Caractérisation de la signature transcriptionnelle chez des femmes québécoises avec une histoire familiale de cancer du sein
Mémoire
Marie-Christine Pouliot
Maîtrise en médecine moléculaire Maitre ès sciences (M.Sc.)
Québec, Canada
Caractérisation de la signature transcriptionnelle chez
des femmes québécoises avec une histoire familiale
de cancer du sein
Mémoire
Marie-Christine Pouliot
Sous la direction de :
Francine Durocher, directrice de recherche
iii
RÉSUMÉ
Au Canada, 5 à 10% des cas de cancer du sein sont des cancers héréditaires provenant de familles à risque élevé de développer la maladie. Cependant, la majorité des cas héréditaires ne sont pas encore caractérisés. L’épissage alternatif est reconnu pour être impliqué dans le développement du cancer. L’analyse de la signature transcriptionnelle d’individus atteints et non-atteints de cancer du sein pourrait révéler des transcrits impliqués dans la susceptibilité ou le développement de ce type de cancer.
La technique «RNA sequencing» a été utilisée pour établir le profil transcriptionnel de femmes provenant de familles à risque élevé. L’ARN a été extrait des lignées de lymphocytes immortalisés provenant de 117 femmes de familles dites à risque élevé, c’est-à-dire porteuses d’une mutation délétère dans le gène BRCA1, BRCA2 ou sans mutation BRCA1/2. Une analyse Anova suivie d’un test Bonferroni et d’un test de Scheffé ont été utilisés pour identifier les transcrits significativement et différentiellement exprimés entre les différents groupes.
Au total, 95 transcrits correspondant à 85 gènes sont significatifs (p-value <0.01). Selon les signatures transcriptionnelles, il est possible de séparer les groupes BRCA1/2 du groupe BRCAX. Un enrichissement dans les sentiers métaboliques au niveau de la signalisation comme EIF2, IL-3 et mTOR a été obtenu. De plus, 28 transcrits sont différentiellement exprimés entre les femmes BRCAX atteintes et non atteintes. L’identification de transcrits différentiellement exprimés permettrait d’identifier des individus ayant une susceptibilité plus élevée de développer un cancer du sein.
iv
ABSTRACT
In Canada, 5 to 10% of breast cancer cases are inherited and come from high-risk families. However, the majority of hereditary breast cancer is not yet characterized. Alternative splicing (AS) is a mechanism known to be involved in cancer development. The analysis of transcriptome in high-risk breast cancer individuals affected with breast cancer or not could reveal transcripts implicated in breast cancer susceptibility and development.
RNA-seq technology was used to characterize the transcriptome in French Canadian families with high risk of breast and ovarian cancer. RNA extracted from immortalized lymphoblastoid cell lines of 117 women (affected or unaffected) and issued from BRCA1, BRCA2 or non-BRCA1/2 (BRCAX) families was used. Anova and Bonferroni tests followed by Scheffé test were performed to detect significantly and differentially expressed transcripts within these groups.
In total, 95 transcripts corresponding to 85 genes were significant (p-value <0.01). Hierarchical clustering based on transcriptional data allowed distinctive subgrouping of BRCA1/2 subgroups from BRCAX individuals. Enrichment in signaling pathways such as EIF2, IL-3 and mTOR was obtained. Furthermore, 28 transcripts were differentially expressed between BRCAX affected and unaffected women. The identification of differentially expressed transcripts could allow identifying individuals with a high susceptibility for breast cancer.
v
TABLE DES MATIÈRES
RÉSUMÉ ... iii
ABSTRACT ... iv
TABLE DES MATIÈRES ... v
LISTE DES TABLEAUX ... vii
LISTE DES FIGURES ... viii
LISTE DES ABRÉVIATIONS UTILISÉES ... ix
DÉDICACE ... xiii
REMERCIEMENTS ... xiv
AVANT-PROPOS ... xv
INTRODUCTION ... 1
1. Le cancer du sein ... 1
1.1. Les gènes de susceptibilité dans le cancer du sein ... 2
1.1.1. Gènes à risque élevé ... 3
1.1.1.1. BRCA1/2... 4
1.1.2. Gènes à risque faible et modéré ... 9
1.1.3. BRCAX ... 10
2. L’épissage ... 11
2.1. L’épissage alternatif ... 12
2.1.1. La régulation de l’épissage alternatif ... 14
2.1.2. L’épissage alternatif dans le cancer ... 15
2.1.2.1. La régulation de l’épissage alternatif dans le cancer ... 16
3. Séquençage à haut débit ... 17
3.1. «RNA sequencing» ... 18
3.1.1. «RNA sequencing» dans le cancer ... 20
4. Problématique ... 21 5. Objectifs ... 22 CHAPITRE 1 ... 23 Résumé ... 24 Abstract ... 25 Introduction ... 27 Results ... 29 Discussion ... 35
Materials and methods ... 42
Legends to Figures ... 45
Acknowledgements ... 46
Disclosure of Potential Conflicts of interest ... 46
Grant support ... 46
References ... 47
DISCUSSION / LIMITES DE L’ÉTUDE ... 76
CONCLUSION ... 80
BIBLIOGRAPHIE ... 82
ANNEXE ... 96
Abstract. ... 98
vi
DNA Methyltransferases and Methylation of CpG islands ... 99
DNA Methylation as Biomarker for Breast Cancer Detection ... 101
Breast Cancer Treatment Through Regulation of Methylation ... 104
Conclusion ... 107
vii
LISTE DES TABLEAUX
Table 1 : Transcripts specifically associated with each group and their corrected p-value... 66 Table 2 : The 15 most significant canonical pathways enriched for significantly and
differentially expressed transcripts. ... 72 Table 3 : Overrepresented functions, network and diseases for significantly and
differentially expressed transcripts. ... 73 Table 4 : The most significant canonical pathways enriched for significantly and
differentially expressed transcripts between BRCAX affected and BRCAX unaffected individuals. ... 74 Table 5 : Overrepresented functions, network and diseases for significantly and
differentially expressed transcripts between BRCAX affected and BRCAX unaffected individuals. ... 75
viii
LISTE DES FIGURES Introduction
Figure 1 : Gènes et loci associés à la susceptibilité de développer le cancer du
sein ... 3
Figure 2 : Fréquence cumulative des cancers du sein selon la mutation retrouvée chez les femmes québécoises. ... 6
Figure 3 : Porteurs BRCA1 et gènes modificateurs. ... 8
Figure 4 : Mécanisme d’épissage avec le splicéosome ... 12
Figure 5 : Différentes options d’épissage alternatif ... 13
Figure 6 : Séquences et éléments régulateurs de l’épissage alternatif. ... 15
Figure 7 : Étapes de la réaction de «RNA sequencing» ... 19
Chapitre 1 Figure 1: Principal component analysis (PCA) on lymphoblastoid cell lines. ... 62
Figure 2 : Hierarchical clustering of the 95 transcripts differentially expressed. .. 63
Figure 3 : Venn diagram of significantly and differentially expressed transcripts compared to BRCAX unaffected individuals. ... 64
Figure 4 : Hierarchical clustering of the 28 transcripts differentially expressed between BRCAX affected and BRCAX unaffected individuals. ... 65
ix
LISTE DES ABRÉVIATIONS UTILISÉES ARN = acide ribonucléique
BRCA1 = BReast Cancer 1 BRCA2 = BReast Cancer 2
BRCAX = individu à risque élevé étant non BRCA1/2 EIF2 = Eukaryotic Initiation Factor 2
IL-3 = Interleukin 3
mTOR = mammalian Target Of Rapamycin AS = Alternative Splicing
RNA = Ribonucleic acid
PCA = Principle Component Analysis GWAS = Genome Wide Association Study TP53 = Tumor Protein 53
ADN = acide désoxyribonucléique
PTEN = Phosphatase and TENsin homolog STK11 = Serine/Treonine Kinase 11
CDH1 = Cadherin 1
BRCT = BRCA1 C terminus
RAD51 = Recombination Protein A SNPs = single nuclotide polymorphisms iCOGS = iSelect genotyping array
BRIP1 = BRca1 Interacting Protein c-terminal helicase 1 CHEK2 = CHEckpoint Kinase 2
PALB2 = Partner And Localizer Of Brca2 ER = Estrogen Receptor
ARNm = Acide ribonucléique messager snRNA = small nuclear ribonucleic acid snRNP = small nuclear ribonucleoprotein BBP = Branchpoint Binding Protein Prp8 = Pre-MRNA Processing Factor 8
x
ISE = intronic splicing enhancer ESE = exonic splicing enhancer ISS = intronic splicing silencer ESS = exonic splicing silencer SR = Sérine/Arginine rich
hnRNPs = heterogeneous nuclear ribonucleoproteins SWI/SNF = SWItch/Sucrose Non-Fermentable
Bcl-X = B-cell lymphoma extra
VEGF-A = Vascular Endothelial Growth Factor A SRSF1/3 = Serine/Arginine-Rich Splicing Factor 1/3 BIM = Bcl-2-Interacting Mediator of cell death
BIN1 = Bridging INtegrator 1
ADNc = acide desoxyribonucléique complémentaire PCR = Polymerase Chain Reaction
HER2 = Human Epidermal growth factor Receptor 2 CDK4 = Cyclin-Dependent Kinase 4
LARP1 = La Ribonucleoprotein Domain Family, Member 1 ADD3 = Adducin 3
PHLPP2 = PH Domain And Leucine Rich Repeat Protein Phosphatase 2 RBFOX2 = RNA Binding Protein, Fox-1 Homolog 2
MBNL1/2 = Muscleblind-Like Splicing Regulator 1/2 GK1 = Glycerol Kinase 1
PPAR = Peroxisome proliferator-activated receptor ATM = Ataxia Telangiectasia Mutated
ATR = Ataxia Telangiectasia And Rad3-Related Protein LCLs = lymphoblastoid cell lines
Ind = individuals
PC1/2 = Principal Component1/2
XRCC6 = X-Ray Repair Complementing Defective Repair In Chinese Hamster Cells 6
xi
HDGF = Hepatoma-Derived Growth Factor IPA = Ingenuity Pathway Analysis
PBMC = Peripheral Blood mononuclear cells SCA28 = Spinocerecellar ataxia 28
RPL29 = Ribosomal Protein L29
PSMF1 = Proteasome Inhibitor Subunit 1 EBV = Epstein-Barr virus
TCGA = The Cancer Genome Atlas
RAP2C = Member of RAS Oncogene Family SPN = Sialophorin
GINS2 = GINS Complex Subunit 2
ESPL1 = Extra Splindle Pole Bodies Like 1, Separase IRF7 = Interferon Regulatory Factor 7
HLA-DPA1 =Major Histocompatibility Complex, Class II, DP Alpha 1 PCDH9 = Protocadherin 9
NCALD = Neurocalcin Delta
NOSIP = Nitric Oxide Synthase Interacting Protein
EIF2AK1 = Eukaryotic Translation Initiation Factor 2 Alpha Kinase 1 TBCB = Tubulin Folding Cofactor B
eNOS = endothelial Nitric Oxide Synthase Ku70 = Eukaryotic Ku heterodimer
NHEJ = Non homologous End Joining HR = Homologous Recombination DSB = Double-Strand Break
RAP = Member of RAS Oncogene Family RAB = Member of RAS Oncogene Family
RT-PCR = Reverse transcription –polymerase chain reaction EMT = Epithelial-mesenchymal transition
ZNF687 = Zinc Finger Protein 687 FGF23 = Fibroblast Growth Factor 23 FGFR = Fibroblast Growth Factor Receptor
xii
PI3K = Phosphoinositide 3-kinase
INHERIT BRCA = Interdisciplinary Health Research Team on BReast CAncer susceptibility
RPMI = Roswell Park Memorial Institute medium RIN = RNA Integrity
TPM = Transcripts Per Million
BOADICEA = The Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm
GO = Gene ontology
KEGG = Kyoto Encyclopedia of Genes and Genomes shRNA = small hairpin RNA
CpG = DNA with a high quantity of the nucleotides G and C next to one another.
