Université de Sherbrooke
Régulation du flot sanguin dans le tissu adipeux sous-cutané
Par Richard Sotorník Programme de physiologie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de philosophiae doctor (Ph.D.)
en physiologie
Sherbrooke, Québec, Canada [Mars, 2018]
Membres du jury d’évaluation
Pr Pedro Miguel Geraldes, Département de physiologie
Pr Rémi Rabasa-Lhoret, Département de nutrition, Faculté de médecine, Université de Montréal
Pr Fernand Gobeil Junior, Département de pharmacologie
Pr Jean-Luc Ardilouze, Département de médecine, Service d’endocrinologie
ii
1R
ÉSUMÉRégulation du flot sanguin dans le tissu adipeux sous-cutané Par
Richard Sotorník Programme de physiologie
Thèse présentée à la Faculté de médecine et des sciences de la santé
en vue de l’obtention du grade de philosophiae doctor (Ph.D.) en physiologie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec,
Canada, J1H 5N4
Le tissu adipeux sous-cutané (TAsc) est le site préférentiel du stockage postprandial des triglycérides (TG). Quand les capacités d’accrétion sont dépassées, le stockage des TG se fait dans des sites ectopiques du TA et dans des tissus non adipocytaires, par exemple foie et muscles, ce qui entraine de multiples dysfonctionnements dans ces organes et tissus, et permet le développement du syndrome d’insulinorésistance.
Chez les sujets obèses, la période postprandiale est caractérisée par des anomalies métaboliques, immunitaires, hormonales, et également par une diminution importante du flot sanguin dans le tissu adipeux (FSTA) sous-cutané. Ce blocage de la perfusion postprandiale du TA a aussi été montrée chez des individus minces qui avaient de très lourds antécédents familiaux de maladies cardiométabolique (obésité, diabète de type 2, maladies cardiovasculaires). Dans cette thèse, on classifiera ces individus comme « non-répondeurs ». À ce jour, peu d’attention a été accordée à ce phénomène.
L’hypothèse qui sous-tend cette thèse est que les anomalies du FSTA sont innées ou primaires et sont impliquées très tôt dans le développement de la résistance à l’insuline (RI), du diabète de type 2 et du syndrome métabolique.
Le but de notre recherche était donc de vérifier si les altérations du FSTA sont présentes chez les personnes saines et minces, mais à très haut risque de développer une RI ou une maladie cardiométabolique. Nous avons aussi cherché à déterminer les facteurs liés à la non-réponse. Pour cela il nous a fallu explorer certains facteurs hormonaux impliqués dans la régulation du FSTA.
Nos résultats montrent que le FSTA est très diminué, à jeun et en postprandial, chez les sujets à haut risque de maladies cardiométaboliques mais encore minces et métaboliquement sains, sans RI. Nous avons aussi montré, pour la première fois, l’effet vasodilatateur du peptide intestinal vasoactif (VIP) dans le TAsc, tout comme le rôle stimulant du système cholinergique dans la régulation postprandiale du FSTA. Cependant, aucun de ces facteurs ne participe au dysfonctionnement du FSTA postprandial chez les non-répondeurs. Des taux répétés de TG plus élevés chez les non-répondeurs et l’association du FSTA avec certains indices de la RI décrits dans la littérature suggèrent que l’altération du métabolisme lipidique suite à la diminution du FSTA puisse servir de médiateur à la détérioration de la sensibilité à l’insuline.
Mots clés : flot sanguin du tissu adipeux, résistance à l’insuline, diabète gestationnel, diabète de type 2, vasoactive intestinal peptide, acétylcholine
iii
2S
UMMARYRegulation of blood flow in subcutaneous adipose tissue By
Richard Sotorník Program of Physiology
Thesis presented at the Faculty of medicine and health sciences for the obtention of Doctor degree diploma philosophiae doctor (Ph.D.) in Physiology, Faculty of medicine and health
sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
Subcutaneous adipose tissue (SCAT) is the preferential site of triacylglycerols (TAG) postprandial disposal. When the buffering capacity of SCAT for lipids is exceeded, TAG are disposed in ectopic adipose tissue depots and in non-adipose tissues, such as liver and muscles. Consequently, multiple dysfunctions of these organs and tissues develop including insulin resistance (IR).
In obese people, the postprandial period is characterized by metabolic, immune and hormonal alterations, but also by severely altered adipose tissue blood flow (ATBF). Nevertheless, significant alteration of postprandial ATBF was also found in lean individuals with highly positive familiar history of cardiometabolic diseases (obesity, type 2 diabetes, cardiovascular diseases). In the thesis, we term them as "non-responders". Up to date, little attention has been payed to this phenomenon.
The underlying hypothesis of this thesis is that alterations in ATBF are inborne or very early and that they participate on the development of IR, type 2 diabetes and metabolic syndrome.
Consequently, the aim of our research was to verify if the alterations in ATBF are present in healthy, normal-weight subjects, but at very high risk for development of IR or cardiometabolic diseases. Simultaneously, we searched for factors linked with non-responsiveness phenomenon. To do this, we examined some hormonal factors in ATBF regulation.
Our results confirm the presence of altered fasting and postprandial ATBF in at high-risk subjects for cardiometabolic diseases, but still lean and metabolically healthy, without IR. For the first time, we have also demonstrated the role of cholinergic system in postprandial ATBF regulation, and vasodilatory effect of vasoactive intestinal peptide (VIP) in SCAT. However, none of these factors takes part in postprandial ATBF dysfunction in non-responders. Higher TAG levels repeatedly found in non-responders and the association of ATBF with some indices of insulin sensitivity described in the literature suggest that alteration of lipid metabolism as a result of low ATBF may mediate deterioration of insulin sensitivity.
Keywords : adipose tissue blood flow, insulin resistance, gestational diabetes, type 2 diabetes, vasoactive intestinal peptide, acetylcholine
iv
3T
ABLE DES MATIÈRES1 Résumé ... ii
2 Summary ... iii
3 Table des matières ... iv
4 Liste des tableaux ... vii
5 Liste des figures ... viii
6 Liste des abréviations ... ix
7 Remerciements ... xii
1 Introduction ... 1
1.1 Structure du tissu adipeux ... 1
1.1.1 Les types de tissus adipeux ... 1
1.1.2 Le lit vasculaire du tissu adipeux ... 3
1.1.3 Le système nerveux du tissu adipeux ... 4
1.2 Résumé des fonctions métaboliques du tissu adipeux blanc ... 5
1.2.1 À jeun ... 5
1.2.2 En postpradial ... 6
1.2.3 Les différences entre les principaux dépôts du tissu adipeux blanc ... 8
1.2.3.1 Les différences en activité lipolytique ... 8
1.2.3.2 Les différences en capacité de captage des lipides ... 9
1.2.4 La dysfonction métabolique du tissu adipeux ... 10
