Analyse préliminaire du rôle des "Ubiquitin specific peptidases" et de l'axe USP7-MDM2-TP53-CDKN1A dans les leucémies myéloïdes aiguës
165
0
0
Texte intégral
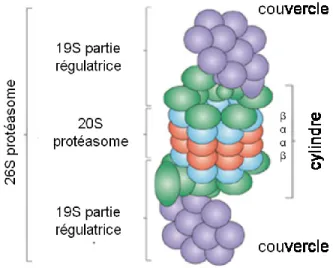
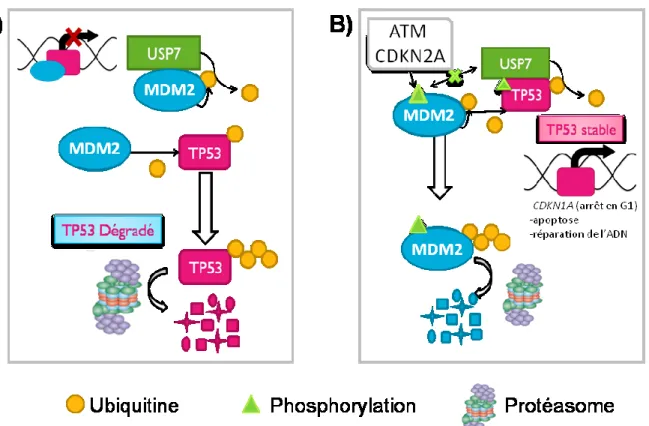
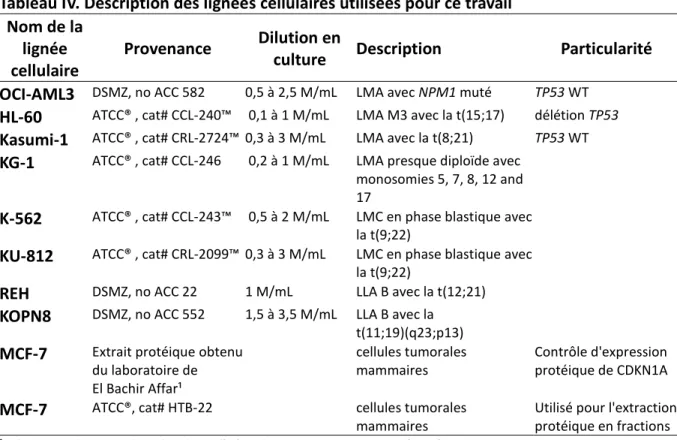
Figure


+7



Documents relatifs