Université de Montréal
Réaction d’amination intramoléculaire de liens C-H à
partir de N-mésyloxycarbamates catalysée par des
complexes de rhodium et d’autres métaux de transition
Synthèse verte d’oxazolidinones
par
Laura Mamani Laparra
Département de Chimie Faculté des Arts et des Sciences
Thèse présentée à la Faculté des études supérieures et postdoctorales en vue de l’obtention du grade de philosophiæ doctor (Ph.D.)
en Chimie
Décembre 2015
Résumé
La réaction d’amination de liens C-H, impliquant la transformation directe d’un lien C-H en lien C-N constitue une approche synthétique d’avenir pour la préparation de composés azotés. L’application de cette stratégie de manière intramoléculaire apparaît comme une approche puissante pour la synthèse de composés hétérocycliques. En particulier, les oxazolidinones, carbamates cycliques à cinq chaînons, constituant une nouvelle classe d’antibiotiques très prometteuse, pourraient être synthétisées par cette méthode.
Il y a moins d’une dizaine d’années, notre groupe de recherche a travaillé sur le développement de méthodologies utilisant des espèces nitrènes métalliques pour l’amination intra et intermoléculaire. Les N-tosyloxycarbamates, en présence d’une base et d’un catalyseur dimère de rhodium (II) tétracarboxylate sont les précurseurs de ces espèces nitrènes métalliques, capables de faire l’insertion de liens C(sp3)-H.
Dans ces travaux de thèse, nous avons travaillé sur le développement d’une méthode plus « verte » d’amination intramoléculaire. Les N-mésyloxycarbamates, plus légers que leurs homologues N-tosyloxycarbamates, ont été identifiés comme d’excellents précurseurs de nitrènes. La méthodologie développée ne nécessite que 3 mol % de dimère de rhodium Rh2(tpa)4 et de 1,5 équivalents de solution aqueuse saturée de K2CO3, le tout dans l’acétate d’éthyle et donne de bons rendements de cyclisation. Une étude de l’étendue réactionnelle a été effectuée, montrant la tolérance et les limitations de notre système catalytique : les hétéroatomes ne posent pas de problèmes hormis l’atome d’azote, qui doit être protégé afin de garantir la transformation. En outre, nous avons constaté que les liens C-H aliphatiques secondaires sont moins réactifs que les liens tertiaires. Après avoir tenté de développer des conditions réactionnelles spécifiques aux liens C-H non activés, nous avons montré la possibilité d’aminer des liens C-H propargyliques de manière chimiosélective ; la triple liaison C-C peut ensuite être dérivatisée efficacement, donnant accès à la formule saturée correspondante ainsi qu’à d’autres motifs.
Dans un désir de substituer les complexes de rhodium par d’autres complexes de métaux plus abondants et moins dispendieux, nous nous sommes tournés, dans un premier temps, vers les
complexes de fer et par la suite, vers les pinceurs de nickel. Les phtalocyanines de fer ont été identifiées comme étant de bons catalyseurs de l’amination intramoléculaire de N-mésyloxycarbamates. Le chlorure de phtalocyanine de fer (III), en présence d’un sel de AgBF4 et de K2CO3, dans le 1,1,2,2-tétrachloroéthane anhydre, permet l’obtention de la 4-phenyloxazolidin-2-one avec 63% de rendement. En outre, il est possible d’atteindre un rendement de 49% à partir du même substrat N-mésyloxycarbamate, par catalyse avec un pinceur de nickel de type POCN, en présence d’un sel de mésylate. Des indices sur le mécanisme des ces deux transformations ont pu être recueillis lors de la courte étude de ces systèmes.
Mots-clés : Amination de liens C-H, N-Mésyloxycarbamates, Oxazolidinones, Dimères de
Abstract
C-H amination reactions, i.e. the direct transformation of a C-H bond into a C-N bond, represents a very promising synthetic approach to prepare nitrogen-containing compounds. The strategy, when applied to intramolecular transformations, represents a powerful method for the preparation of heterocycles. In particular, oxazolidinones (5-membered carbamate heterocycles), which are a novel class of promising antimicrobials, could be easily accessed using C-H amination.
Nearly a decade ago, our research group developed methods for metal nitrene-mediated C-H aminations, for both intra- and intermolecular transformations. N-Tosyloxycarbamates, in the presence of a tetracarboxylate rhodium (II) dimer catalyst and a base, were found to be good precursors to metal nitrenes; the latter being able to perform C(sp3)-H insertions.
In the present thesis, we have worked on developing a “greener” method for C-H amination reactions. N-Mesyloxycarbamates, lighter than their N-tosyloxycarbamate homologues, were identified as nitrene precursors. The methodology requires only 3 mol % of rhodium dimer Rh2(tpa)4 and 1.5 equivalents of an aqueous saturated solution of K2CO3, in AcOEt and provides the cyclized product in good yields. The scope of the reaction was investigated, illustrating the tolerance and limitations of the catalytic system: heteroatoms are not a problem except for nucleophilic nitrogens, which should be protected, in order to allow for efficient transformation. We observed that secondary C-H bonds were less reactive than tertiary ones. After attempting to develop specific conditions for non-activated bonds, we showed that propargylic C-H bonds can be chemoselectively aminated; and the C-C triple bond can easily be further derivatized, allowing for structural diversification.
With regards to replacing rhodium complexes by complexes of other metals, which are more abundant and cheaper, we focused first on iron complexes and then on nickel pincer complexes. Iron phthalocyanin complexes are viable catalysts for the intramolecular C-H amination using N-mesyloxycarbamates. Iron (III) phthalocyanin chloride, along with AgBF4 salt and K2CO3, in anhydrous 1,1,2,2-tetrachloroethane, allows the formation of phenyloxazolidin-2-one in 63% yield. Likewise, the same product can be generated in 49%
yield, when a POCN nickel pincer is used as a catalyst, along with a sodium mesylate salt and a base, in anhydrous DCM. Some mechanistic clues could be collected while studying the catalytic systems.
Keywords: C-H Amination, N-Mesyloxycarbamates, Oxazolidinones, Rhodium dimers, Iron
Table des matières
Chapitre 1 Introduction ... 1
La chimie des nitrènes pour l’amination de liens C-H ... 1
1. 1. La chimie des nitrènes ... 1
1. 1. a. Fonctionnalisation de liens C-H ... 1
1. 1. b. L’amination de liens C-H ... 4
1. 1. c. Les nitrènes ... 4
1. 2. Les réactions d’amination de liens C-H utilisant des nitrènes métalliques ... 12
1. 2. a. Les travaux pionniers ... 12
1. 2. b. Les iminoiodinanes en tant que précurseurs de nitrènes ... 14
1. 2. c. Les azotures en tant que précurseurs de nitrènes ... 31
1. 2. d. Aspects mécanistiques ... 37
1. 2. d. i. Mécanisme d’insertion concertée d’un nitrène singulet dans un lien C-H . 37 1. 2. d. ii. Mécanisme d’abstraction radicalaire d’hydrogène suivie de recombinaison radicalaire ... 42
1. 3. Fondements et objectifs du projet de recherche ... 44
1. 3. a. Les N-sulfonyloxycarbamates comme précurseurs de nitrènes métalliques ... 44
1. 3. b. Aspects mécanistiques ... 45
1. 3. c. Objectifs de thèse ... 48
1. 3. c. i. Les oxazolidinones ... 48
1. 3. c. ii. Problématiques du projet de recherche ... 49
Chapitre 2 Les N-mésyloxycarbamates en tant que précurseurs de nitrènes ... 51
2. 1. Fondements et objectifs du projet ... 51
2. 2. Développement d’une nouvelle méthodologie ... 52
2. 2. a. Nouveau groupe partant : preuve de concept ... 53
2. 2. b. Étude du solvant et du catalyseur de la réaction intramoléculaire ... 54