TSG = Tumor Suppressor Gene
DNMT1/3 = DNA Methyltransferase 1/3
RASSF1A = Ras association domain family 1 isoform A TET = ten-eleven translocation proteins
HYAL2 = hyaluronoglucosaminidase 2 KIF1A = kinesin family member 1A DOK7 = docking protein 7
SFRP1 = secreted frizzled-related protein
ITIH5 = inter-alpha-trypsin inhibitor heavy chain family, member 5 WIF1 = WNT inhibitory factor 1
DKK3 = dickkopf WNT signaling pathway inhibitor 3 LINE1 = long interspersed nuclear element 1
MBD2 = methyl-CpG binding domain protein 2 KDM1B/LSD2 = lysine (K)-specific demethylase 1B P21 = Cyclin-dependent kinase inhibitor
xiii
DÉDICACE
À ma famille, à Simon, Sans vous je n’aurais jamais pu réussir
xiv
REMERCIEMENTS
J’aimerais d’abord remercier Dre Francine Durocher de m’avoir acceptée dans son laboratoire pour faire une maitrise à l’automne 2014. Merci de m’avoir poussée et permis de participer à une grande quantité de congrès tout au long de mon cheminement. Ces opportunités m’ont permis de présenter mon projet de recherche ainsi que mes résultats devant divers publics me donnant une expérience importante dans une carrière scientifique.
Je voudrais aussi remercier toute l’équipe travaillant avec Dre Durocher au cours de ma maitrise. Dr Yvan Labrie, professionnel de recherche, j’aimerais te remercier de m’avoir aidée avec l’écriture de tous mes textes, ton expertise a été grandement appréciée. Frédérick Bouffard, j’aimerais te remercier pour tous tes conseils pour mon projet ainsi que l’expérience que tu m’as transmise en laboratoire. Dre Karine Plourde, j’aimerais te remercier pour tout ce que tu as fait pour faire avancer mon projet en m’aidant avec tous les programmes que j’ai utilisés. Finalement, j’aimerais remercier la professionnelle de laboratoire Geneviève Ouellette pour avoir écouté tous mes problèmes et m’avoir conseillée tout au long de la maitrise.
De plus, j’aimerais remercier toute la plateforme de séquençage du CHUL (Patrick, Annie-Claude, Anne, David, Nicolas) pour tous les moments où vous m’avez fait rire et oublier tous les petits problèmes. Je voudrais aussi vous remercier pour les collations du lundi matin qui m’ont permis de bien commencer chaque semaine. Finalement, j’aimerais remercier toute ma famille pour votre support. Vos encouragements m’ont permis d’apprécier mon projet de recherche encore plus que ce que je souhaitais.
xv
AVANT-PROPOS
L’article du chapitre 1 a été soumis pour publication à la revue Oncotarget. Je suis l’auteure principale de cet article et j’ai fait la grande majorité des analyses bioinformatiques. J’ai également fait toutes les analyses à l’aide des programmes Ingenuity Pathway Analysis et PARTEK. Ces analyses ont permis de mieux comprendre les résultats obtenus, ce qui a mené à la rédaction d’un article scientifique sur le sujet. J’ai aussi fait toutes les figures qui se retrouvent dans l’article ainsi qu’écrit la version initiale du texte de cet article.
Cependant, j’ai eu l’aide de quelques personnes. Le Dr Yvan Labrie m’a aidée dans la correction de mon article et la reformulation de quelques phrases afin d’avoir un article scientifique de meilleure qualité. J’ai aussi eu l’aide de Charles Joly-Beauparlant, étudiant au doctorat, pour m’aider à faire les parties plus complexes des analyses bioinformatiques et pour lesquelles il fallait des connaissances approfondies en la matière. Geneviève Ouellette, la professionnelle de recherche du laboratoire, est en charge des lignées de lymphocytes immortalisés provenant des femmes québécoises à haut risque. Elle a aussi extrait l’ARN et préparé les librairies pour envoyer à la plateforme de séquençage de McGill. Le Dr Jacques Simard est l’investigateur de la banque de lymphocytes immortalisés chez les familles québécoises. Dr Arnaud Droit, le chercheur avec qui Charles Joly-Beauparlant fait son doctorat, a permis la collaboration entre nos deux équipes. Finalement, Dre Francine Durocher m’a permis de faire ma maîtrise dans son laboratoire. Elle a conçu le projet, a supervisé le bon déroulement et est en charge d’approuver la version finale de l’article. Elle a également répondu à toutes mes questions.
1
INTRODUCTION 1. Le cancer du sein
Au cours de l’année 2016, il est estimé que 196 900 Canadiens développeront un cancer et que 78 600 Canadiens vont mourir à la suite d’un cancer [1]. Le cancer est la première cause de mortalité au Canada devant les maladies cardiaques et les maladies cérébro-vasculaires [2]. De plus, la majorité des cancers qui seront diagnostiqués durant l’année seront des cancers du poumon, du sein, colorectal ou de la prostate. Chez les femmes canadiennes, il est estimé que 26% des cas de cancers diagnostiqués durant l’année seront des cas de cancers du sein. La survie à 5 ans après le diagnostic est de 88% pour les femmes diagnostiquées de cancer du sein. Malgré ces statistiques, une femme sur trente décèdera du cancer du sein au cours de l’année au Canada [3]. Le cancer du sein est donc la deuxième cause de mortalité par cancer chez la femme au Canada [1].
Le cancer du sein est une maladie complexe et hétérogène qui a une étiologie multifactorielle. Un grand nombre de facteurs influencent le risque de développer la maladie chez la femme comme les facteurs démographiques tels que l’âge ou l’ethnicité. D’autres facteurs peuvent influencer le risque comme les facteurs hormonaux, les facteurs environnementaux, le style de vie et les caractéristiques physiques. Les facteurs hormonaux sont influencés par la durée des menstruations ou par la prise de contraceptifs oraux. Les facteurs environnementaux et le style de vie sont par exemple, la prise d’alcool ou l’exposition aux rayonnements. La densité mammaire est aussi un facteur très important qui influence la susceptibilité de développer un cancer du sein [4-6]. Finalement, l’histoire familiale a aussi un impact sur le risque [7]. En effet chez certaines familles, la prévalence de développer le cancer du sein est beaucoup plus élevée que dans la population en général. Tous ces facteurs peuvent être combinés pour calculer le risque de développer la maladie. Il a été montré que 50% des cas de cancers du sein qui seront diagnostiqués durant l’année proviennent de seulement 12% de la population ayant un risque de plus de 10% de développer le cancer avant 70 ans. D’autre part, 50% de la population a un risque de moins de 3% de développer le
2
cancer du sein. Cette partie de la population représente seulement 12% des cas de cancer du sein [8].