1.3 Le flot sanguin du tissu adipeux ... 16
1.3.1 Le concept général de la microcirculation ... 16
1.3.2 Dysfonctionnement de la microcirculation ... 18
1.3.2.1 Les mécanismes du dysfonctionnement de la microcirculation ... 18
1.3.2.2 La chronologie du dysfonctionnement de la microcirculation ... 19
1.3.2.3 La localisation du dysfonctionnement de la microcirculation... 19
1.3.3 Évaluation de la microcirculation du tissu adipeux ... 21
1.3.4 Le comportement physiologique du FSTA ... 22
1.3.4.1 Le FSTA à jeun ... 22
1.3.4.2 Le FSTA pendant l’activité physique ... 22
v
1.3.5 La relation du FSTA avec le métabolisme des lipides ... 23
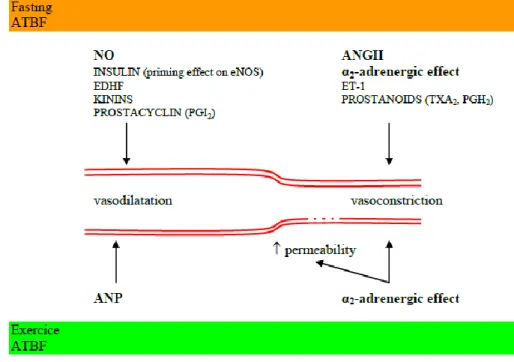
1.3.6 Les facteurs de la régulation du FSTA aujourd’hui ... 25
1.3.6.1 Le FSTA à jeun ... 25
1.3.6.2 FSTA et exercice ... 25
1.3.6.3 FSTA en postprandial ... 26
1.3.6.4 L’insuline et le FSTA ... 27
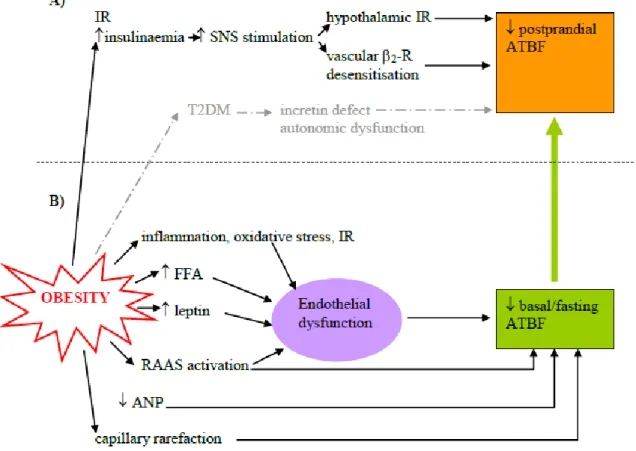
1.3.7 Altérations du FSTA – causes ... 29
1.3.7.1 L’âge ... 29
1.3.7.2 L’obésité... 29
1.3.7.3 La résistance à l’insuline ... 31
1.3.8 Altérations du FSTA – conséquences ... 32
1.3.8.1 L’impact local dans le tissu adipeux ... 32
1.3.8.2 L’impact métabolique ... 33
1.4 La résistance à l’insuline ... 34
1.4.1 L’insuline – son rôle ... 34
1.4.2 L’insuline – signalisation ... 34
1.4.3 La résistance à l’insuline – définition ... 35
1.4.4 La résistance à l’insuline – formes ... 36
1.4.4.1 Anomalies congénitales ... 36
1.4.4.2 Anomalies acquises ... 37
1.4.4.3 Rôle des IRS... 37
1.4.5 La résistance à l’insuline – causes ... 37
1.4.5.1 L’hyperinsulinisme ... 37
1.4.5.2 La lipotoxicité ... 37
1.4.5.3 Le stress du réticulum endoplasmique et le stress oxydatif ... 39
1.4.5.4 L’inflammation subclinique ... 40
1.4.5.5 Les adipokines et autres hormones du TA ... 43
1.4.5.6 La génétique, l’épigénétique ... 47
1.4.5.7 Le microbiome intestinal ... 48
1.4.5.8 D’autres facteurs participants au développement de la RI ... 49
1.4.6 La résistance à l’insuline – des impacts ... 49
1.4.6.1 L’impact sur le métabolisme glucidique ... 50
vi
1.4.6.3 L’impact sur le système cardiovasculaire ... 52
1.4.6.4 Les impacts neuro-hormonales ... 53
1.4.6.5 La RI et le cancer ... 55
1.4.7 La résistance à l’insuline – une récapitulation ... 55
1.5 Le diabète gestationnel ... 56
1.5.1 Définition... 56
1.5.2 La grossesse normale ... 56
1.5.3 Physiopathologie du DG ... 58
1.5.3.1 Diminution de la sensibilité à l’insuline ... 58
1.5.3.2 La sécrétion d’insuline ... 61
1.5.4 La signification du DG ... 63
1.6 Présentation du projet de la thèse ... 66
1.6.1 Problématique ... 66 1.6.2 Hypothèses ... 67 1.6.3 Objectifs ... 67 2 Article 1 ... 68 3 Article 2 ... 81 4 Article 3 ... 108 5 Article 4 ... 129 6 Article 5 ... 148 7 Discussion ... 175
7.1 Si l’on résume cette thèse ... 175
7.1.1 Qu’est-ce qu’un tissu adipeux dysfonctionnel ? ... 175
7.1.2 Quel est le rôle du FSTA et comment est-il régulé ? ... 177
7.2 Contributions ... 178
7.2.1 Dans le domaine de la régulation du FSTA ... 178
7.2.1.1 Le système cholinergique ... 178
7.2.1.2 Le VIP ... 179
7.2.2 Dans le domaine de la non réponse du FSTA ... 181
7.2.3 Dans le domaine du risque cardiométabolique... 181
7.3 Points forts et faibles du projet ... 182
7.4 Perspectives ... 183
8 Liste des références ... 186
vii
4L
ISTE DES TABLEAUXTableau 1. Les facteurs de régulation du FSTA. ... 28
Tableau 2. List of factors known to influence ATBF ... 70
Tableau 3. List of factors considered to have an impact on ATBF ... 88
Tableau 4. Characteristics of participants. ... 119
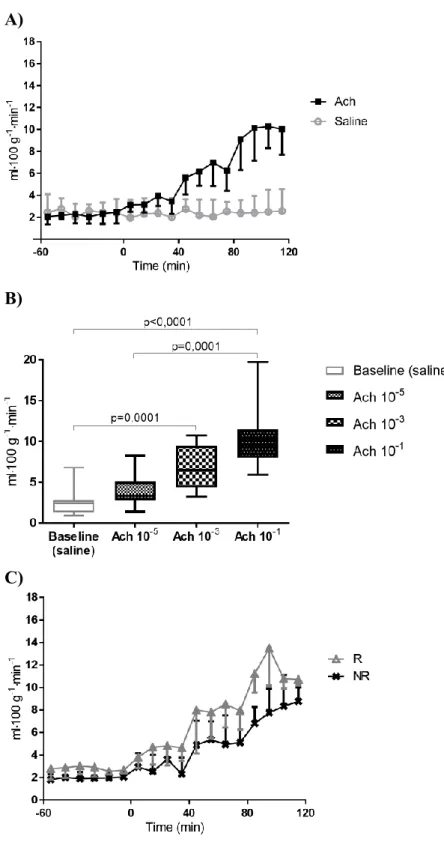
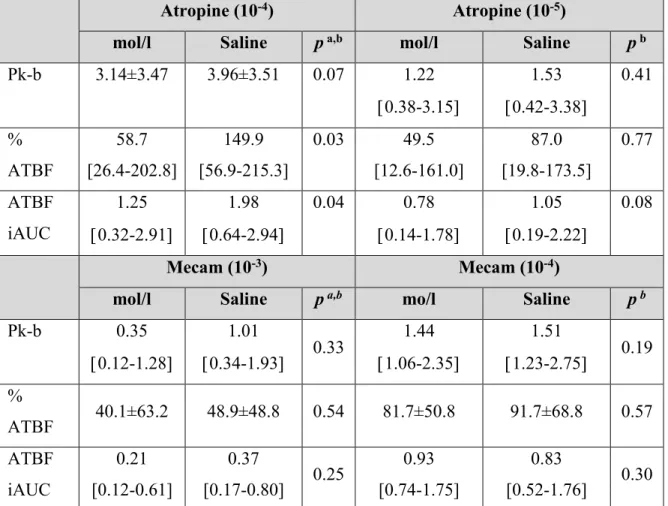
Tableau 5. Effect of Ach on ATBF. ... 120
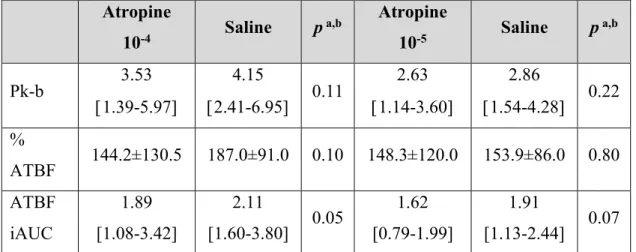
Tableau 6. Effect of 10-4 (n=13) and 10-5 mol/l (n=22) atropine and 10-3 (n=15) and 10-4 mol/l (n=10) Mecam on glucose-stimulated ATBF. ... 123
Tableau 7. Effect of 10-4 (n=10) and 10-5 mol/l atropine (n=14) on glucose-stimulated ATBF in responders only. ... 124
Tableau 8. Characteristics of participants. ... 139
Tableau 9. Characteristics at entry in study. ... 158
Tableau 10. Fasting and post-OGL biochemical characteristics. ... 160
viii
5L
ISTE DES FIGURESFigure 1. Mediscint, Oakfield Instruments, Eynsham, UK. ... 72 Figure 2. Quick-set Infusion Set; Minimed, Medtronic of Canada Ltd., Mississauga,
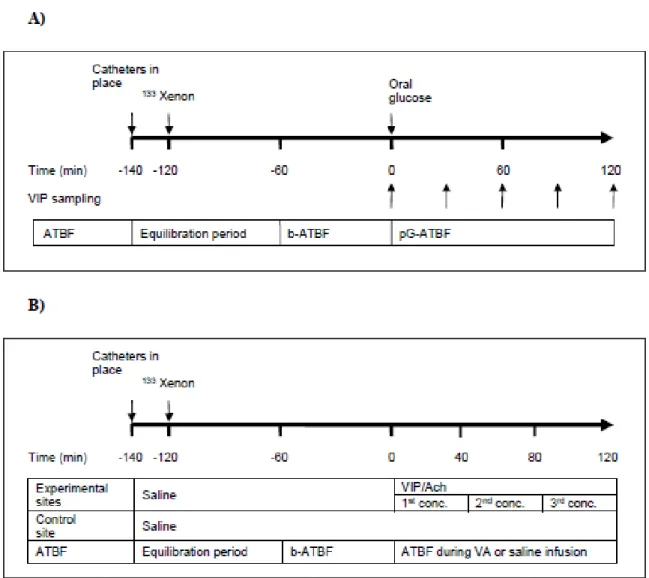
Canada. ... 73 Figure 3. Inset II Infusion Set, Animas Corporation, LifeScan Canada Ltd., Burnaby, British Columbia, Canada. ... 74 Figure 4. ATBF in fasting state or during exercise. Known (in bold) and possible
mechanisms. ... 89 Figure 5. Postprandial ATBF. Known (in bold) and possible mechanisms. ... 90 Figure 6. Possible mechanisms of impacts of obesity on postprandial and basal ATBF. . 103 Figure 7. Time line of microinfusion experiments. ... 116 Figure 8. Effect of acetylcholine (Ach) on ATBF. ... 121 Figure 9. Time line of experiments. ... 135 Figure 10. ATBF after 75 g oral glucose load in responders (R; n = 9) and non-responders (NR; n = 7). ... 140 Figure 11. ATBF response to infusions of three incrementally increasing concentrations of VIP (A-1 and A-2) and Ach (B-1 and B-2). ... 142 Figure 12. Time line of experiments. ... 155 Figure 13. Time course of PG (A), serum insulin (B), C-peptide (C) and NEFA (D) during experiments. ... 162 Figure 14. Fasting and post-OGL ATBF. ... 164 Figure 15. Les causes et conséquences de l’expansion du TA. ... 176
ix
6L
ISTE DES ABRÉVIATIONSACE enzyme de conversion de l’angiotensine / angiotensin-converting enzyme Ach acétylcholine / acetylcholine
ACS acyl-coenzyme A synthétase / acyl-coenzyme A synthetase Acyl-CoA acyl-coenzyme A / acyl-coenzym A
ADN acide désoxyribonucléique / deoxyribonucleic acid
AGL acides gras libres / free, non-esterified fatty acids (FFA, NEFA) AMPc adénosine monophosphate cyclique / cyclic adenosine monophosphate AngII angiotensine II / angiotensin II
ARNm acide ribonucléique messager / messenger ribonucleic acid (mRNA) AT1R récepteur de type 1 pour l’angiotensine II / angiotensin II receptor type 1/
ATGL lipase de TG adipocytaire / adipose triglyceride lipase
(i)AUC aire sous la courbe (incrémentale) / (incremental) area under the curve A-V artérioveineux / arteriovenous
AV agent vasoactif / vasoactive agent (VA)
CETP protéine de transfert des esters de cholestérol / cholesteryl ester transfer protein
CEU real-time contrast-enhanced ultrasound CIR-30 corrected insulin response at 30 minutes
COG charge orale en glucose / oral glucose load (OGL) CRP protéine C réactive / C-reactive protein
DAG diacylglycérol / diacylglycerol
DE dysfonction endothéliale / endothelial dysfunction
DG diabète gestationnel / gestational diabetes mellitus (GDM) DI disposition index
DT1 diabète de type 1 / type 1 diabetes (T1D) DT2 diabète de type 2 / type 2 diabetes (T2D)
EC50 concentration efficace médiane / half maximal effective concentration
