4pb (Sonneveld et al. 2005). On suppose qu’il en serait de même chez l’amandier. Cependant, on ne sait pas encore comment cette complémentarité est maintenue au cours de l’évolution.
Des allèles S très similaires peuvent être retrouvés dans des espèces relativement différentes au sein du genre Prunus. Par exemple, la S-RNase 11 de l’amandier est identique à la S-RNase 1 de Prunus avium (cerisier) (Ortega et al. 2006).
La S-RNase présente cinq régions dont la séquence nucléotidique est relativement bien conservée (C1, C2, C3, RC4 et C5) et une région hypervariable (RHV) (Ushijima et al. 1998) (figure 2.b). Ces régions ont été obtenues par comparaison des séquences peptidiques des différents allèles, puis, ont été reportées sur la séquence nucléotidique. Le gène de la S-RNase possède deux introns. Les régions codantes ont été déterminées par comparaison avec de l’ADNc (ADN complémentaire) obtenu à partir d’une RT-PCR (reverse transcriptase PCR) de l’ARNm de la protéine S-RNase isolée dans le style.
La distance entre les gènes de la S-RNase et de la SFB est très variable selon les allèles.
Les différentes techniques de biologie moléculaire permettant le génotypage s’appuient sur des PCR. Pour cela, il existe pour la S-Rnase des amorces consensus pour chaque région conservée. Lorsque les séquences des allèles sont connues, des amorces spécifiques peuvent également être disponibles.
Comme pour la plupart des gènes, les nouveaux allèles peuvent provenir de l’accumulation progressive de mutations ponctuelles non synonymes.
La comparaison de séquences a montré que certains allèles notés différemment dans un premier temps étaient en fait identiques. En effet, l’allèle S1 est identique aux allèles S16 et S17, l’allèle S4 à S20, l’allèle S5 à S15 et l’allèle S13 à S19 (Ortega et al. 2006).
Une trentaine d’allèles ont été découverts et séquencés à ce jour mais d’autres sont sur le point d’être enregistrés.
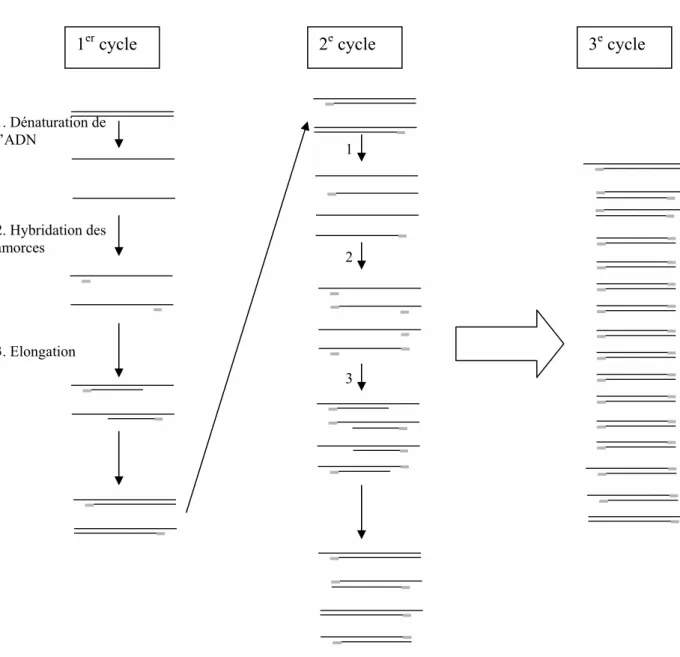
1er cycle 2e cycle 3e cycle
Figure 3 : Représentation schématique des trois premiers cycles d’une PCR
1. Dénaturation de l’ADN 1 2. Hybridation des amorces 2 3. Elongation 3
II/ Matériel et méthodes
1/ Matériel végétal utilisé
Le matériel végétal utilisé pour cette étude est issu de la collection de ressources génétiques de l’amandier de l’INRA d’Avignon et des collections des partenaires du programme européen SAFENUT. Les arbres sont conservés à la fois en conservatoire insect proof (IP) et au champ. Les prélèvements ont eu lieu en priorité dans le conservatoire insect proof.
2/ Obtention de l’ADN
Les feuilles ont été prélevées jeunes, à l’apex, pour qu’il y ait moins de lignine et ainsi faciliter l’extraction. De plus, les jeunes feuilles comptent autant de cellules que les feuilles plus âgées mais pour une moindre surface. Ainsi, pour une même quantité de matière prélevée, on obtient plus de matériel génétique utilisable. Ce prélèvement a été effectué durant Avril et Mai 2008.
Les échantillons ont été conservés en tubes dans l’azote liquide à -196°C, permettant ainsi la destruction des parois cellulaires lors du broyage.
L’extraction de l’ADN a été faite selon le protocole de Patrick Samper (GAFL Avignon) mis au point à partir de celui de S. Santoni (plate-forme INRA Montpellier) (annexe 2).
Les manipulations suivantes réalisées en laboratoire ont été effectuées par Patrick SAMPER (GAFL Avignon) puis Emeline BAPTISTE (IUT d’Avignon). J’ai également participé occasionnellement à ces travaux en laboratoires (PCR, lecture sur gel agarose).
3/ Techniques basées sur des PCR
La réaction de polymérisation en chaîne (PCR) est une technique qui permet de multiplier une région donnée d’un ADN. Celle-ci se déroule en cycle de trois étapes se répétant. La première étape est une dénaturation de l’ADN par une température élevée, c'est-à-dire une séparation des brins d’ADN par l’affaiblissement des liaisons hydrogènes les maintenant ensemble. La seconde étape est l’hybridation d’amorces sur la séquence ADN permise par un abaissement de température. Ces amorces sont des séquences simple brin d’une vingtaine de nucléotides complémentaires de l’ADN. La température d’hybridation est spécifique des amorces utilisées. La troisième étape est la synthèse des brins complémentaires par élongation des amorces par une enzyme ADN polymérase thermorésistante. Cette dernière ajoute à l’amorce les nucléotides, présents dans le milieu, complémentaires du brin matrice. Ces étapes sont répétées de nombreuses fois, jusqu’à obtention d’une quantité d’ADN amplifié suffisante. Après quelques cycles, l’ADN amplifié ne correspond plus qu’à la région délimitée par les amorces. Ces amorces sont donc choisies en fonction de la zone que l’on souhaite amplifier (figure 3).
RHV Peptide signal C1 C2 C3 RC4 C5 Intron1 Intron2 EM-PC2consFD EM-PC3consRD
a. amorces utilisées pour amplifier la région du deuxième intron
RHV Peptide
signal C1 C2 C3 RC4 C5
Intron1
PaConsI-F EM-PC1consRDIntron2
b. amorces utilisées pour amplifier la région du premier intron
RHV Peptide signal C1 C2 C3 RC4 C5 Intron1 Intron2 As1II AmyC5-R c. amorces utilisées pour amplifier la région à séquencer
Les PCR réalisées sur l’ADN extrait du matériel végétal sont effectuées selon le protocole suivant : Colorless GoTaq® Flexi Buffer 5X (Promega) : 5µL
MgCl2 à 25mM (Promega) : 1,5µL DNTP (A, T, C et G) à 4mM : 1µL Eau : 13,4µL Amorce sens à 20µM : 1µL Amorce anti-sens à 20µM : 1µL Taq gudule : 0,1µL Programme PCR appliqué : 95°C, 2 minutes 95°C, 30 secondes (Tm)°C, 45 secondes X34 72°C, 1 minute 72°C, 10 minutes
La température d’hybridation Tm dépend du couple d’amorces utilisé. (annexe 3) Le choix des amorces dépend des manipulations effectuées.
a/ Couple d’amorces EM-PC2consFD/EM-PC3consRD
(figure 4.a)Pour voir le polymorphisme de taille de l’intron 2, on amplifie un fragment contenant l’intron par PCR. Le couple d’amorces utilisé est EM-PC2consFD/EM-PC3consRD (Sutherland et al. 2004). Ces amorces sont dégénérées. C'est-à-dire que pour quelques positions, la base à inclure n’est pas déterminée. Pratiquement, pour chaque position dégénérée, on a un mélange équimolaire de différents types d’amorces. Chaque type d’amorces possède une base possible à cette position. Lorsque plusieurs positions sont dégénérées, toutes les séquences d’amorces possibles sont représentées.