1.1. Les gènes de susceptibilité dans le cancer du sein
Une partie des cancers du sein démontre un regroupement familial et chez ces familles, la maladie est de deux à quatre fois plus présente que dans la population en général. Cette augmentation du risque est expliquée en partie par des facteurs héréditaires. En effet, de 5 à 10% des cancers du sein sont dits héréditaires [9]. Sur la proportion des cancers qui sont dits héréditaires, environ la moitié des cas sont expliqués par des gènes de susceptibilité. D’autre part, il reste encore la moitié des cas héréditaires qui demeurent inexpliqués [10]. Le cancer du sein est une maladie très complexe et hétérogène ce qui explique que plusieurs types d’études ont été effectués pour essayer de trouver de nouveaux gènes de susceptibilité.
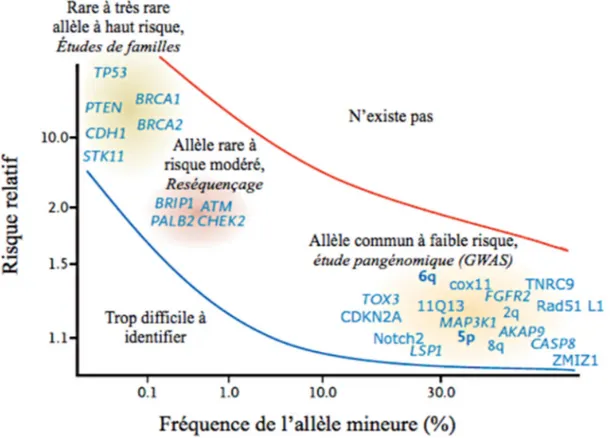
Premièrement, des études d’association ont été réalisées dans des familles pour trouver une partie du génome qui est associée au cancer du sein. Dans ce type d’étude, on évalue la co-transmission d’un marqueur génétique pour une région large du génome. Un séquençage est suivi dans la région identifiée précédemment pour trouver le gène associé à la maladie [11]. En second lieu, des études pan génomique ou «Genome Wide Association Study (GWAS)» ont été effectuées [12]. Ces études utilisent une grande quantité d’individus atteints et leurs contrôles. Un génotypage des polymorphismes pour les cas et pour les contrôles est fait. Les polymorphismes qui sont présents plus fréquemment pour les cas en comparaison avec les contrôles sont étudiés. Ces polymorphismes sont ceux associés à la maladie. Finalement, des études de gènes candidats ont été réalisées en étudiant des gènes qui sont connus pour avoir un effet dans des voies métaboliques qui sont liées au cancer du sein. Ces études sont réalisées à l’aide de méthodes de re-séquençage des gènes d’intérêt. Chacune de ces techniques a permis d’identifier des gènes ayant un risque relatif plus ou moins grand et une fréquence différente dans la population (Figure 1).
3
Figure 1 : Gènes et loci associés à la susceptibilité de développer le cancer du sein
Graphique montrant les gènes ou les régions du génome étant associés avec un risque plus élevé de développer le cancer du sein. La fréquence de l’allèle réfère à la fréquence où la mutation est retrouvée dans la population. Avec les données qui ont été obtenues jusqu’à présent, il est estimé qu’aucun gène ayant une grande fréquence et un grand risque relatif ne sera trouvé (en haut de la courbe rouge). De plus, aucun gène ne sera découvert avec un faible risque et une faible fréquence (en dessous de la courbe bleue). La méthode avec laquelle les gènes ont été découverts est indiquée au-dessus de chaque groupe. Figure tirée et traduite de Foulkes et al [11].
1.1.1. Gènes à risque élevé
Certaines études ont trouvé, à l’aide d’analyses, que certains gènes prédisposent à un risque élevé de développer le cancer du sein. Ces études sont faites lorsque certaines familles ont été reconnues pour avoir un risque plus élevé que le reste de la population de développer une certaine maladie. Avec des microsatellites, on regarde les parties du génome qui sont liées à la maladie. Il y a ensuite une étape de séquençage pour permettre de déterminer le ou les gènes ayant une mutation
4
dans cette partie du génome et qui seraient reliés à la maladie étudiée. Ces études permettent de trouver des mutations dans des allèles rares dans la population. À l’aide de cette méthode, Miki et al. ont trouvé un lien entre une mutation dans le gène BRCA1 et le cancer du sein et de l’ovaire [13]. Peu de temps après, Wooster et al. ont trouvé une relation entre le cancer du sein et une mutation dans le gène BRCA2 [14].
De plus, plusieurs syndromes héréditaires ont été associés au cancer du sein. Premièrement, le syndrome de Li-Fraumeni correspondant à une mutation dans le gène TP53 [15]. La protéine p53 agit comme un suppresseur de tumeur en régulant le cycle cellulaire, la réparation de l’ADN et l’apoptose [16]. Une mutation germinale de ce gène très important augmente donc le risque de développer un cancer, et ce, à un jeune âge. Deuxièmement, le syndrome de Cowden, une mutation germinale du gène PTEN, explique aussi une partie du risque. PTEN est une protéine tyrosine phosphatase qui est impliquée dans le contrôle de la prolifération cellulaire [17]. Les femmes affectées du syndrome de Cowden ont entre 25 et 50% de chance de développer le cancer du sein au cours de leur vie, et ce, de façon précoce [18]. Finalement, le syndrome de Peutz-Jeghers est associé à un risque cumulatif allant jusqu’à 54% pour le cancer du sein [19]. Ce syndrome implique une mutation dans le gène STK11 qui encode une sérine/thréonine kinase qui inhibe la prolifération cellulaire [20]. Par contre, chez les familles canadiennes-françaises à risque élevé de développer le cancer du sein, une mutation dans le gène STK11 ne contribue pas à la susceptibilité [21]. De plus, le gène CDH1 donne un risque élevé de développer le cancer du sein sans être associé à un syndrome. Ce gène code pour la protéine E-cadhérine qui est impliquée dans la suppression de l’invasion des tumeurs [22].
1.1.1.1. BRCA1/2
Le gène BRCA1 code pour une protéine qui joue un rôle dans le maintien de la stabilité génomique. La protéine s’associe avec l’ARN polymérase II et interagit avec des complexes d’histones déacétylase. De cette façon, la protéine BRCA1 joue un rôle dans la transcription, la réparation des bris double-brins et la
5
recombinaison. Cette protéine se retrouve sur le chromosome 17 et contient dans ses 1863 acides aminées un domaine RING et des domaines BRCT [13]. Quant à lui, le gène BRCA2 se retrouve sur le chromosome 13 et contient 3418 acides aminées codant pour des répétitions du motif BRCA [14]. Ces motifs permettent la liaison entre la protéine BRCA2 et RAD51. Tout comme BRCA1, ce gène est impliqué dans le maintien de la stabilité génomique spécifiquement dans la réparation des bris double-brins et la recombinaison homologue [23].
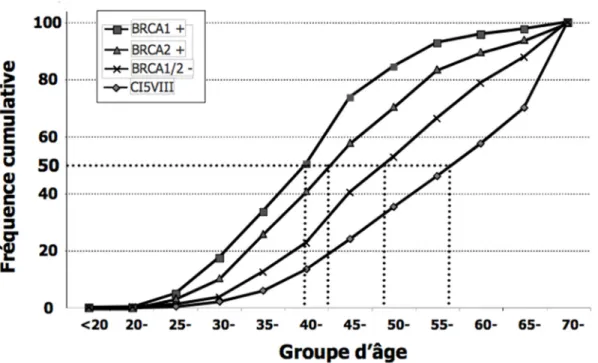
Il a été démontré que les femmes ayant une mutation dans le gène BRCA1 ou le gène BRCA2 ont tendance à développer un cancer du sein plus tôt. Cette comparaison a été faite avec des femmes ayant une histoire familiale et des femmes n’ayant aucune histoire familiale connue (Figure 2) [24]. En effet, 50% des cancers qui se développeront chez les femmes avec une mutation BRCA1 le feront avant l’âge de 40 ans. Par analogie, l’âge pour lequel 50% des cancers du sein seront développés chez les femmes n’ayant aucune histoire familiale est de 56 ans.
6
Figure 2 : Fréquence cumulative des cancers du sein selon la mutation retrouvée chez les femmes québécoises.
Distribution du pourcentage de femmes ayant développé le cancer du sein durant leur vie en fonction de la mutation. Les femmes avec une mutation dans le gène BRCA1 (BRCA1 +) vont développer la maladie plus tôt que les femmes avec une mutation dans le gène BRCA2 (BRCA2 +). Cette observation est la même pour les femmes ayant une histoire familiale sans mutation connue (BRCA1/2 -) et pour les femmes avec un cancer sporadique (CI5VIII). Figure tirée de Simard et al [24].
