Étude du profil des populations cellulaires du sang en
vue d’une application au diagnostic du lupus
érythémateux disséminé
Mémoire
Jennifer Lemieux
Maîtrise en microbiologie
Maître ès sciences (M.Sc.)
Québec, Canada
© Jennifer Lemieux, 2016
Étude du profil des populations cellulaires du sang en
vue d’une application au diagnostic du lupus
érythémateux disséminé
Mémoire
Jennifer Lemieux
Sous la direction de :
André Darveau, directeur de recherche
Sonia Néron, codirectrice de recherche
iii
Résumé
Le lupus érythémateux disséminé (LED) est une maladie auto-immune systémique dont le diagnostic est très complexe. Le clinicien doit baser son diagnostic sur une liste de 11 critères reliés à des observations cliniques et à des mesures sérologiques. Afin de faciliter ce diagnostic, plusieurs groupes recherchent de nouveaux marqueurs biologiques quantifiables. C’est dans ce but que la cytométrie en flux a été utilisée afin de comparer les cellules du sang des patients et celles de sujets sains. La caractérisation exhaustive des sous-populations cellulaires montre que l’expression de HLA-DR est amplifiée chez les patients même si la maladie est inactive. De plus, l’analyse du contenu sérique en cytokines inflammatoires a montré que la quantité de GM-CSF était plus importante chez les patients LED. Nos travaux suggèrent que HLA-DR et GM-CSF pourraient être considérés comme des candidats intéressants dans les études sur le diagnostic du LED.
iv
Table des matières
Résumé ... iii
Table des matières ... iv
Liste des tableaux ... vii
Liste des figures ... viii
Liste des abréviations ... ix
Avant-propos ... xi
1. Introduction ... 1
1.1 Immunité ... 1
1.1.1 Réponse immunitaire et intermédiaires cellulaires ... 1
1.1.1.1 Réponse immunitaire innée ... 1
1.1.1.2 Réponse immunitaire adaptative ... 2
1.1.2 Les lymphocytes B ... 3
1.1.3 Les lymphocytes T ... 4
1.1.4 Messagers ... 5
1.2 Lupus érythémateux disséminé ... 6
1.2.1 Causes ... 6
1.2.2 Diagnostic ... 7
1.2.3 Évaluation thérapeutique ... 8
1.2.4 Traitements ... 11
1.2.5 Cellules impliquées dans la pathogénèse ... 11
1.2.5.1 Modèles de souris ... 11 1.2.5.2 Lymphocytes T ... 12 1.2.5.3 Lymphocytes B ... 13 1.2.6 Cytokines ... 14 1.2.6.1 IL-6 ... 14 1.2.6.2 IL-10 ... 14 1.2.6.3 IL-17 ... 14 1.2.6.4 Interférons ... 15 1.2.6.5 B-cell-activating factor ... 15
v
1.3 Cytométrie en flux et usage clinique ... 16
1.3.1 Human Immunophenotyping Consortium ... 16
1.3.2 Outils d’analyses de données multiparamétriques ... 18
1.3.2.1 SPADE et viSNE ... 18
2. Hypothèse ... 19
3. Objectifs ... 20
4. Méthodologie ... 21
4.1 Source des cellules mononucléées du sang, du sérum et du plasma ... 21
4.1.1 Participants en santé ... 21
4.1.2 Patients LED ... 22
4.2 Analyses phénotypiques ... 23
4.2.1 Marquage extracellulaire ... 23
4.2.2 Anticorps ... 23
4.2.3 Acquisition et analyse des données ... 26
4.2.3.1 Spanning-tree Progression Analysis of Density-normalized Events (SPADE) ... 27
4.3 Dosage des facteurs solubles ... 28
4.3.1 ELISArray ... 28
4.3.2 Essai Bioplex ... 28
4.4 Activation in vitro ... 29
4.4.1 Milieu de culture ... 29
4.4.2 Essais d’activation ... 29
4.4.3 Mesure des facteurs solubles dans les surnageants de culture ... 30
4.5 Analyses statistiques ... 30
5. Résultats ... 32
5.1 Caractéristiques médicales des patients et des participants à l’étude ... 32
5.2 Patron des cytokines ... 35
5.2.1 Facteurs solubles dans le sérum des patients LED ... 37
5.2.3 Sécrétion in vitro ... 43
5.3 Étude comparative de l’effet de la congélation sur les PBMC ... 45
5.3.1 Effets de la congélation sur les lymphocytes et les monocytes ... 45
vi
5.4 Caractérisation du phénotype LED ... 52
5.4.1 Stratégie pour l’établissement des arbres SPADE ... 52
5.4.2 Monocytes, cellules dendritiques et cellules NK ... 54
5.4.3 Lymphocytes B ... 59
5.4.4 Vue d’ensemble des cellules B, NK, DC et monocytes chez les patients LED ... 59
5.4.5 Lymphocytes T ... 64
5.4.5.1 Fine distribution des cellules CD4+ et CD8+ ... 64
5.4.5.2 Visualisation avec SPADE des lymphocytes T ... 66
5.4.5.3 Proportion des cellules T HLA-DR+ ... 74
5.4.6 Lymphocytes T auxiliaires ... 76
5.4.7 Lymphocytes T régulateurs ... 80
6. Discussion ... 84
6.1 Des concentrations sériques de GM-CSF plus élevées chez les patients LED ... 84
6.2 Les cellules des patients LED sont plus activées ... 85
6.2.1 Survol des variations dans les populations ... 86
6.2.2 Expression élevée de HLA-DR ... 86
6.3 Une période de repos de 1h est bénéfique pour les cellules congelées ... 87
7. Conclusion ... 89
vii
Liste des tableaux
Tableau 1.1: Médicaments impliqués dans le DILE. ... 7
Tableau 4.1: Liste des anticorps monoclonaux utilisés pour la caractérisation des PBMC ... 25
Tableau 4.2 : Panels des cibles cellulaires en fonction des marqueurs de surface. ... 26
Tableau 5.1 : Caractéristiques médicales des patients atteints de LED. ... 34
Tableau 5.2 : Niveau des cytokines dans le sérum de patients sélectionnés. ... 42
Tableau 5.3 : Effet de la congélation sur la distribution des sous-populations de lymphocytes T 51 Tableau 5.4 : Disposition des arbres SPADE pour les participants en santé. ... 53
Tableau 5.5 : Disposition des arbres SPADE pour les patients LED. ... 53
Tableau 5.6 : Distribution des monocytes, des cellules DC et NK, et des lymphocytes B chez les patients. ... 63
Tableau 5.7 : Distribution des sous-populations de lymphocytes T chez les patients. ... 65
viii
Liste des figures
Figure 1.1 : Étapes de la réponse immunitaire adaptative. ... 2
Figure 1.2 : Critères de diagnostic du LED ... 8
Figure 1.3 : Critères d’évaluation de l’activité de la maladie selon le SLEDAI. ... 10
Figure 1.4 : Sous-populations cellulaires des PBMC. ... 17
Figure 5.1 : Niveau des cytokines inflammatoires dans le sérum des patients. ... 35
Figure 5.2 : Patron de sécrétion des cytokines inflammatoires dans le sérum des patients ... 37
Figure 5.3 : Niveau de RANTES suite à l’activation in vitro des PBMC. ... 42
Figure 5.4 : Effet de la congélation sur la distribution des cellules mononucléées. ... 44
Figure 5.5 : Effet de la congélation sur la distribution des cellules mononucléées. ... 46
Figure 5.6 : Variation des populations de lymphocytes T CD4+ et CD8+ avant et après congélation ... 47
Figure 5.7 : Délinéation des monocytes, des cellules dendritiques et des cellules NK. ... 53
Figure 5.8 : Expression de HLA-DR sur les monocytes, DC et NK de participants en santé. ... 55
Figure 5.9 : Expression de HLA-DR sur les monocytes, DC et NK des patients LED. ... 56
Figure 5.10 : Délinéation des cellules mémoires dans les lymphocytes B CD19+ ... 58
Figure 5.11 : Expression de CD38 sur des lymphocytes B CD19+ des participants en santé. ... 59
Figure 5.12 : Expression de CD3 sur des lymphocytes B CD19+ des patients LED. ... 60
Figure 5.13 : Délinéation des sous-populations de lymphocytes T CD3+. ... 65
Figure 5.14 : Expression de CD38 sur des lymphocytes T CD3+ de participants en santé ... 66
Figure 5.15 : Expression de CD38 sur des lymphocytes T CD3+ de patients LED. ... 67
Figure 5.16 : Expression de HLA-DR sur les lymphocytes T CD3+ de participants en santé. ... 70
Figure 5.17 : Expression de HLA-DR sur les lymphocytes T CD3+ de patients LED ... 71
Figure 5.18 : Délinéation des sous-populations de lymphocytes T auxiliaires. ... 75
Figure 5.19 : Expression de HLA-DR sur les lymphocytes T auxiliaires de participants en santé. ... 76
Figure 5.20 : Expression de HLA-DR sur des lymphocytes T auxiliaires des patients LED. ... 77
Figure 5.21 : Délinéation des lymphocytes T régulateurs ... 79
Figure 5.22 : Expression de HLA-DR sur les lymphocytes T régulateurs des participants en santé. ... 80
ix
Liste des abréviations
ACD : Acid–Citrate-Dextrose
ACR : American College of Rheumatology ADNsb : Acide désoxyribonucléique simple brin ADNdb : Acide désoxyribonucléique double brin AF-488 : Alexa-Fluor 488