2. 2. c. Étude de la base utilisée ... 58
2. 2. d. Criblage de catalyseurs dimères de rhodium (II) ... 61
2. 2. e. Optimisation fine d’autres facteurs ... 63
2. 3. a. Voie de synthèse des N-mésyloxycarbamates ... 66
2. 3. b. Étendue réactionnelle de la transformation ... 75
2. 3. b. i. Insertions dans des liens C-H benzyliques ... 75
2. 3. b. ii. Insertions dans des liens C-H α à des cycles hétéroaromatiques ... 81
2. 3. b. iii. Insertions dans des liens C-H α à un atome d’oxygène ... 86
2. 3. c. Conclusions sur l’étendue réactionnelle ... 88
2. 4. Conclusions et perspectives ... 89
Chapitre 3 Développement de conditions réactionnelles pour l’amination intramoléculaire de liens C-H non activés ... 90
3. 1. Introduction et objectifs du projet ... 90
3. 1. a. Précédents dans la littérature ... 90
3. 1. a. i. Catalyse impliquant une activation du lien C-H par un métal ... 91
3. 1. a. ii. Catalyse faisant intervenir des espèces nitrénoïdes ... 92
3. 1. b. Mise en évidence des difficultés et objectifs du projet ... 97
3. 2. Optimisation des conditions réactionnelles ... 98
3. 2. a. Étude du groupement partant ... 98
3. 2. b. Étude du catalyseur ... 101
3. 2. c. Étude du solvant ... 102
3. 2. d. Étude de la base ... 103
3. 2. e. Levier d’amélioration : influence du catalyseur sur la sélectivité de la réaction 106 3. 3. Synthèse de nouveaux ligands de type triarylacétate ... 108
3. 3. a. Objectifs ... 108
3. 3. b. Synthèse des ligands ... 109
3. 3. c. Synthèse des dimères de rhodium correspondants ... 113
3. 3. d. Conclusion et perspectives ... 117
3. 4. Insertion de nitrènes dans des liens C(sp3)-H propargyliques ... 117
3. 4. a. Précédents dans la littérature ... 118
3. 4. b. Parallèle avec les substrats allyliques ... 120
3. 4. c. Insertion dans un lien C-H propargylique : Optimisation ... 122
3. 4. e. Conclusion ... 129
3. 5. Conclusions ... 129
Chapitre 4 Étude de complexes de fer et de nickel (II) et (III) pour promouvoir l’amination intramoléculaire de liens C-H ... 135
4. 1. Mise en contexte et objectifs du projet ... 135
4. 2. Catalyse par les complexes de fer ... 136
4. 2. a. Précédents dans la littérature et aspects mécanistiques ... 137
4. 2. a. i. Catalyseurs portant des ligands de type hème ... 137
4. 2. a. ii. Catalyseurs portant des ligands de type non-hème ... 141
4. 2. b. Développement d’une méthodologie avec des complexes de fer ... 147
4. 2. c. Conclusions et perspectives ... 156
4. 3. Catalyse par les complexes de nickel ... 159
4. 3. a. Précédents dans la littérature et aspects mécanistiques ... 160
4. 3. b. Les pinceurs de nickel ... 163
4. 3. c. Évaluation de quelques pinceurs POCN de nickel dans la réaction intramoléculaire d’amination de liens C-H à partir de N-mésyloxycarbamates ... 166
4. 3. d. Conclusions sur les pinceurs de nickel ... 176
4. 4. Conclusion et perspectives ... 178
Conclusion ... 179 Bibliographie et notes ... I Partie expérimentale ... XIV Procédures décrivant la synthèse des composés décrits aux chapitres 2 et 3 ... XVII Procédures décrivant la synthèse des composés décrits au chapitre 4 ... LXXXVIII
Liste des schémas
Schéma 1. Stratégie traditionnelle en synthèse organique ... 2
Schéma 2. Mécanismes de sphère interne et externe de fonctionnalisation de lien C-H ... 3
Schéma 3. Réactivité des nitrènes singulet et triplet ... 6
Schéma 4. Les précurseurs de nitrènes métalliques ... 8
Schéma 5. Réactions d'amination et d'aziridination intermoléculaires avec la bromamine-T ... 9
Schéma 6. Réactions d'insertion de nitrènes dans des liens C-H de manières intra- et intermoléculaires ... 12
Schéma 7. Hétérocycles accessibles par la méthode d'amination intramoléculaire de Du Bois ... 19
Schéma 8. Fonctionnement du système réactionnel de Du Bois ... 19
Schéma 9. Application de la stratégie d'amination de sulfamates dans la synthèse de (+)-saxitoxine ... 21
Schéma 10. Mécanisme supposé de formation de la cétone à partir d'un carbamate dérivé d'alcool secondaire ... 24
Schéma 11. Application de la méthodologie d'amination intramoléculaire de He dans la synthèse totale de dérivés de la welwitindolinone C ... 25
Schéma 12. Formation de benzimidazoles et de quinazolones par amination intramoléculaire d'arylamines catalysée par la porphyrine Fe(F20TPP)Cl ... 29
Schéma 13. Synthèse de 1,3-diamines par amination intramoléculaire d'azotures de sulfamoyle suivie de l'ouverture des sulfamides cycliques correspondants ... 33
Schéma 14. Mécanisme de l'insertion concertée d'un nitrène singulet dans une liaison C-H .. 38
Schéma 15. Les chemins réactionnels possibles pour l'amination d'un substrat "horloge radicalaire" ... 40
Schéma 16. Mécanisme radicalaire en deux étapes d'amination intramoléculaire à partir d'un nitrène métallique à l'état triplet ... 42
Schéma 17. Mécanisme proposé par Kim Huard pour la réaction d'amination intramoléculaire à partir de N-tosyloxycarbamates catalysée par des dimères de rhodium carboxylates ... 47
Schéma 19. Étendue réactionnelle de l'amination de liens C-H intramoléculaire de
N-tosyloxycarbamates dans les conditions de Kim Huard132 ... 52
Schéma 20. Synthèse des N-tosyloxycarbamates en deux étapes à partir de l'alcool132 ... 66
Schéma 21. Synthèse des composés N-sulfonyloxycarbamates dérivés d'alcools secondaires145 ... 67
Schéma 22. Synthèse de l'alcool 74146 ... 68
Schéma 23. Synthèse de l'alcool 79147 ... 69
Schéma 24. Synthèse de l'alcool racémique (±)-81 à partir du Naproxen ... 70
Schéma 25. Synthèse de N-mésyloxycarbamates substitués par des groupements aromatiques ... 71
Schéma 26. Réaction parasite de l'addition de l'hydroxylamine ... 72
Schéma 27. Synthèse du substrat 95 par activation de l'imidazole ... 73
Schéma 28. Synthèse de N-mésyloxycarbamates divers et variés ... 74
Schéma 29. Réaction d'amination de liens C-H intramoléculaire avec des substrats possédant des cycles aromatiques riches en électrons ... 75
Schéma 30. Réaction d'amination de liens C-H intramoléculaire avec d'autres N-mésyloxycarbamates possédant des cycles aromatiques ... 78
Schéma 31. Réaction d'amination intramoléculaire de liens C-H α à des cycles hétéroaromatiques ... 83
Schéma 32. Activation C-H catalysée par le palladium pour la formation d'hétérocycles ... 91
Schéma 33. Amination de liens C-H tertiaire, secondaire et primaire de sulfamates primaires par le système de White ... 95
Schéma 34. Plusieurs chemins réactionnels possibles à partir des nitrénoïdes de rhodium .. 105
Schéma 35. Voie rétrosynthétique menant à l'acide p-méthoxyphénylacétique 149 ... 109
Schéma 36. Approche vers la synthèse du composé 149 ... 110
Schéma 37. Synthèse du composé 149 ... 111
Schéma 38. Synthèses infructueuses des ligands 156 et 157 ... 113
Schéma 39. Synthèse du substrat propargylique 169 ... 122
Schéma 40. Synthèse du N-mésyloxycarbamate 172 ... 123
Schéma 41. Synthèse de l'alcool 178 ... 125
Schéma 43. Mécanisme réactionnel proposé pour l'amination intramoléculaire de liens C-H à
partir de N-mésyloxycarbamates catalysée par un dimère de rhodium ... 130
Schéma 44. Mécanisme élucidé par DFT de l'amination intramoléculaire de liens C-H
aromatiques à partir d'azotures catalysée par Fe(F20TPP)Cl ... 141
Schéma 45. Mécanisme proposé pour l'amination de liens C-H catalysée par le complexe de
fer Ad2Fe de Betley ... 143
Schéma 46. Mécanisme élucidé par DFT de la réaction en cascade catalysée par FeBr2, menant à des indoles à partir d'azotures ... 147
Schéma 47. Cycle catalytique proposé pour l'amination de liens C-H de