La région amplifiée est celle allant de la région conservée C2 à la région conservée C3. La taille des fragments obtenue est observée après migration des produits PCR sur gel d’agarose (1,2%). On cherche à voir des différences de taille sur les introns car ceux-ci peuvent subir plus de modifications que les exons sans incidence sur la protéine. Les introns présentent donc entre eux un polymorphisme beaucoup plus marqué.
Le polymorphisme de taille est observé après séparation des produits PCR par électrophorèse sur gel d’agarose.
b/ Couple d’amorces PaConsI-F/EM-PC1consRD
(figure 4.b)On amplifie par PCR la région allant du peptide signal à la région C1 à l’aide du couple d’amorces dégénérées PaConsI-F/EM-PC1consRD (Ortega et al. 2005). Celle-ci contient le premier intron. La taille du fragment amplifié étant inférieure à 500 paires de bases pour la majorité des allèles, on a pu utiliser un séquenceur à capillaires pour plus de précision. En effet, le séquenceur ne permet pas de déterminer des tailles de fragments au-delà de cette taille mais, pour ceux pour lesquels c’est possible, la précision est proche de la paire de base. Pour permettre la détection par le séquenceur à capillaires ABI (plate-forme de génotypage haut débit de l’INRA de Clermont-Ferrand), on ajoute
de l’HEXA, un fluorochrome, sur les amorces. Les résultats sont ensuite lus sur le logiciel Genemapper® (Applied Biosystems) sous forme de pics.
Pour ces premiers couples d’amorces, la taille des fragments amplifiés selon les allèles est connue jusqu’à l’allèle S29 (Ortega et al. 2005).
c/ Amorces spécifiques d’allèles
(figure 4.c)Pour vérifier les génotypes établis à l’aide des méthodes précédentes, on utilise des PCR avec un couple d’amorces spécifiques de la séquence nucléotidiques de l’allèle à confirmer. Cette méthode s’appuie sur la séquence nucléotide et pas seulement sur un polymorphisme de taille et présente donc une plus grande validité des résultats. En effet, ces amorces n’amplifieront un fragment que pour l’allèle concerné. Les résultats se lisent sous forme de présence/absence de l’allèle testé.
Un certain nombre d’amorces spécifiques est déjà disponible dans la bibliographie : Sf, S1, S2, S5, S7, S8, S9, S10, S23 (Channuntapipat et al. 2003) et S3 (Ma et al. 2001).
D’autres ont été créées durant le stage, pour des Tm proches de 60°C, à partir des logiciels Primer3 et BLAST réunis dans l’outil Primer-BLAST disponible sur le site Internet de NCBI. Cela permet, à la fois, de créer les amorces et de vérifier leur spécificité en les comparant aux séquences des allèles enregistrés.
Pour cela, on choisit des séquences de référence pour chaque allèle la plus complète possible et donc on connaît la région amplifiée.
Les amorces sont commandées au laboratoire Eurofins MWG Operon.
Pour toutes ces manipulations, le grand nombre d’individus à génotyper oblige à se contenter d’une seule répétition. Seules les variétés de référence sont répétées.
4/ Séquençage
a/ Clonage
Pour certains individus dont on ne peut déterminer le génotype, on a effectué un séquençage. Pour cela, on a utilisé le couple d’amorces As1II/AmyC5-R (Tamura et al. 2000) amplifiant de la région C1 à C5. Les produits PCR ainsi obtenus ne permettent de séquencer que les exons 2 et 3 mais ont une taille permettant d’espérer un séquençage sans amorces intermédiaires. En effet, le gène S-RNase a une taille de plusieurs milliers de paires de bases et la méthode de séquençage utilisée par le laboratoire ne permet de déterminer que jusqu’à 500pb selon la qualité de l’ADN. L’utilisation de ces amorces permet, tout de même, de garder une grande partie de la séquence d’ADN codant pour la S-RNase et permet donc de déclarer des allèles différents de ceux déjà publiés.
L’utilisation de l’amorce PaConsI-F aurait permis de connaître toute la séquence de la S-RNase. Mais, beaucoup de fragments auraient été trop long pour un séquençage et auraient nécessité des amorces intermédiaires.
Avant d’être séquencés, les fragments amplifiés ont été clonés (protocole complet disponible en
annexe 4). Le clonage consiste en l’insertion de l’ADN à séquencer dans un vecteur afin de le
multiplier. Celui-ci permet d’obtenir un fragment plus long que la séquence que l’on souhaite séquencer et donc obtenir celle-ci en totalité, sans perte des bases proches des amorces As1II/AmyC5-R utilisées pour amplifier le fragment. Cela s’explique par le fait que les amorces utilisées pour le séquençage sont propres au vecteur de clonage et encadrent le fragment d’ADN à séquencer.
Les individus choisis, pour le séquençage, sont les individus problématiques. Le clonage étant une méthode lourde à mettre en œuvre, les individus sont sélectionnés pour séquencer le maximum de nouveaux allèles. La priorité est donnée aux cultivars français. Par souci d’efficacité, on a déterminé, d’après les résultats précédemment obtenus sur gel avec ces amorces, les individus pour lesquels on obtient une amplification pour les deux allèles. Par contre,dans notre étude, l’allèle S8 n’est pas amplifié avec As1II/AmyC5-R. On a donc également pris les individus ayant l’allèle S8 et ne présentant qu’une seule bande correspondant à l’allèle inconnu. Une seule bande suffit puisqu’elle correspond à l’allèle inconnu.
Pour le clonage, on utilise le vecteur pGEM T. Celui-ci est un plasmide de 3015pb et fait partie du Kit pGEM-T Easy Vector System. Lorsque la ligation fonctionne, dans chaque vecteur, un seul fragment obtenu par PCR à partir de l’ADN extrait s’insère. Ce fragment peut s’insérer dans un sens comme dans l’autre.
Le mélange de ligation contient 2µL de produits PCR, 1µL d’eau UP stérile, 5µL de tampon de ligation 2X, 1µL de pGEM vector et 1µL de T4 DNA ligase. L’incubation se fait pendant 14 heures à 4°C.
Pour ce clonage, on emploie des bactéries Escherichia coli. Celles utilisées sont dites thermocompétentes car une hausse brutale de température suffit à leur faire intégrer les plasmides, recombinants ou non. La transformation se fait lors d’un passage de la glace à un bain marie à 42°C où elles restent pendant 30 secondes.