APC : Allophycocyanin
BAFF : B cell-activating factor
BAFF-R : B cell-activating factor receptor BCMA : B-cell maturation antigen
BCR : B-cell receptor
BILAG : British Isles Lupus Assessment Group BPFM : Bovine protein free medium
BSA : Bovine serum albumin BV421 : Brilliant Violet 421 CCR : CC-chemokine receptor
CD : Cluster of differenciation, marqueur de surface CMH : Complexe majeur d'histocompatibilité
CHUS : Centre hospitalier universitaire de Sherbrooke CXCL : CXC-chemokine ligand
CXCR : CXC-chemokine receptor DC : Dendritic cell
DILE : Drug-induced lupus erythematosus DMSO : Diméthysulfoxyde
DNAse : Deoxyribonucléase
DPBS : Dulbecco's Phosphate-Buffered Saline
ECLAM : European Consensus Lupus Activity Measurement ELISA : Enzyme-linked immunosorbent assay
FBS : Fetal bovine serum
FCB : Fluorescent Cell Barcoding FDA: Food and Drug Administration FMO : Fluorescent Minus One FoxP3 : Forkhead box P3
GM-CSF : Granulocyte-macrophage colony-stimulating factor HIC : Human Immunophenotyping Consortium
HIV : Human immunodeficiency virus HLTV : Human T-lymphotropic virus HSA : Human serum albumin
x
Ig : Immunoglobuline
IGF : Insulin-like growth factor IL : Interleukine
IMDM : Iscove’s modified Dulbecco’s medium ITS : Insuline, transferrine et sélénium
IVIg : Immunoglobulines intraveineuses LDL : Low-density lipoprotein
LED : Lupus érythémateux disséminé LP-1 : Mélange lipidique défini LPS : Lipopolysaccharide
MFI : Mean fluorescence intensity
MRL/lpr : Murphy Roths Large/lymphoproliferation
NIAID : National Institute of Allergy and Infectious Disease NFκB : Nuclear factor-B
NK : Natural killer
NZB : New Zealand Black NZM : New Zealand Mixed NZW : New Zealand White
PAMP : Pathogen-associated molecular pattern PBMC : Peripheral blood mononuclear cells PMA : Phorbol 12-myristate 13-acetate PE : Phycoerythrin
PE-Cy7 : PE-cyanine 7
PerCP-Cy5.5 : Peridin-chlorophyll protein-cyanine 5.5 PRR : Pattern recognition receptor
RANTES : Regulated on Activation, Normal T Cell Expressed and Secreted SLAM : Systemic Lupus Erythematosus Activity Measure
SLE : Systemic lupus erythematosus SLEDAI : SLE disease activity index
SLICC : Systemic Lupus International Collaborating Clinics
SPADE : Spanning-tree Progression Analysis of Density-normalized Events Syk : Spleen tyrosine kinase
TACI : Transmembrane activator and CAML interactor TCR: T cell receptor, récepteur des lymphocytes T Th : T helper, lymphocyte T auxiliaire
TMB : 3,3′,5,5′-Tetramethylbenzidine TLR: Toll-like receptor
Treg : Regulatory T cell, lymphocyte T régulateur V450 : Violet 450
xi
Avant-propos
Ces belles années de maîtrise à Héma-Québec n’auraient pas été possibles sans ma directrice de recherche, Dr. Sonia Néron, qui m’a d’abord acceptée dans l’équipe en tant que stagiaire et, par la suite, en tant qu’étudiante à la maîtrise. Je tiens à la remercier pour ses bons conseils, pour sa grande disponibilité et surtout pour ses encouragements à persévérer. Je voudrais également remercier mon co-directeur, André Darveau, pour ses idées et ses commentaires pertinents dans l’avancement de ma maîtrise.
Je voudrais remercier les autres membres de mon comité d’encadrement, Fatiha Chandad et Caroline Gilbert, pour leurs précieux conseils et pour l’intérêt porté à mon projet de maîtrise. Je tiens également à remercier Éric Boilard d’avoir accepté à la dernière minute de faire partie de mes examinateurs.
Ce projet de maîtrise n’aurait pas été possible sans la collaboration de Dr Gilles Boire, de ses collaborateurs et de son équipe au Centre hospitalier universitaire de Sherbrooke. Je remercie aussi Mme Marie-Ève Allard, infirmière coordonnatrice de Héma-Québec, pour son efficacité. Je tiens à remercier tout spécialement les patients de Dr Boire et les donneurs de plaquettes du Centre Globule à Place Laurier pour leur participation à cette étude.
Je tiens aussi à remercier les membres de l’équipe du Dr. Néron (autant actuels qu’anciens) qui ont contribué à ma réussite : Annie Roy, Carl Simard, Marc Cloutier, Philippe Nadeau, Guillaume Bonnaure, Rayelle Itoua Maïga, Catherine Gervais St-Amour et David Gaumond. Je ne voudrais surtout pas oublier de remercier tous les autres étudiants pour les moments de réflexion, mais aussi pour les moments de divertissement.
Évidemment, je ne serais peut-être pas rendue où je suis en ce moment sans le support inconditionnel et les encouragements de mes amis et de ma famille. Je pense plus spécifiquement à ma mère qui m’a toujours rappelé l’importance de ne pas abandonner, en particulier lorsqu’on est si près du but.
1
1. Introduction
1.1 Immunité
1.1.1 Réponse immunitaire et intermédiaires cellulaires
1.1.1.1 Réponse immunitaire innée
Les pathogènes qui franchissent les épithéliums, première barrière physique contre les infections, peuvent ensuite coloniser les tissus et établir un foyer infectieux. Lorsque les pathogènes traversent cette barrière, c’est la réponse immunitaire innée qui prend la relève. Les pathogènes portent des motifs moléculaires appelé PAMPs (pathogen-associated molecular patterns) qui sont des motifs hautement conservés présents sur les membranes des pathogènes [1]. Les cellules du système immunitaire reconnaissent ces PAMPs grâce à des récepteurs appelés pattern
recognition receptors (PRRs), dont font partie les récepteurs de type Toll (TLR, Toll-Like Receptors) [2]. Ces derniers sont localisés à la surface des cellules immunitaires ou à l’intérieur
de la cellule, sur la paroi des endosomes, ce qui permet la reconnaissance des motifs microbiens comme l’ADN. Ainsi, la liaison des cellules immunitaires et des agents infectieux provoque la phagocytose de ces derniers grâce aux macrophages, cellules dérivant des monocytes [3]. Cette étape donne une réponse immédiate qui ne prend que quelques heures, c’est la phase innée. Par la suite, c’est la réponse innée induite qui est déclenchée. Une réaction inflammatoire se produit en raison de l’interaction entre le pathogène et les phagocytes. L’inflammation joue un rôle important dans l’immunité puisqu’elle induit la coagulation, elle aide à la réparation des tissus et elle permet d’attirer diverses molécules et cellules sur le site de l’infection. Ainsi, cette phase permet la migration de cytokines, de neutrophiles et de monocytes pour combattre l’infection [4]. Chez l’humain, d’autres acteurs cellulaires sont impliqués dans la réponse innée comme les basophiles, les éosinophiles, les cellules dendritiques et les cellules tueuses (natural killers ou NK). Un autre mécanisme important lors de la réponse innée est le système du complément. Ce dernier est un système composé de protéines plasmatiques et qui peut être activé par l’une des trois voies suivantes : la voie classique, la voie des lectines et la voie alternative [5]. Bien que la défense innée permette l’élimination constante de pathogènes, certains résistent et doivent, par la suite, s’attaquer à la réponse immunitaire adaptative.
2
1.1.1.2 Réponse immunitaire adaptative
C’est grâce à l’immunité innée que peut commencer la réponse immunitaire adaptative puisque ce sont les cellules dendritiques immatures qui, une fois un pathogène phagocyté, vont se déplacer dans les ganglions lymphatiques et présenter les antigènes microbiens aux lymphocytes T [6]. La réponse adaptative fait appel à des récepteurs d’antigènes spécialisés pour éliminer les pathogènes et, contrairement à la réponse immunitaire innée, elle se produit plus tardivement puisqu’elle implique un réarrangement génique V(D)J. Cette réponse permet donc une élimination plus spécifique du pathogène. De plus, lorsqu’un antigène est rencontré à plusieurs reprises, la mémoire immunologique se développe, ce qui permet une réponse immunitaire plus rapide et plus efficace à chaque nouvelle infection avec ce même antigène [7].