N-mésyloxycarbamates catalysée par la phatlocyanine de fer FePcCl ... 157
Schéma 48. Réaction du complexe imido de nickel 209 avec un alcyne terminal ... 161
Schéma 49. Amination du lien C-H de l'éthylène par un nitrène de nickel possédant un ligand
NHC ... 162
Schéma 50. Réaction stoechiométrique de l'imido [Me3NN]Ni=NAd 218 avec des substrats benzyliques ... 163
Schéma 51. Synthèses des ligands POCN et des complexes de Ni(II) correspondants ... 176
Schéma 52. Proposition de cycle catalytique pour la réaction rhodo-catalysée d'amination
Liste des tableaux
Tableau 1. Influence du dimère de rhodium sur le rendement et l'excès énantiomère de la
réaction d'amination de liens C-H intramoléculaire de sulfamates ... 17
Tableau 2. Système chimiosélectif de Schomaker utilisant des complexes d'argent :
phénanthroline pour l'aziridination ou l'amination de substrats carbamates homoalléniques ... 26
Tableau 3. Étude du groupement partant pour la réaction d'insertion intramoléculaire dans une
lien C-H de N-sulfonyloxycarbamates catalysée par un dimère de rhodium132 ... 53
Tableau 4. Solubilité de certains dimères de rhodium dans les solvants étudiés pour la
réaction d'amination de liens C-H intramoléculaire ... 55
Tableau 5. Étude du catalyseur et du solvant de la réaction d'amination intramoléculaire à
partir de N-mésyloxycarbamates ... 56
Tableau 6. Optimisation de la base dans l'acétate d'éthyle pour la réaction d'amination de liens
C-H intramoléculaire à partir de N-mésyloxycarbamates ... 59
Tableau 7. Second criblage de dimères de rhodium(II) pour la réaction d'amination de liens
C-H intramoléculaire ... 62
Tableau 8. Optimisation de la température de la réaction d'amination de liens C-H
intramoléculaire et de la fraîcheur des réactifs ... 64
Tableau 9. Étude de deux bases dans l'acétate d'éthyle pour l'amination intramoléculaire de
liens C-H ... 65
Tableau 10. Essais de déprotection du groupement TBS du substrat 75 ... 68
Tableau 11. Optimisation des conditions réactionnelles pour le substrat 89 ... 85
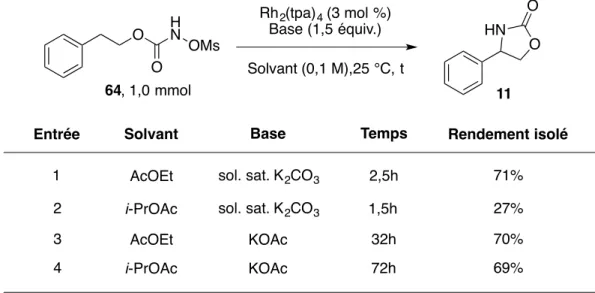
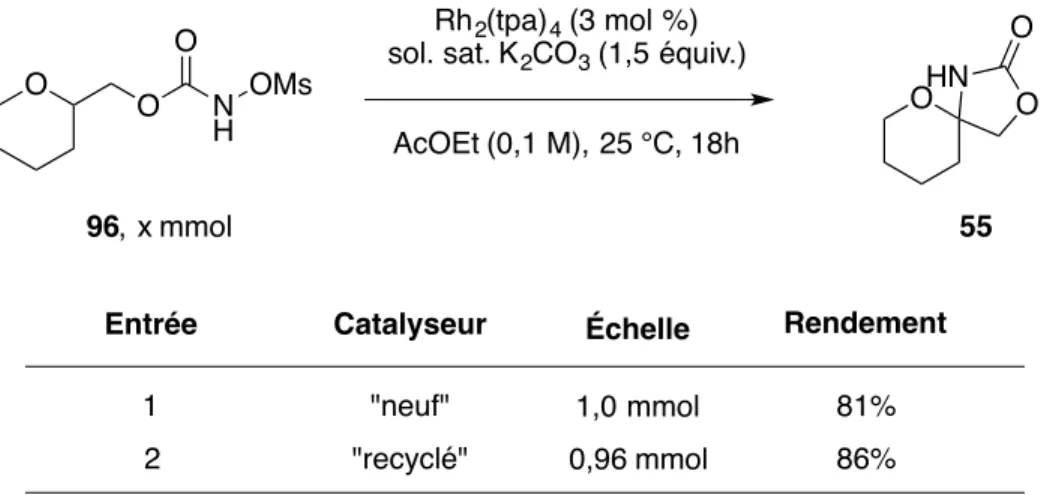
Tableau 12. Recyclabilité du catalyseur Rh2(tpa)4 dans la réaction d'amination de liens C-H intramoléculaire de composé 96 ... 87
Tableau 13. Étude des groupements de type sulfonate pour la réaction d'amination
intramoléculaire de liens C-H non activés ... 100
Tableau 14. Étude des catalyseurs dimères de rhodium(II) dans la réaction d'amination
Tableau 15. Étude des solvants de type acétate dans la réaction d'amination intramoléculaire
de liens C-H non activés à partir de N-mésyloxycarbamates ... 102
Tableau 16. Étude de la base dans la réaction d'amination intramoléculaire de liens C-H non
activés à partir de N-mésyloxycarbamates ... 104
Tableau 17. Influence du catalyseur sur la sélectivité de l'insertion de
N-tosyloxycarbamates132 ... 107
Tableau 18. Influence du catalyseur sur la sélectivité de l'insertion de N-mésyloxycarbamates
... 107
Tableau 19. Essais de préparation du dimère de rhodium 146 ... 114
Tableau 20. Amination intramoléculaire de liens C-H propargyliques par Schomaker et al.
... 119
Tableau 21. Amination de liens C-H à partir d'azotures de sulfamoyle
bishomopropargyliques106 ... 120
Tableau 22. Courte étude des conditions réactionnelles de l'amination intramoléculaire de
liens C-H propargyliques ... 124
Tableau 23. Courte optimisation des conditions réactionnelles de l'amination du substrat 180
... 126
Tableau 24. Optimisation des conditions réactionnelles pour la réduction de 173 par le
catalyseur de Lindlar ... 128
Tableau 25. Énergies de liaison et rendements d'amination intramoléculaire de liens C-H non
activés, propargyliques, benzyliques et allyliques159 ... 132
Tableau 26. Énergies de liaison et rendements d'amination intramoléculaire de liens C-H non
activés et α à un oxygène159 ... 133
Tableau 27. Variation du rendement d'amination intermoléculaire de l'adamantane en fonction
de la porphyrine de fer (III) utilisée ... 138
Tableau 28. Étendue réactionnelle de l'amination de liens C-H intramoléculaire de sulfamates
catalysée par le complexe de fer quinquepyridine ... 145
Tableau 29. Criblage de quelques complexes de fer pour la réaction d'amination de liens C-H
intramoléculaire à partir de N-mésyloxycarbamates ... 148
Tableau 30. Criblage de quelques sels d'argent et de sodium dans la réaction d'amination
Tableau 31. Criblage de solvants pour la réaction d'amination intramoléculaire, à l'abri de la
lumière ... 150
Tableau 32. Influence du ligand phtalocyanine et du degré d'oxydation du complexe de fer sur
le rendement de la réaction d'amination intramoléculaire, à l'abri de la lumière ... 151
Tableau 33. Essais de quelques complexes pinceurs POCOP, NCN et SCS de nickel dans la
réaction d'amination de liens C-H intramoléculaire ... 167
Tableau 34. Essais de quelques pinceurs de nickel POCN dans la réaction d'amination de liens
C-H intramoléculaire ... 169
Tableau 35. Influence de l'anion sur la catalyse de la réaction d'amination de liens C-H
intramoléculaire ... 172
Tableau 36. Influence d'additifs sur le rendement de la réaction d'amination de liens C-H
Liste des figures
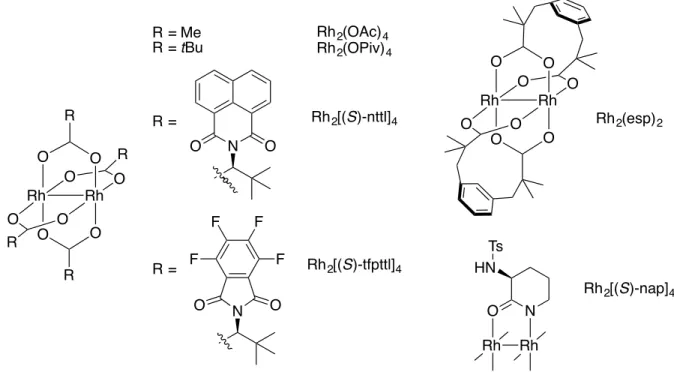
Figure 1. Structures et configurations électroniques des états singulet et triplet des nitrènes ... 5
Figure 2. Structures des nitrènes de carbamoyle, de sulfamoyle et de phosphoryle ... 7
Figure 3. Structures d'un nitrène métallique ... 8
Figure 4. Structure du catalyseur (OC)Ru(salen) 4 de Katsuki ... 11
Figure 5. Structures des chlorures de tétraphénylporphyrines de fer et de manganèse et du dimère de rhodium tétraacétate ... 14
Figure 6. Structure du ligand meso-tetrakis(pentafluorophényl)porphyrine F20TPP ... 16
Figure 7. Structures de dimères de rhodium utilisés dans l'amination de liens C-H intramoléculaire à partir de sulfamates primaires ... 18
Figure 8. Types de liens C-H réagissant de manière satisfaisante dans les conditions réactionnelles d'amination intramoléculaire de Du Bois ... 20
Figure 9. Structure de la porphyrine chirale de ruthénium [Ru(Por*)(CO)] de Che ... 23
Figure 10. Espèces catalytiques proposées pour l'amination et l'aziridination intramoléculaire ... 26
Figure 11. Structure du chlorure de phtalocyanine de fer (III) ... 28
Figure 12. Structure de la phtalocyanine Mn(tBuPc)Cl ... 29
Figure 13. Structures des catalyseurs de fer non-hèmes développés par le groupe de Che pour l'amination de liens C-H ... 30
Figure 14. Structure de la porphyrine de cobalt (II) Co(P1) développée par le groupe de Zhang ... 33
Figure 15. Structure du complexe dipyrrométhène de fer de Betley ... 36
Figure 16. Structure du dimère de ruthénium Ru2(hp)4Cl ... 43
Figure 17. Structure du Linézolide ... 49
Figure 18. Liens C-H et O-H susceptibles de réagir sur les solvants AcOEt, i-PrOAc, EtOH et MeOH ... 55
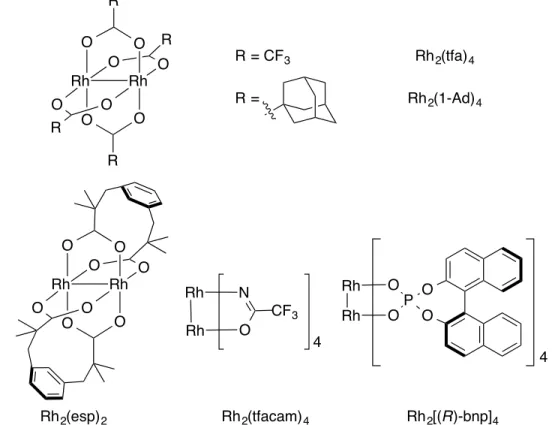
Figure 19. Structures de quelques dimères de rhodium(II) tétracarboxylates ... 56
Figure 20. Structures de plusieurs dimères de rhodium(II) évalués dans la réaction d'amination de liens C-H intramoléculaire ... 61
Figure 21. Échelle de pKa des acides conjugués des ligands des dimères de rhodium évalués
dans la réaction d'amination de liens C-H intramoléculaire ... 63
Figure 22. Analyse DSC-TGA du composé 64 ... 74
Figure 23. Structure du 2-éthylhexanoate de sodium EH-Na ... 80
Figure 24. Énergies de liaisons de liens C-H secondaire et tertiaire159 ... 88
Figure 25. Structure du ligand bathophénanthroline ... 93
Figure 26. Structures des catalyseurs Ru(tpfpp)(CO) et Fe(qpy)(MeCN)2(ClO4)2 ... 94
