Analyse des profils de méthylation de l’ADN des
spermatozoïdes de taureaux péri-pubères
Mémoire
Simon Lambert
Maîtrise en sciences animales
Maître ès Sciences (M.sc)
Québec, Canada
iii
Résumé court
La pression de sélection pour la reproduction des bovins laitiers se traduit par l'utilisation d’animaux de plus en plus jeunes. Bien que la manipulation hormonale puisse accélérer la puberté, les gamètes et les embryons qui en résulteraient pourraient être touchés par le contexte endocrinien différent apporté par ces manipulations. De plus, l’information portée par le gamète mâle dépasse largement la qualité ses gènes. En effet, les marques portées par les histones ainsi que les patrons de méthylation amenés à l’ovocyte par le spermatozoïde ont un rôle clé à jouer pour le développement de l’embryon qui sera formé. Le but de cette étude est donc d’observer les modifications possibles dans la méthylation de l’ADN des gamètes mâles au début et à la fin de la puberté .
Une analyse épigénétique des patrons de méthylation de l'ADN sur la semence bovine d’animaux âgés de 10 vs 12 vs 16 mois a été effectuée par un procédé direct, c’est-à-dire via la plateforme EDMA d’EmbryoGENE. L’analyse d’un même taureau à différent âge n’a jamais été faite et pourrait pointer vers certains locus spécifiques permettant de déterminer à quel âge la maturité épigénétique est atteinte. Nos analyses ont démontré que des différences significatives entre les profils de méthylations des spermatozoïdes provenant d'animaux âgés de 10 mois peuvent être observées lorsque comparées aux profils d’animaux âgés de 16 mois. Les différences observées entre 12 et 16 mois ne sont pas significatives. Nos conclusions sont donc que le sperme provenant d’animaux âgés de 10 mois n’a pas un profil de méthylation adulte. Cette situation semble s’être corrigée lorsque l’animal est âgé de 12 mois.
iv
Résumé long
L’industrie bovine est un secteur de grande importance au Canada. En effet, à elle seule l’industrie laitière canadienne représentait un chiffre d’affaires de 15.7 milliards de dollars pour l’année 2013 (commission canadienne du lait). L’industrie laitière canadienne est reconnue pour la qualité génétique supérieure de son cheptel bovin, de même que pour ses programmes génétiques laitiers (c’est à dire les programmes de contrôle laitier et les programmes d’enregistrement du bétail laitier et de la classification pour le type). Le Canada détient 41 % du marché mondial d’exportation (2008) de bovins reproducteurs de race pure et d’embryons. La race holstein est la race laitière la plus importante (92 % du cheptel laitier). Les semences de bovins laitiers canadiens ont été exportées vers 84 pays différents principalement vers les États-Unis, les Pays-Bas, le Japon et l’Espagne.
Le Canada est à l’avant-garde des nouvelles technologies en matière de génétique laitière. Grâce au génotypage, les généticiens peuvent déterminer le profil d'ADN d'un animal et produisent actuellement des évaluations génomiques pour plus de 60 caractères différents. Depuis août 2009, le Réseau laitier canadien (RLC) publie des évaluations génomiques qui combinent la valeur génomique directe (VGD) d’un animal avec son évaluation génétique traditionnelle. Pour rester compétitif, le Canada doit rester à l’affut des nouvelles technologies qui permettent de discerner les meilleurs animaux reproducteurs.
La demande grandissante pour des animaux de qualité pousse l’industrie à utiliser les animaux de plus en plus jeunes. Il est reconnu que la qualité des gamètes des mâles âgés de moins de 16 mois est inférieure à celle des animaux matures. À ce jour il n’est pas clair si l’utilisation de gamètes immatures peut avoir un impact sur le phénotype des animaux issus de ces gamètes. La génétique permet de donner
v
une information clé qu’an au potentiel reproducteur d’un animal et le génome d’un animal ne varie pas avec l’âge, toutefois il est possible que l’information épigénétique transmise par un gamète imparfait ait un impact négatif sur la descendance. L’étude des patrons épigénétique ainsi que le développement de méthode fiable pour récolter ces informations s’impose.
Le réseau embryoGENE a développé une plateforme d’analyse épigénétique permettant d’établir un profil de méthylation de l’ADN à la grandeur du génome. Une lame de micro array comportant plus de 420 000 sondes combinées à une plateforme de bio-informatique permet une résolution sans précédent de l’épigénome bovin.
Afin de déterminer si le méthylome des gamètes d’un animal évolue avec le temps. Quatre taureaux ont été récoltés alors qu’ils étaient âgés de 10, 12 et 16 mois. Les échantillons récoltés à 10 et 12 mois ont été comparés à l’échantillon mature (16 mois) afin de déterminer si les profils de méthylation ont varié dans le temps.
Un total de 2604 sites différentiellement méthylés ont été détectés entre les échantillons de 10 mois à ceux de 16 mois. Cela démontre que les spermatozoïdes provenant de taureaux âgés de 10 mois n’ont pas un profil mature. Aucune différence significative au niveau des méthylations n’a pu être détectée lorsque l’on compare les échantillons de 12 mois à ceux de 16 mois.
L’importance de ces différences pour l’embryon est méconnue. Il est toutefois connu que des altérations dans les marques épigénétiques portées par l’embryon soient à la source de plusieurs pathologies ou défauts embryonnaires. Il est raisonnable de soupçonner que l’utilisation de sperme immature puisse représenter un risque potentiel pour l’embryon.
vi
Table des matières
Résumé court ... iii
Résumé long ... iv
Table des matières ... vi
Liste des figures ... ix
Liste des abréviations ... x
Chapitre 1 ... 1
Introduction ... 1
Structure et fonction de la chromatine ... 1
Histone ... 3
Méthylation des histones ... 3
Acétylation des histones ... 6
Méthylation de l’ADN ... 7
Dnmt 1 ... 8
Dnmt 3 ... 9
Déméthylation ... 10
Gènes à empreinte parentale ... 13
Modification post traductionnelle ... 14
Transposons ... 16
Spermatogénèse ... 18
Cellules germinales primordiales ... 19
Établissement de l’empreinte parentale ... 21
Spermatogonie ... 22
vii
Condensation du génome ... 28
Histone persistante... 30
ARN livré à l’ovocyte ... 31
Matrix Attachement Region (MARs) ... 32
Fécondation ... 34 Transition protamine-histone ... 34 Déméthylation ... 35 Asymétrie ... 36 Évolution de la chromatine ... 38 Inactivation du chromosome X ... 39 EGA ... 40 Morula ... 43 Chapitre 2 ... 62 Abstract ... 63 Introduction ... 64
Material and Method ... 67
DNA Extraction ... 67
Methylation Analysis... 68
Fragment selection by ligation-mediated PCR ... 69
Sample labeling and hybridization ... 69
Array data analysis ... 70
Validation of differentially methylated probes using bisulfite sequencing ... 71
Result ... 72
viii
Differentially methylated region ... 75
CpG Island ... 77
Chromosomal region analysis ... 78
Validation of micro-array result ... 80
Discussion ... 82
Conclusion ... 86