Figure 1.1: Étapes de la réponse immunitaire adaptative. Résumé des différentes étapes
menant à la mémoire immunologique. Cette figure a été adaptée de http://gopixdatabase.com/adaptive+immunity+diagram.
3
Les cellules du système immunitaire adaptatif sont les lymphocytes T et les lymphocytes B [8]. Les lymphocytes T sont impliqués dans la réponse à médiation cellulaire. Les lymphocytes T CD8+ ont des propriétés cytotoxiques qui induisent la destruction des pathogènes. Lorsqu’elles
sont à l’état naïf, ces cellules sont activées par la présentation de complexe peptide-CMH de classe I. Les lymphocytes T CD4+ sont plutôt activés par des complexes impliquant le CMH de
classe II. Une fois activées, ces cellules peuvent activer les macrophages, contribuer à la production d’anticorps par les lymphocytes B ou inhiber les réponses des lymphocytes T au besoin. Pour leur part, les lymphocytes B sont des acteurs importants dans l’immunité humorale grâce aux anticorps qu’ils produisent. Ils sont également impliqués dans la présentation antigénique comme les cellules dendritiques et les macrophages. En plus de la recombinaison des gènes, cette réponse immunitaire repose aussi sur le principe de l’expansion clonale qui permet la prolifération des lymphocytes spécifiques aux antigènes rencontrés [9, 10].
1.1.2 Les lymphocytes B
Les cellules hématopoïétiques pluripotentes de la moelle osseuse se divisent en progéniteurs lymphoïdes communs et en progéniteurs myéloïdes communs. C’est à partir de la lignée lymphoïde que se forment les lymphocytes B, mais également les lymphocytes T et les cellules NK [11]. La différenciation des lymphocytes B a lieu dans la moelle osseuse. Une fois différenciées, ces cellules matures, mais encore naïves, prennent la circulation sanguine pour se rendre dans les organes lymphoïdes périphériques comme les ganglions lymphatiques, la rate et les tissus lymphoïdes associés aux muqueuses [12]. C’est à ces endroits que les lymphocytes sont activés suite à la rencontre avec leur antigène spécifique.
Avant d’en arriver à l’étape du lymphocyte B mature, il est important de mentionner que le progéniteur lymphoïde commun est, en fait, le précurseur de la cellule la plus précoce, la cellule pro-B. Grâce aux interactions avec les cellules stromales, les progéniteurs B peuvent réarranger leurs gènes d’immunoglobulines. La production de la chaîne lourde est le premier réarrangement génique qui se produit chez la lignée B [13]. Son association à des chaînes légères de substitution donne lieu au récepteur de la cellule pré-B, ce qui envoie le signal que la production de la chaîne lourde μ est réussie. Le réarrangement génique de la chaîne lourde peut donc arrêter et fait place au réarrangement des gènes des chaînes légères. Une fois ces dernières produites, elles s’associent à la chaîne lourde pour ainsi former des immunoglobulines de surface fonctionnelles,
4
les IgM. Le stade des cellules B immatures est maintenant atteint [14]. Le récepteur d’antigène nouvellement formé est soumis à un test d’autoréactivité [15]. Si les cellules ne réagissent pas aux autoantigènes, elles peuvent quitter la moelle osseuse pour devenir des cellules B matures. Les cellules naïves entrent dans les ganglions lymphatiques où elles peuvent rencontrer leur antigène spécifique [16]. Leur activation a lieu lorsqu’elles se lient également à un lymphocyte T activé par le même antigène. Certains lymphocytes B activés peuvent migrer dans un follicule primaire pour former un centre germinatif [17]. C’est à cet endroit que les lymphocytes B vont proliférer et continuer leur différenciation. Certains vont devenir des lymphocytes B mémoires alors que les autres vont devenir des cellules sécrétrices d’anticorps, les plasmocytes.
À chaque stade de différenciation cellulaire, des marqueurs de surface permettent la distinction des différentes sous-populations de lymphocytes B. Les marqueurs de surface CD19 et CD20 sont les plus précoces puisqu’ils sont exprimés dès le stade pro-B précoce et tardif respectivement [18]. Ils sont également présents tout au long du développement de la cellule sauf sur les plasmablastes dans le cas du CD20. Le marqueur CD24 est fortement exprimé (CD24hi)
sur les lymphocytes B transitionnels, qui sont en fait des cellules immatures qui transitent de la moelle osseuse à un organe lymphoïde périphérique pour terminer leur développement en cellules matures naïves [19]. Il est possible de distinguer les cellules mémoires grâce au marqueur CD27, alors que les cellules naïves ne l’expriment pas à leur surface [20]. Le marqueur CD38, quant à lui, est exprimé à la surface des plasmablastes, en plus d’être fortement exprimé sur les lymphocytes B transitionnels [21]. Il est possible de reconnaître les plasmocytes à l’aide du marqueur CD138 [22]. Les lymphocytes B peuvent également exprimer des immunoglobulines de surface comme IgD [23]. Elles se retrouvent particulièrement sur les cellules matures naïves, mais certaines cellules mémoires l’expriment également.
1.1.3 Les lymphocytes T
Alors que la maturation des lymphocytes B a lieu dans la moelle osseuse, celle des lymphocytes T se fait plutôt dans le thymus où les gènes du récepteur y sont réarrangés. À cette étape, les gènes de la chaîne β sont réarrangés pour former un pré-TCR, correspondant à une association de la chaîne β et de la chaîne préTα. Par la suite, la sélection positive a lieu; les récepteurs des cellules T immatures qui reconnaissent les molécules du CMH du soi transmettent un signal de survie [24]. Il existe également une étape de sélection négative pour les cellules qui réagissent
5
fortement avec les antigènes du soi. Un signal est transmis pour éliminer ces cellules autoréactives. Par la suite, les cellules naïves simples positives CD4+ ou CD8+ migrent dans les
organes lymphoïdes périphériques pour continuer la différenciation des sous-populations lymphocytaires.
Le répertoire lymphocytaire T est composé de plusieurs sous-populations dont les différentes fonctions permettent une meilleure protection contre les infections. On retrouve les lymphocytes T CD4+ auxiliaires dont la fonction est principalement d’aider les lymphocytes B dans la
production d’anticorps suite à une réponse à un antigène [25]. Il existe 3 sous-populations qui peuvent se distinguer par leur fonction mais également par leurs marqueurs extracellulaires : TH1
(CXCR3+CCR6-), T
H2 (CXCR3-CCR6-) et TH17 (CXCR3-CCR6+) [26]. Les lymphocytes T
CD4+ régulateurs (Treg) inhibent la réponse des cellules T en produisant des cytokines ayant un
pouvoir inhibiteur. Ces cellules possèdent entre autres le marqueur CD25. D’autres études montrent que ces cellules portent également les marqueurs CCR4 et FoxP3 et qu’elles expriment faiblement le marqueur CD127 (CD127lo) [27, 28]. Les prochaines sous-populations sont
présentes autant chez les lymphocytes T CD4+ que chez les CD8+. Les cellules naïves
(CD45RA+CCR7+) n’ont pas encore rencontré leur antigène spécifique alors que les cellules
effectrices (CD45RA+CCR7-) ont déjà fait la rencontre avec l’antigène et peuvent ainsi exercer
leurs fonctions immunes pour éliminer le pathogène [29]. Les cellules mémoires contribuent à la mémoire immunologique puisqu’elles ont un niveau de sensibilité plus élevé et répondent plus rapidement à un antigène que les cellules naïves. Il existe les cellules mémoires centrales (CD45RA-CCR7+) et les cellules mémoires effectrices (CD45RA-CCR7-) [30]. Les premières se
situent davantage dans les organes lymphoïdes pour proliférer et générer de nouvelles cellules effectrices alors que les secondes se retrouvent dans les tissus périphériques pour agir de façon immédiate.
1.1.4 Messagers
Au cours de la réponse immunitaire, des cytokines sont sécrétées par les macrophages activés par la reconnaissance d’agents infectieux. D’autres cellules peuvent également contribuer à la production de cytokines comme les lymphocytes T auxiliaires et régulateurs. Les cytokines sont de petites protéines qui contribuent de différentes façons pour combattre une infection. En effet, l’action autocrine de certaines permet de changer le comportement de la cellule sécrétrice alors
6
que l’action paracrine se concentre sur les cellules voisines [31]. Ces molécules jouent un rôle important dans l’inflammation en stimulant la migration et l’activation des cellules. Selon leur fonction, certaines cytokines peuvent être qualifiées de pro-inflammatoires ou d’anti-inflammatoires.