Par ailleurs, le risque de développer un cancer du sein est beaucoup plus élevé chez les femmes ayant une mutation BRCA1/2 comparativement au risque de cancer du sein sporadique. Effectivement, les femmes ayant une mutation dans le gène BRCA1 et BRCA2 ont un risque de développer le cancer du sein au cours de leur vie de 40 à 87% et de 40 à 84%, respectivement. Une prévalence pour le cancer de l’ovaire est aussi rapportée pour ces gènes, étant de 16 à 68% pour BRCA1 et de 11-27% pour BRCA2 [25-33]. De plus, il a été démontré que la pathologie des tumeurs BRCA1 diffère de celle des tumeurs BRCA2 et diffère également de celle des tumeurs sporadiques [34]. D’autre part, les tumeurs
7
BRCA1/2 sont aussi différentes des tumeurs non BRCA1/2. En effet, les cancers du sein reliés à une mutation dans le gène BRCA1 et BRCA2 sont en majorité des cancers de plus haut grade que ceux reliés à une histoire familiale mais non reliés à une mutation dans ces gènes [35]. Hedenfalk et al. ont démontré qu’avec le profil d’expression génique des tumeurs, il est possible de différencier les porteuses d’une mutation BRCA1, les porteuses BRCA2 et les patients avec une tumeur sporadique [36]. Par la suite, le profil d’expression génique pour les cellules normales a été regardé. Cette comparaison a été faite entre les porteuses d’une mutation BRCA1 et les contrôles qui sont des personnes sans histoire familiale ou non atteintes. Une différence d’expression a été remarquée après l’irradiation [37]. De plus, il est possible de calculer le risque absolu de développer le cancer du sein pour chaque femme et de classer ces femmes en centile avec le génotype d’autres polymorphismes (SNPs) connus (Figure 3).
8
Figure 3 : Porteurs BRCA1 et gènes modificateurs.
Le risque absolu de cancer du sein selon l’âge des porteuses de mutations BRCA1 classé par centile de la distribution des génotypes des SNPs. Le risque est basé sur l’association de loci de susceptibilité identifiés par GWAS pour 1q32, 10q25.3, 19p13, 6q25.1, 12p11, TOX3, 2q35, LSP1, RAD51L1, TERT. Les femmes porteuses faisant partie du 5% dits à faible risque auraient entre 10 et 20% de risque de développer la maladie à l’âge de 50 ans. Au contraire, les femmes faisant partie du 5% dits à haut risque auraient un risque absolu allant de 50 à 80% pour le même âge. Figure traduite de Couch et al. 2013 [38].
Cancer
du
sein
Âge
Risque
absolu
Moyenne 5‐95% 10‐90% Min, max20
30
40
50
60
70
80
0.0
0.2
0.4
0.
6
0.8
1.0
9
1.1.2. Gènes à risque faible et modéré
Plusieurs gènes donnant une susceptibilité faible de développer le cancer du sein ont été identifiés à l’aide d’études pangénomiques. Michailidou et al. ont trouvé plus de 90 loci donnant un risque relatif faible de développer la maladie à l’aide d’iCOGS [12]. Cette technologie est une puce développée par Illumina étudiant plus de 200 000 polymorphismes. Plusieurs projets utilisent cette puce, permettant ainsi d’obtenir de l’information sur un très grand nombre de cas de cancer du sein ou contrôle. Ces loci à faibles risques peuvent expliquer jusqu’à 23% des cas de cancers du sein héréditaires [39]. Ces mutations sont dites communes, car elles sont souvent retrouvées dans la population. De plus, un individu peut avoir plusieurs loci mutés et le risque relatif de développer la maladie peut être cumulé. Pour ce qui est des gènes à risque modéré, quelques gènes ont été trouvés à l’aide du re-séquençage. Cette technique consiste à séquencer les gènes qui sont dans les mêmes cascades que les gènes déjà connus pour avoir un impact dans le cancer. Ces gènes peuvent augmenter le risque de développer la maladie d’environ deux fois. Premièrement, BRIP1 est un gène codant pour une hélicase impliquée dans le sentier de la réparation de l’ADN dans le complexe de Fanconi [40]. Seal et al. ont trouvé cinq mutations germinales différentes dans ce gène pour les familles à haut risque de développer le cancer du sein donnant un risque relatif modéré pour le cancer du sein [41]. Ensuite, CHEK2 est une protéine kinase du cycle cellulaire impliquée dans la réparation de l’ADN [42]. Meijers-Heijboer et al. ont démontré qu’une mutation dans ce gène se retrouve plus souvent dans les familles à risque élevé que chez des individus sains [43]. PALB2 est une protéine qui interagit avec la protéine BRCA2 et aide à sa stabilité. De la même façon que CHEK2, une mutation dans cette protéine se retrouve plus souvent chez les familles à risque élevé [44]. Par contre, certains gènes à fréquence modérée sont encore discutés dans la communauté scientifique. En effet, il a été démontré que le gène CHEK2 et PALB2 ne contribuent pas à une importante partie de la susceptibilité au cancer du sein chez les familles québécoises [21]. Aussi, une
10
méta-analyse a été effectuée et suggère qu’il faut considérer un manque de résultats concluants pour certains gènes qui ont été étudiés [45].
1.1.3. BRCAX
Lorsqu’il est question de familles BRCAX, on parle en fait de toutes les familles à risque élevé de développer le cancer du sein et qui n’ont pas de mutation délétères dans le gène BRCA1 et le gène BRCA2. Ce groupe est prédominant dans les familles québécoises démontrant de nombreux cas de cancers du sein. En effet, à ce jour, plus de la moitié des cas de cancers héréditaires ne sont pas encore expliqués. Plusieurs études ont été réalisées afin d’identifier des gènes responsables de ce regroupement familial. De plus, différentes techniques ont été utilisées pour essayer de démystifier ce groupe. Aloraifi et al. ont utilisé le séquençage à haut débit sur 312 gènes pour essayer de trouver des mutations associées avec le cancer du sein [10]. Il a aussi été démontré que les tumeurs de familles non-BRCA1/2 sont de grades plus faibles que les tumeurs BRCA1, BRCA2 et sporadiques [35]. De plus, Fernández-Ramires et al. ont employé une biopuce d’ADN pour étudier l’expression de gènes reliés au cancer [46]. Ils ont été capables de différencier les tumeurs selon le type moléculaire de la tumeur sans pour autant être capable de différencier les cas BRCA1, des cas BRCAX et des cas sporadiques. Le marquage immunohistochimique a permis de déterminer que les tumeurs BRCAX sont de grades plus faibles que les tumeurs sporadiques [47]. Cette étude a aussi permis de séparer les tumeurs BRCAX en deux groupes selon le statut du récepteur d’estrogène (ER). Ces deux groupes peuvent être divisés en cinq sous-groupes qui sont très similaires à ceux des tumeurs sporadiques. Avec la signature transcriptionnelle, une classification moléculaire semblable a pu être obtenue pour le groupe non-BRCA1/2 (BRCAX) [48]. Finalement, avec le profil d’expression génique, il a été possible de séparer les individus BRCAX des individus avec une mutation dans le gène BRCA1 ou BRCA2 [49].
11
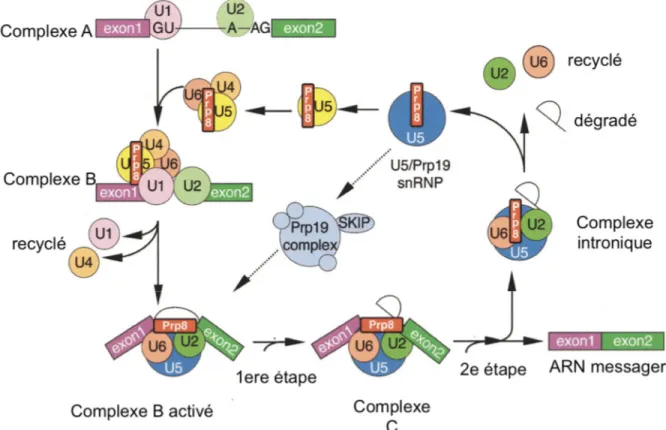
2. L’épissage
Dans les cellules, les gènes contiennent des introns et des exons qui sont transcrits par l’ADN polymérase. Cette étape forme l’ARN pré-messager qui doit ensuite être épissé pour former l’ARN messager (ARNm). Cet ARNm sera ensuite traduit en protéine. L’épissage des gènes se fait par le splicéosome (Figure 4). La machinerie d’épissage consiste en environ 150 protéines et 5 petits ARN nucléaires (snRNA) U1, U2, U3, U4, U5 [50,51]. Ces ARNs sont complexés avec des protéines appelées petites ribonucléoprotéines nucléaires (snRNPs) [52]. Au cours de l’épissage, U1 va reconnaitre le site d’épissage de la jonction entre l’exon et l’intron qui se trouve en 5’ de l’intron [53]. La plupart du temps, cette jonction est définie par une séquence GU à la fin de l’exon. Une sous-unité U2AF va se lier à l’extrémité 3’ de l’intron à un site riche en pyrimidines et ayant une séquence AG qui est le site d’épissage 3’. Cette sous-unité va aider la protéine BBP à se lier au site d’embranchement ce qui forme le complexe précoce ou le complexe E. Ensuite, U2 va se lier au site d’embranchement et il va y avoir libération de BBP et formation du complexe A. Par la suite, un complexe U4, U6, U5 se joint au complexe A et il y a un réarrangement pour former le complexe B et rapprocher les deux sites d’épissage [54]. Durant cette étape, U2AF est libéré. L’étape suivante consiste au relâchement de U1, et U6 prend sa place sur le site d’épissage 5’. U4 est ensuite relâchée et il y a interaction entre U2 et U6 ce qui forme un site actif et permet la catalyse de la réaction de transestérification. Au cours de cette réaction, l’extrémité 5’ de l’intron va s’attacher à la base A du site d’embranchement et un groupement 3’-OH est formé à l’extrémité 3’ du premier exon. La deuxième réaction de transestérification est aidée par U5 et le groupement 3’-OH fera la réaction sur le site d’épissage 3’. Cette réaction fera finalement une jonction entre les deux exons et il y aura libération de l’intron ce qui forme un transcrit [55]. Après la libération de l’intron, celui-ci est dégradé et les composants du splicéosome sont relargués et recyclés pour faire une nouvelle réaction d’épissage [56]. Cette réaction est très importante, car elle permet de conserver l’intégrité des protéines qui seront formées à partir de l’ARN messager (ARNm).