EDH hyperpolarisation endothélium-dépendante / endothelium-dependent hyperpolarisation
EDHF endothelium-derived hyperpolarising factor
EDTA acide tétraacétique diamine éthylène / ethylene diamine tetraacetic acid eNOS oxyde nitrique synthase endothéliale / endothelial nitric oxide synthase ET-1 endothéline-1 / endotheline-1
ERK extracellular signal regulated kinase
FABP protéine de liaison des acides gras / fatty acid binding protein FAT fatty acid translocase ou CD36
FATPs protéines de transport des acides gras / fatty acid transport proteins FFM masse maigre / fat free mass
FM masse grasse / fat mass
FSTA flot sanguin du tissu adipeux / adipose tissue blood flow (ATBF) FSV fraction stromovasculaire / stromovascular fraction (SVF) G-3-P glycérol-3-phosphate / glycerol-3-phosphate
GIP glucose-dependent insulinotropic polypeptide GLUT transporteur de glucose / glucose transporter
x GMPc guanosine monophosphate cyclique / cyclic guanosine monophosphate HbA1c hémoglobine glyquée / glycated hemoglobin
HDL lipoprotéines de haute densité / high density lipoproteins
HGPO hyperglycémie provoquée orale / oral glucose tolerance test (OGTT) HOMA-IR homeostatic model assessment for insulin resistance
11βHSD1 11β-hydroxystéroïde déshydrogénase 1 / 11β-hydroxysteroid dehydrogenase type 1
IADPSG International Association of the Diabetes and Pregnancy Study Groups IDL lipoprotéines de densité intermédiaire / intermediate density lipoproteins IGF-1 insuline-like growing factor-1
IKK inhibitor kappa B kinase IL-6 interleukine 6 / interleukin 6
IMC indice de masse corporelle / body mass index (BMI)
IMTG triglycérides intramusculaires / intramuscular triglycerides IRS-1, 2 insulin receptor substrates-1, 2
ISIAT insulin sensitivity index in adipose tissue
ISIBelf(Mats) Belfiore (Matsuda) insulin sensitivity index
ISSI-2 insulin sensitivity-secretion index-2 JNK c-Jun HN2-terminal kinase
LCFA AGL avec une chaîne longue / long-chain fatty acids LDL lipoprotéines de basse densité / low density lipoproteins LHS lipase hormonosensible / hormone-sensitive lipase (HSL) LPL lipoprotéine lipase / lipoprotein lipase
MAPKK mitogen-activated protein kinase kinase MAPK mitogen-activated protein kinases
MEC matrice extracellulaire / extracellular matrix (ECM)
MEGJ jonctions communicantes myoendothéliales / myoendothelial gap junctions MODY maturity onset diabetes of the young
mTOR mammalian target of rapamycin
NADPH nicotinamide adénine dinucléotide phosphate / nicotinamide adenine dinucleotide phosphate
NAFLD stéatose hépatique non alcoolique / non-alcoholic fatty liver disease NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells NO oxyde nitrique / nitric oxide
NR non-répondeur / non-responder PGI2 prostacycline / prostacycline
PI3K phosphoinositide 3-kinase / phosphatidylinositol-3-kinase PKB, C, G protéine kinase B, C, G / protein kinase B, C, G
PNA peptide natriurétique atrial / atrial natriuretic peptide (ANP) pO2 pression de l’oxygène
PPARγ récepteur activé par les proliférateurs de peroxysomes / peroxisome proliferator-activated receptor
R répondeur / responder
RE réticulum endoplasmique / endoplasmic reticulum RI résistance à l’insuline / insulin resistance (IR)
xi SHBG globuline liant les hormones sexuelles / sex hormone-binding globulin SCV système cardiovasculaire
sICAM-1 serum intercellular adhesion molecule-1
SM syndrome métabolique / metabolic syndrome (MS) SNC système nerveux central / central nervous system (CNS)
SNPS système nerveux parasympathique / parasympathetic nervous system (PSNS) SNS système nerveux sympathique / sympathetic nervous system
SOPK syndrome des ovaires polykystiques / polycystic ovary syndrome (PCOS) SRA(A) système rénine-angiotensine (-aldostérone) / renin-angiotensin(-aldosteron)
system (RA(A)S))
TA tissu adipeux / adipose tissue (AT)
TAsc tissu adipeux sous-cutané / subcutaneous adipose tissue (SCAT) TAV tissu adipeux viscéral / visceral adipose tissue (VAT)
TEP tomographie par émission de positrons / positron emission tomography (PET) TG triglycérides / triacylglycerols (TAG)
TGRL lipoprotéine riche en triglycérides / triglyceride-rich lipoproteins TNFα tumor-necrosis factor α
TT tour de taille / waist circumference (WC)
VIP peptide intestinal vasoactif / vasoactive intestinal peptide
VLDL lipoprotéines de très basse densité / very low density lipoproteins VSMC cellules musculaires lisses vasculaires / vascular smooth muscle cells WHR rapport taille-hanche / waist to hip ratio
xii
7R
EMERCIEMENTSJ’aimerais tout d’abord remercier mes directeurs de recherche : le Pr Jean-Luc Ardilouze et le Pr Jean-Patrice Baillargeon.
Je remercie le Pr Ardilouze pour son enseignement, son enthousiasme contagieux pour la science et pour m’avoir initié à la recherche. Merci pour ton intérêt et ton amitié, ta compréhension intuitive, ta droiture, ta générosité. Merci d’avoir été beaucoup plus qu’un directeur de thèse.
Je remercie le Pr Baillargeon pour avoir bonifié mes articles par ses connaissances de chercheur et de statisticien. Merci pour toutes les discussions sur mes résultats et pour votre soutien, surtout en l’absence du Pr Ardilouze.
Je voudrais également remercier tous les membres de l’équipe du Pr Ardilouze pour leur accueil, leur compréhension, leur patience et leur soutien professionnel et personnel. Spécialement Julie Ménard qui, d’entrée de jeu, a supporté mon français et plusieurs différences culturelles. Merci pour ton aide très pratique et pour ton optimisme incessant! Merci énormément à Pascal Brassard pour son amitié, son humour et son aide dans mes premiers pas d’étudiant au Ph.D. Merci à Maude Gagnon-Auger pour m’avoir initié aux mystères du flot sanguin dans le tissu adipeux et pour m’avoir accompagné dans le labyrinthe scientifique! D’énormes mercis à l’infirmier Mario Houde et aux infirmières Maude Gérard, Marie-Josée Gosselin et Georgette Proulx qui ont été des collaborateurs très professionnels, expérimentés, efficaces et amicaux pendant les longues journées au Centre de recherche. Je tiens aussi à remercier toute l’équipe du Centre de recherche du CHUS, dont Caroll-Lynn Thibodeau, Diane Lessard et tous les autres ayant contribué de près ou de loin au bon cheminement de mon projet. Je voudrais remercier Lucie Bouffard toujours prête à effectuer tous les dosages demandés. Un merci tout spécial à mon copain d’étude Samuel Leblanc dont l’intérêt et la spontanéité n’ont jamais cessé de m’étonner et de m’encourager à persévérer. Un grand merci également à tous les volontaires qui ont participé à l’étude et sans qui rien n’aurait été possible.
Merci à mon épouse chérie pour avoir passé toutes les difficultés reliées à notre déménagement à l’étranger et pour avoir créé un milieu extraordinaire pour toute notre famille. Je la remercie, ainsi que toute ma famille et mes amis pour leur soutien, leur amour inconditionnel et leurs prières incessantes!