1.2 Lupus érythémateux disséminé
Normalement, les cellules du système immunitaire défendent l’organisme des attaques contre son intégrité, c’est-à-dire des infections, des cellules provenant d’autres individus, des cellules infectées ou cancéreuses, ayant des caractéristiques aberrantes, toutes considérées comme des antigènes du non-soi. En temps normal, des mécanismes d’auto-tolérance sont présents afin d’empêcher l’organisme de réagir contre les antigènes du soi. Par contre, un débalancement du système immunitaire peut entraîner ce dernier à s’attaquer aux autoantigènes et provoquer ce que l’on appelle les maladies auto-immunes [32]. Ces dernières peuvent être divisées en deux catégories : les maladies spécifiques à un organe et les maladies auto-immunes systémiques [33]. Le lupus érythémateux disséminé (LED) est une maladie auto-immune systémique caractérisée par un déséquilibre du système immunitaire affectant plusieurs organes. Cette maladie est aussi associée à des poussées de l’intensité des symptômes (périodes de la maladie active, « flares ») et à des rémissions. Environ 50 000 Canadiens seraient atteints de cette maladie dont 90% sont des femmes [34]. Les personnes les plus à risque sont les femmes entre 15 et 45 ans, en particulier les Afro-Américaines, les Asiatiques et les Hispaniques [35].
1.2.1 Causes
En général, les causes des maladies auto-immunes sont complexes et dans la majorité des cas elles ne sont pas encore bien établies. C’est le cas pour le lupus érythémateux disséminé. Par contre, il existe plusieurs facteurs de risque qui pourraient jouer un rôle dans le développement de la maladie comme l’hérédité, l’exposition à la lumière ultraviolette et le stress [36].
Les hormones et les chromosomes sexuels ont également un rôle important à jouer dans le développement de la maladie puisque 9 fois plus de femmes sont atteintes de la maladie. En effet, il a été démontré chez un modèle murin du lupus, induit au pristane, que la maladie était plus susceptible de se développer chez les souris XX contrairement aux souris XY- (Modèles de
7
hormones sexuelles, en particulier l’estrogène, dans la pathogénèse de la maladie en utilisant deux autres modèles murins de la maladie soit les NZB/NZW et les MLR/lpr (Modèle de souris; section 1.2.5.1) [36, 38]. Ils ont démontré que le taux de survie chez les femelles stériles (ayant subi l’ablation des ovaires) est augmenté contrairement aux mâles castrés. De plus, le fait d’administrer des suppléments d’androgène aux femelles et aux mâles castrés améliore l’état de la maladie alors que l’administration d’estrogène le détériore [38].
D’un autre côté, depuis la première description connue du LED, il a été rapporté que plus de 80 médicaments induisent le lupus (drug-induced lupus erythematosus, DILE) [39, 40] incluant des antibiotiques, en particulier ceux de la classe des sulfonamides [41], des anticonvulsants [42], des antiinflammatoires non-stéroïdiens [43] et des estrogènes. En fait, les médicaments causant le DILE sont regroupés en trois catégories selon la probabilité de causer la condition [44] (Fig. 1.2). Le premier groupe concernant les médicaments dont le rôle pathologique est avéré selon des études contrôlées. Le deuxième groupe montre une association possible basée sur des cas rapportés provenant de plusieurs cohortes alors que le troisième groupe ne montre qu’une possible association selon quelques cas documentés.
Tableau 1.1: Médicaments impliqués dans le DILE. Adapté de [44].
Avéré Probable Possible Cas récents
Hydralazine Sulfasalazine Antibiotiques Infliximab Procainamide Anticonvulsivants Agents anti-inflammatoires
non-stéroïdiens
Etanercept Isoniazide Anti-thyroïdiens Antihypertenseurs Interleukine-2
Méthyldopa Statines Lithium Zafirlukast
Quinidine Terbinafine Interférons Clobazam Minocycline Pénicillamine Sels d’or Tocainide Chlorpromazine Agents fluorouraciles Lisinopril
Hydrochlorthiazide Bupropion
1.2.2 Diagnostic
Le diagnostic de la maladie est assez complexe et se fait selon les critères révisés de l’American
College of Rheumatology (ACR) [45, 46]. Ces derniers comprennent des critères cliniques et des
marqueurs sérologiques. En fait, un résultat positif pour au moins 4 des 11 critères constitue un diagnostic confirmé de lupus. Ces critères sont : un érythème malaire, un érythème discoïde, la
8
photosensibilité, des ulcères oraux, l’arthrite, une sérite, des problèmes rénaux, neurologiques, hématologiques, immunologiques et la présence d’anticorps antinucléaires (Fig. 1.2). Malgré tout, la maladie est souvent difficile à diagnostiquer car elle est très variable en fonction du temps. Il est possible que le patient ne montre pas de symptômes apparents de la maladie le jour de la consultation ou qu’un diagnostic erroné soit posé par le médecin. De plus, les symptômes peuvent prendre des années avant de se faire ressentir [36].
Figure 1.2: Critères de diagnostic du LED. Illustration tirée de la référence [47].
1.2.3 Évaluation thérapeutique
Différentes méthodes ont été mises au point au fil des ans pour l’évaluation thérapeutique des maladies systémiques comme le LED. Deux concepts importants doivent être pris en considération dans la conception de scores. La première est l’activité de la maladie qui correspond principalement aux manifestations réversibles alors que la seconde est la sévérité et correspond aux lésions qui sont irréversibles [48]. Plusieurs scores ont été proposés comme le SLEDAI (Systemic Lupus Erythematosus Disease Activity Index) [45, 46], le BILAG (British
Isles Lupus Assessment Group) [49, 50], le SLAM (Systemic Lupus Erythematosus Activity Measure) [51] et l’ECLAM (European Consensus Lupus Activity Measurement) [52]. Parmi ces
différents scores, le BILAG et le SLEDAI sont les 2 indices les plus fréquemment utilisés. À l’origine, le BILAG évaluait les critères associés aux 4 semaines qui précédaient l’examen. Il était composé de 86 critères divisés selon 9 domaines tels que les signes généraux, les atteintes
9
cutanées, neurologiques, articulaires, cardiaques, respiratoires, rénales et hématologiques [49]. C’est un indice qui détecte bien les poussées de la maladie, mais il ne peut être considéré pour un score global. Le SLEDAI est l’autre score très répandu pour l’évaluation thérapeutique. Bien que plusieurs modifications aient été proposées, la version du groupe SELENA (Safety of Estrogens
in Lupus Erythematosus, National Assessment) est sans aucun doute la plus utilisée [53]. Il est
possible de déterminer l’activité de la maladie en fonction de la présence ou non de 24 critères (Fig. 1.3). Les manifestations sont prises en considération si elles sont présentes le jour de la consultation ou dans les 10 jours précédents. Parmi ces critères, 16 sont des manifestations cliniques alors que les autres concernent des résultats d’analyses de laboratoire. Le SLEDAI peut évaluer les systèmes touchés et peut déterminer l’amélioration ou non de la condition. Contrairement au BILAG, il ne peut être utilisé domaine par domaine, mais il est plutôt utilisé pour évaluer l’ensemble global. Bien que ces indices se servent d’éléments hématologiques, il pourrait être intéressant d’introduire de nouveaux marqueurs biologiques quantifiables, comme l’activité cellulaire.
10
Figure 1.3 : Critères d’évaluation de l’activité de la maladie selon le SLEDAI. Tirée de
11
1.2.4 Traitements
Actuellement, très peu de traitements sont disponibles pour soigner spécifiquement le LED. Ceux qui sont à la disposition des patients sont très variés et ne sont pas curatifs, ils atténuent les symptômes de la maladie pour ainsi améliorer la qualité de vie des patients. D’ailleurs, puisque ces symptômes sont très variables d’une personne à l’autre, les traitements sont personnalisés et adaptés selon les besoins des patients. Les traitements utilisés incluent les anti-inflammatoires, les corticostéroïdes, les antipaludiques, les immunosuppresseurs, les anticoagulants et les anticorps monoclonaux [54]. D’ailleurs, le Benlysta, un anticorps monoclonal dirigé contre le facteur soluble BAFF (B-cell activating factor) (voir section 1.2.6.5) est le premier traitement approuvé par la Food and Drug Administration (FDA) depuis plus de 50 ans [55]. D’autres études sont en cours afin de développer des traitements plus spécifiques en utilisant les cytokines, les lymphocytes B et les lymphocytes T comme cibles thérapeutiques [56-58].