12
Figure 4 : Mécanisme d’épissage avec le splicéosome
Mécanisme d’épissage pour obtenir l’ARN messager. Les premières et deuxièmes étapes sont les étapes de transestérification. Prp8 est une protéine du splicéosome. Figure adaptée de RICHARD J. GRAINGER and JEAN D. BEGGS [57].
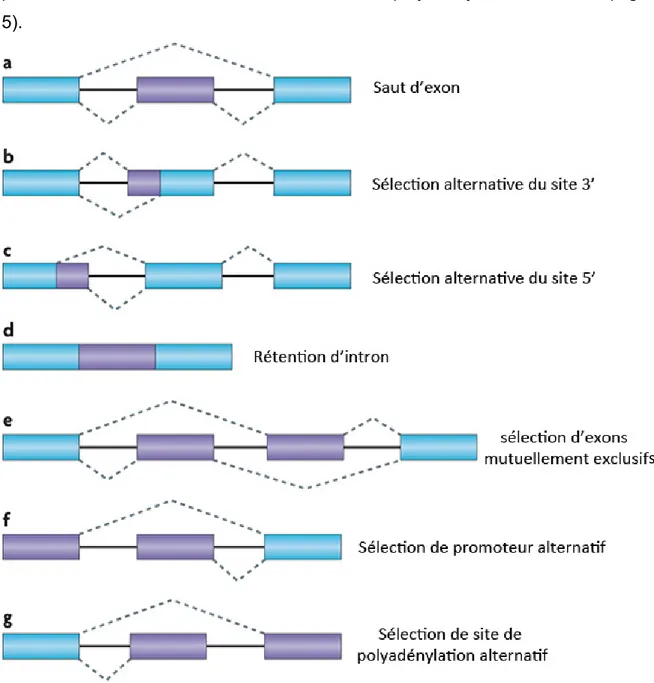
2.1. L’épissage alternatif
Selon le dogme général, un gène est équivalent à une protéine. Par contre, avec la complétion du projet du génome humain, seulement 20 000 à 25 000 gènes codant pour une protéine seraient présents chez l’humain [58]. En fait, il a été démontré en 1977 que, dans la cellule, un mécanisme d’épissage alternatif se produit [59]. L’épissage alternatif est un mécanisme qui permet une diversité protéique des cellules en produisant plusieurs transcrits à partir d’un même gène [60]. On parle de transcrit pour chaque ARN codant ou non codant produit dans la cellule. Chez l’humain, plus de 90% des gènes multi-exoniques forment plus d’un transcrit [61-63]. Il y a plusieurs types d’épissage alternatif qui peuvent se produire dans la cellule : le saut d’exon, la sélection alternative du site d’épissage en 3’ ou en 5’, la rétention d’introns, la sélection d’exons mutuellement exclusifs, la sélection de
13
promoteurs alternatifs et la sélection de sites de polyadénylation alternatifs (Figure 5).
Figure 5 : Différentes options d’épissage alternatif
Les différentes options peuvent être séparées en quatre grands groupes. Premièrement, le saut d’exon (a) qui équivaut à 40% des évènements d’épissage alternatif [64,65]. Deuxièmement et troisièmement, la sélection alternative du site d’épissage en 3’ (b) ou en 5’ (c) correspondant à 18,4% et 7,9% des évènements d’épissage, respectivement. Finalement, la rétention d’introns (d) équivalant à moins de 5%. La sélection d’exon mutuellement exclusif (e), la sélection de promoteur alternatif (f) et la sélection de site de polyadénylation alternatif (g) sont des évènements très rares chez les eucaryotes. Dans cette figure, les rectangles en bleu correspondent aux exons constitutifs et les rectangles en mauve, aux régions alternativement épissées. Les lignes sont les introns et les pointillés sont les options d’épissage alternatif. Figure traduite de HADAS KEREN et al [52].
14
Il se peut aussi qu’un ARN pré-messager subisse plus d’une de ces options, donnant ainsi une très grande variabilité de transcrits pouvant être formés à partir d’un gène donné. Toutes ces modifications peuvent aussi changer le cadre de lecture et affecter la protéine produite. Ces changements peuvent causer la dégradation de la protéine en créant un codon stop prématuré [66]. De plus, l’épissage alternatif est un mécanisme tissu spécifique, c’est-à-dire que certains transcrits qui sont formés sont spécifiques à un tissu ou même à un moment durant le développement [67-69]. Par contre, ce ne sont pas tous les variants d’épissage alternatif qui produisent des ARNms codants pour des protéines. Par exemple, certains transcrits peuvent former des longs ARNs non codant ou même former des ARNms avec un codon stop précoce. En fait, environ 60% des variants produisent des isoformes de la protéine [70].
2.1.1. La régulation de l’épissage alternatif
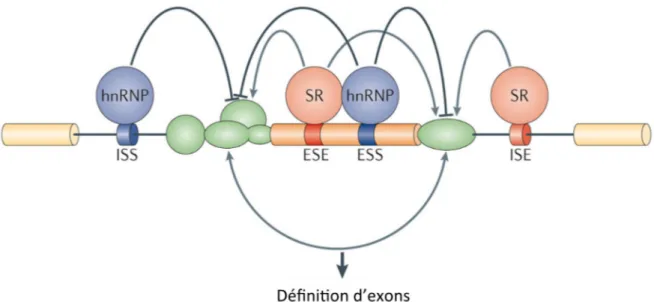
Plusieurs éléments régulateurs positifs et négatifs affectent la formation du splicéosome et donc la formation de certains transcrits [71]. Ce sont des éléments qui sont riches en purine et qui sont proches des séquences épissées [51]. Des protéines peuvent s’attacher à ces séquences présentes dans l’ARN. Elles peuvent se lier à des séquences spécifiques dans les parties introniques ou exoniques pour amplifier (ISE et ESE) ou réprimer l’épissage (ISS et ESS) tel que présenté dans la figure 6 [72]. Les protéines SRs sont des protéines qui s’attachent aux séquences amplificatrices des exons et des introns. Ces protéines activent l’épissage constitutif et alternatif en recrutant les composants du splicéosome [73]. Les protéines hnRNPs sont aussi des protéines qui s’attachent à l’ARN. Il existe environ 20 types de hnRNPs différents chez l’humain, allant de A1 à U [74]. Ce sont des protéines régulant négativement l’épissage alternatif, sauf quelques exceptions. Plusieurs mécanismes d’action sont utilisés par ces protéines pour affecter l’épissage des gènes. Les hnRNPs peuvent former des polymères sur les exons [75], bloquer le recrutement des protéines formant le splicéosome [76,77] ou en formant une boucle avec des exons au complet [72,78].
15
Par contre, contrairement aux protéines SRs, certains hnRNPs peuvent affecter de façon positive la formation du splicéosome selon la position sur l’ARN [79]. Effectivement, il peut y avoir la formation d’un complexe entre hnRNPs permettant de rapprocher les sites d’épissage et donc de favoriser la formation du complexe du splicéosome. L’épissage alternatif peut aussi être influencé par la transcription. En effet, une vitesse lente d’élongation peut affecter l’épissage en permettant de reconnaitre un site d’épissage faible. De cette façon, il serait possible d’inclure un exon alternatif par exemple [80]. De plus, des facteurs comme SWI/SNF peuvent créer des pauses dans le mécanisme d’élongation ce qui permettrait d’inclure des exons alternatifs [81].
Figure 6 : Séquences et éléments régulateurs de l’épissage alternatif.
L’épissage est régulé par des séquences présentes dans l’ARN pré-messager. Ces séquences peuvent être dans l’exon pour amplifier (ESE) ou réprimer (ESS), ou dans l’intron pour encore une fois amplifier (ISE) ou réprimer (ISS) l’épissage. Il y a deux types de protéines qui s’attachent principalement à ces séquences ; les protéines SRs (rouge) et hnRNPs (bleu). Ces protéines ont comme cible la régulation du splicéosome qui s’associe aux exons (vert). Figure traduite de ALBERTO R. KORNBLIHTT et al [82].