Conclusion ... 90
ix
Liste des figures
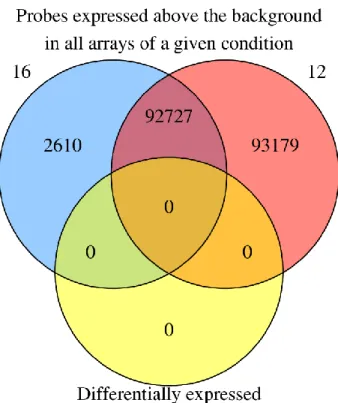
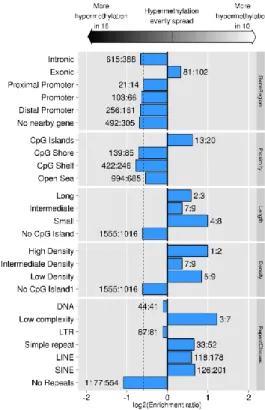
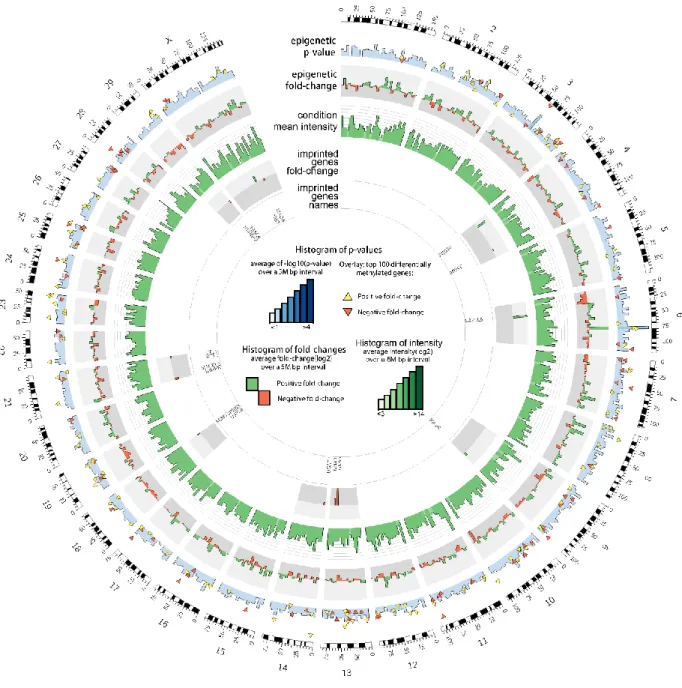
Figure 1.1 - Cycle de déméthylation proposé par Huang et Rao 2012 ... 12 Figure 1.2 -Résumé des facteurs paracrines régissant la méiose. En bleus sont des facteurs paracrines qui favorisent le renouvellement de la population cellulaire en inhibant la différentiation et l’entrée dans la méiose. Les rouges représentent les facteurs stimulant la différentiation. En pourpre on retrouve les facteurs favorisant la différentiation, mais qui empêche l'entrée dans la méiose. Les verts sont les facteurs favorisant la méiose(Rossi and Dolci 2013). ... 24 Figure 1.3 - Résumé des modifications sur les histones subvenant pendant les différentes phases de la spermatogénèse(Payne and Braun 2006). ... 25 Figure 1.4 - Représentation graphique de la structure finale de la chromatine dans le spermatozoïde mature (W. Steven Ward 2010). ... 34 Table 2.1- List of primer used for pyrosequencing ... 72 Figure 2.1 - Number of probes above the background in all array of a give condition ... 74 Figure 2.2 - Number of probes above the background in all array of a give condition ... 75 Figure 2.3 - Combined enrichment of selected probes in genic regions. ... 76 Figure 2.4 - Circular plot of the epigenetic analysis of the comparison of the samples collected from bulls aged of10 months and the bulls aged of 16 months 79 Figure 2.5 - results of pyrosequencing analysis ... 82
x
Liste des abréviations
% Pourcent
5CaC 5-carboxylcytosine 5fC 5-formylcytosin
5hmC 5-hydroxyméthylcytosine 5meC 5-méthylcytosine
Aal Aligned spermatogonia ADN Acide désoxyribonucléique apr Apaired spermatogonia ARN Acide ribonucléique
ARNm Acide ribonucléique messager as Spermatogonie type 1
BER Base excision repair
Blimp 1 B-lymphocyte maturation induced protein 1 BMP4 Bonemorphogenicprotein 4
Bp Paires de bases
CGI Ilot de CpG
Cdx2 Caudal type homoebox 2
CpG Cytosine phosphoryllée suivit d'une guanine Dmnt1 Méthyltransférase ADN
Dmnt1a Méthyltransférase 1a ADN Dmnt1o Méthyltransférase 1o ADN Dmnt3a Méthyltransférase 3a ADN Dmnt3b Méthyltransférase 3b ADN Dmnt3l Méthyltransférase 3l ADN
DMR Région différentiellement méthylé EGA Embryon genome activation Elf5 E74 like factor 5
xi
Fe2+ Fer II
FGF9 Fibroblastgrowth factor 9 FSH Folliclestimulating hormone GLP Glucagon like peptide
h Heure H1 Histone 1 H1t Histone cluster 1 H2A Histone 2A H2B Histone 2B H3 Histone 3 H3.1 Histone 3 variante 1 H3.2 Histone 3 variante 2 H3.3 Histone 3 variante 3 H3K4 Histone 3 lysine 4
H3K4me2/me3 Histone 3 lysine 4 diméthylé ou triméthylé H3K9 Histone 3 lysine 9
H3K9me2/me3 Histone 3 lysine 9 diméthylé ou triméthylé H3K9me2 Histone 3 lysine 9 diméthylé
H3K9me3 Histone 3 lysine 9 triméthylé H3K14 Histone 3 lysine 14
H3K27 Histone 3 lysine 27
H3K27me1 Histone 3 lysine 27 monométhylé H3K27me2 Histone 3 lysine 27 diméthylé H3K36 Histone 3 lysine 36
H3K36me1/me2 Histone 3 lysine 36 monométhylé ou diméthylé H3K39me2/me3 Histone 3 lysine 39 diméthylé ou triméthylé H3K64me3 Histone 3 lysine 64 triméthylé
H3K79 Histone 3 lysine 79
H4 Histone 4
H4Ac5 Histone 4 acétylé sur lysine 5 H4K5 Histone 4 lysine 5
xii H4K8 Histone 4 lysine 8
H4K12 Histone 4 lysine 12 H4K20 Histone 4 lysine 20
H4K20me1 Histone 4 lysine 20 monométhylé H4K20me3 Histone 4 lysine 20 triméthylé
H5 Histone 5
HAT Histone acétyle transférase HDAC1 histone déacétylase 1 HDAC2 histone déacétylase 2 HDAC3 histone déacétylase 3 HDAC8 histone déacétylase 8
HDACi Inhibiteur de l’histone déacétylase
hnRNPU Heterogenousnuclearribonucleoprotein U
HOX Homeoticgenes
HP1 Heterochromatinprotein 1
HP1α Heterochromatinprotein 1paralog α HP1β Heterochromatinprotein 1paralog β IAP Inhibitor of apoptosis
ICM Innercell mass
Igf2r Insulin like growth factor 2 receptor
Jmjc Jumonji C K5 Lysine 5 K8 Lysine 8 K9 Lysine 9 K12 Lysine 12 K14 Lysine 14 K15 Lysine 15 K16 Lysine 16 K18 Lysine 18 K20 Lysine 20 K23 Lysine 23
xiii KDM lysine déméthylase
KDM1A lysine déméthylase 1A KDM2A lysine déméthylase 2A KDM 3 lysine déméthylase 3 KDM 3A lysine déméthylase 3A KDM4A lysine déméthylase 4A KDM 5 lysine déméthylase 5 KDM6A lysine déméthylase 6A KDM 6 lysine déméthylase 6 KDM7A lysine déméthylase 7A
KL Kit ligand
LH Luteinizing hormone
LINE Long interspersednuclearelement LTR Long terminal repeat
MARs Matrix attachment region
miARN Micro- ARN
MuERV-L Murine endogenousretrovirus-like NER Nucleotid excision repair
Nm Nanomètre
Oct4 Octamer binding factor 4 PGC Primordial germcell PHF8 PHD fingerprotein 8
PiARN ARN interagissant avec piwi PN0 Pronucléus étape 0
PN1 Pronucléus étape 1 PN4 Pronucléus étape 4 PN5 Pronucléus étape 5
PRC2 Polycomb Repressive Complex 2 PRMT5 Protein arginine methyl transferase 5
RASGRF1 Ras-protein specific guanine nucleotide releasing factor 1 RISC RNA-induced silencing complex
1
Chapitre 1
Introduction
Par définition, l’épigénétique correspond au domaine ce qui étudie les modifications héritables modulant l’expression des gènes sans toutefois en changer la séquence de nucléotides. Les cellules d’un organisme contiennent toutes le génome et peu importe du type de cellule, la séquence de nucléotide qui le compose reste le même. Pourtant, certains organismes sont composés d’une grande diversité de cellules. La différenciation cellulaire est possible grâce aux modifications épigénétiques. Les marques épigénétiques sont mises en place pendant le développement, le phénomène permet de mettre en place le profil de transcription spécifique. Puisque les modifications sont héritées par les cellules filles après une mitose, les profils sont maintenus dans la lignée cellulaire. Dès les premiers stades de la vie, ces marques sont d’une importance capitale. En effet, la moindre erreur dans la mise en place des patrons épigénétique peut compromettre la viabilité de l’organisme. C’est pourquoi l’étude de la dynamique des marques épigénétiques dans l’embryon est d’une importance capitale. L’épigénome de l’embryon est majoritairement dicté par l’ovocyte, et bien que son impact soit plus limité, par le spermatozoïde. Le présent projet vise à déterminer si les patrons de méthylation dans les cellules de spermatozoïde ont tendance à varier avec la maturation de l’animal et à déterminer dans quelle mesure cette variabilité à un impact sur le méthylome des embryons.
Structure et fonction de la chromatine
La structure de la chromatine est un facteur déterminant de l’expression génique. Les modifications épigénétiques altèrent cette structure. L’unité de base de la structure de la chromatine est le nucléosome, ce dernier est formé d’un octamère d’histone autour duquel sont enroulées environ 146 paires de bases d’ADN. Ils peuvent être précisément positionnés ou distribués aléatoirement. Ce
2
positionnement à un impact direct sur l’accessibilité à l’ADN, ainsi un nucléotide positionné sur un promoteur inhibera la transcription du gène.