1.2.5 Cellules impliquées dans la pathogénèse
1.2.5.1 Modèles de souris
Plusieurs modèles murins du lupus ont permis l’avancement des connaissances sur la maladie. Certains modèles sont classés dans la catégorie des modèles murins classiques de lupus spontané tels que le modèle NZB/W F1 et ses modèles dérivés, le MRL/lpr et le BXSB/Yaa [59]. Le modèle NZB/W F1 est l’hybride F1 entre New Zealand Black (NZB) et New Zealand White (NZW). Ces souris sont largement utilisées puisqu’elles développent un phénotype s’apparentant aux patients LED, ce qui inclut un sérum riche en autoanticorps antinucléaires, une lymphodénopathie, une splénomégalie et des problèmes rénaux importants [60]. Un fort biais est présent chez ce modèle puisque les femelles développent plus souvent la maladie. Les modèles dérivés NZM2328 et NZM2410 proviennent de la souche de New Zealand Mixed (NZM), née d’une combinaison entre NZB/W F1 et NZW suivie d’une série de recombinaisons [61-63]. Ces modèles ont permis d’approfondir les analyses génétiques de la maladie notamment en identifiant des loci et des gènes impliqués dans la pathogénèse de la maladie tels que les loci Sle1-3 et le gène Fcgr2b [62]. Le modèle MRL/lpr provient d’une combinaison de plusieurs souches telles que LG/J, C3H/Di, C57BL/6 et AKR/J [60]. Les caractéristiques associées à ce modèle sont une accumulation de lymphocytes T CD4-CD8- et B220+ et un sérum riche en autoanticorps
12
lpr affectant la transcription de Fas cause un défaut d’apoptose chez les lymphocytes B et T de ce modèle [65]. Le modèle BXSB/Yaa provient d’une recombinaison entre une F1 (C57BL6/J x SB/Le) et SB/Le. Dans ce modèle, la maladie se développe de façon accélérée chez les mâles, ce qui est expliqué par l’élément Y-linked autoimmune accelerator (Yaa) [66, 67]. Alors que ces derniers modèles murins montrent un développement spontané de la maladie en raison de facteurs génétiques, d’autres sont des modèles où le lupus est induit par des facteurs environnementaux. Un modèle induit est préparé par des injections intrapéritonéales de pristane chez des souris BALB/c, ce qui provoque la production d’autoanticorps et de complexes immuns menant à des problèmes rénaux [68]. Ce modèle murin a permis de comprendre l’influence des cytokines sur la production d’autoanticorps. En effet, les souris BALB/c IL-6 -/- ont permis de comprendre que
les anticorps dirigés contre les ADNsb, les ADNdb et la chromatine sont hautement dépendant de l’IL-6 [69]. C’est aussi le seul modèle animal qui présente la signature d’interférons typique au LED. Finalement, le modèle induit par la maladie du greffon contre l’hôte (GvHD) se développe chez des souris semi-allogéniques qui reçoivent une greffe de lymphocytes [70]. Ainsi, les cellules greffées réagissent contre l’hôte, alors que l’hôte est tolérant à la greffe. Les modifications apportées aux cellules du donneur ou à celles de l’hôte peuvent être facilement étudiées par cytométrie en flux puisque les lymphocytes T du donneur passent par des étapes d’activation et d’expansion à la suite de la greffe. Ces études effectuées sur des modèles murins ont permis l’avancement des connaissances reliées à la pathogénèse du LED en plus d’identifier des cibles thérapeutiques potentielles.
1.2.5.2 Lymphocytes T
Les lymphocytes T sont grandement impliqués dans la pathogénèse du LED puisque leurs propriétés sont affectées. En effet, ces cellules ont tendance à promouvoir l’inflammation et la sécrétion de cytokines puisqu’elles sont dans un état d’hyperactivation [71]. Ce phénomène peut s’expliquer par différents facteurs. Tout d’abord, les molécules CD3ζ, par lesquelles passe la signalisation intracellulaire induite lorsque le TCR (T-cell receptor) se lie à un antigène, se trouvent en quantité réduite [72]. Dans le cas de ces lymphocytes T hyperactifs, la signalisation intracellulaire transite plutôt par la chaîne FcRγ, favorisant ainsi le recrutement de Syk (spleen
tyrosine kinase) [73]. Ce changement résulte donc d’un apport plus important de calcium qui
13
le radeau lipidique de la membrane a tendance à s’agréger à un pôle de la cellule avant la liaison du TCR à son antigène [74].
En plus de leur état d’hyperactivation, certaines sous-populations de lymphocytes T sont débalancées comparativement aux cellules de sujets sains. Il a été mentionné que les lymphocytes T CD8+ sont plus nombreux, mais l’activité cytotoxique de ces cellules est diminuée [75, 76]. De
plus, le nombre de cellules doubles négatives (CD4-CD8-) est plus important. Les patients LED
ont également plus de cellules TH17 et des niveaux sériques plus élevés en IL-17 [77]. Les
cellules T régulatrices (Treg), une sous-population de lymphocytes CD4+, sont nécessaires au
maintien de la tolérance immunologique. Chez les patients LED, les cellules Treg sont présentes en faible quantité [78-80]. Ceci pourrait s’expliquer par le fait que la production d’IL-2, cytokine essentielle au développement de ces cellules, est également affectée. Les connaissances acquises sur les lymphocytes T poussent donc les recherches vers de nouvelles cibles thérapeutiques.
1.2.5.3 Lymphocytes B
Les lymphocytes B occupent un rôle important dans le développement et l’évolution du LED, et ce, à plusieurs niveaux. Les lymphocytes B produisent des cytokines jouant un rôle clé dans l’état inflammatoire de la maladie. Parmi celles-ci, on retrouve l’IL-4, IL-6, IL-10 et IFN-γ [81]. Plusieurs de ces cytokines se retrouvent en quantité augmentée dans le sérum des patients LED et le niveau de celles-ci peut même être proportionnel à l’activité de la maladie mesurée sur l’échelle SLEDAI. De plus, le rôle des lymphocytes B comme présentateurs d’antigènes et comme sécréteurs de cytokines a été démontré avec le modèle murin MRL/lpr. En effet, l’élimination de cette population permet l’élimination de la maladie [82]. Ce sont également les lymphocytes B qui produisent les autoanticorps présents dans le sang des patients LED contribuant à la pathogénèse de la maladie. La présence de ces autoanticorps, comme les anti-ADNdb, est utilisée comme facteur déterminant dans le diagnostic de la maladie. Il a longtemps été démontré qu’une signalisation plus forte du BCR (B-cell receptor), due à une surexpression de la molécule CD19, pouvait mener à l’auto-immunité [83]. Par contre, une nouvelle étude a montré le contraire, expliquant qu’un faible signal du BCR permet aux cellules auto-réactives d’échapper au contrôle de la sélection négative [84] promouvant ainsi l’auto-immunité.
14
1.2.6 Cytokines
Des études ont mis en évidence que le sérum de patients LED pouvait contenir des concentrations de cytokines plus élevées que celles des sujets sains [56]; dans certains cas elles étaient proportionnelles à l’activité de la maladie, ce qui en fait des cibles thérapeutiques potentielles. En effet, plusieurs études cliniques de phases I, II et III sont en cours afin de trouver de nouveaux traitements visant ces cibles potentielles. Cette stratégie est considérée comme une voie très importante puisque le débalancement des cytokines contribue fortement à l’état d’inflammation et à la perte de tolérance immune observés chez ces patients.
1.2.6.1 IL-6
L’interleukine-6 est une cytokine dont la concentration sérique est plus importante chez des patients LED que chez des sujets sains [56]. Cette protéine stimule la croissance des lymphocytes B et leur production d’immunoglobulines. Elle joue également un rôle important dans le processus d’inflammation en contribuant à la différenciation des cellules TH17 et en inhibant la
différenciation des cellules Treg [85]. Des études cliniques utilisant l’IL-6 comme cible thérapeutique sont en cours avec le tocilizumab [86]. Cette molécule est un anticorps monoclonal humanisé ciblant le récepteur soluble et membranaire de l’IL-6 pour empêcher sa liaison [85]. Ce traitement a montré des résultats prometteurs dans une étude de phase I chez des patients LED en diminuant l’activité de la maladie [87].
1.2.6.2 IL-10
Des concentrations plus élevées d’IL-10 sont retrouvées dans le sérum de patients LED en plus d’être reliées proportionnellement à l’activité de la maladie [88]. Les rôles joués par l’IL-10 dans la pathogénèse de la maladie se situent au niveau de l’activation des lymphocytes B et de son effet suppresseur sur les cellules présentatrices d’antigènes (APC) et sur les lymphocytes T [89]. Une étude clinique pilote a montré des résultats intéressants lors d’injection d’anticorps monoclonaux anti-IL-10 à des sujets humains [90]. Les résultats montraient une amélioration des lésions cutanées, des douleurs articulaires et une diminution du score SLEDAI.