Je remercie finalement le Pr Rémi Rabasa-Lhoret, le Pr Pedro Miguel Geraldes, le Pr Fernand Gobeil Junior et mon directeur de recherche, le Pr Jean-Luc Ardilouze pour le temps et l’attention consacrés à l’évaluation de ma thèse.
1
1I
NTRODUCTIONDans cette introduction, nous commencerons par décrire la structure du tissu adipeux (TA) et ses fonctions métaboliques. Ensuite, nous détaillerons ce que l’on sait de la régulation de la perfusion du TA et des techniques utilisables pour l’étudier, sujets qui font l’objet de deux articles (publiés). Ces chapitres sont essentiels pour comprendre le pourquoi de nos études sur le système cholinergique et le peptide intestinal vasoactif (VIP, vasoactive intestinal peptide), deux éléments de la régulation du flot sanguin du tissu adipeux (FSTA), le cœur de cette thèse, et qui ont permis de soumettre deux autres articles. Nous aborderons ensuite le sujet difficile de la résistance à l’insuline (RI), pour justifier la partie la plus originale de notre travail. Nous poserons dans un autre des articles (soumis) la question de la préséance des anomalies du FSTA sur la RI, question paradoxale puisqu’aujourd’hui les anomalies du FSTA sont vues comme l’une des multiples facettes du syndrome de résistance à l’insuline (syndrome métabolique, SM), certainement pas comme un signe avant-coureur. Cette étude a comme modèle clinique le diabète gestationnel (DG) et donc, dans la troisième partie de cette introduction, nous disserterons sur ce type de diabète.
1.1 Structure du tissu adipeux 1.1.1 Les types de tissus adipeux
Le tissu adipeux représente environ 20 % de la masse corporelle chez l’homme et 30 % chez la femme en bonne santé et de poids normal (Deurenberg et al., 2001). Le TA blanc constitue le type dominant et se retrouve majoritairement ( 80 %) dans l’hypoderme (Chien et al., 1975), dans l’abdomen (Thomas et al., 1998), en périvasculaire et entourant les organes (Fitzgibbons et Czech, 2014; Sahin et al., 2015). Le TA intra-abdominal est situé en intra- et en rétropéritonéal. Il est aussi appelé TA viscéral (TAV). Il représente 10 à 20 % du TA chez l’homme et 5 à 10 % chez la femme (Lee et al., 2013). Le TA brun représente une faible part du TA chez l’homme adulte avec une histologie et des fonctions différentes du TA blanc. Dans l’organisme, les adipocytes bruns peuvent former de petits dépôts périvasculaires ou rétrocervicaux (Tam et al., 2012b) ou être dispersés dans le TA blanc (Cedikova et al., 2016). Pour plus de détails sur le TA brun, voir (Tam et al., 2012b).
2 Le TA blanc est formé d’adipocytes. L’adipocyte mature est une cellule sphérique contenant une gouttelette lipidique occupant environ 95 % de son volume. Par conséquent, la contenance lipidique du TA oscille autour de 85 à 90 %. Le reste de la cellule est formé d’un noyau plat situé en périphérie du cytoplasme, où se retrouvent quelques mitochondries, et du réticulum endoplasmique (Cedikova et al., 2016). La taille d’un adipocyte varie entre 20 et 200 μm. Un autre groupe cellulaire du TA blanc, appelé la fraction stromovasculaire (FSV), comprend des préadipocytes, des cellules endothéliales et leurs précurseurs, des péricytes, des cellules immunitaires (macrophages, lymphocytes, neutrophiles) et des cellules souches multipotentes. Quantitativement, les cellules de la FSV prédominent sur les adipocytes dans un rapport d’environ 3 à 4:1 (Lee et al., 2013).
La matrice extracellulaire (MEC) est une autre partie importante du TA. Chaque adipocyte est entouré d’une MEC épaisse appelée « basal lamina – lame basale » (Mariman et Wang, 2010). La MEC est composée de protéines structurales (collagène, surtout de type IV) et de différentes sortes de protéines d’adhésion (fibronectine, laminine, élastine, protéoglycane). Les protéines structurales sont nécessaires pour une architecture appropriée conditionnant la résistance mécanique du TA, l’intégrité de l’adipocyte et la différenciation des préadipocytes. Des protéines d’adhésion jouent également un rôle dans la différenciation des préadipocytes et la bonne communication entre la MEC et les adipocytes. Les protéoglycanes fonctionnent comme une réserve de molécules participant aux processus biologiques du TA, par exemple les facteurs de croissance (transforming growth factor - facteur de croissance transformant type ) ou les métalloprotéases matricielles (Divoux et Clément, 2011).
D’un point du vue macroscopique, le TA blanc est segmenté en lobules entourés de septas fibreux. Le dépôt principal du TA se trouve dans l’hypoderme de l’abdomen. Le TA sous-cutanée (TAsc) abdominal est divisé en deux couches par le fascia superficiel.
Les lobules de la couche supérieure (organisés en rayons de miel) sont relativement uniformes. Les lobules sont séparés les uns des autres par des septas bien définis qui assurent la forte résistance mécanique, l’élasticité de la couche supérieure du TAsc et la stabilité vis-à-vis de la peau (Lancerotto et al., 2011).
La couche inférieure se distingue par une moins bonne organisation des lobules qui sont plus plats et moins bien délimités. Les septas fibreux sont également moins bien définis.
3 Cette structure permet au TAsc une certaine mobilité vis-à-vis des muscles situés en dessous, recouverts par le fascia profond. Bien que la couche superficielle du TAsc se déploie de façon continue de l’abdomen au tronc et aux membres, le fascia superficiel s’attache au centre et plus bas aux structures ligamentaires (linea alba, le ligament inguinal) et à la crête de l’os pelvien et ferme ainsi la couche profonde du passage à la partie inférieure du corps (Lancerotto et al., 2011).
Sur le tronc, le TAsc est fait de deux couches organisées de la même façon. Sur les fesses ou les cuisses, il est divisé en plusieurs couches du fascia superficiel (Abu-Hijleh et al., 2006).
1.1.2 Le lit vasculaire du tissu adipeux
Le développement du TA dépend du développement concomitant de la microcirculation. Des études ont montré un rapport étroit entre les précurseurs des cellules adipogéniques et angiogéniques, et une coordination temporelle et spatiale entre adipogenèse et angiogenèse (Hausman et Richardson, 1982). Le début de l’accumulation des lipides dans les adipocytes est relié au développement de vastes réseaux capillaires (Hausman et Richardson, 1983). Durant ce développement, des capillaires du TA subissent des transformations structurelles facilitant le transport transendothélial des lipides (Crandall et al., 1997).
Dans le TA mature, le réseau capillaire est très dense. Des artérioles terminales d’un diamètre d’environ 20 μm avec des anastomoses nombreuses sont connectées par des shunts artério-veineux à des veinules collectrices d’un diamètre d’environ 25 μm (Myrhage et al., 1973). De ces shunts se déploie un réseau capillaire dense qui entoure probablement chaque adipocyte (Ballard et al., 1974; Gersh et Still, 1945). Le diamètre capillaire oscille entre 6 et 7 μm (Gersh et Still, 1945). Le ratio entre le nombre de capillaires et d’adipocytes, c’est-à-dire le flot sanguin basal rapporté à un seul adipocyte, reste relativement constant indépendamment de la taille de l’adipocyte (Crandall et al., 1997; Di Girolamo et al., 1971). Les capillaires du TA sont bordés par un endothélium attaché sur une membrane basale continue (Ballard, 1978) qui est entourée d’un tissu conjonctif lâche comprenant de fines fibres collagènes, des fibroblastes et quelques mastocytes.
Le sang dans le TAsc abdominal provient de l’artère épigastrique profonde (branches supérieure et inférieure) qui, par l’intermédiaire de quelques perforantes, approvisionnent la
4 peau et le TAsc. À la perfusion de la peau et de l’hypoderme abdominal participe aussi l’artère épigastrique superficielle, branches supérieure et inférieure (Rozen et al., 2008). Les deux systèmes s’anastomosent par l’intermédiaire de perforantes, et par les côtés avec les artères intercostales, sous-costales et lombaires, ainsi qu’avec l’artère iliaque superficielle circonflexe (Rozen et al., 2008). La vascularisation du TAsc a probablement un caractère bimodal : la couche profonde du TAsc est plus vascularisée que la couche supérieure. La zone intermédiaire autour du fascia superficiel est vascularisée par des branches terminales de vaisseaux provenant des couches superficielles et profondes (El-Mrakby et Milner, 2003). L’espace interstitiel est drainé par les systèmes veineux et lymphatique. Des veinules collectrices forment un plexus situé à la surface des lobules (Ryan et Curri, 1989). Dans les septas interlobulaires aussi bien que dans la zone de transition entre la peau et le TA, on trouve des veinules et des vaisseaux lymphatiques (Ryan, 1995). Comparativement à la paroi des capillaires sanguins, la paroi des capillaires lymphatiques est plus épaisse (Ryan, 1995) et leur diamètre est de 10-60 μm (Leak, 1976). Leur membrane basale est fenestrée, voire manquante (Hogan et Unthank, 1986). Les vaisseaux lymphatiques sont ancrés dans la MEC environnante par des fibres élastiques (Leak et Burke, 1968).