Le degré de compaction des fibres de chromatines peut varier. Lorsque la fibre a une épaisseur de 11nm, la chromatine est dans un état d’euchromatine, cette configuration est trancriptionellement active. L’hétérochromatine correspond à la configuration compacte, les fibres font environ 32 nm d’épaisseur. La densité des nucléotides fait en sorte que la machinerie enzymatique ainsi que les agents potentiellement dommageables n’ont pas accès à l’ADN (Morales eit al. 2001). La
transcription est donc inactive. L’hétérochromatine est divisée en deux catégories. L’hétérochromatine constitutive correspond à des portions de chromosomes inactives dans toutes les cellules. Il s’agit principalement de séquences répétées dont les plus grandes parties se trouvent à proximité des centromères et télomères. Elle est établie très tôt dans le développement des animaux dans un contexte de reprogrammation des marques épigénétiques sur tout le génome. L’hétérochromatine facultative contient des régions codantes pouvant adopter les caractéristiques structurales et fonctionnelles de l’hétérochromatine. L’inactivation du chromosome x en est un excellent exemple. Les mécanismes épigénétiques ont un contrôle direct sur la structure et la fonction de la chromatine. Une protéine hautement conservée nommée HP1 (Heterochromatin protein 1) joue un rôle déterminant dans la répression de l’expression génique, elle est d’ailleurs une constituante intégrale de l’hétérochromatine. La protéine peut être recrutée par les méthylations sur les histones lors de la formation de l’hétérochromatine. La protéine se retrouve également dans l’euchromatine lorsque certains gènes spécifiques dans la région doivent être réprimés, elle est alors recrutée par des protéines à doigt de zinc. Tout dépendant du contexte chromosomique, un homodimère HP1 peut avoir une fonction activatrice (Singh and Georgatos 2002). La configuration de la chromatine est un processus dynamique, en réponse à des signaux cellulaires, l’hétérochromatine peut s’ouvrir afin de permettre l’expression génique. Il a été démontré que certains facteurs de l’environnement pouvaient modifier l’expression des gènes par le biais de changements dans la chromatine de régions promotrices
3
de gènes, des éléments régulateurs de gènes soumis à empreinte ou des éléments transposables adjacents aux gènes avec épiallèle métastable(Niemann et al. 2010). Les épiallèles métastables sont des locus régulés par une modification épigénétique (souvent une méthylation sur l’ADN) particulièrement sensible à une variable. La mise en place de méthylation à ces endroits est un évènement incertain dont la probabilité est influencée par l’environnement.
Histone
Les histones sont des protéines que l’on retrouve dans les noyaux cellulaires, il s’agit du principal constituant protéique de la chromatine. Elles comportent toutes un domaine globulaire avec 3 hélices α et deux feuillets β. Les queues des histones sont riches en résidu d’acide aminé basique comme la lysine et l’arginine, ce qui lui confère une charge positive permettant l’interaction avec les charges négatives portées par les groupements phosphates de l’ADN. Les histones peuvent donc s’associer à l’ADN en enroulant ce dernier autour d’elle, cela a pour effet de compacter l’ADN et mène à la formation du nucléosome. Les octamères d'histones sont constituées de 2 unités de chacune des histones fondamentales (cœur) : H2A, H2B, H3 et H4. Les histones H1 et H5 agissent comme histones de liaisons, elles induisent le rapprochement de l'ADN de liaison limitant leurs mouvements et scellant ainsi le complexe nucléoprotéique. Les histones sont sujettes à plusieurs modifications telles l’acétylation, la méthylation, l’ubiquitination, la sumoylation et la phosphorylation. Ces modifications ont tout un impact direct sur l’expression des gènes se trouvant sur la chromatine qui y est lié. La méthylation et l’acétylation sont les deux altérations les plus documentées et les plus répandus dans le génome.
Méthylation des histones
La méthylation est catalysée par les histones méthyltransférases, cet enzyme utilise la S-Adenosyl- Methione (SAM) comme donneur de groupement
4
méthyle.Chaque résidu lysine de l’histone peut accepter jusqu’à 3 groupements méthyles alors que les résidus arginine sont limités à 2 groupements méthyles(Rivera and Ross 2013). Il est estimé que les queues des histones ont 9 points de méthylation possible permettant un total de 262 144 combinaisons. Chacune de ces combinaisons peut dicter un patron de régulation spécifique. La caractérisation des effets de la méthylation est donc une tâche particulièrement complexe (Bannister, Schneider, and Kouzarides 2002). Toutefois, il a été constaté que lorsque la chromatine est dans un état d’hétérochromatine (configuration répressive), certaines méthylations étaient persistantes et ont donc été associées avec les marques de répression. C’est le cas de la méthylation du groupe H3K9me3 qui est catalysée par l’histone méthyltransférase SUV39H chez l’humain (D’Alessio and Szyf 2006). Les versions mono et di méthylé de H3K9 sont retrouvées dans les zones d’euchromatine chez les mammifères. Elles sont impliquées dans la répression de gènes plus spécifiques. Cette modification mène au recrutement de la HP1, cette dernière recrute l’enzyme responsable de la méthylation de l’ADN menant ainsi à la répression de la transcription et à la formation d’hétérochromatine constitutive (Bártová et al. 2008). En réponse à certains signaux cellulaires, cette protéine peut être phosphorylée afin de changer la configuration de la chromatine et ainsi permettre l’expression génique (Atsushi Shimada and Murakami 2010).Les marques de méthylation H3K9me3 sont pratiquement toujours accompagnées de méthylation sur l’ADN. Les méthylations sur H4K20 sont également associées à l’hétérochromatine. Il a été démontré que SUV4-20h1 et SUV-20h2, deux protéines situées dans les régions péri-centriques et qui contiennent un domaine SET, étaient responsables de cette modification. Ces protéines peuvent être recrutées par HP1 et donc agir de concert avec les méthylations H3K9. Il a été démontré que la marque H4K20me1 varie durant le cycle cellulaire. Ce mécanisme serait indispensable pour le contrôle du cycle cellulaire (Karachentsev et al. 2005). Une autre marque de méthylation répressive estH3K27. Les di et tri- méthylation sur H3K27 sont catalyser par un complexe protéique appelé PRC2 qui possède une activité méthyltransférase sur les histones. Les marques H3K27me3/me2 peuvent recruter PRC2 lors de la division cellulaire, ces marques sont donc héritées par les cellules filles (Margueron
5
et al. 2009).À l’inverse, dans les zones de transcription active, il est fréquent d’observer une triméthylation de H3K4, H3K36 et H3K79 (Pedersen and Helin 2010). Ces marques peuvent servir de sites de liaison à plusieurs protéines impliquées dans la transcription. L’enzyme responsable de la mise en place des marques H3K4 et H3K36 serait associé à la polymérase à ARN II. Il est donc possible qu’elles soient établies lorsque le gène est activé. Dans le cas de H3K4 il s’agit de Set1. Les marques H3K4me2/me3 sont enrichies sur le côté 5’ des gènes activement transcrits. Le mécanisme de positionnement est méconnu.
Il est pertinent de souligner que la méthylation est traditionnellement associée à des modifications très stables et pratiquement immuables. La réversion de l’état méthylé a non méthylé est possible seulement par dilutions, via les divisions cellulaires (Byvoet 1972; Duerre and Lee 1974). Toutefois, l’état dynamique des patrons de méthylation des histones est maintenant connu. Plusieurs mécanismes menant à la déméthylation des histones ont été recensés. Il existe deux catégories de déméthylases pour les histones. La première protéine à démontrer une activité de déméthylation sur les histones de mammifère fut la KDM1A, une amine oxydase de la famille protéique KDM1, dans un complexe de répression transcriptionnelle. Des études in vitro confirment que l’enzyme a une activité de déméthylation spécifique sur H3K4(Y. Wang et al. 2009). Cette réaction nécessite la formation d’un intermédiaire imine qui exige une paire d’électrons libres dans l’atome d’azote du méthyle-lysine, cette étape limite l’action de l’enzyme aux mono et diméthylation, elle n’a pas d’effet sur H3K4me3. Un dérèglement dans la croissance et la différenciation cellulaire a été observé chez des souris KDM1A déficient (J. Wang et al. 2009). Cette même étude démontre qu’il existe des liens entre KDM1A et les DNMT (enzyme responsable de la déméthylation sur l’ADN) et que la déficience en KDM1A a un impact négatif sur la méthylation de l’ADN. La seconde catégorie de déméthylase pour les histones utilise un domaine JmjC qui contient une histone déméthylase (JHDMs) pour catalyser la réaction. JHDMs convertie le méthyle-lysine en hydroxyméthyle qui est ensuite libéré sous forme de formaldéhyde en utilisant du Fe2+ et de l’α-cétoglutarate comme cofacteurs en présence l’oxygène. Cette
6
stratégie lui permet de retirer tous les stades de méthylation (me1/me2/me3). La structure de quatre enzymes de cette catégorie est à ce jour connue. KDM4A déméthyle H3K9me2/me3 et H3K39me2/me3. PHF8 et KDM7A déméthylent tous deux H3K9me2 et H3K27me2. KDM2A est spécifique pour H3K36me1/me2 (Klose, Kallin, and Zhang 2006; Chen et al. 2006). Plusieurs autres enzymes sont soupçonnés d’avoir une activité de déméthylation sur les histones c’est-à-dire les familles protéiques KDM3, KDM5 et KDM6 (Pedersen and Helin 2010).