Figure 27. Structure du squelette n-hexane choisi pour l'optimisation des conditions réactionnelles de l'amination intramoléculaire de liens C-H non activés ... 98
Figure 28. Quelques carboxylates et leurs pKas approximatifs dans l'eau ... 103
Figure 29. Structures des dimères de rhodium ciblés ... 108
Figure 30. Analyse LC-MS du mélange de dimères de rhodium ... 115
Figure 31. Essais de solubilité du précipité vert turquoise dans les solvants usuels ... 115
Figure 32. Édifices supramoléculaires faisant intervenir des a) ligands équatoriaux, b) ligands axiaux et c) les deux ... 116
Figure 33. Énergies de dissociation de liens C-H allylique, propargylique et alkyle secondaires159 ... 118
Figure 34. Structure de la phtalocyanine FPcFe(III)Cl utilisée par Zhou et al. ... 139
Figure 35. Structure de Fe(salen)Cl ... 148
Figure 36. Structure de NaBArF4 ... 149
Figure 37. Structure de la phtalocyanine de fer per-ClPcFe(II) ... 152
Figure 38. Structure de la naphtalocyanine de fer NcFe(II) ... 153
Figure 39. Structure générale de complexes métalliques de type pinceur ... 164
Figure 40. Plusieurs types de complexes pinceurs de nickel ... 164
Figure 41. Structures des complexes pinceurs de nickel utilisés dans l'amination de liens C-H intramoléculaire ... 168
Figure 42. Structures des complexes POCN de nickel essayés dans la réaction d'amination de liens C-H intramoléculaire ... 170
Figure 43. Relation entre le pKa des ligands azotés des complexes POCN et le rendement d'amination de liens C-H ... 171
Liste des sigles et abréviations
Ac acétyle acac acétylacétonate Ad adamantyle Ar aryle As p-methoxybenzènesulfonyle Bn benzyle Boc tert-butoxycarbonyle i-Bu isobutyleCAM molybdate d’ammonium et sulfate cérique CCM chromatographie sur couche mince
CDI 1,1’-carbonyldiimidazole cod cyclooctadi-1,5-ène
COSY spectroscopie de corrélation homonucléaire (Correlation Spectroscopy)
d doublet
d jour (lat. diem)
DCE 1,2-dichloroéthane DCM dichlorométhane
DEPT 135 (Distortionless Enhanced Polarization Transfer)
DFT Théorie de la fonctionnelle de la densité (Density Functional Theory) DMF N,N-diméthylformamide
DSC (Differential Scanning Calorimetry) EH-Na 2-éthylhexanoate de sodium
ee excès énantiomérique équiv. équivalent molaire
ESI-MS Spectrométrie de masse par ionisation par électronébuliseur (Electronspray
Ionization Mass Spectrometry)
esp ligand espino
et al. et collaborateurs
GF groupement fonctionnel
h heure
HMPA Hexamethylphosphoramide
hp 2-hydroxypyridine
HRMS Spectrométrie de masse de haute résolution
Hz hertz
Imid. Imidazole
IR infrarouge
M mol/L
mol % pourcentage molaire
Ms methanesulfonyl MW micro-ondes (microwave) NBS N-Bromosuccinimide Nc naphtalocyanine Ns p-nitrobenzènesulfonyl oct octanoate
PMA acide phosphomolybdique
Pc phtalocyanine
Ph phényle
phen phénanthroline
Piv Pivaloyle
ppm partie par million
i-Pr isopropyle qpy quinquepyridine
RMN Résonance Magnétique Nucléaire
sat. saturée
sol. solution
t.a. température ambiante TBS tert-butyldiméthylsilyle
TEMPO (2,2,6,6-tétraméthylpipéridin-1-yl)oxy Tf trifluorométanesulfonyle
tfa trifluoroacétate
TGA analyse thermogravimétrique (Thermogravimetric Analysis) THF tétrahydrofurane
TM tamis moléculaire
TOF temps de vol (Time of Flight)
Tol toluène
TON nombre d’échanges (Turnover number) tpa triphénylacétate
tr temps de rétention Ts p-toluènesulfonyle
UdeM Université de Montréal
UV ultra-violet
Remerciements
L’aboutissement de ces travaux de thèse n’aurait pas été possible sans le soutien et l’aide de nombreux protagonistes. J’espère n’oublier personne dans cette brève liste de remerciements.
Tout d’abord, j’aimerais remercier ma directrice de recherche, la Pr. Hélène Lebel, pour m’avoir accueillie dans son laboratoire de recherche. Je lui suis reconnaissante pour la confiance et le temps qu’elle m’a accordés tout au long de ma formation dans son laboratoire. Ses conseils et son accompagnement m’ont été d’une grande aide dans mon cheminement, que ce soit scientifiquement ou professionnellement parlant.
J’aimerais aussi remercier les membres de mon jury, les Prs. Collins, Hanessian et Jubault pour la lecture attentive de mon manuscrit de thèse ainsi que pour leurs commentaires enrichissants.
Je suis très reconnaissante à la faculté des études supérieures et postdoctorales de l’université de Montréal pour l’octroi d’une bourse d’excellence pour mes études doctorales.
Ensuite, je tiens à exprimer ma gratitude aux membres du personnel permanent de l’Université de Montréal. Notamment, Sylvie Bilodeau, Antoine Hamel, Cédric Malveau et Minh Tan Phan Viet du centre régional de résonance magnétique nucléaire de l’UdeM, pour leur aide et leurs conseils précieux. J’aimerais remercier le centre régional de spectrométrie de masse, et en particulier Marie-Christine Tang pour la qualité et la diligence de son travail. Les considérations techniques étant partie intégrante d’un parcours de PhD en chimie, je remercie l’équipe de l’atelier mécanique pour m’avoir consacré beaucoup de son temps, en particulier, Louis Beaumont, Martin Lambert, Jean-François Myre et Cédric Ginart. Je voudrais également exprimer mes remerciements au personnel d’enseignement, qui est pour beaucoup
dans la qualité de la formation donnée à l’UdeM. Merci à Denis Deschênes pour son aide et ses précieux conseils, à Huguette Dinel pour sa sympathie et sa bonne humeur légendaires. Merci également à Gaëtan Caron et Kevin Delorme. Enfin, mes remerciements vont à l’équipe administrative du département de chimie ainsi qu’aux bibliothécaires de la section chimie.
Dans le cadre de mes travaux, j’ai eu la chance de collaborer avec un grand nombre d’étudiants et de chercheurs en général. Notamment, j’ai travaillé en collaboration avec le groupe du Pr. Zargarian. Je tiens à remercier ce dernier pour le soin qu’il a mis à m’aider dans ma recherche et le temps qu’il m’a consacré pour des discussions scientifiques éclairantes. Merci également aux membres de son groupe qui ont fait la synthèse des composés pinceurs de nickel que j’ai utilisés : Jean-Philippe Cloutier, Richard Declercq, Dr. Jingun Hao, Sébastien Lapointe, Berline Mougang-Soumé et Boris Vabre.
Je voudrais remercier les membres des groupes Charette, Collins, Hanessian, Schmitzer, Zargarian et Lubell pour les conversations agréables et les prêts de produits.
Pendant ces années de thèse, j’ai eu la chance de pouvoir interagir quotidiennement avec des collègues sans qui la vie au laboratoire n’aurait pas été aussi haute en couleur. La plupart d’entre eux sont rapidement devenus mes amis et le resteront. Cela commence avec les mentors, pour leurs conseils et leur patience : Dr. Olivier Léogane, Nicolas Lévaray, Dr. Yingdong Lu, Dr. Cédric Spitz, Dr. Saï Sudhir Venkatesh et Dr. Walid Zeghida. J’aimerais aussi remercier tous les étudiants stagiaires ou non, dont le passage dans notre laboratoire a été court mais remarqué : Pauline, Pierre, Jeff, Sébastien, Marion, Claire, Ravi, Charlie, Loïc, Valérie, Marie, Samuel et Maxime. J’ai également eu le privilège de jouer le rôle de mentor pour trois étudiants stagiaires. Prettiny, Matthieu et Nathan, j’ai beaucoup aimé travailler avec vous. Enfin, un merci tout spécial à mes acolytes : Maroua « Triwi » Khalifa, Johan « l’affreux » Bartholoméüs, Henri « M. Net » Piras, Clément « Biscotte » Audubert, Emna « Amouna » Azek, Cendrella « Cici » Maroun et Carl « Loscar » Trudel, pour leur sympathie,
leur bonne humeur, leurs blagues et tout le reste ! Ce fut un plaisir de partager tous ces moments en votre compagnie !