1.1.3 Le système nerveux du tissu adipeux
Le TA blanc est innervé par le système nerveux sympathique (SNS). Des fibres noradrénergiques sont situées dans l’adventice et la média des artères et des veines, mais elles suivent aussi des capillaires jusqu’à l’intérieur des lobules et semblent entrer en contact direct avec quelques adipocytes (Berthoud et al., 2006). Le TA blanc est aussi innervé par des fibres du SNS dont les neurotransmetteurs sont des neuropeptides, notamment le neuropeptide-Y (Giordano et al., 1996). Ces fibres s’adressent surtout aux artérioles et aux artères et co-synthétisent aussi, parfois simultanément, la noradrénaline. La signalisation efférente du TA vers le cerveau est assurée par des nerfs sensoriels qui présentent une immunoréactivité positive pour la substance P et le calcitonin-gene related peptide (Giordano et al., 2006). La présence du système parasympathique dans le TA blanc est peu vraisemblable. Cette problématique est discutée dans l’article No 3.
5 1.2 Résumé des fonctions métaboliques du tissu adipeux blanc
Le TA blanc, avec le parenchyme hépatique et les muscles squelettiques, joue un rôle majeur dans la régulation des flux énergétiques. La fonction principale du foie, à part le stockage limité du glycogène, est d’être un carrefour métabolique assurant la néosynthèse et l’interconversion des composantes du métabolisme glucidique, lipidique et protéinique. Le muscle est surtout responsable de la consommation d’énergie alors que le TA blanc sert de réserve (Coppack et al., 1990). Le stock d’énergie du TA blanc permet deux activités physiologiques significatives : 1) en cas de déficit énergétique et/ou en cas de besoin élevé (à jeun ou pendant une activité physique), le TA blanc fournit à l’organisme des acides gras libres (AGL) riches en énergie. Le glucose est donc épargné au profit des tissus gluco-dépendants et l’organisme est protégé d’un catabolisme excessif de protéines; 2) pendant et après un repas, l’énergie est stockée sous forme de triglycérides (TG) dans le TA et, ce faisant, l’organisme est protégé contre une augmentation prolongée de la lipémie. On estime que le TA stocke 35 % des TG ingérés dans les 6 heures suivant un repas (Coppack et al., 1990). Le stockage est assuré par les dépôts classiquement liés à la régulation du métabolisme lipidique (TAsc, TAV), mais aussi, comme il a été montré récemment, par le TA entourant les organes (Fitzgibbons et Czech, 2014; Marchington et Pond, 1990).
1.2.1 À jeun
À jeun, la lipolyse intracellulaire permet de libérer une quantité plus ou moins considérable d’AGL dans la circulation, selon les besoins (Coppack et al., 1990). Sous l’effet d’hormones lipolytiques (catécholamines, peptide natriurétique atrial /PNA/) il se produit une accumulation d’adénosines monophosphate cycliques (adénosine monophosphate cyclique, AMPc; guanosine monophosphate cyclique, GMPc). Ces adénosines phosphorylent le périlipine-1, l’adipose triglyceride lipase (ATGL) et la lipase hormonosensible (LHS) (Jocken et Blaak, 2008). Les AGL libérés gagnent les muscles squelettiques, le myocarde (-oxydation) et le foie (-oxydation ou synthèse de TG). Pendant l’activité physique, sous l’effet de ces hormones lipolytiques, la lipolyse intracellulaire augmente davantage la libération des AGL dans la circulation pour les muscles squelettiques et le myocarde (Chatzinikolaou et al., 2008). Conjointement à la libération des AGL, le TA libère le glycérol, pierre angulaire de la néoglucogenèse hépatique.
6 À jeun, à part la lipolyse intracellulaire, la lipoprotéine lipase (LPL) capillaire est aussi mise en jeu (Frayn et Humphreys, 2012; Samra et al., 1996a). Par son intermédiaire, les TG transportés par les VLDL (very low density lipoproteins) sont hydrolysés. La majorité des AGL ainsi libérés continuent dans la circulation (nous reprendrons ici le mot anglais spillover) (Ruge et al., 2009). Cependant, la lipolyse intravasculaire ne représente qu’une partie mineure de la production globale d’AGL par le TA à jeun (Frayn, 2002; Karpe et al., 2011). Quoique la majorité des AGL ainsi formés passe dans la circulation, environ 10 % est réestérifié en TG.
À jeun, le TA capte aussi de la circulation des corps cétoniques (hydroxybutyrate et acétylacétate) dont le destin n’est pas complètement élucidé : ils peuvent prendre part dans la lipogenèse de novo mais ils peuvent aussi être oxydés (Frayn et Humphreys, 2012).
1.2.2 En postpradial
En postprandial, la lipolyse intracellulaire est inhibée par l’action de l’insuline sur la phosphodiestérase 3B qui décompose l’AMPc. Il en résulte une suppression significative du taux d’AGL (et de glycérol) dans la circulation avec les niveaux les plus bas observés 90 minutes après un repas (Coppack et al., 1990). L’insuline stimule aussi la transcription de la LPL ainsi que son activité par son effet sur les événements post transcriptionnels et post translationnels (Goldberg et al., 2009). Le glucose participe aussi directement et indirectement, c’est-à-dire par la potentialisons de l’effet de l’insuline, à l’activation de la LPL (Wang et Eckel, 2009).
Après le déplacement de l’adipocyte sur le côté luminal des capillaires, la LPL attaque les TG circulants. Dans ce processus participe l’apolipoprotéine CII présente dans les VLDL et les chylomicrons, et la glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 qui fournit une plateforme pour l’hydrolyse des TG. Il a été démontré que l’extraction maximale des TG dans le TA (c’est-à-dire l’activité maximale de la LPL) 3 à 5 heures après un repas (Coppack et al., 1992) coïncide avec des niveaux maximaux postprandiaux de TG (Coppack et al., 1990; Frayn, 2002). Les TG sont présents surtout dans les lipoprotéines riches en TG (TGRL – triglyceride-rich lipoproteins), les VLDL et les chylomicrons. En postprandial, c’est surtout la concentration de chylomicrons qui augmente (avec un maximum observé 3 à 4 heures après un repas (Bickerton et al., 2007;
7 Potts et al., 1991)). Étant donné que la LPL s’attaque en priorité aux TG présents dans les chylomicrons (Bickerton et al., 2007; Xiang et al., 1999b), la fraction d’extraction des TG dans les chylomicrons atteint jusqu’à 21 à 47 % (Coppack et al., 1990; Ruge et al., 2009) comparativement à d’autres lipoprotéines ( 4 %) (Coppack et al., 1990). La demi-vie biologique de 5 minutes des TG dans les chylomicrons (Grundy et Mok, 1976) est beaucoup plus courte en comparaison avec la demi-vie des TG dans d’autres lipoprotéines ( 120 minutes) (Frayn, 2002). De plus, durant une journée normale comprenant trois repas, l’activité de la LPL est régulée à la hausse, ce qui mène à un métabolisme plus efficace des TG successivement ingérés (Ruge et al., 2009).
L’action de la LPL permet une grande production d’AGL, et ils peuvent être captés par l’adipocyte et réestérifiés en TG. Dans ce processus de captation, deux mécanismes s’appliquent : le transport facilité et le transport non-facilité. Le transport facilité s’effectue par l’entremise des protéines de transport (par exemple FABP – fatty acid binding protein, CD36 ou FAT - fatty acid translocase, ou FATPs - fatty acid transport proteines (Lobo et al., 2007; Schaffer, 2002)), il est saturable et se fait valoir à des concentrations faibles d’AGL (Schaffer, 2002). Le transport non-facilité est basé sur la diffusion d’AGL à travers la membrane plasmique, et il n’est probablement pas saturable. En fonction de l’état métabolique cellulaire, les deux processus sont soit bidirectionnels, soit unidirectionnels vers l’intérieur de la cellule quand le transport est lié à l’estérification des AGL. L’enzyme clé dans ce sens est l’enzyme attachée à la face interne de la membrane plasmique appelée acyl-CoA-synthetase (ACS) qui active les AGL en acyl-CoA et entame ainsi l’estérification des AGL. De cette façon, l’ACS maintient un gradient de concentration des AGL vers l’intérieur de la cellule. On suppose que l’ACS est facteur limitatif de la captation des AGL dans l’adipocyte (Lobo et al., 2007). L’activité de l’ACS et de certaines protéines de transport est, comme la LPL, régulée par l’insuline (Frayn, 2002; Grenier-Larouche et al., 2012; Lobo et al., 2007). L’estérification des AGL est conditionnée par l’approvisionnement du glycérol-3-phosphate (G-3-P) de la glycolyse. Pour ce faire, le TA capte le glucose de la circulation, sous l’influence de l’insuline (Ball et al., 1959; Rodbell, 1964) avec un effet maximum observé 30 à 60 minutes après un repas (Coppack et al., 1990). Ce captage augmente en postprandial environ trois fois et son intensité correspond à l’augmentation de l’insuline (Ruge et al., 2009). Le glycérol libéré par la lipolyse ne peut participer à l’estérification des
8 AGL car le TA ne contient pas la glycérolkinase, une enzyme-clé nécessaire à son activation (Lee et al., 2013).
À part les AGL formés par la lipolyse intravasculaire, le TA capte aussi des AGL directement de la circulation, mais ceci, en comparaison avec les AGL provenant des chylomicrons, n’est pas un processus majeur (Bickerton et al., 2007). Au contraire, et même en postprandial, une partie des AGL s’échappe dans la circulation. Il s’agit d’AGL libérés des chylomicrons, tandis que le spillover des VLDL n’est pratiquement pas détecté (Ruge et al., 2009). En postprandial, le spillover d’AGL peut créer jusqu’à 40 à 50 % de réserve plasmatique d’AGL (McQuaid et al., 2011). Bien que l’efficacité du captage des AGL par le TA (le trapping et la ré-estérification) augmente durant la journée (Ruge et al., 2009) (parallèlement à l’activité de la LPL) et que le spillover s’abaisse, il a été proposé que l’abondante activité de la LPL ne sert pas seulement au captage des AGL dans le TA, mais aide aussi au parenchyme hépatique avec la clairance des chylomicrons et de leurs remnants par la dissociation des TG en AGL (Coppack et al., 1990).