Acétylation des histones
L’acétylation de la queue N-terminale des histones présentes un riche potentiel d’information épigénétique. Elle peut autant être utilisée pour la médiation des changements transitoires de la transcription via la modification du promoteur proximal que pour moduler l’expression génique à long terme. Cette modification est catalysée par les histones acétyle-transférase (HAT) qui utilisent l’acétyle coenzyme A comme donneur de groupement acétyle. L’enzyme est très conservé, elle peut être observée autant chez la levure que chez l’humain. Elle est composée de plusieurs sous-unités catalytiques. L’acétylation d’une histone a pour effet de neutraliser la charge positive de l’acide aminé auquel le groupement acétate s’associe. Ce changement de polarité a pour effet de diminuer l’affinité de la queue de l’histone pour l’ADN, cela ouvre le nucléosome habituellement condensé et permet à la machinerie de transcription de venir en contact avec la matrice d'ADN, ce qui conduit généralement à la transcription des gènes. L’acétylation n’est pas un processus aléatoire, chez les mammifères, H2A peut être acétylé aux lysines 5 et 9, H2B sur K5, K12, K15 et K20, H3 sur K9,K14, K18 et K23, et H4 sur K5, K8, K12 et K16 (Turner 2001). Les résidus lysine sur les histones peuvent être acétylés plusieurs fois, certains peuvent porter jusqu’à quatre groupements. Les principales cibles d’acétylation post traductionnelle sont les lysines situées sur les histones H3 et H4 (Rice and Allis 2001). En général, suite à leurs traductions les histones sont acétylées sur les lysines 5 et 12, puis lors de la formation du nucléosome, elles sont
7
désacétylées ce qui permet de rétablir la charge positive et de permettre la fixation des dimères d’histones. Les histones déacétylases sont des enzymes ayant la capacité de retirer les groupements acétyles liés à la lysine (Rice and Allis 2001). Les HDAC sont sous-divisées en plusieurs catégories en fonctions de leurs domaines catalytiques. Les membres de la classe I, c’est-à-dire HDAC1, HDAC2, HDAC3 et HDAC8, sont présents dans l’ensemble des cellules. Les autres sont tous plus spécifiques à certains tissus. Il est pertinent de mentionner qu’il existe une famille d’enzyme inhibiteur d’histones déacétylases (HDACi). Ces protéines peuvent altérer l’expression génique en maintenant les patrons d’acétylation(Xu, Parmigiani, and Marks 2007). Par contre, les mécanismes permettant à ces enzymes de cibler spécifiquement les gènes restent à ce jour méconnus. L’acétylation d’acides aminés spécifiques est également associée à d’autres processus biologiques comme la réplication de l’ADN (Strahl and Allis 2000) ainsi que l’assemblage du nucléosome (Grunstein 1997). L’acétylation de H3K9 et H3K14 peuvent empêcher la méthylation à ces positions, prévenant ainsi la formation d’hétérochromatine et donc la répression de l’expression des gènes à cette position (Mateescu et al. 2004). Certaines acétylations sont liées à la méthylation sur les histones, cette dernière peut servir de point d’ancrage à l’histone acétyle-transférase.
Méthylation de l’ADN
La méthylation de l’ADN est l’un des premiers mécanismes épigénétiques à avoir été identifié. La méthylation de l’ADN principalement restreinte aux cytosines des dinucléotides CpG. Chez les humains, environ 1% des bases sont des cytosines méthylé, ce qui affecte entre 70% et 80% des dinucléotides CpG. Ce doublet de nucléotides est sous représentés dans le génome, en effet seulement 20% de la fréquence attendue est observé et environ 60% à 90% de ces cytosines sont méthylé en 5e positions pour former 5meC. Toutefois, certains CpG semblent être
8
500 à 2000 répétitions (Bird 1986). Ces ilots, principalement ceux associés avec un promoteur sont hautement conservés par l’évolution.
La méthylation de l’ADN est impliquée dans le degré de compaction de l’ADN et à la formation de l’hétérochromatine. Cette modification épigénétique est donc généralement associée à la répression de l’expression génique. Ce n’est toutefois pas toujours le cas. Le mécanisme moléculaire derrière son action est inconnu. La méthylation de l’ADN est un facteur critique pour l’expression génique, notamment les gènes à empreinte, la répression d’éléments transposable, l’inactivation du chromosome X ainsi que le développement et la reprogrammation de l’embryon (Rivera and Ross 2013). La méthylation se retrouve donc généralement à des endroits prévisibles et la mise en place de cette marque est d’une importance capitale. D’ailleurs, des patrons de méthylations aberrants ont été observés dans le cadre plusieurs pathologies comme la dystrophie musculaire, le lupus ainsi que des cancers.
Dnmt 1
La méthylation de l’ADN est établie et maintenue par les enzymes méthyltransférases de l’ADN (Dmnt). Dnmt1 est un enzyme constitué de 1620 acides aminés, cette molécule à une préférence pour les substrats hémiméthylés. Lors de la synthèse d’ADN, elle est située au niveau de la fourche de réplication, elle est donc généralement associée à des fonctions de maintenances des patrons de méthylation. Il faut mentionner qu’il a été démontré que l’enzyme avait la capacité de générer de nouvelle méthylation in vitro, il n’y a toutefois aucune preuve directe que Dnmt1 est impliqué dans la mise en place de nouvelle méthylation dans un organisme (Chédin 2011).
L’expression de Dnmt1 est unique du fait que son expression est gérée par des promoteurs spécifiques au sexe. Le premier de ces promoteurs induit la traduction
9
d’une version tronquée de l’enzyme appelé Dmnt1o, qui est spécifique à l’ovocyte (Bestor 2000). Dmnt1o s’accumule dans le cytoplasme des ovocytes en croissance. Après fécondation, à l’étape pronucléus (PN0), l’enzyme est d’abord associé au pronucléus maternel. Pour les étapes PN1 à PN 4, Dnmt1o s’associe également avec le pronucléus paternel, elle y reste même durant les vagues de déméthylation. À l’étape PN5, l’enzyme est également associé aux 2 pronucléi, cela suggère qu’elle a été exportée dans le cytoplasme (Rivera and Ross 2013). Dmnt1 reste cytoplasmique avant l’activation du génome, puis entre et sort des noyaux lorsque l’embryon est constitué de 8 cellules. Après l’implantation, Dnmt1 est remplacé par sa forme somatique et est localisé presque exclusivement dans les noyaux des cellules somatiques (Mertineit et al. 1998). Le promoteur et exon (exon 1s) responsable de la traduction de la Dnmt1 est actif dans toutes les cellules somatiques, il est toutefois modéré dans des conditions d’arrêt de prolifération. Une expression massive du gène a été rapportée dans des cellules tumorales (el-Deiry et al. 1991), et une mutation du gène dans un embryon mène à un développement anormal et ultimement la mort.
Dnmt 3
Trois enzymes ont été recensés dans cette famille, Dnmt3a, Dnmt3b et Dnmt3l. Dnmt3a et Dnmt3b ont toutes deux une structure relativement similaire comportant un site de liaisons a l’ADN ainsi qu’un domaine catalytique méthyltransférase. Leurs structures ressemblent davantage à une méthyltransférase d’origine bactérienne plutôt qu’à Dnmt1 (Chédin 2011).
Ces molécules ont un niveau d’expression élevé dans les cellules souches, les cellules germinales en développement ainsi que les cellules embryonnaires, suite aux vagues massives de déméthylation, bref à des étapes où les patrons de méthylation sont posés (Handa and Jeltsch 2005). Contrairement à Dnmt1, elles ne démontrent aucune préférence pour les substrats hémiméthylés. Ces enzymes ont
10
donc un profil approprié pour la mise en place de nouvelle méthylation. Dnmt3b est impliqué dans la mise en place de méthylation péricentrique dans les régions d’hétérochromatines. Son recrutement est effectué par une triméthylation H3K9 (Lehnertz et al. 2003).
Dnmt3l est une version tronquée de Dnmt3a et Dnmt3b. Individuellement, elle n’a pas d’activité catalytique, il s’agit d’un facteur de régulation. Elle s’associe avec Dnmt3a afin de stabiliser la conformation active de ce dernier lors de son activité catalytique. Il a été démontré que chez la souris, cette enzyme n’était pas nécessaire au développement de l’embryon, bien qu’une mutation dans ce gène mène à des retards dans la croissance embryonnaire et a des structures anormales au niveau du placenta, les animaux déficients en Dnmt3l sont viables. Toutefois, des animaux issus de femelles déficientes en Dnmt3l ne sont pas viables et les mâles présentant ce phénotype sont simplement stériles (Bourc’his et al. 2001; Hata et al. 2002).En effet, cette inactivation mène à une altération des patrons de méthylation des gènes à empreintes dans les cellules germinales. L’inactivation conditionnelle de Dnmt3a mène exactement au même phénotype que les animaux déficients en Dnmt3l, cela suggère que les deux sont nécessaires pour la méthylation des gènes à empreinte.