2.1.2. L’épissage alternatif dans le cancer
Une dérégulation de l’expression génique peut être une cause dans la formation des cancers chez l’individu. En effet, il a été démontré que plusieurs processus
16
peuvent influencer l’expression. La méthylation de l’ADN est un des mécanismes qui a un impact sur l’expression des gènes. De plus, il a été démontré qu’une hyper ou hypométhylation peut être associée avec le cancer du sein [83]. L’épissage alternatif est une autre forme de régulation de l’expression génique qui se produit dans la cellule et qui peut être dérégulée, causant ainsi un problème [84]. Il a été démontré qu’une dérégulation de l’épissage alternatif est impliquée dans 15 à 50% des maladies chez l’humain [85]. De plus, il a été prouvé qu’il y a moins d’épissage alternatif qui se produit dans le cancer que dans les cellules normales [86]. L’épissage alternatif peut avoir un impact au niveau de plusieurs processus qui peuvent influencer la croissance tumorale. Un bon exemple est l’épissage alternatif du gène Bcl-X dont la forme longue a une fonction anti-apoptotique et la forme courte a une fonction pro-anti-apoptotique [87]. Donc, la production de l’isoforme long pourrait aider à la croissance des cellules cancéreuses. Un autre bon exemple est l’épissage du gène VEGF-A. Une des isoformes du gène a une fonction angiogénique alors qu’une autre isoforme a une fonction anti-angiogénique [88]. Une étude a été faite sur le cancer du sein en étudiant des évènements d’épissage alternatif sur des gènes associés à ce cancer. Les auteurs ont été capables de séparer les tissus sains des tissus cancéreux à l’aide des 41 meilleurs évènements d’épissage alternatif [89].
2.1.2.1. La régulation de l’épissage alternatif dans le cancer
Des protéines amplificatrices comme SRSF3 peuvent être surexprimées et réguler l’épissage de gènes importants dans la cellule comme p53 [90]. Puisque p53 est une protéine qui joue un rôle dans la régulation du cycle cellulaire, elle est donc une protéine qui a une fonction de suppresseur de tumeur [15,66]. SRSF1 est une autre protéine de régulation qui peut influencer le cancer. En effet, SRSF1 régule l’épissage alternatif de plusieurs gènes et donc, la surexpression de cette protéine pourrait donner un avantage à la croissance tumorale [66]. En fait, SRSF1 favorise l’expression de transcrits ayant une fonction anti-apoptotique des gènes BIM et BIN1 [91]. Les protéines hnRNPs sont aussi impliquées dans le développement du cancer [92]. La protéine hnRNP A1 a été associée à plusieurs cancers. Dans le
17
cancer du poumon, sa surexpression est associée à la prolifération tumorale [93]. Il a aussi été démontré que la diminution de l’expression des protéines hnRNPs A2/B1 dans la lignée cellulaire MDA-MB-231 réduit l’invasion cellulaire et donc la prolifération cellulaire [94].
3. Séquençage à haut débit
Les technologies de séquençage à haut débit ont été utilisées avec le projet du génome humain. Le premier génome humain à être séquencé, a pris plusieurs années et des millions de dollars et a été réalisé avec le séquençage de Sanger [95]. Le séquençage à haut débit est une méthode qui est apparue durant les années 2000. Ces nouvelles méthodes de séquençage permettent d’obtenir un grand nombre d’informations sur l’ADN, l’ARN ou les protéines, et ce, à faible coût. Ces techniques s’effectuent rapidement, ce qui a aidé au projet de séquençage de 1000 génomes humains [96,97]. Ces nouvelles techniques permettent de faire des analyses systématiques en transcriptomique, protéomique, épigénomique et en métagénomique. Le but de ces techniques est d’obtenir de l’information sur tout le génome et non seulement sur quelques gènes [98]. La plus grande sensibilité du séquençage à haut débit comparativement au séquençage Sanger permet de trouver des mutations dans des petites portions du génome [99]. Un des avantages du séquençage à haut débit est la possibilité de regarder les mutations, les réarrangements chromosomiques et la variation du nombre de copies en une seule plateforme [100,101]. Ces techniques de séquençage à haut débit permettent de comprendre des mécanismes moléculaires dans la cellule et sont donc de plus en plus importantes dans la recherche [98,102]. Un exemple de la compréhension de certains mécanismes moléculaires a été démontré par Huber-Keener et al. Ils ont regardé les gènes différentiellement exprimés entre les cellules cancéreuses du sein qui sont résistantes ou non au tamoxifène. Cette étude permet donc de déterminer les gènes impliqués dans le mécanisme du développement de la résistance à cet agent dans les cellules [103]. Une autre étude du même genre a permis de trouver des gènes associés avec une cytotoxicité reliée au traitement avec des agents chimiothérapeutiques sur des
18
lignées de lymphocytes immortalisés [104]. Le séquençage à haut débit a aussi permis d’obtenir de l’information sur le processus d’épissage alternatif qui se produit dans la cellule [105].
3.1. «RNA sequencing»
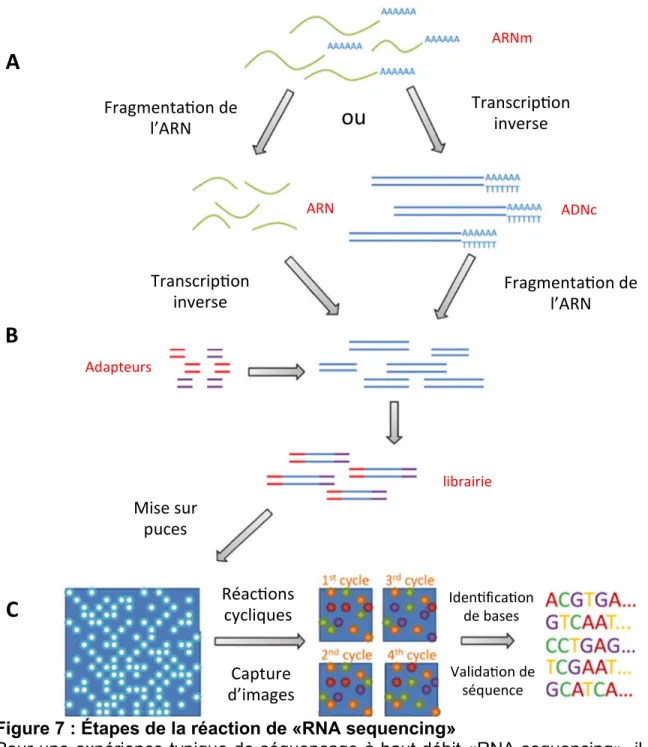
Cette technique est une méthode de séquençage à haut débit utilisée pour faire l’analyse transcriptomique. Tel que présenté à la figure 7, cette méthode consiste en l’extraction de l’ARN pour ensuite faire la fragmentation de celui-ci et la production d’ADN complémentaire (ADNc) [106]. Ces deux dernières étapes peuvent aussi être inversées (figure 7a). Des séquences adaptatrices ont ensuite été ajoutées aux fragments pour former une librairie (figure 7b). Ces séquences adaptatrices permettent aux fragments de s’attacher à une puce par complémentarité. Les fragments sont ensuite amplifiés à l’aide d’une polymérase pour avoir une copie de chaque brin attachée à la puce. Puis, les brins sont dénaturés et le fragment de départ est enlevé. La formation de pont PCR avec des séquences complémentaires aux séquences adaptatrices permet une amplification des fragments. Cette amplification crée des milliers de regroupements d’ADNc provenant de tous les fragments de départ. Les fragments de sens inverses sont par la suite enlevés et seulement les brins restants sont séquencés [96]. La séquence adaptatrice sert d’amorce au séquençage et des bases fluorescentes entrent en compétition pour s’attacher aux fragments. Seules les bases complémentaires vont pouvoir s’attacher. Une lumière excite alors la base et une couleur spécifique est émise par la base ajoutée pour chaque regroupement. Ce mécanisme est produit de façon cyclique et la couleur émise est enregistrée par la machine et des fragments de séquences connues sont obtenus (figure 7c). Les mêmes étapes sont ensuite faites pour le brin inverse. Tous les fragments de séquence obtenus sont alors alignés pour former des contigs. Les contigs sont ensuite alignés sur le génome de référence. L’alignement à l’aide de cette méthode permet de déterminer les transcrits qui sont présents dans la cellule et permet aussi de déterminer l’importance de ces transcrits [107].
19
Figure 7 : Étapes de la réaction de «RNA sequencing»
Pour une expérience typique de séquençage à haut débit «RNA sequencing», il y a extraction d’ARN qui est rétro-transcrit pour former une librairie de longueurs égales. Figure traduite de LIN WANG et al [106].
Plusieurs avantages et inconvénients sont reliés à cette technologie. En effet, plusieurs avantages sont remarquables en comparant avec les biopuces d’expression génique étudiant le transcriptome humain [107]. Premièrement, le problème de choix des sondes utilisées pour chaque expérience ne se retrouve pas avec la technologie de «RNA-sequencing», ce qui évite de passer à côté
A
C
B
ou
Transcrip oninverse Fragmenta onde l’ARN Transcrip on inverse Fragmenta onde l’ARN ARNm ARN ADNc librairie Adapteurs Réac ons cycliques Capture d’images Iden fica on de bases Valida on de séquence Misesur puces20
d’informations importantes [108]. De plus, il n’y a pas de saturation du signal possible, ni de problème de détection affecté par l’hybridation croisée [109,110]. D’ailleurs, la technique de RNA-seq permet une détection d’un signal qui est beaucoup plus faible que ce que les biopuces permettent et il est possible de voir une différence dans l’expression plus facilement [111]. Cette méthode permet aussi de découvrir de nouveaux transcrits ce qui est un avantage [112]. À l’inverse, certains désavantages sont aussi reliés avec cette technologie comme le fait qu’il faut une grande quantité de matériel biologique au départ [107]. De plus, le coût par échantillon est plus faible pour les biopuces que pour le RNA-seq [111]. Finalement, il faut une grande capacité de stockage de l’information ainsi que plusieurs outils de traitement des données obtenues ce qui peut créer un biais [113].