1.2.3 Les différences entre les principaux dépôts du tissu adipeux blanc
L’accumulation de la graisse diffère en fonction de la race, de l’âge et du sexe (Lee et al., 2013). Les femmes ont généralement une plus grande adiposité (en pourcentage de TA dans l’organisme) avec une accumulation préférentielle dans la région glutéofémorale sous-cutanée. Chez les hommes, l’accumulation du TA blanc prédomine dans la région abdominale (sous-cutanée et viscérale) (Geer et Shen, 2009). Parmi ces dépôts, il existe plusieurs différences fonctionnelles importantes (Tchkonia et al., 2013).
1.2.3.1 Les différences en activité lipolytique
Le TAV est considéré comme un tissu ayant une activité lipolytique élevée, suivi par le TAsc abdominal et finalement par le TAsc glutéofémoral (Arner, 1995; Mårin et al., 1992). Bien que la concentration d’AGL soit comparable dans le sang veineux du TA omental et du TAsc abdominal (Karpe et Tan, 2005) quand on tient compte du flot sanguin (Viljanen et al., 2009), la lipolyse semble être plus élevée dans le TAV. Cependant, si on considère le volume de chacun des dépôts adipeux, la participation du TAV sur la production totale d’AGL à jeun est seulement de 6 à 19 %, possiblement plus élevée chez les obèses (Jensen, 1995; Nielsen
9 et al., 2004). Chez les sujets sains, la participation du TAV dans l’afflux d’AGL directement vers le foie est aussi peu importante (5 à 10 %) (Nielsen et al., 2004). Bien que cette portion puisse dépasser 30 % chez des sujets ayant beaucoup de graisse viscérale, le TAsc reste la principale source d’AGL même pour le parenchyme hépatique (Nielsen et al., 2004).
Si on l’exprime en grammes de TA, l’activité lipolytique est généralement plus élevée dans la moitié supérieure du corps que dans la région glutéofémorale, surtout chez les hommes. Dans le même ordre d’idées, le TAsc fémoral est plus résistant à l’effet lipolytique de l’adrénaline (Manolopoulos et al., 2012). La participation du TAsc abdominal à la production totale d’AGL à jeun est approximativement de 55 à 58 % comparativement à la région fémorale (28 %) (Jensen, 1995; Meek et al., 1999).
En postprandial, il y a une suppression substantielle de la lipolyse dans tous les dépôts. Cependant, il a été démontré que le TA fémoral est le plus sensible à l’effet antilipolytique de l’insuline (Jensen, 1995), tandis que le TAV y est le plus résistant (Meek et al., 1999).
En résumé, pour ce qui est de la lipolyse, le plus actif des dépôts de TA est le TAV, suivi par le TAsc abdominal et finalement par le TAsc glutéofémoral. Néanmoins, quantitativement, la principale source d’AGL pour l’organisme provient du TAsc de la moitié supérieure du corps.
1.2.3.2 Les différences en capacité de captage des lipides
Des études ex vivo sur les adipocytes ont montré que chez les sujets minces, l’activité de la LPL était plus élevée dans les adipocytes fémoraux comparativement aux adipocytes abdominaux (Romanski et al., 2000). La comparaison entre le TAsc abdominal et le TAV a montré que l’activité de la LPL était comparable chez les hommes (Boivin et al., 2007), mais qu’elle était supérieure dans les adipocytes sous-cutanés chez les femmes (Tchernof et al., 2006).
Des études in vivo ont apporté des conclusions quelque peu différentes. Ces études sont basées soit sur l’administration de lipides marqués suivie d’une biopsie du TA, soit par le calcul de la différence artério-veineuse (A-V) des lipides. L’ingestion de TG marqués pendant un repas par des sujets sains a montré un captage plus efficace des AGL par le TAV comparativement au TAsc abdominal, avec une faible participation du TAsc fémoral (Jensen
10 et al., 2003; Romanski et al., 2000). La comparaison des tissus sous-cutanés a confirmé un captage plus efficace dans la région abdominale que dans la région glutéofémorale (Votruba et Jensen, 2006). Après avoir pris en considération le volume des différents dépôts, il a été montré que la clairance des TG en postprandial avait surtout lieu dans la région abdominale (~ 57 %), avec une participation comparable du TAV (~ 17 %) et du TAsc glutéofémoral (~ 16 %) (Jensen et al., 2003). La différence A-V des chylomicrons marqués chez les sujets minces a montré que le captage des TG est beaucoup plus élevé dans le TAV des hommes (~ 50 % de tous les chylomicrons) que dans celui des femmes (~ 13 %) (Nguyen et al., 1996). En postprandial, la captation directe d’AGL circulants a été plus efficace dans le TAsc abdominal que fémoral chez les hommes et les femmes (Koutsari et al., 2012).
À jeun, des AGL administrés par voie intraveineuse se sont accumulés en priorité dans le TAV par rapport au TAsc abdominal, et ce chez les deux sexes (Ali et al., 2011). Le stockage des AGL circulants dans le TAsc est généralement plus efficace chez les femmes et il y a une différence reliée au genre entre les dépôts sous-cutanés : les hommes stockent les AGL en priorité dans la région abdominale. Par contre, chez les femmes, les choses sont débattues : selon Shadid et al. (2007), il n’y a aucune différence (Shadid et al., 2007) tandis que pour Koutsari et al. (2011), le stockage serait plus élevé dans la région glutéale (Koutsari et al., 2011). Le stockage préférentiel des AGL dans le TA de la partie inférieure du corps chez les femmes a également été démontré pendant l’activité physique (Koutsari et al., 2012). La clairance des VLDL administrés par voie intraveineuse a été comparable dans les deux dépôts sous-cutanés chez les femmes (Nellemann et al., 2010).
Il semble donc que le TAV, sur le plan fonctionnel, capte davantage d’AGL que le TAsc. Ce taux de captation élevé s’accompagne d’une activité lipolytique intensifiée, deux facteurs qui indiquent que le TAV est métaboliquement plus actif que le TAsc. En revanche, comme dans le cas de la lipolyse, d’un point du vue quantitatif, le site majeur du captage des lipides est le TAsc de la région abdominale.
1.2.4 La dysfonction métabolique du tissu adipeux
L’obésité est la principale cause de la RI, du diabète de type 2 (DT2) et des maladies cardiovasculaires (Mlinar et Marc, 2011). Pour quantifier l’obésité, on utilise l’indice de masse corporelle (IMC). Cependant, l’IMC ne permet pas de déterminer la distribution de la
11 graisse corporelle (Britton et Fox, 2011). Il a été démontré que l’accumulation de graisse dans la région abdominale (distribution centrale, masculine) apporte un risque cardiométabolique élevé comparativement à l’accumulation dans la région glutéofémorale (distribution périphérique, gynoïde) (Kahn et al., 2001; Tchkonia et al., 2013) qui aurait au contraire un rôle plutôt protecteur (Snijder et al., 2005). Chez les sujets avec obésité centrale, on suppose que la cause principale du profil métabolique défavorable et du risque cardiométabolique élevé est l’accumulation de graisse viscérale. Les individus avec une augmentation du TAV présentent, pour un niveau donné d’adiposité, un plus grand risque de la RI et du SM (Després et Lemieux, 2006). Bien que le TAsc ait également été relié à des composantes du SM (obésité centrale, intolérance au glucose, dyslipidémie, hypertension artérielle) indépendamment de la graisse intra-abdominale, l’association entre le TAV et profil métabolique à risque reste la plus forte (Demerath et al., 2008; Fox et al., 2007).
L’augmentation du volume du TAV signifie afflux augmenté d’AGL dans la circulation portale (Nelson et al., 2007; Nielsen et al., 2004). Même si le TAsc demeure la source dominante des AGL, aux niveaux systémique et porte (Nielsen et al., 2004), chez les obèses le TAV peut devenir une source importante d’AGL pour le parenchyme hépatique, ce qui a des conséquences métaboliques néfastes (Jensen, 2006) : augmentation de la gluconéogenèse hépatique et de la résistance hépatique à l’insuline (Wueest et al., 2016) ce qui entraîne une augmentation de la formation des TGRL (Després et Lemieux, 2006). Le TAV est aussi une source majeure de plusieurs cytokines pro-inflammatoires comme l’interleukine-6 (IL-6) ou la protéine C réactive (CRP) dans la circulation portale (Lee et al., 2013). Ces cytokines participent à l’état inflammatoire chronique accompagnant le SM (Fontana et al., 2007).