Le plasmide pGEM T utilisé possède une origine de réplication ORI permettant sa multiplication dans une bactérie, un gène de résistance à l’antibiotique ampicilline et le gène lacZ issu de l’opéron lactose (annexe 5). Le gène de résistance à l’ampicilline permet de sélectionner, par ajout d’ampicilline dans le milieu de culture, les bactéries ayant intégré le vecteur. Le site de ligation se situe dans la séquence du gène lacZ. L’insertion d’un fragment d’ADN à cet endroit rend ce gène non fonctionnel. Or, ce gène code pour la β-galactosidase qui est une enzyme participant à l’hydrolyse de substrat tel que le X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) donnant ainsi un produit bleu. Ainsi, les bactéries possédant un plasmide dans lequel un fragment s’est inséré apparaissent blanches et celles dont le plasmide n’a pas subi d’insertion de fragment sont bleues. La synthèse de cette enzyme nécessite dans le milieu un inducteur de l’opéron lactose tel que l’IPTG. Cela permet de sélectionner les bactéries avec un plasmide ayant intégré un fragment PCR.
La culture des bactéries se fait sur milieu Lb Agar. Celui-ci contient de l’ampicilline, de l’IPTG et du X-gal.
En vue du séquençage, une PCR de vérification d’amplification est effectuée pour déterminer les colonies bactériennes possédant le fragment à séquencer. Pour chaque allèle potentiel à séquencer,
deux colonies sont envoyées au séquençage. Celui-ci est effectué par le laboratoire Eurofins MWG Operon.
Comme l’on est en présence de séquences longues, le séquençage se fait à partir de deux amorces: l’amorce sens T7 et l’amorce sens Sp6, propres au vecteur. Dans ce cas, les termes sens et anti-sens correspondent au anti-sens du plasmide puisque le fragment d’intérêt peut s’insérer aléatoirement dans un sens ou l’autre.
b/ Logiciels utilisés
Les séquences reçues sont alignées dans le logiciel Bioedit Sequence Alignement Editor version 7.0.9.0 (Hall 1999) avec les amorces As1II/AmyC5-R pour connaître le sens d’insertion du fragment PCR dans le vecteur. Si la séquence s’aligne parfaitement avec l’amorce sens As1II, alors la séquence est dans le sens conventionnel. Lorsque la séquence s’aligne avec l’amorce anti-sens AmyC5-R, on la transforme en son brin complémentaire inversé à l’aide du logiciel Bioedit.
Les séquences sont ensuite regroupées par identité et alignées entre elles par la méthode ClustalW (Thompson et al. 1994) disponible dans le logiciel Bioedit. A l’aide de ce dernier, des ajustements manuels sont faits lorsque l’alignement automatique n’est pas satisfaisant. Seule la région comprise entre les amorces As1II et AmyC5-R est conservée. Celle-ci est comparée aux séquences déjà enregistrées dans les bases de données NCBI et EMBL à l’aide de l’outil Nucleotide blast du site internet de NCBI en utilisant l’algorithme Megablast. Cet algorithme permet de trouver les séquences les plus proches de celle entrée et donc les allèles enregistrés les plus proches. Dans le cas d’une correspondance avec un allèle enregistré, l’alignement est vérifié avec Bioedit.
Si la séquence entrée n’est pas un allèle connu mais qu’elle présente des similarités avec les séquences d’autres allèles S-RNase, on la traduit en protéine. Pour cela, il est possible d’aligner la séquence obtenue par séquençage sur une séquence d’allèle connu dont les introns ont été déterminés et d’en déduire les introns de notre séquence par comparaison. Lorsque l’alignement obtenu n’est pas suffisamment bon, la détection d’introns est effectuée à l’aide de Splice predictor (http://deepc2.psi.iastate.edu/cgi-bin/sp.cgi) avec comme espèce de référence Arabidopsis. Pour certains cas où les introns sont trop différents et gênent l’alignement, le programme Est2genome disponible dans Emboss (Rice et al. 2000) permet un alignement des régions codantes et donc indirectement la présence d’introns. Les introns sont enlevés manuellement et la séquence restante traduite en séquence protéique par Bioedit.
La séquence protéique partielle ainsi obtenue est, ensuite, comparée aux protéines enregistrées à l’aide de l’outil Protein blast de NCBI.
Pour les nouveaux allèles, des amorces spécifiques sont créées, comme précédemment, à partir de la séquence nucléotidique.
III/ Résultats
1/ Observation du polymorphisme de taille
Allele Allele identique variété de référence taille 1
er intron
observée
taille 1er intron publiée
(Ortega et al. 2005) taille 2
e intron publiée 0 Sf Lauranne (R916) 418 417 850 1 16, 17 Ferragnès (R486) 750 2 Cristomorto (R210) 276 275 450 3 Ferragnès (R486) 339 339 900 4 20 Ai (R269) 408 620 5 15 Texas (R270) 293 330 6 Ramillette 362 361 570 7 Nonpareil (R645) 361 360 1720 8 Nonpareil (R645) 411 410 2230 9 Primorskii (R613) 348 348 1560 10 Ferrastar (R800) 311 300 11 Marcona (R185) 418 416 400 12 Marcona (R185) 206 205 1300 13 19 Atocha (R881) 387 386 1080 14 Jordanolo* 1050 18 Padre 300 299 350 21 Rumbetta 431 430 500 22 Atocha (R881) 408 407 1130 23 Belle d’Aurons (R216) 382 380 690 24 Fournat de Brezenaud (R307) 383 381 875 25 La Mona 328 327 850 26 Avellanera Gruesar 370 369 3000 27 Garrigues (R882) 380 378 1360
28 Fina del Alto* 312 340
29 Fina del Alto* 363
30 cinquanta vignali*
31 Totsol*
32 Taiatona*
33 Muel*
34 Pané-Barquets*
35 Planeta de les garrigues (R1516) 372 *variété absente de la collection INRA
Tableau 1 : Taille constatée des allèles pour les régions du 1er intron et du 2e intron
a/ Amplification de la région du deuxième intron
L’utilisation des amorces EM-PC2consFD et EM-PC3consRD, amplifiant la région du deuxième intron, permet de déterminer une partie des génotypes des cultivars étudiés. Cependant, la séparation sur gel d’agarose manque de précision et plusieurs allèles présentent des bandes de taille proche et ne sont pas différenciables. C’est le cas des allèles S5 (330pb), S10 (300pb), S18 (350pb) et S18 (340pb) et des allèles Sf (850pb), S3 (900pb), S24 (875pb) et S25 (850pb). Ainsi, si cette
technique est rapide et efficace, elle ne permet pas le génotypage de tous les cultivars. Une méthode complémentaire est nécessaire dans beaucoup de cas.
b/ Amplification de la région du premier intron
L’utilisation du séquenceur à capillaires permet de différencier les tailles des fragments amplifiés à la paire de base près. Pour les variétés de référence, les résultats obtenus sur séquenceur à capillaires sont décalés d’une ou deux paires de bases par rapport aux résultats publiées par E. Ortega en 2005 (tableau 1) sauf pour S3 et S9 où il n’y a pas de décalage constaté. Cela est dû au calibrage de la machine. Les tailles à attribuer aux allèles sont donc déterminées à partir des cultivars de référence.
Cependant, plusieurs allèles n’ont pas été pas amplifiés par le couple d’amorces utilisé : S1, S4, S5, S10 et S14.
D’autres allèles, S14 et de S28 à S34, n’ont pas pu être référencés car la variété de référence était absente de la collection INRA. Pour l’allèle S35, on a déterminé une taille de 372 à partir de la variété de référence Planeta de les garrigues (R1516).
Par ailleurs, certains allèles ne sont pas différenciables par leur taille, celle-ci étant identique. C’est des allèles Sf (418pb) et S11 (418pb).
c/ Résultats obtenus à partir du polymorphisme de taille
Les informations fournies par les deux méthodes ont permis la détermination d’une grande partie des génotypes. Seuls 9 génotypes ne donnent de résultats pour aucun allèle et 60 que pour un seul allèle. Cela est, en partie, dû au fait que plusieurs allèles ne sont pas amplifiés par les couples d’amorces utilisés. Pourtant, certains doutes subsistent et plusieurs génotypes montrent des contradictions entre les résultats de ces méthodes. On oriente donc le choix des méthodes de détermination des génotypes à utiliser par la suite vers des méthodes se basant sur la séquence nucléotidique des allèles.