3.1.1. «RNA sequencing» dans le cancer
La technique de «RNA sequencing» permet d’étudier le transcriptome pour chaque individu. Il est donc possible de trouver les transcrits différentiellement exprimés entre ceux-ci. Depuis l’apparition de cette technologie, un grand nombre d’études ont été réalisées pour étudier la différence dans le transcriptome entre des individus atteints et non atteints de cancer à l’aide de la méthode «RNA sequencing». Jakhesara et al. ont étudié la différence d’épissage alternatif entre des cancers buccaux et des tissus normaux [114]. À l’aide du programme web DAVID [115], ils ont pu faire une analyse fonctionnelle des gènes différentiellement exprimés ce qui leur a permis de mieux comprendre le grand nombre de données qui étaient à leur disposition [115,116]. Eswaran et al. ont regardé les transcrits différentiellement exprimés dans le cancer du sein [117,118]. Ils ont utilisé le tissu de trois types de cancer du sein; le cancer du sein triple négatif, le cancer du sein non triple négatif et le cancer du sein HER2 positif. Avec leur approche, ils ont pu confirmer plusieurs gènes qui avaient été rapportés dans la littérature comme étant différentiellement épissés dans le cancer du sein tels que CDK4, LARP1, ADD3 et PHLPP2 [119-123]. D’autres études du même genre ont été faites sur plusieurs types de cancers pour trouver les variants d’épissage alternatif qui sont
21
différentiellement exprimés entre les patients atteints et non atteints [124-126]. Une étude a aussi été effectuée par Danan-Gotthold et al. sur un grand nombre de patients atteints et non atteints pour huit types de cancers différents [127]. Ils ont trouvé que des régulateurs spécifiques de l’épissage comme RBFOX2, MBNL1/2 et GK1 seraient responsables du changement d’épissage alternatif, et ce, pour plusieurs types de cancers. Yang et al. ont utilisé des valeurs de «RNA sequencing» pour faire l’analyse des gènes et sentiers impliqués dans un type de cancer du rein [128]. Ils ont pu confirmer certains sentiers métaboliques qui étaient rapportés dans la littérature comme étant associés avec ce cancer, tel que le sentier signalétique de PPAR.
4. Problématique
Le cancer du sein est une maladie très complexe et son développement est influencé par un grand nombre de facteurs. Il est donc difficile de prédire la susceptibilité de chaque individu. Une partie du risque est héréditaire et chez ces familles le risque est plus facile à calculer. Par contre, tel que mentionné précédemment, plus de la moitié de cas de cancers du sein héréditaires ne sont pas encore associés à une mutation conférant une susceptibilité accrue à la maladie. Pour ces familles, il est donc plus difficile de prédire si l’individu sera atteint ou non de la maladie au cours de sa vie.
Afin de palier à cette problématique, l'étude de l'expression génique pourrait s'avérer être une technique efficace pour trouver des gènes de prédisposition. Ainsi, l’étude du transcriptome chez ces individus serait donc une excellente alternative afin de trouver des transcrits impliqués dans le développement de la maladie. En effet, il a été démontré que l’épissage alternatif est une modification post-transcriptionnelle qui est héréditaire [129]. Il devient par conséquent possible d’étudier le transcriptome et de trouver des transcrits qui sont associés avec une susceptibilité de développer la maladie chez les familles à risque élevé de développer le cancer du sein.
22
5. Objectifs
L’objectif de ce projet est d’identifier des transcrits conférant une susceptibilité au cancer du sein chez des familles québécoises à haut risque à l’aide de la technique de «RNA sequencing». Ces transcrits pourraient ainsi être utilisés comme biomarqueurs. Les objectifs spécifiques de ce projet sont :
1. Extraire l’ARN de lymphocytes immortalisés des patientes BRCA1/2/X chez des femmes atteintes et non atteintes.
2. Déterminer les transcrits qui sont significativement et différentiellement exprimés entre ces individus.
3. Comprendre les mécanismes et fonctions moléculaires associés à ces transcrits significatifs.
4. Sélectionner certains transcrits qui pourraient être utilisés comme biomarqueurs de prédisposition au cancer du sein héréditaire chez des familles québécoises à haut risque pour des études ultérieures.
23
CHAPITRE 1
Transcriptional Signature of lymphoblastoid cell lines of BRCA1, BRCA2 and
non-BRCA1/2 High Risk Breast Cancer Families
Marie-Christine Pouliot, Yvan Labrie, Charles Joly-Beauparlant, Geneviève Ouellette, Jacques Simard, Arnaud Droit and Francine Durocher
*CHU de Québec Research Centre-Université Laval, Department of Molecular Medicine, Québec, Canada, G1V4G2
*Correspondence: Francine Durocher
CHU de Québec Research Centre, Laval University 2705 Laurier Blvd, Bloc R4778
Québec (Québec) G1V4G2
Francine.Durocher@crchudequebec.ulaval.ca Phone 418 525-4444, ext. 48508
Fax 418 654-2278
Running Title: Whole-gene expression profile in high-risk breast cancer families
Accession code: Gene expression data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE87080
24
Résumé
Approximativement 25% des cas de cancer du sein héréditaires associés avec une importante histoire familiale peuvent être expliqués par une mutation dans BRCA1, BRCA2 ou d’autres gènes à pénétrance plus faibles. Le reste des familles à haut risque peuvent être classées comme des familles BRCAX (non BRCA1/2).
L’expression génique impliquant l’épissage alternatif représente un mécanisme bien connu régulant l’expression de multiples transcrits qui pourraient être impliqués dans le développement du cancer. En utilisant le séquençage à haut débit d’ARN, l’analyse du transcriptome a été entrepris pour potentiellement trouver des transcrits impliqués dans la susceptibilité ou le développement du cancer du sein.
L’ARN a été extrait de lignées cellulaires de lymphocytes immortalisés pour 117 femmes (atteintes et non-atteintes) provenant de familles BRCA1, BRCA2 et BRCAX. L’analyse Anova a révélé un total de 95 transcrits, correspondant à 85 gènes différents, qui sont différentiellement exprimés (p-value corrigé par Bonferroni < 0.01) entre ces groupes. Un regroupement hiérarchique a permis une séparation distinctive entre les sous-groupes BRCA1/2 et BRCAX. 67 transcrits peuvent distinguer les individus BRCAX des individus BRCA1/2 et 28 peuvent distinguer les individus BRCAX atteintes des non atteintes.
À notre connaissance, ceci est la première étude identifiant des transcrits différentiellement exprimés dans des lignées cellulaires de lymphocytes provenant des principales catégories de sous-groupes du cancer du sein liés à une mutation, c’est-à-dire BRCA1, BRCA2 et BRCAX. De plus, quelques transcrits peuvent distinguer les individus BRCAX atteints des non atteints, ce qui pourrait représenter des cibles thérapeutiques potentielles pour le traitement du cancer du sein.
25
Abstract
Approximately 25% of hereditary breast cancer cases associated with a strong familial history can be explained by mutations in BRCA1 or BRCA2 and other lower penetrance genes. The remaining high-risk families could be classified as BRCAX (non-BRCA1/2) families.
Gene expression involving alternative splicing represents a well-known mechanism regulating the expression of multiple transcripts, which could be involved in cancer development. Thus using RNA-seq methodology, the analysis of transcriptome was undertaken to potentially reveal transcripts implicated in breast cancer susceptibility and development.
RNA was extracted from immortalized lymphoblastoid cell lines of 117 women (affected and unaffected) coming from BRCA1, BRCA2 and BRCAX families. Anova analysis revealed a total of 95 transcripts corresponding to 85 different genes differentially expressed (Bonferroni corrected p-value <0.01) between those groups. Hierarchical clustering allowed distinctive subgrouping of BRCA1/2 subgroups from BRCAX individuals. 67 transcripts could discriminate BRCAX from BRCA1/BRCA2 individuals while 28 discriminate affected from unaffected BRCAX individuals.
To our knowledge, this represents the first study identifying transcripts differentially expressed in lymphoblastoid cell lines from major classes of mutation-related breast cancer subgroups, namely BRCA1, BRCA2 and BRCAX. Moreover, some transcripts could discriminate affected from unaffected BRCAX individuals, which could represent potential therapeutic targets for breast cancer treatment.
26
Key words: Hereditary breast cancer, high-risk BRCA1/2/X families, gene expression, RNA-Seq, lymphoblastoid cell lines
27
Introduction
In 2015, breast cancer represented 26% of all cancer cases among Canadian women and was the second leading cause of cancer death with 14% [1]. Like every common cancer, breast cancer shows some degree of familial clustering [2]. Those high-risk families having multiple cases of breast or ovarian cancer are associated with a higher risk of developing breast cancer during their lifetime than other families [3]. It is thought that approximately 10-15% of breast cancer cases are hereditary and associated with mutations in BRCA1 or BRCA2 genes and some other genes having high to moderate penetrance such as TP53,
PTEN, ATM, CHEK2, PALB2 and BRIP1 and ATR, which account for approximately 5% of
the risk [4-10]. Common variants have also been identified in additional susceptibility loci and would explain a further ~16% of the 2-fold familial risk of breast cancer [11]. Among our French Canadian cohort, 24% of high-risk breast cancer families were found to be carriers of a deleterious BRCA1 or BRCA2 mutation [12].