Pendant mon séjour à Montréal, j’ai aussi pu rencontré des personnes exceptionnelles en dehors du laboratoire. Merci aux deux meilleures coloc’ au monde : Jacynthe et Marie-Jeanne. Merci énormément à Audrey pour sa gentillesse, son humour et son cousin ! Un grand merci à mes belles amies de Paris : Maryam, Sarah, Sherley et Émilie !
Finalement, je voudrais adresser toute ma gratitude à mes proches, pour leur soutien et leur tendresse inconditionnels tout au long de ces années. Merci à mon cousin Vladimir, à qui je dédie tout ce travail. Merci à Émilie, Julien, mon frère Jérôme et mes parents Marie-Annick et Nolasco. Je vous aime.
Chapitre 1 Introduction
La chimie des nitrènes pour l’amination de liens C-H
1. 1. La chimie des nitrènes
1. 1. a. Fonctionnalisation de liens C-H
Lorsqu’un étudiant en chimie suit ses premiers cours d’introduction en chimie organique, on lui enseigne qu’il existe des fonctionnalités réactives les unes vis-à-vis des autres et que les alcanes n’en font pas réellement partie. Cette impression d’ « inutilité » ou plus justement, d’ « inertie » de la liaison carbone-hydrogène a persisté assez longtemps dans le monde des chimistes organiciens de synthèse, principalement à cause de l’énergie élevée de cette liaison (entre 80 et 105 kcal/mol). Comme chacun sait, les molécules organiques sont composées de chaînes d’atomes de carbones, décorées d’atomes d’hydrogènes et d’autres atomes comme l’oxygène, l’azote, le soufre ou le phosphore (que l’on nomme hétéroatomes). Les liens C-H sont nombreux et de natures diverses au sein d’une même molécule. Cependant, leur réactivité n’a été exploitée que très récemment (depuis environ une quarantaine d’années). La stratégie de fonctionnalisation d’un lien C-H est intéressante du point de vue synthétique car c’est une méthode directe, qui permet de diminuer le nombre d’étapes d’une synthèse.1 En effet, la transformation d’un lien C-H en groupement fonctionnel GF1 est une voie plus courte (Équation 1) et plus attrayante que la transformation d’un groupement fonctionnel GF2 en GF1, fonctionnalité qui aura dû être installée préalablement (Schéma 1). Cette stratégie est particulièrement intéressante pour le domaine de la synthèse totale de molécules complexes,
1 Godula, K.; Sames, D. Science 2006, 312, 67.
2 Gutekunst, W. R.; Baran, P. S. Chem. Soc. Rev. 2011, 40, 1976. 3 Dick, A. R.; Sanford, M. S. Tetrahedron 2006, 62, 2439.
lorsqu’il s’agit de développer des voies de synthèse économiques en atomes et comportant le minimum d’étapes.2
Schéma 1. Stratégie traditionnelle en synthèse organique
Cependant, étant donné le grand nombre de liens C-H présents sur les structures organiques, cette stratégie de fonctionnalisation directe n’est viable que si elle est parfaitement sélective vis-à-vis du lien C-H visé. De manière plus générale, cette stratégie n’est intéressante que si elle laisse intactes les fonctionnalités environnantes au lien C-H ciblé.1
Il existe deux méthodes complémentaires pour la fonctionnalisation des liens C-H : l’activation de ce dernier (c’est-à-dire le clivage) par un complexe métallique, et l’utilisation d’intermédiaires organométalliques d’énergie élevée (carbènes ou nitrènes métalliques). Ces deux méthodes diffèrent par leurs mécanismes, qui peuvent être illustrés de manière simple, au Schéma 2.
2 Gutekunst, W. R.; Baran, P. S. Chem. Soc. Rev. 2011, 40, 1976.
C H C GF1 (1)
C GF2 C GF1
Schéma 2. Mécanismes de sphère interne et externe de fonctionnalisation de lien C-H
Le premier illustré, mécanisme de « sphère interne », implique le clivage du lien C-H par un complexe métallique en premier lieu, puis la fonctionnalisation de l’intermédiaire formé. Le mécanisme de « sphère externe » repose, quant à lui, sur la formation d’un intermédiaire métallique possédant un degré d’oxydation élevé et un ligand prêt à réagir; puis, sur la réaction de ce ligand activé avec le lien C-H visé.3 Ces deux mécanismes étant très différents, la sélectivité, et plus précisément, la chimiosélectivité (on entend ici, sélectivité dans la nature du lien C-H) des transformations dépend fortement du mécanisme qui est impliqué. Sans exposer toutes les conditions réactionnelles qui ont été développées pour des transformations empruntant un mécanisme de « sphère interne » (que l’on appelle communément activation de liens C-H), nous nous contenterons de dire que ces méthodes sont complémentaires en sélectivité des méthodes empruntant un mécanisme de « sphère externe ».4
3 Dick, A. R.; Sanford, M. S. Tetrahedron 2006, 62, 2439.
4 Pour quelques revues choisies sur l’activation de liens C-H par des métaux de transition, voir : a) Shilov, A. E.; Shul'pin, G. B. Chem. Rev. 1997, 97, 2879 ; b) Crabtree, R. H. J. Chem.
Soc., Dalton Trans. 2001, 2437 ; c) Labinger, J. A.; Bercaw, J. E. Nature 2002, 417, 507 ; d)
Kakiuchi, F.; Chatani, N. Adv. Synth. Catal. 2003, 345, 1077 ; e) Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. Eur. J. 2010, 16, 2654 ; f) Wencel-Delord, J.; Droge, T.; Liu, F.; Glorius, F. Chem. Soc. Rev. 2011, 40, 4740 ; g) Qiu, G.; Wu, J.
Org. Chem. Front. 2015, 2, 169 ; h) Yang, L.; Huang, H. Chem. Rev. 2015, 115, 3468.
C H M C M C X
Mécanisme de "sphère interne"
M M X C H C XH
Mécanisme de "sphère externe"
X est un carbone ou un hétéroatome.
1. 1. b. L’amination de liens C-H
L’amination de liens C-H, c’est-à-dire la transformation d’un lien C-H en lien C-N, est une réaction qui a suscité beaucoup d’intérêt de la part des chimistes de synthèse. D’une part, l’azote est un élément omniprésent dans les molécules naturelles, ainsi que dans beaucoup de molécules synthétiques possédant une activité biologique. D’autre part, les méthodes permettant de générer des liens carbone-azote, quoique nombreuses, supposent la présence de groupements fonctionnels réactifs sur les substrats de départ. Parmi ces méthodes, on peut citer les plus employées : la substitution nucléophile utilisant un nucléophile azoté,5 l’amination réductrice,6 l’hydroamination d’alcènes ou d’alcynes7 ou encore la réaction de couplage de Buchwald-Hartwig.8 L’une des méthodes de choix pour effectuer une réaction d’amination est l’insertion d’une espèce nitrène dans un lien carbone-hydrogène. Aussi utile qu’elle soit, cette stratégie synthétique n’a pas été aussi étudiée et développée que ses homologues oxygénées et carbonées.
1. 1. c. Les nitrènes
Les nitrènes sont des composés organiques neutres possédant un atome d’azote monovalent. Ce sont des espèces hautement réactives possédant un caractère électrophile car elles ne possèdent que six électrons de valence (deux électrons provenant d’une liaison sigma, deux autres constituant son doublet non-liant, et deux autres électrons).9 Ces espèces chimiques peuvent donc se trouver dans deux états quantiques différents, se trouvant en équilibre. Dans
5 a) Marcotullio, M.; Campagna, V.; Sternativo, S.; Costantino, F.; Curini, M. Synthesis 2006,
2006, 2760 ; b) Motokura, K.; Nakagiri, N.; Mizugaki, T.; Ebitani, K.; Kaneda, K. J. Org. Chem. 2007, 72, 6006 ; c) Green, J. E.; Bender, D. M.; Jackson, S.; O'Donnell, M. J.;
McCarthy, J. R. Org. Lett. 2009, 11, 807.
6 Alinezhad, H.; Yavari, H.; Salehian, F. Curr. Org. Chem. 2015, 19, 1021.
7 Muller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795. 8 Correa, A.; Bolm, C. Top. Organomet. Chem. 2013, 46, 55.
9 Moody, C. J. (1991). Oxidation by nitrene insertion. Dans Trost, B. M. ; Fleming, I.
l’état singulet, tous les électrons sont appariés dans trois orbitales moléculaires alors que dans l’état triplet, on retrouve deux électrons non-appariés (Figure 1). Pour les nitrènes libres, c’est l’état triplet qui est l’état fondamental.10 Ces configurations électroniques distinctes ont une incidence très importante sur la réactivité de l’espèce nitrène considérée. En effet, les nitrènes à l’état singulet réagissent dans des réactions impliquant le mouvement de deux électrons, typiquement des réactions d’insertion dans des liens C-H, des réarrangements, ou d’aziridination. Les nitrènes à l’état triplet, quant à eux, réagissent comme des diradicaux, typiquement dans des réactions d’aziridination, d’abstraction radicalaire d’hydrogène ou encore de réarrangement (Schéma 3).