2/ Amorces spécifiques
Pour vérifier un allèle précédemment attribué à un cultivar, on effectue une PCR avec des amorces spécifiques de cet allèle. Les cultivars sélectionnés pour ce test sont ceux dont la taille d’un fragment du 1er intron est égale ou proche de celle attendue pour cet allèle. Dans les publications, des couples d’amorces spécifiques pour une dizaine d’allèles (Sf, S1, S2, S3, S5, S7, S8, S9, S10 et S23) sont disponibles. Ceux-ci ont permis de confirmer près de 200 présences d’allèles. Cependant, les résultats obtenus avec le couple d’amorces spécifiques de l’allèle S10 sont incohérents avec les résultats attendus. En effet, Alnem-1 (R1107) (S18S24), bien que ne possédant pas l’allèle S10, est apparu positif.
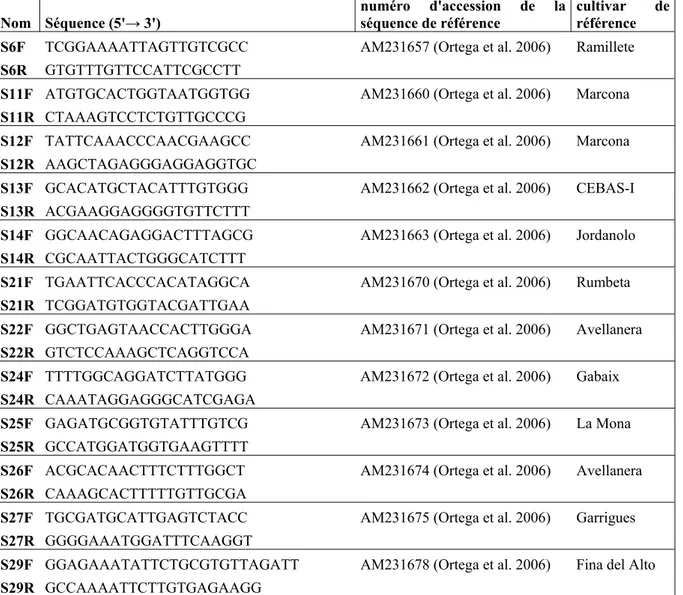
Pour déterminer avec certitude un maximum de génotypes, on a créé des amorces spécifiques pour les allèles dont la séquence était enregistrée dans les bases de données et pour lesquels aucun couple d’amorces spécifiques n’avait été développé (tableau 2).
Nom Séquence (5'→ 3')
numéro d'accession de la séquence de référence
cultivar de référence
S6F TCGGAAAATTAGTTGTCGCC AM231657 (Ortega et al. 2006) Ramillete
S6R GTGTTTGTTCCATTCGCCTT
S11F ATGTGCACTGGTAATGGTGG AM231660 (Ortega et al. 2006) Marcona
S11R CTAAAGTCCTCTGTTGCCCG
S12F TATTCAAACCCAACGAAGCC AM231661 (Ortega et al. 2006) Marcona
S12R AAGCTAGAGGGAGGAGGTGC
S13F GCACATGCTACATTTGTGGG AM231662 (Ortega et al. 2006) CEBAS-I
S13R ACGAAGGAGGGGTGTTCTTT
S14F GGCAACAGAGGACTTTAGCG AM231663 (Ortega et al. 2006) Jordanolo
S14R CGCAATTACTGGGCATCTTT
S21F TGAATTCACCCACATAGGCA AM231670 (Ortega et al. 2006) Rumbeta
S21R TCGGATGTGGTACGATTGAA
S22F GGCTGAGTAACCACTTGGGA AM231671 (Ortega et al. 2006) Avellanera
S22R GTCTCCAAAGCTCAGGTCCA
S24F TTTTGGCAGGATCTTATGGG AM231672 (Ortega et al. 2006) Gabaix
S24R CAAATAGGAGGGCATCGAGA
S25F GAGATGCGGTGTATTTGTCG AM231673 (Ortega et al. 2006) La Mona
S25R GCCATGGATGGTGAAGTTTT
S26F ACGCACAACTTTCTTTGGCT AM231674 (Ortega et al. 2006) Avellanera
S26R CAAAGCACTTTTTGTTGCGA
S27F TGCGATGCATTGAGTCTACC AM231675 (Ortega et al. 2006) Garrigues
S27R GGGGAAATGGATTTCAAGGT
S29F GGAGAAATATTCTGCGTGTTAGATT AM231678 (Ortega et al. 2006) Fina del Alto
S29R GCCAAAATTCTTGTGAGAAGG
Tableau 2 : Amorces spécifiques d’allèles connus créées
Les amorces spécifiques pour les allèles S4, S18 et S28 n’ont pas pu être trouvées. En effet, pour l’allèle S4, les amorces données par NCBI peuvent s’hybrider parfaitement sur l’allèle S9. De plus, malgré des mésappariements de quelques paires de bases, ces amorces risquent également d’amplifier les allèles S1, S8, S12, S25 et S32. De même, les amorces pour S18 peuvent amplifier les allèles S21, S22 et S24. Les amorces pour S28 peuvent amplifier les allèles Sf, S1, S11, S12, S18, S21, S22 et S24. Ces problèmes sont dus à des séquences relativement courtes avec moins de possibilités de sites d’hybridation différents pour les amorces et des délétions importantes dans les allèles par rapport aux autres allèles. Cela permet de comprendre que, le plus souvent, on puisse trouver des amorces spécifiques pour chacun de ces autres allèles puisque celles-ci peuvent s’hybrider sur des régions de la séquence présente seulement chez ces allèles. L’impossibilité de créer des amorces spécifiques s’explique aussi par la ressemblance des séquences dont les différences avec l’allèle que l’on souhaite confirmer consistent en des substitutions ponctuelles espacées dans la séquence. Celles-ci permettent de différencier les allèles d’après leur séquence
Ligne de dépôt Sens de migration de l’ADN Bande spécifique
_
+ + + + + + +
Echelle Les « + » désignent les génotypes positifs aux amorces et donc possédant l’allèle S13. Les « - » désignent les génotypes ne possédant pas l’allèle S13.Figure 5 : Résultats lus sur gel d’agarose des amorces spécifiques de S13, exemple d’amorces optimisées. Sens de migration de l’ADN Bande spécifique
_
_
+
_
+ +
_
+
+
+ + + +
+ +
EchelleLes « + » désignent les génotypes positifs aux amorces et donc possédant l’allèle S6. Les « - » désignent les génotypes ne possédant pas l’allèle S6.
Figure 6 : Résultats lus sur gel d’agarose des amorces spécifiques de S6, exemple d’amorces optimisées.
mais les amorces PCR peuvent, elles, supporter un mésappariement de quelques paires de bases et donc amplifier un fragment qui apparaîtra sur le gel d’agarose de migration. Selon les cas, la taille du fragment serait différente mais le manque de précision des migrations sur gel d’agarose rendrait difficile une éventuelle vérification du nombre de paires de bases.
Les individus sur lesquels les amorces spécifiques ont été essayées sont ceux possédant l’allèle concerné ou, éventuellement, dont l’amplification du 1er intron donne un fragment de taille proche au séquenceur à capillaires.