La diminution du potentiel de réplication des adipocytes est une des principales raisons de la dysfonction du TAV en situation d’obésité (Tchkonia et al., 2013). En cas d’apport excessif d’énergie, l’hypertrophie des adipocytes viscéraux produit une perturbation des processus métaboliques, un déséquilibre hormonal et des cytokines, l’ensemble menant à l’apoptose précoce des adipocytes (Lafontan, 2014; Mårin et al., 1992). Ces mécanismes stimulent l’infiltration du TAV par des macrophages (Lee et al., 2013) qui est, chez les obèses, deux fois plus élevée dans le TAV que dans le TAsc (Cancello et al., 2006). Le taux
12 d’infiltration du TAV se rapporte aussi à la dysfonction hépatique et aux changements métaboliques délétères comprenant la RI (Cancello et al., 2006).
Pour résumer, il semble que le TAV n’est pas primordialement destiné à une accumulation des TG à long terme et que cette accumulation n’est que le reflet du dysfonctionnement du TAsc (Després et Lemieux, 2006). Le TAsc est désigné dans la littérature comme le dépôt physiologique, classically nonectopic (Britton et Fox, 2011; Grenier-Larouche et al., 2012; Mlinar et Marc, 2011; Tchkonia et al., 2013). Ainsi, lorsque la capacité de stockage du TAsc est altérée, les AGL sont dirigés vers les dépôts ectopiques, tels que le TAV, la graisse péricardique, ou périvasculaire, ou périrénale (les dépôts désignés classically ectopic) (Britton et Fox, 2011). Les individus capables d’accumuler des TG dans la graisse sous-cutanée, surtout dans la région glutéofémorale, sont longtemps protégés contre l’expansion des dépôts ectopiques et leurs impacts métaboliques et cardiovasculaires négatifs (Lemieux, 2004; Votruba et Jensen, 2006). De même, il a été montré que chez les hommes avec une expansion du TAV, une quantité élevée de graisse sous-cutanée abdominale diminue la probabilité de développement du SM (Demerath et al., 2008). Le traitement avec des glitazones (des agents pharmacologiques qui agissent sur des récepteurs nucléaires appelés peroxisome proliferator-activated receptors γ /PPAR γ/; par leur effet sur l’expression et répression de gènes spécifiques, ils augmentent le captage des AGL dans les adipocytes) a causé un accroissement de l’adiposité en général, mais surtout du TAsc abdominal, alors que le TAV était diminué, et que l’on notait une amélioration de la RI hépatique et systémique (Miyazaki et al., 2002). En revanche, la diminution de la graisse sous-cutanée abdominale par la liposuccion chez des personnes obèses n’a pas entraîné l’effet positif attendu sur les paramètres métaboliques et les facteurs de risque cardiovasculaires (Klein et al., 2004). Pourtant, l’atrophie ou la dystrophie du TAsc sont reliées à des altérations métaboliques de l’obésité : développement rapide de la RI, du DT2, de la stéatose hépatique et de la dyslipidémie avec des TG élevés. La gravité de ces altérations corrèle avec le degré de diminution du TA (Simha et Garg, 2006).
Simultanément avec le stockage dans les dépôts ectopiques, et suite à la saturation des mécanismes de stockage, les AGL sont dispersés dans des tissus non-adipocytaires tels que le foie, le muscle, le myocarde ou les cellules du pancréas. En association avec des composantes locales déficientes, par exemple l’oxydation des AGL altérée
(Grenier-13 Larouche et al., 2012; Stinkens et al., 2015) ou la diminution de la perfusion musculaire (Labbé et al., 2011), le dysfonctionnement de ces organes mène, entre autres, vers la RI, hépatique ou systémique, ou un défaut de sécrétion de la cellule β. L’impact néfaste du stockage excessif des AGL dans les tissus non-adipocytaires est désigné par le terme lipotoxicité (Frayn, 2002).
L’accumulation des lipides dans les tissus non-adipocytaires est due à l’augmentation du captage des AGL dont la source peut être soit des AGL circulants, soit des TG. Des niveaux élevés d’AGL à jeun et/ou une altération de leur métabolisme postprandial ont été démontrés chez les personnes avec un DT2 (Annuzzi et al., 2004; Arner et Rydén, 2015; Labbé et al., 2011; Normand-Lauzière et al., 2010; Riemens et al., 2000). Néanmoins, ces constatations ne s’appliquent pas à tous les sujets DT2, en particulier si le diabète est bien contrôlé (Rivellese et al., 2004; Søndergaard et al., 2012). Ce désaccord est encore probablement plus important en cas d’obésité simple (c’est-à-dire obésité, sans manifestation clinique telles qu’hypertension, diabète, dyslipidémie, etc.). Quelques études sur l’obésité simple ont montré des niveaux élevés d’AGL à jeun (Arner et Rydén, 2015; Nielsen et al., 2004) mais d’autres travaux, y compris des méta-analyses, n’ont pas confirmé ces conclusions (Karpe et al., 2011). Des études de clamp ont comparé des sujets obèses, des sujets DT2 et des sujets contrôles minces. On a ainsi montré un taux d’apparition artérielle des AGL pour des concentrations faibles d’insuline plus élevé chez les sujets obèses et les sujets DT2. Cependant, ceci n’était pas causé par la pire suppressibilité de la lipolyse intracellulaire, mais par le spillover augmenté des AGL durant la lipolyse intravasculaire en raison des niveaux de TG augmentés (Riemens et al., 2000).
En même temps, il a été démontré qu’avec l’accroissement de l’obésité, la lipolyse par gramme de tissu est régulée à la baisse via la résistance aux catécholamines. Ainsi, l’organisme se protège contre des niveaux toxiques d’AGL (Frayn, 2005; Grenier-Larouche et al., 2012). Quoique ce mécanisme de protection puisse être outrepassé par l’augmentation de la masse grasse (Mitrou et al., 2010; Mittendorfer et al., 2009), même des changements importants d’adiposité ne sont que peu reflétés par des variations des niveaux d’AGL à jeun (McQuaid et al., 2011; Mitrou et al., 2010; Reeds et al., 2006). Il y a aussi des études qui n’ont pas démontré de lien entre les AGL et le degré d’adiposité (Johns et al., 2014).
14 Des tissus peuvent également être exposés aux AGL pendant la période postprandiale à cause de la suppression insuffisante de la lipolyse résultant de la RI (Frayn, 2005), mais la littérature laisse planer des doutes sur cette affirmation (Karpe et al., 2011). Une certaine perturbation de la suppression de la lipolyse a été retrouvée chez des sujets DT2 et ou insulinorésistants (Mook et al., 2004; Søndergaard et al., 2012), mais aucune différence n’a été démontrée chez des sujets obèses vs des sujets minces (McQuaid et al., 2011; Mitrou et al., 2010; Riemens et al., 2000). De surcroît, quelques études ont mis en doute l’altération de la suppressibilité postprandiale de la lipolyse par l’insuline et ce, même chez les personnes diabétiques (Riemens et al., 2000).
Il semble donc que le rôle des AGL dans l’accumulation des TG et la lipotoxicité qui en découle soit négligeable chez les sujets porteurs d’obésité simple (Arner et Rydén, 2015; Johns et al., 2014; Labbé et al., 2011). L’effet néfaste des AGL semble apparaître avec le développement concomitant du SM ou du DT2. Une position particulière peut avoir l’afflux des AGL vers le foie via la circulation portale, aussi bien à jeun qu’en postprandial, du fait de l’expansion du TAV, de la résistance à l’effet antilipolytique de l’insuline et/ou du spillover surabondant des AGL dans le TAV pendant la lipolyse intravasculaire (Nelson et al., 2007).
Le second véhicule des AGL vers les tissus non-adipocytaires, surtout au stade de l’obésité simple, sont les TG (Labbé et al., 2011). On peut supposer que des tissus ayant une activité LPL élevée, par exemple le muscle squelettique ou le myocarde (Wang et Eckel, 2009), mais aussi les cellules du pancréas (Marshall et al., 1999), vont être exposés aux AGL provenant des TG en priorité (Marshall et al., 1999; Wang et Eckel, 2009). Le taux excessif de TG chez les personnes obèses vient de la clairance altérée des TG des chylomicrons, c’est-à-dire la forme postprandiale des TG en circulation; l’effet de l’obésité sur la clairance des VLDL est moins évidente (McQuaid et al., 2011). La clairance insuffisante des chylomicrons entraîne une exposition considérablement prolongée de l’organisme à des niveaux nocifs de TG. La réponse de la LPL à l’insuline après un repas est altérée dans le TA, mais pas dans les muscles squelettiques. Ainsi, le réacheminement des TG à des tissus non-adipocytaires est facilitée (Wang et Eckel, 2009). Au foie, les AGL des chylomicrons incomplètement dégradés sont utilisés pour la resynthèse des TG, et ils circulent à nouveau sous forme de VLDL (Knuth et Horowitz, 2006; McQuaid et al., 2011).