Figure 1. Structures et configurations électroniques des états singulet et triplet des nitrènes
10 Vyas, S. ; Winter, A. H. ; Hadad, C. M. (2013). Theory and computation in the study of nitrenes and their excited-state photoprecursors. Dans Falvey, D. E ; Gudmundstdottir, D.
Nitrenes and Nitrenium Ions (p. 33-76). Hoboken, NJ, USA : John Wiley & Sons, Inc.
R N R N
État singulet État triplet
px
Schéma 3. Réactivité des nitrènes singulet et triplet
L’équilibre entre ces deux états est fortement dépendant du substituant sur le nitrène, puisque la différence d’énergie entre les deux orbitales frontières dépend directement du substituant. Ainsi, les substituants électroattracteurs tendent à faire diminuer l’énergie de l’état singulet. Les nitrènes de carbamoyle, de sulfamoyle et de phosphoryle ont leur état singulet assez bas en énergie même si l’état fondamental est l’état triplet (Figure 2).11 Les réactions parasites, les réarrangements notamment, sont minimisées pour ces nitrènes, favorisant ainsi l’insertion de liens C-H.9 L’étude de ces types de nitrènes a permis le développement de la plupart des méthodologies efficaces d’amination de liens C-H à ce jour, que ce soit en version intra- ou intermoléculaire.
11 Gritsan, N. P. (2013). Properties of carbonyl nitrenes and related acyl nitrenes. Dans Falvey, D. E ; Gudmundstdottir, D. Nitrenes and Nitrenium Ions (p. 481-548). Hoboken, NJ, USA : John Wiley & Sons, Inc.
N Aziridination R' R R 2 R3 R1 N R2 R3 R1 R R' R R' H R R' HN R2 R1 R3 Insertion dans un lien C-H N R2 R1 R3 Réarrangement de type Curtius N R2 R3 R1 Aziridination R' R N R2 R3 R1 R R' Réarrangement de type Curtius Abstraction radicalaire d'hydrogène H NH2 R2 R3 R1 Nitrène singulet Nitrène triplet
Figure 2. Structures des nitrènes de carbamoyle, de sulfamoyle et de phosphoryle
L’utilisation de certains métaux de transition permet de moduler la réactivité des nitrènes. Il a été montré, par l’étude par modélisation théorique de la fonctionnelle de la densité (DFT) des mécanismes de certaines réactions, que des nitrènes métalliques étaient impliqués dans les réactions d’insertion de liens C-H.12,13,14,15,16,17,18 Certains intermédiaires nitrènes métalliques ont pu être isolés19,20,21et d’autres observés par des techniques de spectrométrie de masse utilisant l’ionisation par électronébuliseur (ESI-MS).22,23,24,25 L’intéraction entre une orbitale π du métal et une orbiale π de l’azote serait à l’origine de la stabilisation de l’état singulet, dans le cas du rhodium.12 Ces nitrènes métalliques peuvent être représentés comme à la figure 3.
12 Lin, X.; Zhao, C.; Che, C. M.; Ke, Z.; Phillips, D. L. Chem. Asian J. 2007, 2, 1101. 13 Lin, X.; Che, C. M.; Phillips, D. L. J. Org. Chem. 2008, 73, 529.
14 Harvey, M. E.; Musaev, D. G.; Du Bois, J. J. Am. Chem. Soc. 2011, 133, 17207. 15 Li, J.; Wu, C.; Zhang, Q.; Yan, B. Dalton Trans. 2013, 42, 14369.
16 Zhang, Q.; Wu, C.; Zhou, L.; Li, J. Organometallics 2013, 32, 415. 17 Zhang, X.; Xu, H.; Zhao, C. J. Org. Chem. 2014, 79, 9799.
18 Ren, Q.; Shen, X.; Wan, J.; Fang, J. Organometallics 2015, 34, 1129.
19 Au, S.-M.; Huang, J.-S.; Yu, W.-Y.; Fung, W.-H.; Che, C.-M. J. Am. Chem. Soc. 1999, 121, 9120.
20 Liang, J. L.; Yuan, S. X.; Huang, J. S.; Che, C. M. J. Org. Chem. 2004, 69, 3610.
21 Guo, Z.; Guan, X.; Huang, J. S.; Tsui, W. M.; Lin, Z.; Che, C. M. Chem. Eur. J. 2013, 19, 11320.
22 Perry, R. H.; Cahill, T. J., 3rd; Roizen, J. L.; Du Bois, J.; Zare, R. N. Proc. Natl. Acad. Sci.
U. S. A. 2012, 109, 18295.
23 Liu, Y.; Guan, X.; Wong, E. L.; Liu, P.; Huang, J. S.; Che, C. M. J. Am. Chem. Soc. 2013,
135, 7194.
24 Bagchi, V.; Paraskevopoulou, P.; Das, P.; Chi, L.; Wang, Q.; Choudhury, A.; Mathieson, J. S.; Cronin, L.; Pardue, D. B.; Cundari, T. R.; Mitrikas, G.; Sanakis, Y.; Stavropoulos, P. J.
Am. Chem. Soc. 2014, 136, 11362.
25 Liu, Y.; Chen, G. Q.; Tse, C. W.; Guan, X.; Xu, Z. J.; Huang, J. S.; Che, C. M. Chem. Asian
J. 2015, 10, 100. O N O R O S N R O O O P N R O OR' Nitrène
de carbamoyle de sulfamoyleNitrène
Nitrène de phosphoryle
Figure 3. Structures d'un nitrène métallique
Comme cela a déjà été mentionné plus haut, les nitrènes sont des espèces assez instables, il est donc nécessaire de les générer in situ. Il existe pour cela un certain nombre de précurseurs, permettant, en présence de certains complexes métalliques, de générer les intermédiaires nitrènes métalliques correspondants (Schéma 4).26
Schéma 4. Les précurseurs de nitrènes métalliques
Les haloamines, ne génèrant comme sous-produits que des sels inorganiques, ont été assez peu développées dans des méthodologies de synthèse. La bromamine-T, en particulier, a été
26 Che, C. M.; Lo, V. K. Y.; Zhou, C. Y. (2014). Oxidation by metals (Nitrene). Dans Knochel, P. (dir.), Comprehensive Organic Synthesis II (2ème édition, volume 7, p. 26-85). Amsterdam : Elsevier. R N M R N M R N M R N N N M Azotures M R N X Y M X = Cl-, Br -Y = Na+, NR' 4+ Haloamines R N I R' Iminoiodinanes M Base R H N O S R' O O Composés N-sulfonyloxy N2 IR' R'SO3 -Y+X
-utilisée dans des réactions d’amination de liens C-H27,28,29 et d’aziridination27,30,31 intermoléculaires (Schéma 5).
Schéma 5. Réactions d'amination et d'aziridination intermoléculaires avec la bromamine-T
Les composés sulfonyloxy ont été identifiés, il y a une cinquantaine d’années, comme précurseurs de nitrènes libres, dans des réactions d’insertion de liens C-H32 et d’aziridination33 intermoléculaires. La déprotonation du N-nosyloxycarbamate d’éthyle par une base est à l’origine de l’α-élimination menant au nitrène de carbamoyle libre (Équation 2). Depuis, le groupe de Lebel a développé des méthodologies utilisant la catalyse organométallique avec ce type de réactifs, permettant l’amination intra- et intermoléculaire de liens C-H34,35,36 ainsi que l’aziridination d’alcènes intermoléculaire.34,37 Nous aurons le loisir de nous étendre plus longuement sur le sujet dans la suite de ce chapitre et de cette thèse.
27 Chanda, B. M.; Vyas, R.; Bedekar, A. V. J. Org. Chem. 2001, 66, 30.
28 Harden, J. D.; Ruppel, J. V.; Gao, G. Y.; Zhang, X. P. Chem. Commun. 2007, 4644.
29 Wang, H.; Li, Y.; Wang, Z.; Lou, J.; Xiao, Y.; Qiu, G.; Hu, X.; Altenbach, H.-J.; Liu, P.
RSC Adv. 2014, 4, 25287.
30 Vyas, R.; Gao, G. Y.; Harden, J. D.; Zhang, X. P. Org. Lett. 2004, 6, 1907. 31 Gao, G. Y.; Harden, J. D.; Zhang, X. P. Org. Lett. 2005, 7, 3191.
32 Lwowski, W.; Maricich, T. J. J. Am. Chem. Soc. 1965, 87, 3630. 33 Seno, M.; Namba, T.; Kise, H. J. Org. Chem. 1978, 43, 3345.
34 Lebel, H.; Huard, K.; Lectard, S. J. Am. Chem. Soc. 2005, 127, 14198. 35 Lebel, H.; Huard, K. Org. Lett. 2007, 9, 639.