Les amorces spécifiques des allèles S13, S21, S26 (avec Tm=56°C) et S27 (avec Tm=52,7°C) sont optimisées et utilisables. (figure 5)
Par contre, les amorces spécifiques des allèles S6, S11, S12, S22, S24 et S25 montrent des profils avec plusieurs bandes après séparation sur gel d’agarose (figure 6). En effet, idéalement, une seule bande, correspondant au fragment spécifiquement amplifié, devrait être visible. Ces profils peuvent être dus à une température d’hybridation (Tm) trop faible permettant l’amplification de bandes aspécifiques. Par comparaison des profils avec ceux de témoins négatifs et connaissant la taille de fragment attendue, on peut deviner quelles sont les bandes aspécifiques et celle spécifique. Cela permet de conclure quant à la présence de l’allèle testé.
Seules les amorces présumées spécifiques de S14 et de S19 n’ont pas été testées car trop peu d’individus sont supposés posséder ces allèles pour s’assurer de l’efficacité de ces amorces.
D’une manière générale, les résultats obtenus à l’aide des amorces spécifiques d’allèles sont cohérents et confirment les génotypes précédemment établis. Ceci correspond, dans notre étude, à près de 80 présences d’allèles dans la collection.
3/ Séquençage
Au total 19 clones ont été séquencés. Les résultats de ces séquençages sont présentés par allèle.
a/ allèles séquencées correspondant à des allèles déjà identifiés
Le séquençage a confirmé la présence de deux allèles : l’allèle S24 de Princesse JR (R1046) et l’allèle S18 de Fascionello (1440).
L’allèle S22 a pu être déterminé chez A Trochet (R118). La séquence nucléotidique y est incomplète mais la partie manquante se situe dans le 2ème intron. La protéine ne serait pas différente de la S-RNase 22 même si la séquence nucléotidique était différente à cet endroit.
Certaines séquences obtenues correspondent à des allèles dont la séquence n’est connue que depuis peu. C’est le cas de S33 observé chez Tardive de la Verdière (R195) et chez Avola (R1439). Mais, ceux-ci n’ont pas fait partie de l’étude d’Ortega et les résultats de polymorphisme de taille ne sont pas disponibles. Ces allèles n’ont donc pas pu être identifiés par les précédentes méthodes.
D’autres présentent, en fait, la séquence nucléotidique d’allèles qui ont été enregistrés par Y. Mukai et R-C. Ma en 2004 dans les bases de données ; mais non publiés. Ces allèles sont numérotés de S50 à S64. Ainsi, on a trouvé S50 dans Languedoc 759 (R759), S52 chez Bar-Att3 (BR13), S57 chez Agen (R488), S59 chez Princesse JR (R1046) et S61 chez Turc 101-13 (R1010). La numérotation des bases de données pour ces allèles a été conservée.
Ligne de dépôt Sens de migration de l’ADN Témoin négatif (Ferragnès) Bar-Att3 (BR13)
+ +
+
_
Bande spécifique+
Les « + » désignent les génotypes positifs aux amorces et donc possédant l’allèle S59. Les « - » désignent les génotypes ne possédant pas l’allèle S59.
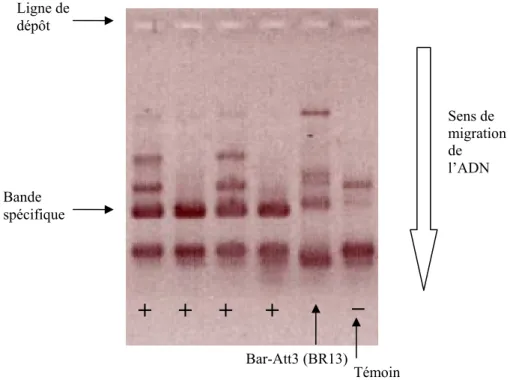
Figure 7 : Résultats lus sur gel d’agarose des amorces spécifiques de S59, exemple de lecture difficile (Bar-Att3 (BR13)). Ligne de dépôt Sens de migration de l’ADN Témoin négatif (Ferragnès)
Figure 8 : Résultats lus sur gel d’agarose des amorces spécifiques de S52, exemple de profils difficilement exploitable.
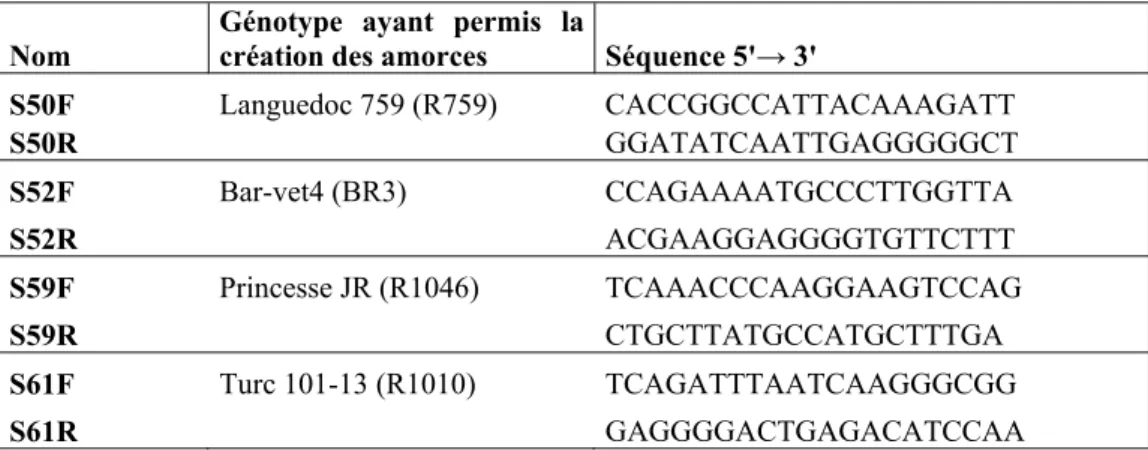
Des amorces spécifiques de ces allèles ont été créées lorsque cela était possible (tableau 3).
Nom Génotype ayant permis la création des amorces Séquence 5'→ 3'
S50F Languedoc 759 (R759) CACCGGCCATTACAAAGATT S50R GGATATCAATTGAGGGGGCT S52F Bar-vet4 (BR3) CCAGAAAATGCCCTTGGTTA S52R ACGAAGGAGGGGTGTTCTTT S59F Princesse JR (R1046) TCAAACCCAAGGAAGTCCAG S59R CTGCTTATGCCATGCTTTGA S61F Turc 101-13 (R1010) TCAGATTTAATCAAGGGCGG S61R GAGGGGACTGAGACATCCAA
Tableau 3 : Amorces spécifiques d’allèles enregistrés non publiés créées
Aucun couple d’amorces pour les allèles S33 et S57 n’a pu être retenu par manque de spécificité. Les amorces spécifiques de S59 et celles de S61 doivent être optimisées mais ont permis d’obtenir des résultats exploitables.
Ainsi, l’allèle S59 est présent avec certitude chez quatre cultivars et sa taille pour le premier intron est de 394pb. Toutefois, Bar-Att3 (BR13), bien que possédant une taille de premier intron de 394, montre un profil douteux avec une bande plus fine et légèrement plus haute que chez les individus positifs. (figure 7)
L’allèle S61 a été trouvé chez quatre cultivars et on peut en déduire que la taille associée du premier intron est de 282pb.