15 Ainsi, l’altération de la clairance postprandiale se projette au fur et à mesure aux niveaux élevés de TG à jeun (Fielding, 2011). Les taux élevés de TG observés d’abord en postprandial et ensuite à jeun ont été trouvés chez les personnes obèses (Annuzzi et al., 2008; McQuaid et al., 2011), diabétiques (Annuzzi et al., 2004; Annuzzi et al., 2008; Søndergaard et al., 2012) et chez des sujets à risque de développement du DT2 (Normand-Lauzière et al., 2010). Des niveaux de TG constamment élevés dans les chylomicrons, les VLDL et leurs remnants (chylomicrons-remnants, lipoprotéines de densité intermédiaire /IDL/) induisent un profil très pro-athérogénique des lipides en circulation (augmentation des TG et des lipoprotéines de basse densité /LDL/ petites et denses, diminution des lipoprotéines de haute densité /HDL/) (Kolovou et al., 2005). L’importance des TG par rapport aux AGL est soulignée chez des sujets avec atrophie complète du TA : leur hypertriglycéridémie les place à haut risque de diabète précoce et d’athérosclérose accélérée, malgré des niveaux normaux d’AGL (Garg, 2000).
De tout ce qui a été mentionné, on peut tirer deux conclusions : 1) la cause fondamentale de l’expansion du TA ectopique (par exemple TAV) et de l’infiltration adipeuse des tissus non-adipocytaires est la défaillance de la capacité de stockage du TAsc (McQuaid et al., 2011) de la région abdominale et du tronc. En conséquence, toute recherche sur ce dépôt lipidique est pertinente pour évaluer les impacts métaboliques et cardiovasculaires de l’obésité (Coppack et al., 1992); 2) les complications métaboliques de l’obésité semblent débuter par des anomalies du métabolisme des TG en période postprandiale (Eliasson et al., 1997; Kolovou et al., 2005). En conséquence, toute recherche sur le métabolisme lipidique postprandial va permettre d’améliorer notre connaissance des stades initiaux des complications cardiométaboliques reliées à l’obésité.
La défaillance de la capacité de stockage, nous l’avons montré, est une des manifestations princeps du dysfonctionnement complexe du TA qui comprend des altération hormonales, immunitaires, structurales et aussi vasculaires (Frayn, 2002; Lafontan, 2014). C’est pourquoi la recherche sur la perfusion du TA est une partie intégrante des études sur la physiologie et la physiopathologie du TA.
16 1.3 Le flot sanguin du tissu adipeux
1.3.1 Le concept général de la microcirculation
Du point du vue fonctionnel, on peut diviser l’arbre artériel en trois : 1) les grandes et moyennes artères, appelées « vaisseaux conducteurs », qui ont un impact minimal sur la résistance vasculaire périphérique; 2) les petites artères, d’un diamètre de 100 à 200 µm, qui ont déjà un impact partiel sur la résistance vasculaire et la régulation de la tension artérielle (par exemple les artères nutritives dans des muscles, qui diminuent la tension intraluminaire de 20 à 35 % (Williams et Segal, 1993)); 3) la microcirculation, artérioles et capillaires, se trouve à l’intérieur des organes, leur diamètre est inférieur à 100 à 150 µm (De Boer et al., 2012; de Wit et Wolfle, 2007). Aux capillaires succèdent les veinules, qui, conceptuellement, font aussi partie de la microcirculation. Cette microcirculation est le siège principal de la résistance vasculaire périphérique et elle est responsable de la perfusion des organes, de l’oxygénation des tissus et des échanges de métabolites.
Un diamètre minuscule des vaisseaux pose un problème méthodologique pour la mesure du micro flot sanguin et donc, pour l’évaluation de la microcirculation. La majorité des études ont porté sur les vaisseaux conducteurs, plus accessibles, et les résultats de ces travaux ont été appliqués à la microcirculation. L’utilisation de techniques plus perfectionnées a permis des lectures directes ou indirectes, en rendant possible la visualisation ou l’évaluation fonctionnelle de la microcirculation. On s’est ainsi aperçu que le comportement et la régulation des vaisseaux à ce niveau ont des particularités propres et importantes (de Wit et Wolfle, 2007).
La perfusion des organes est assurée par la vasodilatation des vaisseaux. Cette vasodilatation est due à une myorelaxation des cellules musculaires lisses vasculaires (vascular smooth muscle cells, VSMC), par effet direct (endothélium-indépendante) sur le VSMC de divers agents, mais surtout par la vasodilatation endothélium-dépendante qui est vue comme indispensable au contrôle hémodynamique (Serban et al., 2010). L’endothélium stimulé va produire des facteurs vasodilatateurs sous l’influence du shear stress et d’une série de substances produites systémiquement ou localement par exemple les catécholamines, l’acétylcholine (Ach), le VIP, la bradykinine, le glucagon-like peptide, et bien d’autres vasodilatateurs.
17 La cellule endothéliale répond par la production de produits actifs, le plus connu et le plus répandu est l’oxyde nitrique (NO), qui diffuse par l’espace intercellulaire dans les VSMC où il active la guanylatcyclase et la production de GMPc qui stimule la protéine-kinase G (PKG) (Serban et al., 2010). La PKG activée stimule plusieurs systèmes effecteurs, soit les canaux calciques, l’hyperpolarisation des VSMC, ou la phosphorylation de la myosin light chain kinase menant à la phosphorylation diminuée de la myosine. Un autre facteur vasodilatateur libéré par les cellules endothéliales est la prostacycline (PGI2), produit de
l’action de la cyclo-oxygénase sur l’acide arachidonique. Sous l’influence de PGI2, l’AMPc
augmente dans les VSMC, ce qui active la protéine-kinase A et les systèmes effecteurs vasodilatateurs, comme sous l’effet du NO. Mais il semble que l’importance de la PGI2 soit
assez secondaire par rapport au NO. Il s’agit plutôt d’un mécanisme vasodilatateur de réserve, mais qui peut devenir important chez sujets avec une biodisponibilité de NO abaissée (Flammer et Lüscher, 2010). Le NO et la PGI2 sont les facteurs vasodilatateurs
principaux dans les vaisseaux conducteurs mais leur importance diminue avec le diamètre des vaisseaux. Les études animales et humaines ont montré qu’à l’échelle de la microcirculation, le blocage de la production du NO ou de la PGI2 affecte peu la
vasodilatation et n’a même aucune influence, d’où l’existence d’un possible troisième facteur. Considérant que l’hyperpolarisation des VSMC est le mécanisme effecteur majeur dans la microcirculation, ce troisième facteur a été nommé « endothelium-derived hyperpolarising factor (EDHF) » (de Wit et Wolfle, 2007). En fait, on s’est rendu compte que le EDHF vaut pour plusieurs facteurs ou processus possibles initiés dans les cellules endothéliales qui mènent à l’hyperpolarisation des VSMC. Et donc le terme « hyperpolarisation endothélium-dépendante (endothelium-dependent hyperpolarisation, EDH) » a été introduit (Feletou, 2016). L’hyperpolarisation des VSMC peut être causée par certaines substances produites ou libérées par les cellules endothéliales, par exemple le potassium, le peroxyde d’hydrogène (Matoba et al., 2002), des produits du cytochrome P450 (par exemple des acides epoxyeicosatrioenoïques) ou peptide natriurétique C (de Wit et Wolfle, 2007). Un autre mécanisme important pour l’hyperpolarisation des VSMC est celui des jonctions communicantes myoendothéliales (myoendothelial gap junctions, MEGJ). Elles assurent la propagation immédiate de l’hyperpolarisation des cellules endothéliales dans la musculature vasculaire. L’importance des MEGJ augmente avec la diminution du
18 diamètre des vaisseaux, car une proximité entre les cellules endothéliales et musculaires s’intensifie. De plus, des jonctions existent aussi entre les cellules endothéliales (Feletou, 2016). On voit donc que les segments de la microcirculation travaillent de façon très coordonnée et qu’après un stimulus, la vasodilatation se propage sur l’arbre artériolaire et capillaire (de Wit et Wolfle, 2007).
1.3.2 Dysfonctionnement de la microcirculation
Les variations de la microcirculation et donc, de la perfusion tissulaire, résultent de changements structuraux et fonctionnels interreliés. Une aberration structurale fondamentale est la raréfaction des vaisseaux dans la microcirculation qui est souvent mais pas uniquement mise en rapport avec la diminution de la disponibilité du NO et le stress oxydatif (Villela et al., 2009). Des changements de fonction se lient à une augmentation insuffisante du flot sanguin, mais aussi à une altération de sa distribution en réponse aux demandes métaboliques des tissus.
1.3.2.1 Les mécanismes du dysfonctionnement de la microcirculation
Étant donné le rôle central de l’endothélium dans la régulation du tonus vasculaire, la dysfonction endothéliale (DE) joue un rôle pathogène majeur pour les gros vaisseaux comme pour la microcirculation (De Boer et al., 2012; Park et al., 2001). La DE est le plus souvent associée à une diminution de la biodisponibilité du NO (le stress oxydatif est impliqué) avec comme conséquence un comportement constricteur, inflammatoire et pro-thrombogénique des cellules endothéliales.
Des anomalies au niveau de l’EDH peuvent aussi, et de façon considérable, contribuer à la DE. Dans des modèles animaux et humains d’hypertension ou de DT2, on a montré des modifications d’expression ou de fonction de plusieurs médiateurs de l’EDH, en particulier les canaux ioniques et les MEGJ (Feletou, 2016). Par exemple, chez les personnes DT2 subissant un pontage aorto-coronarien, on a retrouvé une inactivation des small/intermediate conductance of calcium-activated potassium channels (SKCa/IKCa) qui
sont responsable de l’efflux de potassium et de l’hyperpolarisation des cellules endothéliales et des VSMC (Liu et al., 2015). Il a aussi été établi qu’une altération de l’hyperpolarisation peut se produire dans les VSMC. Le DT2 est relié à une altération des large conductance of