Therefore, susceptibility alleles for more than half of high-risk families remain unknown. A portion of these remnant breast cancer families could be explained among others by modulation of gene expression, which is mainly regulated through methylation or alternative splicing (AS) mechanisms. Indeed, more than 90% of human genes undergo alternative splicing and it is now becoming clear that AS plays an important role in human cancer development [13,14]. RNA sequencing allows a genome-wide expression study of the transcriptome and can likely detect and quantify all coding and non-coding transcripts [15]. The use of RNA sequencing greatly enhanced our understanding of gene expression in cells [16].
Human immortalized lymphoblastoid cell lines (LCLs) provide information on gene expression without having to consider tissue specific expression [17]. LCLs used for the
28
establishment of gene expression or splicing signatures are recognized as a reliable biological material to study a given disease [18-28], and some studies recently showed the heritability of splicing as some exons were spliced in an allele-specific manner [18,29,30]. Moreover, It has been demonstrated that LCLs can be used to study life-course environmental epigenetics [31].
Previous studies attempted to discriminate BRCA1/2, non-BRCA1/2 (BRCAX) and sporadic breast cancers based on gene expression levels and histological tests performed on breast tumor tissue [32-37]. In another study, although several genes or spliced transcripts were identified as differentially expressed in familial cases, they did not allow clusterization of BRCA1, BRCA2 and BRCAX tumor tissues [38].
In this study, we performed RNA sequencing on LCLs coming from BRCA1/2 and BRCAX affected and unaffected individuals coming from high-risk breast cancer families in an attempt to distinguish breast cancer subgroups based on their transcriptome profile. This study revealed several transcripts involved in regulation of translation, apoptosis, cell cycle as well as cell growth and proliferation, which could discriminate BRCAX individuals from BRCA1/2 subgroups.
29
Results
Our French Canadian cohort comprised three major familial breast cancer subgroups namely BRCA1 and BRCA2 carriers as well as BRCAX individuals, i.e. non BRCA1/2 (affected and unaffected). The BRCA1 cases included 25 individuals affected with breast cancer and 11 unaffected women, who were carriers of BRCA1 mutations namely R1443X (22 individuals), 3705insA (2 ind), 2244insA (7 ind), 2953del3+C (2 ind) and three individuals carrying E352X, 4160delAG or 1723del9ins13 mutation, respectively. The BRCA2 subgroup was composed of 31 affected and 18 unaffected individuals carrying 8765delAG (44 ind), E3002K (2 ind), 6503delTT (1 ind), R3128X (1 ind) or 3036del4 mutation (1 ind). The BRCAX subgroup included 16 affected and 16 unaffected individuals, which represented 16 pairs of sisters (1 affected and 1 unaffected per family). It should be noted that the oldest unaffected sister available was purposely selected in BRCAX families. This subgroup of unaffected sisters was used as controls for comparison purpose in the analyses described below.
RNA-Seq analyses generated an average of 68 million reads per sample, and more than 85% of the reads were aligned to the hg19 human reference genome using TopHat (data not shown). As displayed in Supplemental Table 1, out of a total of 173 259 transcripts detected, 95 transcripts (0.05 % of all transcripts) were found to be significantly (p<0.01) and differentially expressed based on the Bonferroni-corrected ANOVA analysis, when considering all four breast cancer subgroups (BRCA1, BRCA2, unaffected and affected BRCAX individuals). All these significant transcripts were encoded by 85 different genes. In addition to the main isoforms (one per gene), 10 mRNA isoforms were considered as
30
alternatively spliced isoforms (11.8%). These significant transcripts included 54 gene isoforms showing a highly significant Bonferroni-corrected p-value (p < 0.001).
Principal component analysis (PCA) of these 95 transcripts identified as differentially expressed among BRCA1-carriers (n=36), BRCA2-carriers (n=49), unaffected BRCAX (n=16) and affected BRCAX (n=16) individuals is presented in Figure 1. The first three principal components of transcriptional variation, accounted for 59.6 % of the total variance. PCA on the full dataset showed that the PC1 component accounted for 46 % of the variance, which is highly informative, while PC2 was also informative compared to the variance explained in the randomized data set.
Unsupervised hierarchical clustering of all BRCA1, BRCA2 and BRCAX individuals was then performed using the 95 significant transcripts. As illustrated in Figure 2, gene expression levels of these significant transcrits allowed to discriminate distinctly BRCA1/2 from BRCAX (unaffected and affected) individuals. However, it was not possible to segregate BRCA1 from BRCA2 individuals as well as affected from unaffected BRCAX individuals. In addition, when considering BRCA1 and BRCA2 individuals, no specific clustering could be observed based on gene mutation or the status of the disease (data not shown).
The ANOVA analysis followed by conservative post hoc Scheffé test, which is appropriate for comparing groups with unequal sample sizes, allowed to potentially identify transcripts discriminating BRCA1, BRCA2 and affected BRCAX individuals from unaffected BRCAX individuals, which were used as controls in this analysis. This analysis revealed 69, 71 and 28 gene isoforms differentially expressed from BRCAX unaffected for BRCA1, BRCA2 and affected BRCAX individuals, respectively (See Supplemental Table 1). It
31
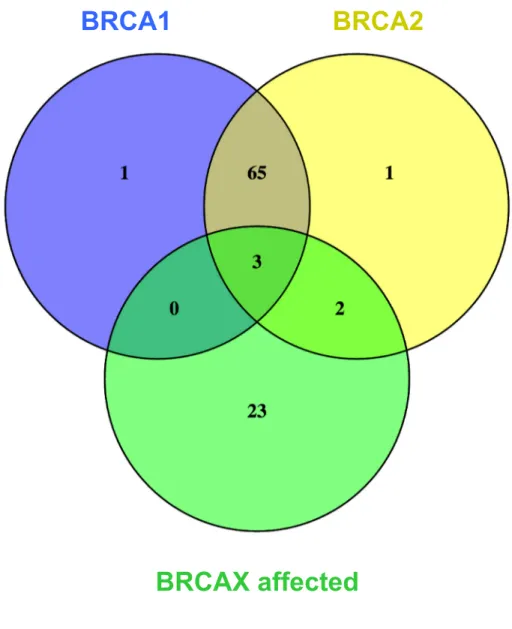
should be noted that the large majority of transcripts identified in BRCA1-carriers were also found in BRCA2-carriers.
As presented in Figure 3, these transcripts were then illustrated in Venn diagrams, which showed that 3 common transcripts (3.2%) were differentially expressed in all three subgroups, when compared to unaffected BRCAX individuals. In addition, a large portion of transcripts (65: 68.4%) was commonly identified in BRCA1 and BRCA2 subgroups, while only 1 transcript was specifically and exclusively associated with BRCA1, and another one (1) different transcript with BRCA2. This illustrated the similarity between BRCA1 and BRCA2-carrier individuals regarding their gene expression profile. On the other hand, 23 gene isoforms were exclusively associated with affected BRCAX individuals and were not different in BRCA1 and BRCA2 subgroups.
In an attempt to further discriminate unaffected and affected BRCAX individuals, hierarchical clustering was then performed using the 28 gene isoforms discriminating both BRCAX subgroups (Figure 4). Although a much better clustering could be observed between both subgroups, these genes could not differentiate distinctly unaffected from affected BRCAX individuals, with 4 affected individuals being located among the unaffected individuals.
Then, Scheffé analysis performed on all 4 subgroups (BRCA1, BRCA2, unaffected and affected BRCAX) combined allowed the identification of specific transcripts, which are exclusively associated with each subgroup. As listed in Table 1, although no specific transcripts were specifically associated with BRCA1 or BRCA2 individuals, we could identify 67 transcripts specifically associated with BRCA1/BRCA2 following combination
32
of both subgroups and compared to BRCAX individuals. In addition, 3 and 28 transcripts showed exclusive association with unaffected and affected BRCAX subgroups, respectively.
Of interest, out of 67 transcripts specifically associated with BRCA1/BRCA2 subgroups combined, 19 transcripts (28%) were involved in DNA repair, cell proliferation, apoptosis and cell cycle, 32 (48%) different transcripts exert an action in transcription, translation as well as mRNA and protein metabolic processes, while 10 transcripts (15%) were involved in immune processes. Regarding the 28 transcripts exclusively associated with BRCAX affected individuals, more than 28% were involved in DNA repair/proliferation/apoptosis/cell cycle mechanisms, and approximately 43% (12 transcripts) were implicated in transcription and translation-related processes. It should be noted that XRCC6, SGSM3 and HDGF transcripts were associated with BRCA1/BRCA2, BRCAX unaffected and BRCAX affected individuals given that their expression was differentially expressed between all three subgroups.
The 85 genes associated with the 95 significant transcripts (95) identified as differentially expressed between BRCA1, BRCA2 and BRCAX (unaffected or affected) individuals were then submitted for pathway and molecular function analyses. Using Ingenuity Pathway analysis, enrichment of several canonical pathways and functions were then identified. It should be noted that mapped genes can be classified in more than one biological process or metabolic process.