1.2.6.3 IL-17
L’interleukine-17 est une autre cytokine pro-inflammatoire dont la concentration sérique est augmentée chez les patients atteints du LED comparativement à des sujets sains [91]. L’effet
15
inflammatoire de cette cytokine se traduit par l’activation de médiateurs et de chimiokines impliqués dans le processus d’inflammation, qui à leur tour, engendrent l’accumulation de neutrophiles. L’IL-17 participe également à la production de cytokines inflammatoires permettant la génération de cellules TH17 [92]. Bien que plusieurs études cliniques aient eu lieu dans des cas
de maladies inflammatoires comme l’arthrite rhumatoïde et le psoriasis, cette cible thérapeutique doit être étudiée dans des cas de LED.
1.2.6.4 Interférons
Des études ont démontré que les patients LED ont des concentrations sériques plus élevées d’IFN-γ et d’IFN-α, qui sont en plus reliées directement à l’activité de la maladie [93-95]. Les cellules dendritiques plasmacytoïdes sont les principaux producteurs d’IFN-α. La sécrétion de cet interféron stimule le processus d’auto-immunité à plusieurs niveaux que ce soit pour l’activation des cellules dendritiques, la différenciation des monocytes, l’élimination des cellules Treg, la différenciation des lymphocytes B ou encore la production d’anticorps [96, 97]. De son côté, l’IFN-γ est un acteur important de l’immunité puisqu’il permet d’activer les macrophages, les cellules NK et les lymphocytes T cytotoxiques en plus de réguler quelques cytokines inflammatoires. Ce sont les cellules NK qui produisent l’IFN-γ au commencement de la réponse immune, puis les lymphocytes T prennent la relève une fois que la réponse immune adaptative est activée [98, 99]. Les interférons et sont aussi utilisées dans le cadre d’études cliniques comme cibles thérapeutiques pour le traitement du LED [100].
1.2.6.5 B-cell-activating factor
La molécule B-cell-activating factor (BAFF) est un important facteur de survie pour les lymphocytes B, en particulier les lymphocytes B transitionnels et matures [101]. BAFF est principalement sécrété par les cellules myéloïdes lors d’une réaction inflammatoire. Cette molécule peut se lier à trois récepteurs : TACI, BAFF-R et BCMA. Chez les patients atteints de LED, la concentration sérique de BAFF est supérieure à celle retrouvée chez des sujets sains et elle est en corrélation avec l’activité de la maladie [102]. Plusieurs agents biologiques ciblant BAFF ont été mis au point au cours des dernières années, mais seul le belimumab s’est retrouvé dans des études cliniques de phase III. Le belimumab est un anticorps monoclonal qui est capable d’inhiber, non pas la forme membranaire de BAFF, mais plutôt la forme soluble de la molécule [103]. Cet anticorps est commercialisé sous le nom de Benlysta (voir 1.2.4).
16
1.3 Cytométrie en flux et usage clinique
La cytométrie en flux est un outil d’analyse très puissant et rapide et comporte des applications cliniques très utiles. En effet, il est possible d’étudier des populations cellulaires du sang comme les érythrocytes, les leucocytes et les plaquettes grâce à la disponibilité d’anticorps dirigés contre des marqueurs de surface et des protéines intracellulaires [104]. Ainsi, la cytométrie en flux peut être utilisée dans le but de détecter des anomalies immunologiques et hématologiques. Par exemple, dans les années 1980, on se servait de ce genre d’analyses pour montrer qu’un contenu anormal en ADN était indicateur de la présence d’un cancer [105]. On peut aussi se servir de cet outil pour détecter des immunodéficiences [106], analyser des clones anormaux de la maladie d’hémoglobinurie paroxystique nocturne [107] et énumérer les cellules hématopoïétiques progénitrices [108]. D’autres études se servent de la méthode du Fluorescent Cell Barcoding (FCB) pour analyser rapidement le profil de phosphorylation dans des échantillons de sang pour le diagnostic de cancer [109-111]. Cette méthode a été utilisée par notre équipe pour caractériser des échantillons de moelle osseuse de patients atteints de myélomes multiples [112]. Ce ne sont là que de brefs exemples qui montrent l’énorme potentiel de la cytométrie en flux pour des applications cliniques.
1.3.1 Human Immunophenotyping Consortium
Le Human Immunophenotyping Consortium (HIC) s’insère dans un projet de grande envergure :
Human Immunolgy Project Consortium. Ce programme a débuté en 2010 au National Institute of Allergy and Infectious Disease (NIAID) et a pour but de créer une banque de données
caractérisant les phénotypes de cellules provenant d’individus sains et atteints de différentes maladies [113]. De plus, ce groupe propose que le phénotype ne se caractérise pas seulement par les proportions des sous-populations cellulaires, mais peut aussi inclure la réponse à des stimuli et leur état d’activation. Leur projet est basé sur l’utilisation de la cytométrie en flux. Afin d’uniformiser la méthode d’analyse, le HIC propose une sélection de marqueurs extracellulaires pour cibler les populations cellulaires du sang [26]. La figure 1.5 montre les différentes populations qui peuvent être ciblées selon les marqueurs qui les caractérisent. Ce consortium a publié une liste de panels composés de 8 anticorps ciblant ces marqueurs et qui peuvent être utilisés pour des analyses de cytométrie en flux.
17
18
1.3.2 Outils d’analyses de données multiparamétriques
1.3.2.1 SPADE et viSNE
Actuellement, les données obtenues par cytométrie en flux sont analysées en utilisant deux paramètres à la fois avec une séquence de régions et de gates qui s’emboitent. Ces stratégies de gating conventionnel sont dites dirigées. Les données brutes comportant des panels de deux ou trois anticorps sont facilement analysées de cette façon. Cependant, plus le nombre de marqueurs analysés augmente, plus le nombre de combinaisons possibles augmente également, ce qui peut facilement créer des centaines de possibilités différentes. Par exemple, un panel de 8 anticorps peut facilement générer plus de 80 diagrammes en deux dimensions (2D). L’analyse de petites populations, rares ou inattendues est d’autant plus ardue, qu’une analyse dirigée peut très bien ne pas permettre de les observer. Dernièrement, l’équipe de Nolan a mis au point un outil d’analyse basé sur des algorithmes et permettant d’analyser des données de cytométrie en flux en regroupant les cellules qui partagent les mêmes caractéristiques phénotypiques [114]. Le programme SPADE (Spanning-tree Progression Analysis of Density-normalized Events) élimine les biais d’interprétation créés par la stratégie dirigée [115]. De plus, ce même groupe a développé viSNE, qui permet d’utiliser la représentation schématique en 2D correspondant à tous les paramètres testés [116]. ViSNE établit la position des cellules qui reflète la proximité des cellules entre elles. De plus, cet outil permet de visualiser directement via la gradation de couleur l’expression de chaque marqueur pour chaque cellule analysée. Le développement de tels outils constitue un avancement dans l’interprétation des données de cytométrie en flux car il permet de visualiser la diversité et l’hétérogénéité des cellules en un seul coup d’œil. Les auteurs proposent d’ailleurs ces outils pour établir des diagnostics cliniques [114, 116].
19
2. Hypothèse
Face à la demande des cliniciens pour trouver des tests spécifiques et quantifiables qui seraient applicables au diagnostic du LED, nous suggérons que l’identification de marqueurs biologiques mesurables grâce aux méthodes de cytométrie en flux pourrait faciliter le diagnostic, le pronostic et le traitement de la maladie.
Notre hypothèse est qu’il serait possible de détecter les différences phénotypiques des cellules du sang de patients atteints de lupus érythémateux disséminé en les comparant avec des cellules provenant d’individus sains.
20
3. Objectifs
1. Criblage des profils de cytokines présentes dans le sérum des patients LED comme indicateur de leur état inflammatoire lors du prélèvement.
2. Détermination de la répartition des sous-populations de lymphocytes, de monocytes et de cellules dendritiques dans le sang des patients.
3. Évaluation de l’impact de la congélation sur la répartition des sous-populations des cellules mononucléées du sang.
21
4. Méthodologie
4.1 Source des cellules mononucléées du sang, du sérum et du plasma
Le processus de recrutement, le protocole de recherche et les formulaires de consentement étaient tous approuvés par le comité d’éthique de la recherche d’Héma-Québec et celui du Centre hospitalier universitaire de Sherbrooke (CHUS). Tous les participants à cette étude ont signé un formulaire de consentement éclairé. L’identité des participants et les informations personnelles recueillies par le médecin ou dans le cadre du don de sang étaient gardées confidentielles.