Les amorces spécifiques des allèles S50 et S52 ne présentent pas de profils interprétables. Aucune bande spécifique commune n’est visible. (figure 8)
b/ allèles présentant une séquence peptidique identifiée et une séquence
nucléotidique différente
Une séquence clonée de Iran 1-4-5 (R1042) code pour une séquence peptidique équivalente à celle de la S-RNase 11 pour la partie séquencée (de C1 à C5). Cependant, la séquence ADN n’est pas parfaitement identique. En effet, la région du deuxième intron présente une importante délétion, supérieure à 100pb, par rapport à la séquence nucléotidique S11 enregistrée.
De même, une séquence nucléotidique de Babatsiko (GR3), bien que différente de celle du cultivar de référence de S33, code pour une même protéine S-RNase 33 sur la région séquencée.
Ligne de dépôt Sens de migration de l’ADN Bande spécifique
_
_
+ +
+
_ _ _
_
+
+
_ _ _
Puit vide Echelle
Echelle
Témoin
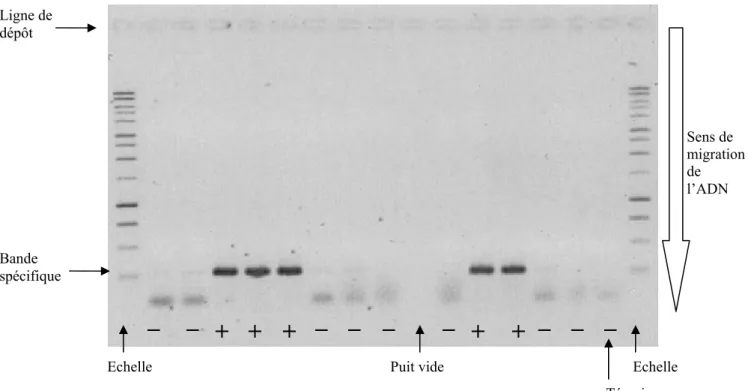
négatif (Ferragnès) Les « + » désignent les génotypes positifs aux amorces et donc possédant l’allèle S68.
Les « - » désignent les génotypes ne possédant pas l’allèle S68.
Figure 9 : Résultats lus sur gel d’agarose des amorces spécifiques de S68, exemple d’amorces d’un nouvel allèle optimisées.
c/ allèles séquencés ne correspondant pas à des allèles déjà identifiés : Nouveaux
allèles
Le clonage et le séquençage ont également permis de découvrir 8 nouveaux allèles. Ceux-ci ont été nommés de S66 à S73.
A partir de ces nouveaux allèles séquencés, on a créé des amorces spécifiques de ces allèles (tableau 4). Cela a été fait pour confirmer la présence de ces allèles dans les génotypes non séquencés ayant un même polymorphisme de taille pour la région du 1er intron.
Nom Génotype ayant permis la création des amorces Séquence 5'→ 3'
S66_F Horvat12 (SV12) CTGCAGGATTCGCAAGAAAT S66_R AGGGGACTGACACATTTTGG S67_F Horvat22 (SV22) GGGTTCGCTATTTGAGACCA S67_R TGGATGCAAACAACCCAGTA S68_F Avola (R1439) GCTATCATTCTGTTCTAATGTCTACG S68_R CCATGTTTGTTCCACTCACG S69_F Pilusedda (R896) AACTCTCCCTTCCGTTTTCA S69_R CATGTTTTTGTCGGATGTGG
S70_F Tardive de la Verdière (R195) TGGGTCGCAATTTAAGGAAA
S70_R AGGGGACTGACACATTTTGG
S71_F Narbonne (R208) CGTGCCCAGGAAAATTTCTA
S71_R AAGTCTTTGCAGGGCTTTGA
S72_F Iran 1-4-5 (R1042) CCACCGGTCACCTGTAGATT
S72_R CAAAGAAAAGAACCGACACCA
S73_F Constantini (R42) et Iran500 (R500) GATTCACCATTTGGGCGTAT
S73_R AGGAGGGGTGTTCTTCCAGT
Tableau 4 : Amorces spécifiques de nouveaux allèles créées
On a ainsi obtenu des amorces spécifiques directement utilisables pour les allèles S67, S68, S69, S70 et S71 à 58°C. Celles spécifiques de S72 et S73 doivent être optimisées. Seules les amorces S66 ne présentent pas de résultats interprétables. En effet, dans ce dernier cas, les profils des individus négatifs, dont le témoin négatif (Ferragnès (R486)), présentent un grand nombre de bandes.
L’allèle S66 a été séquencé à partir de Horvat12 (SV12), clone slovène. Celui-ci correspond à une taille pour la région du 1er intron de 294pb. Les amorces spécifiques n’ayant pas fonctionné, cet
allèle n’a pas pu être confirmé dans d’autres génotypes.
L’allèle S67 a été séquencé à partir de Horvat22 (SV22), un clone de Slovénie. Grâce aux amorces spécifiques, cet allèle a été confirmé chez trois cultivars, dont Horvat22 (SV22), et la taille de 341pb a pu lui être associée.
L’allèle S68 correspond à une taille 1er intron de 382pb. Sa séquence nucléotidique a été déterminée à partir de Avola (R1439), clone de Sicile. L’allèle S23 présente aussi une taille pour la région du premier intron de 382pb. Les amorces spécifiques de l’allèle S68 ont été testées sur les individus ayant une taille de 382pb et négatifs aux amorces spécifiques de S23. 5 génotypes sont apparus positifs pour l’allèle S68, dont Avola (R1439) et 8 négatifs (figure 9).
L’allèle S69 a été séquencé à partir de Pilusedda (R896). Cet allèle présente une taille pour la région du 1er intron de 406pb. Cette taille se retrouve chez 8 cultivars. Avec les amorces spécifiques de S69, cinq sont positifs et trois négatifs. Cependant, ces trois derniers sont tous d’origine grecque contrairement aux autres cultivars testés. Ils possèdent donc probablement d’un autre allèle.
L’allèle S69 de Pilusedda (R896) est nouveau chez l’amandier mais est identique à l’allèle S2 du pêcher (Prunus persica) (AB252417) (Tao et al. 2007). Cela confirme qu’une S-RNase peut être partagée entre différentes espèces du genre Prunus.
L’allèle a été séquencé à partir de Tardive de la Verdière (R195) S70 et correspond à une taille 1er intron de 420pb. Les amorces spécifiques de S70 ont été testées sur les 17 individus possédant un pic à 420pb au premier intron. Tous les génotypes sont apparus positifs ; à l’exception d’un seul génotype : Loudun 28 (LO28).
La séquence de l’allèle S71 a été déterminée à partir de Narbonne (R208). Cet allèle ne donne aucune amplification pour la région du premier intron. Par ailleurs, bien que le couple d’amorces spécifiques de cet allèle fonctionne, il n’a été découvert chez aucun autre cultivar testé.
L’allèle S72 a été séquencé à partir de Iran 1-4-5 (R1042), clone iranien. La taille de cet allèle pour la région du 1er intron est de 422pb. Les amorces spécifiques de l’allèle S72 de Iran 1-4-5 (R1042) fonctionnent mais n’ont pas permis d’identifier cet allèle chez d’autres cultivars.
La séquence de l’allèle S73 a été déterminée à partir de Iran500 (R500), clone iranien, et de Constantini (R42). Cet allèle correspond à une taille pour la région du 1er intron de 318pb. Un
troisième génotype, Horvat17 (SV17), présente une taille de 318pb mais est apparu négatif avec les amorces spécifiques de S73.