36 Huard, K.; Lebel, H. Chem. Eur. J. 2008, 14, 6222.
37 Lebel, H.; Lectard, S.; Parmentier, M. Org. Lett. 2007, 9, 4797.
S N O O Br Na Bromamine-T [M] cat. NHTs [M] cat. N Ts 1 2
Bien que les azotures38 aient été les premières espèces identifiées comme capables de générer des nitrènes libres par décomposition (par chauffage ou par photoexcitation),9,26 ce n’est qu’assez récemment qu’ils ont été utilisés très efficacement comme précurseurs de nitrènes métalliques pour l’amination de liens C-H, l’aziridination d’alcènes ou encore l’amination de composés soufrés.39 Par décomposition, c’est-à-dire en l’absence d’additifs, l’azoture perd une molécule de diazote, seul sous-produit, pour générer le nitrène. C’est cette production de diazote qui confère à certains azotures organiques, notamment les plus légers, un caractère explosif.38 L’équation 3 illustre un exemple de sulfylimination intermoléculaire énantiosélective de sulfures à partir d’azotures de carbamoyle catalysée par un complexe de ruthénium chiral.40
38 Pour une revue récente sur les azotures organiques voir : Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem. Int. Ed. 2005, 44, 5188.
39 Pour une revue couvrant les avancées, jusqu’à 2010, sur les réactions de transfert d’azote catalysées par des métaux de transition à partir d’azotures, voir : Driver, T. G. Org. Biomolec.
Chem. 2010, 8, 3831.
40 Tamura, Y.; Uchida, T.; Katsuki, T. Tetrahedron Lett. 2003, 44, 3301.
NO2 S O O O H N O O N-Nosyloxycarbamate d'éthyle Base N O O O S O O NO2 -BH (2) NO2 S NO2 S N O O
*
5, 99%, 99% ee N3 O O Cl3C (OC)Ru(salen) 4 (2 mol %) TM 4 Å, DCM, t.a., 24h (3) N2(g) CCl3 3Figure 4. Structure du catalyseur (OC)Ru(salen) 4 de Katsuki
Les derniers précurseurs de nitrènes décrits au Schéma 4 sont les iminoiodinanes (pouvant aussi être considérés comme des ylures de iodonium). Le sous-produit généré par la formation du nitrène métallique est une espèce iodoarène. Les premières applications de ces précurseurs de nitrènes pour l’amination de liens C-H41,42,43 et l’aziridination44 ont été rapportées dans les années 1980 et marque le début des réactions de transfert de nitrènes catalysées par des métaux de transition.45 L’équation 4 illustre un des premiers exemples d’aziridination énantiosélective catalysée par un catalyseur bis(oxazoline) de cuivre (I), par le groupe d’Evans.46
41 Breslow, R.; Gellman, S. H. J. Chem. Soc., Chem. Commun. 1982, 1400. 42 Breslow, R.; Gellman, S. H. J. Am. Chem. Soc. 1983, 105, 6728.
43 Mahy, J. P.; Bedi, G.; Battioni, P.; Mansuy, D. Tetrahedron Lett. 1988, 29, 1927.
44 a) Mansuy, D.; Mahy, J.-P.; Dureault, A.; Bedi, G.; Battioni, P. J. Am. Chem. Soc. 1984, 1161. b) Mahy, J.-P.; Bedi, G.; Battioni, P.; Mansuy, D. J. Chem. Soc., Perkin Trans. 2 1988, 1517.
45 Pour des revues sur les réactions d’aziridination et d’amination de liens C-H à partir d’iminodiodinanes, voir : a) Dauban, P.; Dodd, R. H. Synlett 2003, 1571. b) Chang, J. W. W.; Ton, T. M. U.; Chan, P. W. H. Chem. Rec. 2011, 11, 331.
46 Evans, D. A.; Faul, M. M.; Bilodeau, M. T.; Anderson, B. A.; Barnes, D. M. J. Am. Chem.
Soc. 1993, 115, 5328. N N O O Ph Ph Ru CO (R) (R) CO2Me N Ts H CO2Me H (4) PhI NTs (2 équiv.) O N N O Ph Ph CuOTf (5 mol %) (6 mol %) PhH, TM 4Å, 24h, t.a. 6 7
Ayant brièvement décrit les sources de nitrènes qui ont servi au développement de transformations impliquant le transfert de nitrènes métalliques, nous sommes désormais en mesure de nous concentrer sur les réactions d’amination de liens C-H.
1. 2. Les réactions d’amination de liens C-H utilisant des nitrènes métalliques
Il est important de mentionner, qu’à ce jour, un certain nombre de méthodologies permettant d’effectuer cette transformation de manières intra- et intermoléculaire (Schéma 6) ont été développées.26 Cependant, l’emphase sera mise sur les méthodologies de transformations intramoléculaires, cette thèse concernant exclusivement le développement de réactions d’amination intramoléculaire.
Schéma 6. Réactions d'insertion de nitrènes dans des liens C-H de manières intra- et
intermoléculaires
1. 2. a. Les travaux pionniers
Les réactions d’amination de liens C-H ayant lieu grâce à des intermédiaires azotés de degré d’oxydation et de réactivité élevés (comme les nitrènes) peuvent être vues comme homologues
R Y H X N Z n HN Y X R n Z R1 R2 H X N Z Y R3 R1 R2 HN X Y R3 Z X = C(O), SO2, P(O)OR4 Y = O, N, C Z = IAr, N2, OSO2R, Cl, Br Conditions Conditions
aux réactions d’hydroxylation d’alcanes ayant lieu chez le vivant. Les cytochromes P450 constituent une classe d’enzymes capables de catalyser ces transformations.47 Le site actif de ces enzymes étant une porphyrine de fer, de nombreuses méthodologies d’hydroxylation utilisant des métalloporphyrines synthétiques ont été développées.48,49,50 Ainsi, avec du recul, il n’est pas si surprenant de constater que les premiers travaux concluants sur l’amination de liens C-H aient été réalisés en présence de métalloporphyrines. Breslow et Gellman, ont montré, au début des années 1980, qu’il était possible de réaliser l’insertion d’une espèce nitrène (générée en présence de porphyrines de fer, de cobalt ou bien d’un dimère de rhodium à partir d’un iminoiodinane) dans un lien C-H de manières intra- et intermoléculaires,.41,42 La réaction intermoléculaire (Équation 5) montre que les porphyrines de fer(III) et de cobalt(III) sont aptes à promouvoir l’insertion d’un nitrène dans un lien C-H, bien que les rendements obtenus soient relativement faibles. En revanche, la réaction intramoléculaire, à partir d’un précurseur iminoiodinane, est efficacement catalysée par les porphyrines de fer (III) et par le dimère de rhodium tétraacétate (Équation 6), donnant le sulfonamide cyclique 9 dans de bons rendements.
47 Pour des revues sur les cytochromes P450 et les réactions qu’ils sont capables de catalyser, voir : a) Ortiz de Montellano, P. R. Chem. Rev. 2010, 110, 932. b) Denisov, I. G.; Makris, T. M.; Sligar, S. G.; Schlichting, I. Chem. Rev. 2005, 105, 2253. c) Sono, M.; Roach, M. P.; Coulter, E. D.; Dawson, J. H. Chem. Rev. 1996, 96, 2841.
48 Mansuy, D. Coord. Chem. Rev. 1993, 125, 129.
49 Che, C. M.; Lo, V. K.; Zhou, C. Y.; Huang, J. S. Chem. Soc. Rev. 2011, 40, 1950. 50 Lu, H.; Zhang, X. P. Chem. Soc. Rev. 2011, 40, 1899.
PhI NTs H Catalyseur (5 mol %) DCM : Cyclohexane 1 : 1 NHTs Catalyseur Mn(TPP)Cl Fe(TPP)Cl Rendement 6,8% 3,1% excès 1,0 équiv. (5) 8 SO2 NH H SO2
N IPh Catalyseur (5 mol %)
MeCN Catalyseur Mn(TPP)Cl Fe(TPP)Cl Rh2(OAc)4 Rendement 16% 77% 86% 1,0 équiv. (6) 9
Figure 5. Structures des chlorures de tétraphénylporphyrines de fer et de manganèse et du
dimère de rhodium tétraacétate
Mansuy et ses collaborateurs ont observé, lors du développement de méthodes d’aziridination d’alcènes catalysées par des métalloporphyrines, qu’une porphyrine de manganèse permettait d’effectuer l’amination intermoléculaire de liens C-H allyliques de manière chimiosélective.51 Cependant, ce sont les travaux du groupe de Müller, en analogie avec la chimie des carbènes, qui ont permis l’essor des premières méthodologies efficaces utilisant des iminoiodinanes comme précurseurs de nitrènes et des dimères de rhodium comme catalyseurs.52,53 Il est important de noter que la plupart des réactions d’amination ou d’aziridination dont il a été question dans le présent paragraphe sont des transformations intermoléculaires et que le substrat à insérer est généralement utilisé en excès.
1. 2. b. Les iminoiodinanes en tant que précurseurs de nitrènes
De façon contemporaine à Müller et ses collaborateurs, le groupe de Che, a développé plusieurs méthodologies utilisant des iminoiodinanes comme précurseurs de nitrènes métalliques. Axant ses recherches sur la chimie des porphyrines, ce groupe a développé des
51 Mahy, J. P.; Bedi, G.; Battioni, P.; Mansuy, D. Tetrahedron Lett. 1988, 29, 1927.
52 Nägeli, I.; Baud, C.; Bernardinelli, G. E.; Jacquier, Y.; Moraon, M.; Müller, P. Helv. Chim.
Acta 1997, 80, 1087.