4.1.1 Participants en santé
Pour chaque participant en santé, tous les tests de dépistage des maladies transmissibles par le sang (HIV-1, HIV-2, HLTV-I et -II, syphilis, hépatite B et C) se sont avérés négatifs. Dans le processus du don de plaquettes, les leucocytes sont captés dans une chambre de leucoréduction (chambre LRS) grâce au système Trima Accel (Gambro BCT, Lakewood, CO, É.-U). Le sang résiduel qui se trouve dans ces chambres LRS a été utilisé afin d’isoler les cellules mononucléées du sang périphérique (PBMC, peripheral blood mononuclear cells). Les chambres LRS ont été préparées de façon aseptique tel que décrit précédemment [117]. Leur contenu a été extrait et les cellules ont été rincées avec la solution saline tamponnée au phosphate modifié de Dulbecco (DPBS) composée de 10 mM de K2PO4, 136 mM de NaCl (Invitrogen, Burlington, ON, Canada),
8 g/L de glucose (Sigma Aldrich, Oakville, ON, Canada) additionnée de 10 % d’un anticoagulant, l’ACD (acid–citrate-dextrose, Baxter Healthcare Corp., Deerfield, IL, É.-U.). Le sang récupéré a été déposé sur un coussin de Ficoll-Hypaque (GE HealthCare Biosciences, Baie-d’Urfé, QC, Canada), puis a été centrifugé à 1000 x g pendant 8 minutes en absence de frein. La centrifugation a permis la séparation et la récupération des composantes d’intérêt, soit le plasma et les PBMC. Le plasma a été conservé à 4°C jusqu’à l’analyse. Par la suite, les PBMC ont été lavés avec la solution de DPBS-glucose-ACD, ont été centrifugés à 230 x g pendant 8 minutes, puis mis à une densité cellulaire de 10, 50 ou 100 x 106 cellules/ml dans un milieu de congélation
froid composé de 50 % d’IMDM (Iscove’s modified Dulbecco’s medium, Life Technologies, Burlington, ON, Canada), de 40 % de FBS (fetal bovine serum, Life Technologies) et de 10 % de DMSO (diméthysulfoxyde, Sigma-Aldrich). Chaque échantillon était divisé en plusieurs tubes et les cryovials ont été transférés à -80°C dans une boîte CoolCell (Biocision, Mill Valley, CA, É.-U.) qui permet de diminuer la température d’environ 1°C/minute. Le lendemain, soit après
12-22
18h à -80°C, les cryovials contenant les cellules ont été transférés dans des contenants d’azote en phase liquide.
4.1.2 Patients LED
Ce projet impliquant des échantillons de patients LED a été possible grâce à la collaboration entre l’équipe à Héma-Québec et les collaborateurs de Dr Gilles Boire du CHUS. Les participants étaient recrutés dans le cadre d’une visite à leur médecin traitant. Les patients qui acceptaient de participer à cette étude devaient avoir été diagnostiqués selon les critères de l’American College
of Rheumatology pour le LED. De plus, lors de leur visite, le médecin établissait l’activité de la
maladie mesurée selon le SLE Disease Activity Index (SLEDAI), le Systemic Lupus International
Collaborating Clinics (SLICC) et le SLE Activity Measure (SLAM). Une fois recruté, l’infirmière
coordonnatrice complétait une liste des médicaments pris au moment de la visite et un bref questionnaire sur l’état de santé du patient. Tous ces documents étaient fournis pour chaque échantillon relié à notre étude. Dans le cadre de cette étude, 36 échantillons de sang ont été récoltés entre juillet 2005 et décembre 2013 (LED-24 à LED-59).
Les échantillons des patients LED-24 à LED-54, prélevés entre juillet 2005 et avril 2011, avaient été prélevés dans des tubes (< 10 tubes) contenant de l’héparine et transportés dans des boîtes de styromousse avec une cassette réfrigérante pour les maintenir entre 4°C et 8°C. Par la suite, la méthode de transport fut modifiée afin d’obtenir une meilleure récupération en cellules totales. Pour les échantillons LED-55 à LED-59, entre 8 et 15 tubes de sang contenant de l’ACD (anticoagulant-citrate-dextrose BD Biosciences, Mississauga, ON, Canada) ont été prélevés ainsi qu’un tube sans anticoagulant permettant de récupérer le sérum. Les tubes ont été transportés à la température de la pièce dans des boîtes isolées en polystyrène et envoyés par transport rapide au centre de Québec. Tous les tubes ont été traités dans un maximum de 24h après le prélèvement de l’échantillon (incluant le temps de transport). Les tubes ont été ouverts et laissés au repos pendant 10 minutes, puis le contenu a été dilué 1:1 avec une solution de DPBS-glucose additionnée de 2% de sérum bovin fœtal (FBS). Les PBMC ont été isolés et congelés tel que décrit à la section 4.1.1. Le tube sans anticoagulant a été centrifugé à 800 x g pendant 30 minutes en absence de frein, puis le sérum récupéré a été conservé à 4°C jusqu’à l’analyse.
23
4.2 Analyses phénotypiques
4.2.1 Marquage extracellulaire
Habituellement, un tube de chaque échantillon de cellules était décongelé et utilisé pour une série d’analyses de cytométrie en flux. Les tubes de cellules ont été décongelés dans un bain-marie à 37°C jusqu’à ce qu’il ne reste qu’un petit morceau de glace, puis leur contenu a été transféré goutte à goutte dans une solution froide de DPBS-glucose contenant 50% de FBS. Selon la qualité des échantillons, une solution de DNAse (Sigma-Aldrich) pouvait être ajoutée afin de briser les agrégats formés par l’éclatement des cellules. Ce problème a été observé dans 3 échantillons. Les analyses phénotypiques ont été faites à partir de 1 x 106 cellules/panel
d’anticorps. Les cellules ont été centrifugées à 230 x g pendant 6 minutes, puis mises en suspension dans 0,1 ml de PBS (solution de saline tamponnée 10mM). Par la suite, les cellules ont été incubées en présence d’un marqueur de viabilité, l’Alexa-fluor 488 à 1,25 µg/mL (Invitrogen), pendant 30 minutes. Les cellules ont été lavées avec une solution de PBS et mises en suspension dans du PBS contenant 1% (v/v) FBS et 0,01% (p/v) NaN3 (Sigma-Aldrich). Afin
d’éliminer la liaison non-spécifique, du sérum de souris (Jackson Immunoresearch Labs, West Grove, PA, É.-U.) préalablement décomplémenté à 55°C pendant 30 minutes a été ajouté à une concentration finale de 5% (v/v). Les anticorps listés plus bas (section 4.3.2) ont été ajoutés aux cellules selon les instructions des manufacturiers pour une période de 15 à 30 minutes à la température de la pièce (TP) et conservés à la noirceur. Les cellules marquées ont ensuite été fixées à l’aide d’une solution de formaldéhyde à 2% pendant 10 minutes à TP. Les cellules fixées ont été lavées et remises en suspension dans la solution de PBS-FBS-NaN3. Les cellules ont été
conservées à 4°C jusqu’à leur analyses avec le cytomètre en flux. Une série d’essais a été réalisée avec 10 échantillons de chambres LRS afin de déterminer l’impact de la congélation sur la détection des marqueurs. Les cellules ont été analysées avant la congélation, immédiatement après la décongélation et à la suite de périodes de repos de 1h et 24h post-décongélation. Pour les périodes de repos, les cellules étaient remises en suspension dans un milieu IMDM contenant 10% FBS et incubées à 37°C dans un incubateur à CO2.
4.2.2 Anticorps
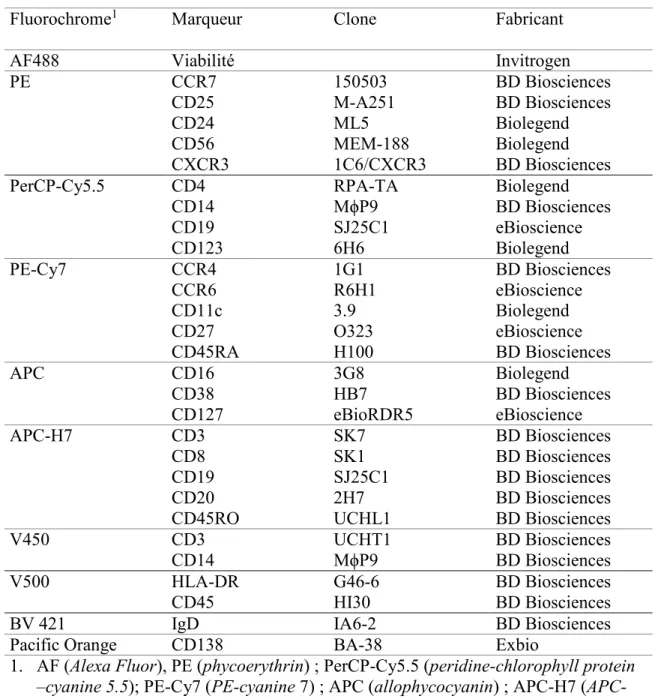
La liste des anticorps utilisés pour réaliser la caractérisation des cellules est présentée au tableau 4.1. Tous les anticorps étaient des IgG et des anticorps monoclonaux de souris couplés à des
24
molécules fluorescentes. Plusieurs populations de cellules ont été ciblées à l’aide d’un panel constitué de 7 anticorps couplés à des fluorochromes différents et permettant de détecter spécifiquement les lymphocytes T, les lymphocytes T régulateurs, les lymphocytes T auxiliaires, les lymphocytes B et un groupe composé de cellules dendritiques, de monocytes et de cellules NK. À une exception près, la composition de ces panels était telle que proposée par le consortium
Human Immunology Project pour la standardisation du phénotypage des cellules humaines [26]
(Tableau 4.2). L’exception étant qu’un anticorps anti-CD138 a été substitué à l’anti-HLA-DR dans le panel ciblant les lymphocytes B pour détecter les plasmocytes. Finalement, une série supplémentaire de marquages extracellulaires a été ajoutée afin de déterminer la fréquence des populations cellulaires majeures de leucocytes soit les lymphocytes B et T, les monocytes et les cellules NK (Tableau 4.2).