IV/ Discussion
1/ Cas des amorces spécifiques S10
Pour résoudre le problème des amorces spécifiques de S10 soulevé par les résultats d’Alnem-1 (R1107), on a vérifié leur spécificité à l’aide de l’outil Primer-BLAST disponible sur le site de NCBI. Il s’avère que les amorces spécifiques de S10 amplifient également l’allèle S24 et produit un fragment de même taille que pour S10 (518pb). Cela peut s’expliquer par le fait que lors de la détermination des amorces spécifiques S10 par Channuntapipat en 2003, l’allèle S24 n’était pas encore connu. Cela explique le résultat positif d’Alnem-1 (R1107) qui possède l’allèle S24. Cependant, un autre problème apparaît : l’allèle S10 ne peut donc pas être confirmé par cette méthode lorsque l’individu à tester possède également l’allèle S24. C’est le cas du clone marocain (LR 510)3 (R910).
2/ Problèmes d’amplification
Le principal problème rencontré lors du génotypage par polymorphisme de taille est l’absence d’amplification de certains allèles et de certains génotypes avec les amorces utilisées. De même, plusieurs allèles étudiés n’ont pas pu être clonés car ils n’étaient pas amplifiés par As1II/AmyC5-R. Il arrive aussi que les séquences soient trop longues pour être séquencées en une fois. Or, le temps a manqué pour définir des amorces intermédiaires et faire tous les clonages nécessaires au séquençage de tous les génotypes indéterminés.
3/ Fiabilité des méthodes
Les résultats du séquençage nous incitent à rester prudent face à la fiabilité des résultats obtenus à partir des méthodes de génotypage basé sur un polymorphisme de taille. En effet, Iran 1-4-5 (R1042) semble posséder un allèle S11, mais le deuxième intron a subi une importante délétion. Si la S-Rnase complète synthétisée était la même que la S-RNase 11, cela nuancerait les résultats obtenus par les méthodes de génotypage s’appuyant sur le polymorphisme de taille de l’ADN génomique. En effet, la longueur des introns pourrait être variable pour un même allèle et donc pourrait induire des erreurs de génotypages s’appuyant sur ce polymorphisme de taille pour des individus dits sauvages, par opposition à ceux fréquemment cultivés et sélectionnés. En effet, ces méthodes reposent sur la conservation des introns dans un allèle. Or, pour des génotypes sauvages, il semble que cette conservation ne soit pas vérifiée. De même, une même protéine S-RNase 33 pourrait être obtenue à partir de deux séquences différentes, l’une de Tardive de la Verdière (R195) et l’autre de Babatsiko (GR3), sur la partie correspondant à la région séquencée. Ces deux séquences présentent également une taille pour la région du 1er intron différente.
Par ailleurs, les amorces spécifiques d’allèles qui ont servies à confirmer les génotypes ne permettent de vérifier que la présence des sites d’hybridation de celles-ci et éventuellement la taille du fragment compris entre elles. De plus, les nouvelles amorces spécifiques créées n’ont vu leur spécificité évaluée que par rapport aux allèles enregistrés dans les bases de données. Or, plusieurs nouveaux allèles ont été découverts au cours de l’étude et il en existe probablement d’autres à découvrir. De plus, certaines amorces spécifiques nécessitent l’optimisation de leur température d’hybridation. Si l’optimisation n’est pas possible, de nouvelles amorces devront être trouvées selon d’autres critères de choix, parmi lesquels une différence de Tm faible entre les deux amorces d’un même couple. Pour certains nouveaux allèles, on ne peut pas toujours faire des amorces spécifiques. On suppose donc que, comme pour les allèles déjà connus, une taille de premier intron donnée correspond à un même allèle. Cela devra toutefois être vérifié par la suite.
Le séquençage, bien qu’étant la meilleure méthode, souffre du manque de la partie allant du peptide signal à la région conservée C1. La protéine produite par prévision de cette séquence est alors amputée de son premier exon. L’interprétation des résultats est alors soumise à une vérification ultérieure. Pour cela, la partie manquante devra être séquencée.
Allèle NB % Allèle NB % 1 63 10,71% 26 3 0,51% 8 31 5,27% 50 3 0,51% 7 27 4,59% 66 3 0,51% 9 27 4,59% 67 3 0,51% 0 25 4,25% 327 3 0,51% 5 24 4,08% 388 3 0,51% 3 22 3,74% 393 3 0,51% 27 20 3,40% 22 2 0,34% 24 18 3,06% 73 2 0,34% 70 16 2,72% 211 2 0,34% 12 15 2,55% 326 2 0,34% 2 14 2,38% 328 2 0,34% 345 13 2,21% 348 2 0,34% 6 12 2,04% 364 2 0,34% 18 11 1,87% 378 2 0,34% 57 11 1,87% 379 2 0,34% 23 10 1,70% 391 2 0,34% 25 10 1,70% 426 2 0,34% 434 10 1,70% 14 1 0,17% 382 9 1,53% 35 1 0,17% 4 8 1,36% 71 1 0,17% 69 8 1,36% 72 1 0,17% 10 7 1,19% 252 1 0,17% 11 7 1,19% 290 1 0,17% 13 6 1,02% 307 1 0,17% 68 6 1,02% 318 1 0,17% 401 6 1,02% 332 1 0,17% 21 5 0,85% 361 1 0,17% 59 4 0,68% 370 1 0,17% 61 4 0,68% 389 1 0,17% 372 4 0,68% « NB » correspond au nombre de variétés possédant l’allèle
« % » correspond à la fréquence allélique constatée dans notre étude
Malgré les réserves précédemment citées, une majorité de résultats obtenus est valables. En effet, beaucoup de cultivars sont génétiquement proches de ceux couramment utilisés et les méthodes reposant sur un polymorphisme de taille sont applicables. De plus, les méthodes utilisées donnent souvent des résultats concordants et le risque d’erreur de génotypage en est donc diminué.
4/ Cas des allèles des clones Iran 1-4-5 (R1042) et de Narbonne (R208)
Les deux nouveaux allèles S71 et S73, issus respectivement de Iran 1-4-5 (R1042) et de Narbonne (R208), présentent un deuxième intron qui, contrairement au cas général, ne présente aucune similarité et donc de parenté avec les deuxièmes introns des autres allèles. Les deuxièmes introns de ces deux allèles n’ont également pas de points communs entre eux. Cela est d’autant plus inattendu que les introns bien qu’extrêmement variables présentent, même au sein des différentes espèces du genre Prunus, une certaine parenté. Ces deux allèles seraient donc issus de populations très éloignées des autres d’un point de vue phylogénétique. Cela ne serait pas aberrant dans la mesure où Iran 1-4-5 (R1042) vient, comme son nom l’indique, d’Iran et Narbonne (R208) a une origine inconnue (découvert sur une aire d’autoroute). Ces individus pourraient donc avoir des parentés sauvages.
V/ Conclusion
Cette étude a permis de génotyper 224 cultivars et donne une bonne vision d’ensemble de la diversité allélique de l’allèle S de la collection (tableau 5). Nous avons constaté que le gène S-RNase présente une grande diversité génétique avec un grand nombre d’allèles différents et une séquence ADN très variable. Par ailleurs, la distribution des allèles ne semble pas structurée géographiquement.
L’allèle Sf, supposé lié à une auto-compatibilité, n’a été découvert dans aucun cultivar. Seuls les variétés déjà connues pour le posséder ont été confirmées.
D’autre part, cette étude a permis de s’apercevoir que le couple d’amorces spécifiques de l’allèle S10 amplifie de manière identique l’allèle S24 et qu’il est préférable de réévaluer fréquemment la spécificité de ce type d’amorces lorsque de nouveaux allèles sont découverts.
Huit nouveaux allèles ont été découverts et séquencés lors de cette étude. Enfin, des amorces spécifiques d’allèles ont été créées pour 17 allèles dont 7 nouveaux.