Déméthylation
Généralement, la méthylation est associée à des patrons épigénétiques très stables qui perdureront durant toute la vie de l’organisme comme les gènes à empreinte ou l’inactivation d’un chromosome X chez les femelles. Toutefois, depuis quelques années, certaines observations démontrent que dans certains contextes spécifiques, il est possible que les patrons de méthylation soient perdus ou altérés. La déméthylation a principalement lieu durant le développement de l’organisme. Ce processus peut être local ou global (Roldán-Arjona and Ariza 2000). Chez les mammifères, le génome subit de vague de déméthylation globale, une première fois
11
dans les cellules germinales primordiales et la seconde prend place après la fécondation, avant l’implantation de l’embryon(Rivera and Ross 2013).
Deux principaux mécanismes de déméthylation ont été identifiés. La déméthylation passive réfère à la perte des groupements méthyles sur l’ADN via l’inhibition ou l’absence de Dnmt1 pendant plusieurs séquences successives de réplication de l’ADN. De cette façon les cellules filles ne présentent pas les patrons de méthylation portés par la cellule mère(Wu and Zhang 2010). Ce processus est donc relativement long. La déméthylation active permet une déméthylation beaucoup plus rapide de l’ADN. Toutefois, ce processus n’est pas aussi bien compris et plusieurs mécanismes sont proposés.
Les enzymes BER (base excision repair) sont présentes durant la vague de déméthylation qui a lieu durant le développement préimplantatoire. Récemment, un lien a été démontré entre le processus de déméthylation et la présence de l’enzyme (Hajkova et al. 2010). Ce mécanisme suggère une déamination qui résulterait en conversion de 5MeC en thymine, qui serait réparée à la prochaine réplication via le recrutement des glycosylase appropriées. Également compte tenu la concentration relative de la protéine durant les vagues de déméthylation les enzymes NER (nucléotids excision repair) sont également des candidats possibles(Wu and Zhang 2010). Ces deux enzymes ont un mode d’action similaire. Premièrement, la cytosine méthylée doit subir une déamination ce qui la transformera en thymine. La cytosine déaminase et les méthyltransférase à ADN sont des enzymes qui ont la capacité de catalyser cette réaction, toutefois, aucune preuve directe n’a permis de confirmer que ces candidats sont impliqués dans la déméthylation. Une fois que la cytosine méthylée a été transformée, les mécanismes de réparation BER ou NER peuvent ensuite réparer le mésappariement en remplaçant la thymine par une cytosine non méthylée.
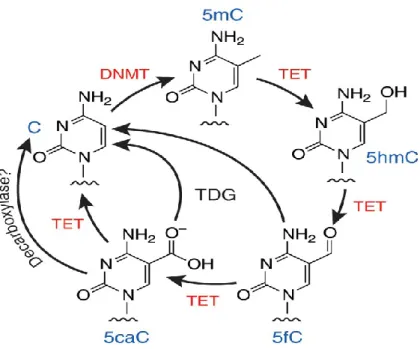
Récemment les enzymes ten-eleven translocation (TET) ont été identifiées. Elles sont capables d’hydroxyler le groupement méthyle lié à l’ADN (Kriaucionis and
12
Heintz 2009; Tahiliani et al. 2009). Toutefois, la conséquence de l’hydroxylation de 5meC en 5hmC n’est pas claire. Il a été démontré que l’oxydation en étape de 5hmC donnait 5fC et 5caC. Ce dernier peut être reconnu et excisé par une glycosylase TDG ou une décarboxylase(He et al. 2011; Huang and Rao 2012). D’autre part, certaines études démontrent que certaines régions du génome des mammifères sont enrichies en 5hmC. Cette base serait donc potentiellement une marque épigénétique en soi et non seulement un intermédiaire l’ADN (Kriaucionis and Heintz 2009; Tahiliani et al. 2009). Il est pertinent de mentionner que certaines protéines s’associant avec l’ADN méthylé ne reconnaissent pas les sites oxydés (Jin, Kadam, and Pfeifer 2010).
13
Gènes à empreinte parentale
Chez les mammifères euthériens ainsi que les marsupiaux, les allèles parentaux n’ont pas toujours une activité équivalente pour un gène donné. Il s’agit là de gènes à empreinte. Ils sont généralement disposés en groupes de 3 à 12 gènes, mais certains peuvent être seuls. La méthylation de l’ADN est le mécanisme moléculaire clé des gènes à empreinte en modulant l’expression des gènes ciblés. Les marques de méthylation diffèrent entre l’œuf et le spermatozoïde et l’héritage de différentes marques épigénétiques mène à une expression génique différente. Ces gènes sont généralement résistants à la déméthylation, après la fécondation, malgré la déméthylation globale, les marques sont maintenues sur les chromosomes en réplication du nouvel organisme en développement. Dans la différenciation des cellules germinales primordiales, les marques ne sont pas rétablies au même moment. Ces cellules sont maintenues naïves et l’empreinte est mise en place à un moment ultérieur. Dans les cellules somatiques, l’empreinte est plus stable, elle peut toutefois être modifiée durant le développement.
Lorsque des gènes à empreinte sont transcrits, l’expression de ceux-ci dépend de l’origine parentale (Plasschaert and Bartolomei 2014). Ils sont également d’une importance capitale pour le développement adéquat de l’embryon. La démonstration que les gènes à empreintes étaient d’une importance capitale pour le développement embryonnaire a d’abord été faite sur des embryons uniparentaux. Il a été observé que la présence seule du matériel génétique dans un zygote était insuffisante pour produire des embryons viables. En effet, pour les embryons androgéniques, des tissus embryonnaires sont manquants alors que pour les embryons parthénogénique, des tissus du placenta sont manquants (Barton, Surani, and Norris 1984; McGrath and Solter 1984). Cela suggère que les empreintes mâles et femelles sont complémentaires et nécessaires pour le bon fonctionnement d’un organisme.
14
L’un des cas les mieux documentés de gènes à empreinte régulant la croissance embryonnaire est le cas de Igf2. Il s’agit d’un gène à empreinte paternel stimulant la croissance embryonnaire. Un gène à empreinte maternel, Igf2r, code pour un
inhibiteur du facteur de croissance paternel. La balance entre les deux antagonistes est primordiale pour une croissance embryonnaire normale, une défaillance de ce mécanisme mène à une taille anormale du fœtus (Leighton et al. 1995; Ludwig et al. 1996). L’expression de plusieurs autres gènes à empreintes semble être coordonnée dans plusieurs tissus à des stades spécifiques de développement. L’étendue de cette corégulation reste à déterminer et son rôle exact est à ce jour encore nébuleux (Varrault et al. 2006; Arima et al. 2005).
Modification post traductionnelle
Comme son nom l’indique, les petits ARN non codants sont des transcrits de petites tailles. Ils ont une taille variant entre 19 et 25 nucléotides et ils ne codent pour aucune protéine. Une cellule de mammifère contient en moyenne 50 000 petits ARN. Ils sont très bien conservés par l’évolution tant chez les animaux que chez les plantes, ils sont donc généralement perçus comme des composantes clés de la régulation génique. En effet, malgré leurs petites tailles, ils sont tout à fait fonctionnels. Ils représentent une façon indirecte de moduler l’action d’un gène via des modifications post traductionnelle.
Les petits ARN non codants peuvent être divisés en trois grandes catégories. Les miARN (micro-ARN) et les siARN (short interfering ARN) sont deux catégories qui ont beaucoup de points en commun. Les miARN et les siARN partage la même voie de synthèse. Ils sont tous deux transcrits par la polymérase à ARN II. Dans le noyau, ce petit ARN primaire est clivé par une ribonucléase, Drosha, pour donner un précurseur qui contient une structure en épingle à cheveux qui sera exportée dans le cytoplasme. Le siARN/miARN est alors excisé du pré ARN par les enzymes dicer. Pour être fonctionnel, l’ARN précurseur est incorporé au complexe RISC
(RNA-15
induced silencing complex). Ce complexe peut interagir avec l’ARN mature et ainsi inhiber la traduction de l’ARN ciblé. Tout dépendant le degré de complémentarité entre le complexe RISC et l’ARN ciblé, 2 résultats sont possibles. Dans le cas des siARN, la complémentarité doit être parfaite. Dans ce cas, l’ARN ciblé sera clivé et dégradé. Les miARN sont moins sélectifs. Ils peuvent inhiber la traduction de l’ARN cible même lorsque la complémentarité est partielle, ils peuvent donc cibler une plus grande diversité de transcrits. Les mécanismes entourant ce processus sont à ce jour mal compris (Stefani and Slack 2008). Les deux catégories de petits ARN diffèrent également de par leur origine. Les siARN peuvent être d’origine virale ou être le produit de la transcription de séquence répétée du génome. Les miARN sont le produit d’ARN doubles brins qui sont codés par un gène. Une autre différence majeure entre les siARN et les miARN réside dans la composition de leurs extrémités, les siARN sont plus variables et hétérogènes, alors que les miARN présentent plus de similarité entre eux (Carthew and Sontheimer 2009).