53 Müller, P.; Baud, C.; Nägeli, I. J. Phys. Org. Chem. 1998, 11, 597.
N N N N Ph Ph Ph Ph N N N N Ph Ph Ph Ph Fe Cl Mn Cl III III Fe(TPP)Cl Mn(TPP)Cl O Rh Rh O O O O O O O Rh2(OAc)4
méthodes d’amination de liens C-H intermoléculaires induites par des porphyrines de ruthénium et de manganèse, dont certaines sont chirales.54,55 Toutefois, ce n’est qu’au début des années 2000, qu’une avancée majeure dans le domaine a été faite, éliminant le problème posé par la préparation et l’utilisation d’iminoiodinanes. En effet, les iminoiodinanes se décomposent lentement à l’air à température ambiante, donnant l’espèce azotée primaire correspondante, et de manière explosive à plus haute température.42 Ils doivent donc être entreposés sous atmosphère inerte à basse température. De plus, des problèmes de solubilité de ces composés dans les solvants usuels sont souvent observés, sans compter les problèmes d’isolement des dits composés ainsi que des soucis de reproductibilité des procédures de préparation.45a
Comme il a été mentionné plus haut, en 2000, le groupe de Che, a mis au point une méthode de génération in situ de l’espèce iminoiodinane, à partir de plusieurs sulfonamides et de l’oxydant diacétoxyiodobenzène PhI(OAc)2, permettant l’amination de liens C-H intermoléculaire catalysée par des porphyrines de ruthénium et de manganèse (Équation 7).56
54 Au, S.-M.; Huang, J.-S.; Yu, W.-Y.; Fung, W.-H.; Che, C.-M. J. Am. Chem. Soc. 1999, 121, 9120.
55 Zhou, X.-G.; Yu, X.-Q.; Huang, J.-S.; Che, C.-M. Chem. Commun. 1999, 2377. 56 Yu, X.-Q.; Huang, J.-S.; Zhou, X.-G.; Che, C.-M. Org. Lett. 2000, 2, 2233.
H
PhI(OAc)2 TsNH2
[Mn(F20TPP)Cl] (1 mol %)
DCM, 40 °C, 2h
NHTs
1,00 équiv. 1,25 équiv. 1,50 équiv.
(7)
Figure 6. Structure du ligand meso-tetrakis(pentafluorophényl)porphyrine F20TPP
Très peu de temps après, le groupe de Du Bois, a rapporté la même découverte, permettant cette fois-ci, l’amination intramoléculaire de précurseurs carbamates primaires, catalysée par des dimères de rhodium (Équation 8).57 Ce même groupe a développé la réaction analogue avec des dérivés sulfamates, également catalysée par des dimères de rhodium.58 Il a été observé que la transformation s’effectuait de manière stéréospécifique, cependant, nous n’exposerons les aspects mécanistiques de ces transformations qu’ultérieurement.
Le développement d’un dimère de rhodium possédant des ligands carboxylates bidentates, le ligand esp, nommé ainsi en l’honneur de la doctorante qui l’a conçu, Christine Espino, a permis d’améliorer grandement l’activité catalytique du dimère de rhodium et d’augmenter sa
57 Espino, C. G.; Du Bois, J. Angew. Chem. Int. Ed. 2001, 40, 598.
58 Espino, C. G.; Wehn, P. M.; Chow, J.; Du Bois, J. J. Am. Chem. Soc. 2001, 123, 6935.
N HN N NH F20TPP F F F F F F F F F F F F F F F F F F F F O O NH2 PhI(OAc) 2 O HN O H 10, 1,0 équiv. 1,4 équiv. Rh2(tpa)4 (5 mol %) MgO (2,3 équiv.) DCM, 40 °C (8) 11, 74%
robustesse.59 Des réactions énantiosélectives utilisant des dimères de rhodium portant des ligands carboxylates chiraux ont été développées dans la foulée de ces avancées (Tableau 1).60,61 Notamment, le catalyseur Rh2[(S)-nap]4, possédant un ligand carboxamidate chiral, développé par le groupe de Du Bois, permet l’amination énantiosélective très efficace des sulfamates.62
Tableau 1. Influence du dimère de rhodium sur le rendement et l'excès énantiomère de la
réaction d'amination de liens C-H intramoléculaire de sulfamates
59 Espino, C. G.; Fiori, K. W.; Kim, M.; Du Bois, J. J. Am. Chem. Soc. 2004, 126, 15378. 60 Fruit, C.; Müller, P. Helv. Chim. Acta 2004, 87, 1607.
61 Hashimoto, S.; Yamawaki, M.; Kitagaki, S.; Anada, M. Heterocycles 2006, 69, 527. 62 Zalatan, D. N.; Du Bois, J. J. Am. Chem. Soc. 2008, 130, 9220.
R
O S H2N
O O Oxydant, BaseCatalyseur
HN S O R
O O
*
Solvant, TempératureEntrée Catalyseur Oxydant Base Rendement % ee
1 2 3 4 5 6 R Ph Me Me Me Ph Ph Rh2(OAc)4 (2 mol %) PhI(OAc)2 (1,1 équiv.) MgO (2,3 équiv.) 90%a -a DCM, 40 °C; b DCM, TM 4Å, - 20 °C; c Benzène, 23 °C; d DCM, TM 3Å, 23 °C. Rh2(OPiv)4 (1 mol %) PhI(OAc)2 (1,1 équiv.) MgO (2,3 équiv.) 15%a -Rh2(esp)2 (1 mol %) PhI(OAc)2 (1,1 équiv.) MgO (2,3 équiv.) 90%a -Rh2[(S)-nttl]4 (3,5 mol %) PhI(OAc)2 (1,5 équiv.) MgO (2,5 équiv.) 97%b 21% Rh2[(S)-tfpttl]4 (2 mol %) PhI(OAc)2 (1,1 équiv.) MgO (2,3 équiv.) 87%c 10% Rh2[(S)-nap]4 (2 mol %) PhIO (1,2 équiv.) - 85%d 92%
Figure 7. Structures de dimères de rhodium utilisés dans l'amination de liens C-H
intramoléculaire à partir de sulfamates primaires
En comparant les produits cycliques obtenus à partir des composés carbamates et sulfamates, on constate que la taille du cycle azoté généré est différente. En effet, les carbamates forment préférentiellement des cycles à cinq chaînons, les oxazolidinones, alors que les sulfamates (et les composés sulfonés en général) donnent des cycles à six chaînons, les oxathiazinanes. La raison à cela est géométrique : les liaisons S-O et S-N sont assez longues (1.58 Å) et l’angle O-S(O2)-N mesure 103 ° pour un sulfamate primaire. Ces caractéristiques sont très proches de celles de l’hétérocycle correspondant.58 D’autres hétérocycles peuvent également être formés en utilisant la méthodologie de Du Bois (dérivé aminé primaire en présence d’une source d’iode hypervalent et d’un catalyseur dimère de rhodium), comme cela est illustré sur le schéma 7.57,63,58,60,64,65
63 Kim, M.; Mulcahy, J. V.; Espino, C. G.; Du Bois, J. Org. Lett. 2006, 8, 1073. 64 Kurokawa, T.; Kim, M.; Du Bois, J. Angew. Chem. Int. Ed. 2009, 48, 2777. 65 Olson, D. E.; Du Bois, J. J. Am. Chem. Soc. 2008, 130, 11248.
Rh Rh O O O O O O O O R R R R R = Me
R = tBu RhRh22(OAc)(OPiv)44
R = N O O Rh2[(S)-nttl]4 R = N O O F F F F Rh 2[(S)-tfpttl]4 Rh Rh O O O O O O O O Rh2(esp)2 N O HN Ts Rh Rh Rh2[(S)-nap]4
Schéma 7. Hétérocycles accessibles par la méthode d'amination intramoléculaire de Du Bois
La méthodologie développée par Du Bois mérite qu’on s’attarde quelque peu sur son fonctionnement et ses applications car c’est celle qui a été la plus largement utilisée parmi les méthodologies intramoléculaires disponibles. Comme cela a été mentionné, le précurseur de nitrène, l’iminoiodinane est généré in situ (bien qu’il n’ait jamais été observé) et permet ensuite la formation du nitrène de rhodium, suivi de son insertion dans un lien C-H (Schéma 8). Il est important de noter qu’une fois le nitrène de rhodium formé, celui-ci, pouvant se trouver dans l’état triplet ou singulet, peut soit s’insérer dans un lien C-H, soit faire l’abstraction radicalaire d’un hydrogène et donc donner le produit de départ. L’utilisation d’un léger excès d’oxydant permet donc d’obtenir de meilleures conversions. Cependant, lors de la formation de l’iminoiodinane, deux équivalents d’acide acétique sont formés, c’est pourquoi un peu plus de deux équivalents de base MgO sont nécessaires au bon fonctionnement de la réaction.
Schéma 8. Fonctionnement du système réactionnel de Du Bois
En ce qui concerne l’étendue réactionnelle de la transformation, elle s’étend aux liens C-H que l’on qualifie d’activés, c’est-à-dire les liens possédant en α une fonctionnalité ou une
R X H2N Y R X HN Y X = O, Y = O X = NR', Y = O X = NH, Y = NR' R X Y S H2N O O HN X Y S O O X = CH2, Y = O X = CH2, Y = NBoc X = CH2, Y = CH2 X = NR', Y = O cat. Rh2(O2CR'')4 PhI(OAc)2 MgO Solvant R NH2 PhI(OAc)2 2 AcOH R N IPh PhI R N M R1 R2 H R1 R2 HN R
abstraction radicalaire d'hydrogène [M]