Dans l’avenir, les génotypes déterminés cette année devront être confirmés au niveau phénotypique par croisements contrôlés en champ. En effet, cela reste la seule méthode entièrement fiable de détermination d’un allèle puisque la définition d’un allèle est liée à un comportement en verger et non à une séquence nucléotidique.
En ce qui concerne le programme SAFENUT, la diversité allélique du gène S-RNase devra être associée à d’autres critères de diversité génétique, tels que la diversité évaluée par marqueurs microsatellites, afin de pouvoir sélectionner les individus à conserver dans la core-collection.
Bibliographie
Sites Internet
SplicePredictor Online. SplicePredictor. http://deepc2.psi.iastate.edu/cgi-bin/sp.cgi. Site de l'INRA. www.inra.fr
Articles
Bošković, R., K.R. Tobutt, H. Duval, I. Batlle, F. Dicenta, et F.J. Vargas. 1999. A stylar
ribonuclease assay to detect self-incompatible seedlings in almond progenies. Theoretical and Applied Genetics 99: 800-810.
Channuntapipat, C., M. Wirthenson, S.A. Ramesh, I. Battle, P. Arús, M. Sedley, et G. Collins. 2003. Identification of incompatibility genotypes in almond (Prunus dulcis Mill.) using specific primers based on the introns of the S-alleles. Plant Breeding 122: 164-168. Grasselly, C., et G. Olivier. 1976. Mise en évidence de quelques types autocompatibles parmi les
cultivars d'amandier (P. amygdalus batsch) de la population des Pouilles. Annales de l'amélioration des Plantes 26(I): 107-113.
Hall, T.A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95-98.
Hanada, Toshio, Kyoko Fukuta, H. Yamane, et Tomoya Esumi. 2009. Cloning and Characterisation of a Self-compatible Sf Haplotype in Almond [Prunus dulcis (Mill.) D.A.Webb.syn. P. amygdalus Batsch] to Resolve Previous Confusion in Its Sf-RNase Sequence. HortScience 44(3): 609-613.
Hua, Z.-H., A. Fields, et T.-h. Kao. 2008. Biochemical Models for S-RNase-Based Self-Incompatibility. Molecular Plant 1(4): 575-585.
Kodad, O., R.i.c. Socias, A. Sánchez, et M. Olivieira. 2009. The expression of self-incompatibility in almond may not only be due to the presence of the Sf allele. The American Society for Horticultural Science 134: 221-227.
Ma, R.-C., et M.M. Olivieira. 2001. Molecular cloning of the self-incompatibility genes S1 and S3 from Almond (Prunus dulcis) cv. Ferragnès. Sexual Plant Reproduction 14: 163-167. Ortega, E., R. Bošković, D.J. Sargent, et K.R. Tobutt. 2006. Analysis of S-RNase alleles of almond
(Prunus dulcis): characterization of new sequences, resolution of synonyms and evidence of intragenic recombinaison. Molecular Genetic Genomics 276: 413-426.
Ortega, E., B.G. Sutherland, F. Dicenta, R. Bošković, et K.R. Tobutt. 2005. Determination of incompatibility genotypes in almond using first and second intron consensus primers: detection of new S alles and correction of reported S genotypes. Plant Breeding 124: 188-196.
Rice, P., I. Longden, et A. Bleasby. 2000. EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics 16(6): 276-277.
Sonneveld, T., K.R. Tobutt, S. P. Vaughan, et T.P. Robbins. 2005. Loss of Pollen-S Function in Two Self-compatible Selections of Prunus avium Is Associated with Deletion/Mutation of an S Haplotype-Specific F-Bax gene. The Plant Cell 17: 37-51.
Sutherland, T. P. Robbins, et K. R. Tobutt. 2004. Primers amplifying a range of Prunus S-allele. Plant Breeding 123: 582-584.
Sutherland, B.G., K.R. Tobutt, et T.P. Robbins. 2008. Trans-specific S-RNase and SFB alleles in Prunus self-incompatibility haplotypes. Molecular Genetic Genomics 279: 95-106. Tamura, M., K. Ushijima, H. Sassa, H. Hirano, R. Tao, T.M. Gradziel, et A.M. Dandekar. 2000.
Identification of self-incompatibility genotypes of almond by allele-specific PCR analysis. Theoretical and Applied Genetics 101: 344-349.
Tao, R., A. Watari, T. Hanada, T. Habu, H. Yaegaki, M. Yamagushi, et H. Yamane. 2007. Self-compatible peach (Prunus persica) has mutant versions of haplotypes found in self-incompatible Prunus species. Plant Molecular Biology 63: 109-123.
Thompson, J.D., G.D. Higgins, et T.J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Research submitted.
Ushijima, K., H. Sassa, R. Tao, H. Yamane, A.M. Dandekar, T.M. Gradziel, et H. Hirano. 1998. Cloning and characterisation of cDNAs encoding S-RNases from almond (Prunus dulcis): primary structural features and sequence diversity of the S-RNases in Rosaceae. Molecular Genetic Genomics 260: 261-268.
Glossaire
ADNc : acide désoxyribonucléique complémentaire de l’ARNm. Il ne contient que les exons
c'est-à-dire les régions codantes.
ADNg : acide désoxyribonucléique génomique. C’est le support de l’information génétique. Il
contient les exons, les introns et les régions intergéniques.
Amorces PCR : oligonucléotides d’une vingtaine de paires de base s’hybridant sur la séquence
d’ADN permettant l’amplification lors d’une PCR. Ces amorces s’utilisent en couple. L’une est dite sens (ou forward) car l’élongation, à partir de celle-ci, se fait dans le sens de lecture du gène et se situe au début de la région à amplifier. L’autre est dite anti-sens (ou reverse) et se situe à la fin.
Amorces PCR dégénérées : mélanges d’amorces PCR qui diffèrent par uniquement quelques
paires de bases définies.
ARNm : acide ribonucléique messager. C’est le support de la traduction en protéine.
Auto-incompatibilité gamétophytique : auto-incompatibilité pollinique due à l’interaction entre le
génotype diploïde de la fleur et le génotype haploïde du pollen.
Core-collection : collection de ressources génétiques dont le but est d’avoir un maximum de
diversité génétique dans un minimum d’individus.
Cultivar : ensemble d’un individu et de ses clones obtenus par multiplication végétative. Dans le
cas de l’arboriculture, la sélection ne conduit pas à l’obtention de lignées pures et une variété correspond à un cultivar.
Epissage : étape de la maturation des ARNm au cours de laquelle les introns sont éliminés.
Exon : région conservée lors de l’épissage de l’ARNm ou la région de l’ADN génomique
correspondante. L’ensemble des exons représente la région codante.
Intron : région non conservée lors de l’épissage de l’ARNm ou la région de l’ADN génomique
correspondante.
Locus : emplacement sur le génome d’un gène ou d’une fonction.
PCR : Polymerase Chain Reaction. C’est une réaction permettant l’amplification en grande quantité
d’une région désignée (comprise entre deux amorces) de l’ADN.
Peptide : assemblage d’une succession d’acides aminés, ces derniers étant les unités de base de la
construction des protéines.
RNase : enzyme dégradant les ARN.
SFB : protéine S F-box. C’est l’élément du pollen impliqué dans l’auto-incompatibilité pollinique
gamétophytique.
S-RNase : RNase qui dégrade l’ARN du tube pollinique dans le cas d’une auto-incompatibilité.
Annexes
Table des annexes
Annexe 1 : Tableau des résultats du génotypage Annexe 2 : Protocole d’extraction de l’ADN Annexe 3 : Tableau des amorces utilisées Annexe 4 : Protocole de clonage