Les piARN sont les plus longs des petits ARN non codants avec une taille variant de 20 à 35 nucléotides. Ils sont particulièrement abondants dans les gonades animales. Ils s’associent avec les protéines de type PIWI pour former des complexes impliqués dans le contrôle épigénétique des transposons (Seto, Kingston, and Lau 2007). Contrairement aux deux autres catégories de petits ARN, la voie de synthèse des piARN n’est pas dépendante de l’enzyme dicer. Chez la souris, 3 types d’homologues PIWI ont un haut niveau d’expression dans les gonades males; Miwi, Miwi 2 et Mili. Ils sont tous nécessaires pour la synthèse d’embryons viable (Aravin et al. 2006). Une dysfonction dans la synthèse des piARN mène à une diminution des méthylations H3K9me3, il est donc possible que ce mécanisme guide le processus de méthylation.
À la fin de la maturation de l’ovocyte ainsi qu’au début du développement embryonnaire, l’expression des petits ARN est très importante. En effet, après la fécondation le génome de l’embryon est éteint, il n’y a pas de transcription. L’œuf est donc dépendant des transcrits maternel emmagasiné dans le cytoplasme
16
pendant la maturation de l’ovocyte. Les miARN joue un rôle critique dans la régulation de la traduction de ces transcrits. En effet, les ovocytes dont l’expression de l’enzyme dicer a été supprimée sont incapables de survivre à la première division cellulaire (Tang et al. 2007). La contribution des miARN paternel a également été mise en évidence. Six miARN absents dans l’ovocyte mature, mais présent dans les spermatozoïdes et dans les embryons ont été identifié chez la souris. Ces miARN ont un impact sur le premier clivage, notamment sur l’orientation de ce dernier (W.-M. Liu et al. 2012a). Après l’implantation de l’embryon les miARN ont un rôle primordial notamment dans la différenciation cellulaire (L. Song and Tuan 2006). Les siARN, quant à eux, semblent avoir un rôle majeur dans les débuts du développement embryonnaire des mammifères. En effet, sans les siARN il est simplement impossible de générer des ovocytes (Suh and Blelloch 2011).
Transposons
Chez l’humain, les transposons occupent environ 45% du génome, toutefois, ils ne représentent que 1 à 2 % des séquences exoniques. Les transposons sont caractérisés par leurs capacités à se mobiliser dans le génome. À leurs découvertes, ils étaient considérés comme des éléments nuisibles, des parasites du génome. Depuis, leur contribution dans l’évolution a été mise en évidence. En effet, ces éléments ont le potentiel d’influencer l’évolution du génome de plusieurs façons. Ils peuvent altérer la fonction d’un gène en s’y insérant, réarranger les chromosomes ou carrément apporter de nouveaux gènes pour l’organisme. D’ailleurs, certains points tournant dans l’évolution correspondent à l’intégration de certains transposons. Le génome des primates est d’ailleurs le seul à contenir les éléments de type alu (Oliver and Greene 2009).
Leurs capacités à ce mobilisé les rends menaçant pour l’intégrité du génome, car ils peuvent causer des brisures doubles brins, des mutations d’insertion et des réarrangements chromosomiques (Goodier and Kazazian 2008). Ils sont donc
17
généralement contrôlés par divers mécanismes épigénétiques. Cette capacité de mobilisation peut toutefois être bénéfique, car elle permet de stimuler de la variabilité. En effet, dans les cellules primordiales ainsi que dans le développement des neurones, les transposons sont brièvement activés. Cet évènement contribue à l’individualité de ces cellules permettant de les rendre uniques (Muotri et al. 2007). Chez la souris, les éléments transposables ont également un rôle à jouer dans l’embryogenèse. En effet, à l’état de zygote, MERVL (un transposon de type LINE) serait indispensable pour la régulation de la totipotence (Macfarlan et al. 2012).
Les éléments transposables sont divisés en deux catégories qui peuvent être vulgairement décrites comme élément copier/collé (classe I) ou couper/collé (classe II). Les éléments de la classe I, également appelée rétrotransposon, sont copiés en utilisant un intermédiaire ARN, c’est-à-dire que l’ADN est transcrit en ARN pour être ensuite rétro transcrit en ADN, ce n’est qu’après ces étapes qu’ils peuvent être réintégrés dans L’ADN. Ce mécanisme est similaire à celui utilisé par les rétrovirus. Cette classe de transposons est sous-divisée en 3 groupes, les éléments avec LTR (long terminal repeat) sont des transposons flanqués de répétitions de séquence. Ils sont sous-divisés en fonction de la lignée de virus ou de pseudo virus duquel ils descendent. Les deux autres groupes, de la classe I sont dits non LTR, c’est-à-dire qu’ils ne sont pas flanqués de séquences répétées. Il s’agit des LINE (long interspersed elements) et des SINE (short interspersed elements). Les LINE sont les éléments les plus répandus dans le génome humain, ils en constituent 17%. Toutefois, plus de 99% de ceux-ci ne sont plus en mesure de se mobiliser (Doucet et al. 2010). Ils codent pour une transcriptase inverse et sont transcrit par la polymérase à ARN II. Les SINE ne possèdent pas le gène de la transcriptase, ils sont donc dépendants de la transcriptase provenant d’un autre rétrotransposon. Ils sont transcrits par la polymérase à ARN III. Les transposons de classe II moins communs dans le génome et ne représente que 2% de la totalité des éléments transposable. Leurs transpositions est catalysé par les transposases et leur insertion peut être spécifique ou non(Leslie A. Pray 2008).
18
Spermatogénèse
Le spermatozoïde est la cellule la plus petite et la plus cytologiquement différenciée chez les mammifères. D’une apparence proche de celle d’une cellule somatique au début de la spermatogenèse, la cellule germinale mâle va, au moyen d’un processus hautement sophistiqué, atteindre une organisation finale entièrement dédiée à sa mission qui est de conduire le lot chromosomique paternel au sein de l’ovule pour la féconder. Afin de pouvoir accommoder l’ADN paternel dans la minuscule tête du spermatozoïde, un remodelage complet de la chromatine est nécessaire.
La gamétogénèse chez le mâle est un processus complexe et bien détaillé qui ultimement mène à la génération de spermatozoïde. Chez le taureau, 2 à 3 milliards de spermatozoïdes sont générés chaque jour, chez l’homme, ce nombre se situe à moins de 200 millions. Le processus est divisé en plusieurs étapes.
La spermatocytogénèse représente la multiplication des spermatogonies pour former les spermatocytes primaires. La spermatogénèse est le processus de production des spermatozoïdes qui a lieu dans les tubules séminifères des testicules. Elle englobe le phénomène qui forme des spermatides à partir des cellules germinales primaires, les spermatogonies. La spermatocytogénèse est la multiplication des spermatogonies pour former les spermatocytes primaires. La spermiogenèse est la phase finale du processus, il s’agit de la spécialisation des spermatides nouvellement créée en spermatozoïdes matures.
Basée sur la biogenèse des structures du spermatozoïde ainsi que sur la progression de la condensation du bagage nucléaire, la spermiogenèse peut être divisée en plusieurs étapes, chez le rat elle comporte 19 étapes, 16 chez la souris et 14 chez les primates (Noblanc, Kocer, and Drevet 2014). Chez le taureau, la
19
spermiogenèse peut être divisée 4 grandes phases qui sont sous-divisées pour un total de 14 étapes (Barth and Oko 1989).
Cellules germinales primordiales
Dans les embryons mâles et femelles, certaines cellules de l’épiblaste entrent dans la lignée germinale pour devenir des cellules germinales primordiales (PGC), ces cellules seront les cellules souches de la gamétogenèse dans le spécimen adulte. Pour atteindre cet état, l’expression des gènes de nature somatique doit être supprimée. Le processus est régi par des mécanismes paracrines. BMP4 (Bone morphogenetic protein 4) est connue pour induire la formation des cellules germinales primordiales. Pendant la différenciation cellulaire, l’expression du gène Sox2 est nécessaire pour l’induction de la transcription du récepteur tyrosine kinase KIT. L’activation de ce dernier par son ligand est critique pour la prolifération et la migration des PGC. Son expression sera réprimée lorsque la migration sera complétée(Rossi and Dolci 2013). Les cellules précurseures des PGC réagissent également au signal extra embryonnaire BLIMP 1 (B-lymphocyte maturation induced protein 1) (Ying, Qi, and Zhao 2001).L’effet exact de BLIMP 1 sur ces cellules n’est pas clair, il est toutefois connu que cette protéine possède un domaine histone méthyltransférase. Il a également été documenté que BLIMP 1 pouvait s’associer à PRMT 5, une histone arginine-méthyltransférase plus tard (Deshpande et al. 1999). Les modifications épigénétiques ont aussi leur rôle à jouer dans la différentiation des cellules germinales primordiales.