25
Tableau 4.1: Liste des anticorps monoclonaux utilisés pour la caractérisation des PBMC
Fluorochrome1 Marqueur Clone Fabricant
AF488 Viabilité Invitrogen
PE CCR7 150503 BD Biosciences
CD25 M-A251 BD Biosciences
CD24 ML5 Biolegend
CD56 MEM-188 Biolegend
CXCR3 1C6/CXCR3 BD Biosciences
PerCP-Cy5.5 CD4 RPA-TA Biolegend
CD14 MϕP9 BD Biosciences CD19 SJ25C1 eBioscience CD123 6H6 Biolegend PE-Cy7 CCR4 1G1 BD Biosciences CCR6 R6H1 eBioscience CD11c 3.9 Biolegend CD27 O323 eBioscience CD45RA H100 BD Biosciences APC CD16 3G8 Biolegend CD38 HB7 BD Biosciences CD127 eBioRDR5 eBioscience APC-H7 CD3 SK7 BD Biosciences CD8 SK1 BD Biosciences CD19 SJ25C1 BD Biosciences CD20 2H7 BD Biosciences
CD45RO UCHL1 BD Biosciences
V450 CD3 UCHT1 BD Biosciences
CD14 MϕP9 BD Biosciences
V500 HLA-DR G46-6 BD Biosciences
CD45 HI30 BD Biosciences
BV 421 IgD IA6-2 BD Biosciences
Pacific Orange CD138 BA-38 Exbio
1. AF (Alexa Fluor), PE (phycoerythrin) ; PerCP-Cy5.5 (peridine-chlorophyll protein
–cyanine 5.5); PE-Cy7 (PE-cyanine 7) ; APC (allophycocyanin) ; APC-H7 (APC-cyanin H7). ; BV 421 (Brilliant violet 421).
26
Tableau 4.2 : Panels des cibles cellulaires en fonction des marqueurs de surface. Adapté de [26].
Couleur1
Marqueurs Lymphocytes T2
Lymphocytes
B DC, Mɸ et NK Populations majeures3
Totaux T reg T aux
FL1 AF 488 Viabilité FL2 PE CCR7 CD25 CXCR3 CD24 CD56 CD56 FL3 PerCP* CD4 CD4 CD4 CD19 CD123 CD14 FL4 PE-Cy7 CD45RA CCR4 CCR6 CD27 CD11C CD19 FL5 APC CD38 CD127 CD38 CD38 CD16 CD16 ou CD19 FL6 APC-H7 CD8 CD45RO CD8 CD20 CD3, CD19, CD20 NA FL7 V450 CD3 CD3 CD3 CD138 CD14 CD3
FL8 V500 HLA-DR HLA-DR HLA-DR IgD HLA-DR CD45
1. AF (Alexa Fluor); PerCP* (PerCP-Cy 5 .5)
2. Les lymphocytes T mémoires et naïfs (Totaux), les sous-populations de lymphocytes T régulateurs (reg) et auxiliaires (aux; TH1, TH2, TH17).
3. Les cellules dendritiques (DC) les cellules NK et les monocytes (Mɸ). 4. Les lymphocytes T et B, les cellules NK et les monocytes.
4.2.3 Acquisition et analyse des données
L’acquisition des données a été effectuée avec le cytomètre en flux CyFlow ML et le logiciel FlowMax 3.0 (Partec, Münster, Allemagne). Les débris ont été exclus des analyses en délimitant une région basée sur la taille (FSC) et la granularité (SSC) attendues des cellules. Cette région a servi de paramètre afin de faire l’acquisition de 50 000 à 70 000 évènements correspondant à des cellules pour chaque panel de marquage réalisé. Pour chaque échantillon de cellules, un témoin de cellules non marquées a été utilisé afin d’éliminer l’autofluorescence. De plus, pour chaque panel, des tubes contrôles FMO (fluorescent minus one) [118] ont été utilisés pour déterminer l’emplacement des quadrants pour séparer les cellules positives et les cellules négatives pour un marqueur donné en fonction du fluorochrome utilisé. Des billes de compensation (Compbeads; BD Biosciences) ont été préparées pour chaque fluorochrome et utilisées pour calculer et éliminer le chevauchement entre les différents fluorochromes. Les analyses ont toujours été réalisées sur les données compensées. À la suite de l’acquisition, l’analyse des données a été effectuée avec le logiciel FCS Express version 4 (De Novo Software, Los Angeles, CA, É.-U.).
27
4.2.3.1 Spanning-tree Progression Analysis of Density-normalized Events (SPADE)
Les données brutes ont aussi été analysées afin de minimiser l’impact des limites imposées par l’utilisation de quadrants et le concept classique des cellules positives ou négatives pour un marqueur. Le logiciel SPADE a été mis au point par l’équipe de Garry P. Nolan [109] et permet l’analyse multiparamétrique de données brutes de cytométrie en flux ou de cytométrie de masse en utilisant directement les valeurs compensées. Il est possible de travailler avec SPADE en passant par Cytobank, une plateforme de partage et d’analyse de données de cytométrie. Cet outil peut être utilisé de façon complémentaire au logiciel FCS Express afin de visualiser globalement les cellules selon toutes les cibles utilisées dans chaque panel. SPADE utilise des algorithmes complexes pour regrouper en nœuds les cellules ayant des caractéristiques communes selon leurs marqueurs de surface. SPADE permet de visualiser une arborisation représentative de l’intensité de l’expression des différents marqueurs extracellulaires. SPADE permet aussi d’établir un ratio à partir de l’intensité moyenne de fluorescence (MFI) de fichiers de référence. Dans cette étude, le fichier de référence utilisé était constitué de la moyenne de tous les échantillons de participants en santé (21). Par la suite, chaque analyse pour chacun des échantillons de patients LED ou de participants en santé a été comparée à cette arborisation moyenne/référence afin de visualiser les similitudes et les écarts.
Les évènements analysés dans FCS express ont d’abord été restreints par une région dessinée autour des cellules vivantes en excluant les cellules positives pour le marquage Alexa-fluor 488. Ces fichiers de données (cellules vivantes) et la matrice de compensation ont été exportés à la plateforme Cytobank afin de construire une arborisation prédéterminée de 100 nœuds. Une fois créée, cette arborisation a été utilisée afin de visualiser l’intensité de fluorescence (échelle de couleurs) pour chaque marqueur de tous les panels (Tableau 4.2), et ce, pour chaque échantillon de cellules de patients et de participants en santé. Les six panels utilisés ont générés à eux seuls 42 pages de données regroupant chacun les 18 ou 21 échantillons de patients LED ou de participants en santé respectivement.

![Tableau 1.1: Médicaments impliqués dans le DILE. Adapté de [44].](https://thumb-eu.123doks.com/thumbv2/123doknet/6347193.167332/18.918.106.773.635.851/tableau-médicaments-impliqués-dile-adapté.webp)
![Figure 1.2: Critères de diagnostic du LED. Illustration tirée de la référence [47].](https://thumb-eu.123doks.com/thumbv2/123doknet/6347193.167332/19.918.107.768.312.610/figure-critères-diagnostic-led-illustration-tirée-référence.webp)

![Figure 1.4 : Sous-populations cellulaires des PBMC. Cette figure a été traduite de [26]](https://thumb-eu.123doks.com/thumbv2/123doknet/6347193.167332/28.918.113.769.100.924/figure-populations-cellulaires-pbmc-figure-traduite.webp)
![Tableau 4.2 : Panels des cibles cellulaires en fonction des marqueurs de surface. Adapté de [26]](https://thumb-eu.123doks.com/thumbv2/123doknet/6347193.167332/37.918.98.849.150.556/tableau-panels-cibles-cellulaires-fonction-marqueurs-surface-adapté.webp)