Chez la souris, cette différentiation commence lorsque l’embryon a 7.25 jours (E7.25). À cette étape, des patrons de méthylations de H3K9me2 et H3K27me3, marque associée à la répression de l’expression, sur tout le génome sont similaires à ceux des cellules somatiques. À E7.5, H3K9me2 commence à s’effacer. À l’inverse, la marque de répression H3K27 commence à être surreprésentée à partir d’E8.25 pour être à son maximum à E9.5. Ce phénomène a possiblement comme
20
objectif de maintenir la structure de la chromatine en dépit de la perte de H3K9me2 (Sasaki and Matsui 2008).Certaines études ont démontré que certains gènes clés permettant la totipotence des cellules souches coïncident avec les positions de H3K27me3 et H3K4me3 (Spivakov & Fisher, 2007).En effet, la marque H3K27me3 est une marque répressive qui stimule la compaction de la chromatine. À l’inverse, la marque H3K4me3 est une marque associée avec la promotion de la transcription. Cette configuration bivalente permet une fine régulation de l’expression d’un gène, soit en permettant la transcription seulement lorsque les signaux adéquats sont présents. De cette façon, le gène est maintenu dans un état d’équilibre entre l’expression ou la répression(Voigt, Tee, and Reinberg 2013).La structure bivalente de leur chromatine n’est pas très différente des PGC et de ce qui est observé dans les cellules souches. L’ADN subit une déméthylation générale qui se produit en 2 étapes. À E6.5, le niveau de méthylation de l’ADN est similaire à ce qui peut être observé dans les cellules somatiques. À E9.5, environ 30% des ilots CpG sont méthylé(Guibert, Forné, and Weber 2012). Une première vague de déméthylation aurait donc lieu dans cette période. Cette déméthylation est très globale affectant les promoteurs, les îlots CpG, les introns, les exons ainsi que les séquences intergéniques (Deaton and Bird 2011). La déméthylation à cette étape serait principalement passive, en effet, à E9.5 un grand nombre de brins hémiméthylé ont été détecté (Seisenberger et al. 2012). La migration des précurseurs des PGC vers la future région génitale a lieu à E8.0.Ce n’est qu’après cette migration que la deuxième vague de déméthylation a lieu, c’est-à-dire entre E11.5 et E13.5, ce moment coïncide avec l’effacement des marques épigénétique des gènes à empreinte. Chez la souris, les gonades mâles et femelles deviennent morphologiquement différentes à E12.5.À E13.0, les cellules germinales mâles entrent dans leur phase d’arrêt mitotique G1. C’est à E13.5 que le niveau de méthylation de l’ADN est à son plus bas pour atteindre 0.63% chez la femelle et 0.77% chez le mâle. À cette étape, seulement 0.3% des ilots CpG ont conservé leurs marques de méthylation. (L. Wang et al. 2014). C’est à cette étape que les marques des gènes à empreintes sont effacées. Toutefois, certaines régions de l’ADN sont activement maintenues méthylées. Dans les régions intergéniques,
21
certains LTR gardent leurs profils épigénétiques. Pendant longtemps il a été cru que les IAP restaient également méthylés, toutefois le contraire a été observé chez la souris(L. Wang et al. 2014)
L’empreinte des gènes à expression paternelle sera rétablie à partir de E.14.5. Le taux de méthylation global sera rétabli à 50% à E16.5. Les variations dans la méthylation de l’ADN ne semblent pas modifier le profil transcriptionnel de ces cellules. C’est-à-dire qu’on y retrouve approximativement le même nombre de gènes ayant une forte, moyenne ou une basse expression que dans les cellules somatiques (Seisenberger et al. 2012). Il a été démontré que DNMT3A et DNMT3L jouent un rôle crucial dans la mise en place de ces nouvelles méthylations et que DNMT3B n’a que très peu d’influence dans le processus (Kaneda et al. 2004). Ces méthylations seront maintenues pendant le reste du développement des cellules germinales. Des marques épigénétiques doivent rapidement être misent en places afin contrôler les transposons. En effet, les cellules germinales sont les seules à pouvoir transmettre de l’information génétique à la génération suivante. Afin d’éviter les mutations dues à l’insertion de séquence d’ADN indésirable dans un gène, les transposons, qui ont la capacité de se mobiliser dans le génome, doivent être strictement contrôlés (Kaneda et al. 2004).Chez la souris, un piARN, Miwi2 localise les séquences cible afin de guider les méthylations sur l’ADN (Pezic et al. 2014). Il a été démontré que DNMT3L était nécessaire à cette étape. L’absence de cet enzyme mène à l’activation des rétrotransposons LTR et, par conséquent, à des anomalies pendant la méiose et ultimement a l’induction de l’apoptose (Bourc’his and Bestor 2004).
Établissement de l’empreinte parentale
La méthylation des gènes à empreintes débute dans les gonocytes qui sont arrêtés en phase G1 de la mitose, bref avant le début de la méiose. Elle est achevée après la naissance de l’organisme, la production de spermatozoïdes mature ainsi que
22
plusieurs cycles de division cellulaire seront complétés avant la fin du processus. La méthyltransférase DNMT3A catalyse la méthylation de l’ADN à cette étape. Son facteur de régulation DNMT3L est également requis pour la mise en place de l’empreinte. DNMT3L interagit avec DNMT3A et a un impact sur la structure de l’enzyme augmentant ainsi son affinité pour l’ADN(Jia et al. 2007).La structure de la chromatine au moment de l’acquisition de l’empreinte est connue pour avoir un certain impact sur le processus. Les marques H3K4me diminuent l’affinité de l’ADN pour DNMT3A interférent ainsi avec la mise en place de méthylation sur l’ADN. Toutefois, l’étendue de l’impact de cette marque est nébuleuse (Delaval et al. 2007). Il n’est pas clair si les marques H3K9me3, qui sont connues pour stimuler la méthylation de l’ADN, ont un rôle à jouer dans l’établissement de l’empreinte parentale. Le mécanisme permettant de guider ces nouvelles méthylations est méconnu. Certaines hypothèses ont toutefois été avancées. DNMT3L est un lien significatif entre la mise en place de l’empreinte parentale et le contrôle des éléments parasites, le mécanisme réside donc possiblement dans le système de défense de l’hôte (Barlow 1993). Il a aussi été avancé que la séquence d’ADN elle-même serait impliquée dans le guidage des marques de l’empreinte (Jia et al. 2007). L’hypothèse des marques sur les histones est également considérée (Ooi et al. 2007; Ciccone et al. 2009). Certains croient que la réponse réside plutôt dans l’activité transcriptionnelle de ces régions pendant les étapes d’établissement des marques (Chotalia et al. 2009). Bref, il est possible que toutes ces réponses soient bonnes, il reste toutefois beaucoup de travail à faire pour le démontrer.
Spermatogonie
Ces cellules sont dérivées des gonocytes, qui sont dérivés des PGC après que celles-ci aient migré dans les crêtes génitales dans l’embryon. De 0 à 6 jours après la naissance de l’individu, les gonocytes se transforment progressivement en cellules souches de la spermatogénèse, les spermatogonies. Situées dans les tubules séminifères de l’individu adulte, ces cellules sont responsables de la haute
23
productivité de la spermatogénèse, elles se divisent par mitose et assurent un renouvellement de la population de cellules destinées à devenir des spermatozoïdes (J. Lee and Shinohara 2011). Au départ les cellules de Sertoli, à proximité des spermatogonies sont immatures et la méiose de ces dernières est inhibée. Les cellules de Sertoli sont responsables de l’inhibition de la méiose via la production de facteurs paracrines. FGF9 est l’un des facteurs les mieux connus pour accomplir cette fonction. Ce dernier est connu pour augmenter l’expression de NANOS, un ARN empêchant la méiose par régulation post traductionnelle. Avant l’initiation de la méiose, le profil épigénétique de ces cellules est particulier, les spermatogonies non différentiées manquent généralement d’hétérochromatine et les histones sont hypométhylées, il y a notamment les méthylations sur H3K27me1, H4K20me1 et H3k9me2 qui sont manquantes(Payne and Braun 2006).
À la puberté de l’individu, la spermatogénèse est entamée. Dans leur processus de différenciation, les spermatogonies passent par plusieurs phases qui sont contrôlées par des mécanismes paracrines. La FSH serait la principale hormone impliquée dans l’induction de la spermatogénèse et la LH serait responsable de la production d’androgènes.
24
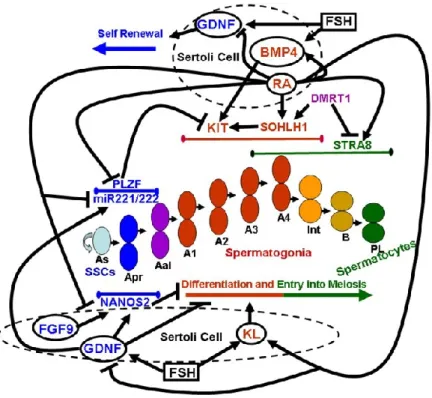
Figure 1.2 -Résumé des facteurs paracrines régissant la méiose. En bleus sont des facteurs paracrines qui favorisent le renouvellement de la population cellulaire en inhibant la différentiation et l’entrée dans la méiose. Les rouges représentent les facteurs stimulant la différentiation. En pourpre on retrouve les facteurs favorisant la différentiation, mais qui empêche l'entrée dans la méiose. Les verts sont les facteurs favorisant la méiose(Rossi and Dolci 2013).
Les cellules de types As sont des cellules capables de se renouveler continuellement. Les types Apr et Aal sont des intermédiaires entre le type non différentié As et les cellules de types A1. L’apparition des A1 coïncide avec un regain de l’expression du récepteur tyrosine kinase KIT. Ce récepteur permet de réagir au signal KL, émis par les cellules de Sertoli. Ce signal stimule la différentiation ainsi que la prolifération cellulaire. Ce changement mène également à une augmentation de l’expression des protéines Dnmt 3a et 3b, de ce fait, le nombre de méthylations sur l’ADN subit également une augmentation (Shirakawa et al. 2013).Des changements majeurs surviennent aussi sur les marques épigénétiques sur les histones comme le démontre la figure suivante.
25
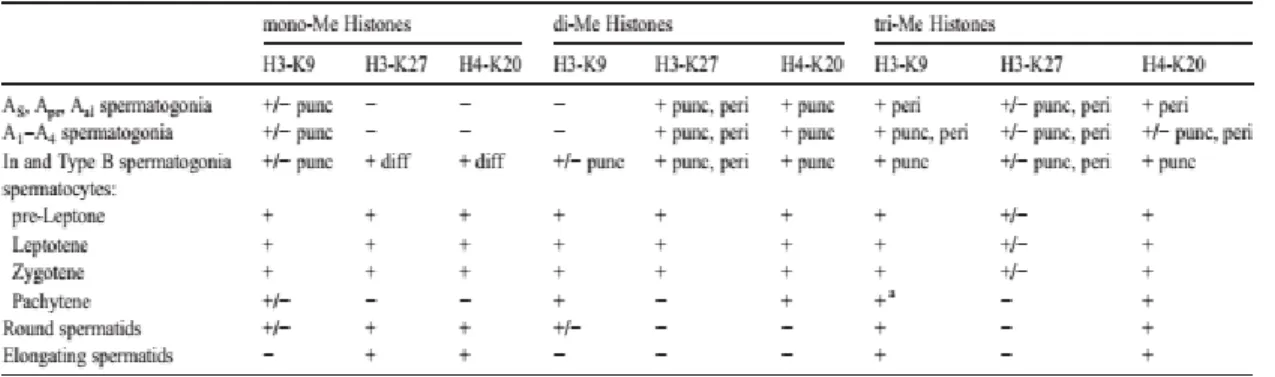
Figure 1.3 - Résumé des modifications sur les histones subvenant pendant les différentes phases de la spermatogénèse(Payne and Braun 2006).
Les modifications sur l’histone H3K9 sont directement reliées à des organisations chromosomales à grande échelle et la formation du chromocentre. Ces structures peuvent déterminer le destin d’une cellule donnée et fixer son expression génique(Bártová et al. 2008)Les méthylations H3K9me2 deviennent plus fréquentes avec la différentiation des spermatogonies, cette modification est catalysée par les enzymes G9a et GLP chez la souris, cette marque est généralement associée avec la répression génique. Il a été démontré avec des inactivations ciblées de gènes que les processus de méthylation de l’ADN et de méthylation sur les histones sont indépendants l’un de l’autre, ils sont toutefois synchronisés avec le retour de l’expression de KIT, les mécanismes entourant ce phénomène sont méconnus (Shirakawa et al. 2013). Quelque mitose suivant ces changements, les cellules se sont davantage différentiées en spermatogonie de type B. Cette étape marque la fin de la spermatogénèse et le début de la méiose.
26 Spermatocytes primaires
Le résultat de la méiose est la transformation d’une cellule diploïde en cellules haploïde, les spermatides. Ce processus est sous-divisé en cinq étapes distinguables au microscope de par la disposition de la chromatine. Au début de la méiose, les spermatocytes entreprennent la réplication de leur chromosome pendant laquelle des variantes d’histones spécifiques au testicule sont incorporées (TH2B).À la fin du préleptotène, les chromosomes sont composés de 2 chromatines sœurs, liés par un complexe protéique : la cohésine. Le leptotène est l’étape ou les chromosomes se condensent via la désacétylation et la méthylation des histones. Les marques de mono, di et triméthylation sur H3K9, H3K27, H4K20 sont très nombreuses a cette étape(Payne and Braun 2006).Cette étape de méthylation semble être impliquée dans le contrôle du pairage des chromosomes homologues et des cassures d’ADN double brin que cela implique (Sasaki and Matsui 2008). De plus, les cassures permettent l’insertion d’une autre variante d’histone γH2A.X. À cette étape, les marquent H4Ac5 sont ajouté afin de permettre l’ouverture de la chromatine et de préparer la réparation de L’ADN (Buard et al. 2009). Vient ensuite le stade zygotène, cette phase débute avec la formation du complexe synaptonémal le long des chromatides sœurs. Les chromosomes homologues sont ensuite pairés. Le stade pachytène est le moment où les recombinaisons homologues ont lieu permettant l’échange d’information génétique entre chromosomes homologues. Simultanément, une série de variantes d’histones sont incorporées : THA2, TH2B, H1t et H3.3 et la chromatine est ouverte afin de permettre la transcription massive. Localement, ces changements sont facilités par l’ubiquitination de H2A et l’acétylation de H3K4 et H3K9 (Kota and Feil 2010; Noblanc, Kocer, and Drevet 2014).La plupart des marques de méthylation, ajoutées au leptotène, ont disparu, à l’exception de H3K9me2/me3 qui restent constantes. Une lacune dans ces méthylations signifie l’entré en apoptose de la cellule très tôt après l’entrée dans la méiose (Payne and Braun 2006).
27
Par contraste, les chromosomes sexuels, qui n’ont pas été pairés, subissent une extinction traductionnelle afin d’éviter des recombinaisons non homologues (Kelly and Aramayo 2007). Lors de cette étape, une perte massive des histones H3.1 et H3.2 a lieu. Elles sont remplacées par les histones spécifiques aux chromosomes sexuels H3.3. D’autres variantes d’histone apparaissent temporairement sur les chromosomes sexuels, mais ils sont très vite remplacés, il est possible qu’ils servent d’intermédiaire pour la mise en place de H3.3. (Greaves et al. 2006; van der Heijden et al. 2007). Après l’inactivation de XY, les méthylations H3K4me2 et H3K4me3 sont respectivement sur et sous représentés (Sasaki and Matsui 2008). Des méthylations H3K9me2 font leurs apparitions afin de recruter HP1. Ces marques, menant à la formation d’hétérochromatine, persisteront jusqu’à ce que les histones soient remplacées. Malgré la configuration répressive des chromosomes sexuels, des miARN originaire du chromosome X font leurs apparitions à cette étape. Le mécanisme permettant ce phénomène ainsi que l’effet de ces miARN est à ce jour incertain (R. Song et al. 2009). Pendant le processus, les méthylations H3K27me1 et H3K27me3 sont complètement absentes des chromosomes XY (Namekawa et al. 2006). À ce jour il n’est pas clair si les piARN jouent un rôle dans la formation de l’hétérochromatine des chromosomes sexuels (F. Wang et al. 2006; Holmes and Cohen 2007). Lorsque les cassures double brins sont réparées, le diplotène commence, le complexe synaptonémal est désassemblé et les chromosomes sont prêts à être séparés. La diakinèse est l’étape finale ou les deux cellules filles sont séparées. Après la méiose, l’expression de plusieurs gènes situés sur les chromosomes sexuels est rétablie (P. J. Wang, Page, and McCarrey 2005).
Les spermatocytes secondaires sont le résultat de la première méiose. Ils restent en prophase seulement quelques heures, ils entreprennent immédiatement la seconde méiose. Cette étape marque la fin de la spermatogenèse et le début de la spermiogenèse.
Au début de la spermiogenèse, les cellules sont au stade de spermatides rondes haploïdes. À la fin, les cellules seront des spermatozoïdes matures très spécialisés.