THESE
Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac Secteur de recherche : Chimie théorique, physique, analytique et industriel
Présentée par : Charly Mve Mfoumou
Piégeage du dioxyde de carbone sur solides à base de zéolithe faujasite X : adsorption seul, en mélange binaire
et/ou en présence d'eau ; étude en thermodésorption
Directeur(s) de Thèse : Samuel Mignard, Thomas Belin Soutenue le 05 décembre 2012 devant le jury
Jury :
Président Christine Travers Professeur - Institut français du pétrole Rapporteur Cécile Vallières Professeur - Université de Nancy
Rapporteur Igor Bezverkhyy Chargé de recherche CNRS - Université de Dijon Membre Samuel Mignard Professeur des Universités - Université de Poitiers Membre Thomas Belin Maître de conférences - Université de Poitiers
Membre François Jérôme Directeur de recherche CNRS - Université de Poitiers
Pour citer cette thèse :
Charly Mve Mfoumou. Piégeage du dioxyde de carbone sur solides à base de zéolithe faujasite X : adsorption seul,
en mélange binaire et/ou en présence d'eau ; étude en thermodésorption [En ligne]. Thèse Chimie théorique,
physique, analytique et industriel. Poitiers : Université de Poitiers, 2012. Disponible sur Internet <http://theses.univ-poitiers.fr>
Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
(Faculté des Sciences Fondamentales et Appliquées) (Diplôme National - Arrêté du 7 août 2006)
Ecole Doctorale : Science pour l’Environnement Gay Lussac.
Secteur de Recherche : Chimie Théorique, Physique, Analytique et Industriel
Présentée par :
MVE MFOUMOU Charly
************************
PIEGEAGE DU DIOXYDE DE CARBONE SUR SOLIDES A BASE DE ZEOLITHE FAUJASITE X :
ADSORPTION SEUL, EN MELANGE BINAIRE ET / OU EN PRESENCE D’EAU; ETUDE EN THERMODESORPTION
************************ Directeurs de Thèse :
Samuel MIGNARD, Professeur, Université de Poitiers
Thomas BELIN, Maître de Conférences, Université de Poitiers ************************
Soutenue le 05 décembre 2012 devant la Commission d’Examen
************************
JURY
Cécile VALLIERES, Professeur, LRGP – Nancy Rapporteurs Igor BEZVERKHYY, Chargé de Recherches CNRS, ICB-Dijon
François JEROME, Chargé de Recherches CNRS, Université Poitiers
Samuel MIGNARD, Professeur, Université de Poitiers Examinateurs Thomas BELIN, Maître de Conférences, Université de Poitiers
!"# $ % & '( () * % + + # , + + + # % - # . % ) / + & +0 *1 2 3 4 +0 2 52 3 + + + . % 6 7 %% 8 ++ % + . % & ++ ) / + ( *9' # % : ; + # ++ % % , < + - ) / % $ + + 9*55 ' ' $ ='>9' ?@44 ++ % , $ + A %% ) / + $ ; B /' 2 ' ++ % % + % , < - ) ' # , + 1 * C# C + - # % A . % + # + . + + & ) +- + + : ( ='5 # + + - # % + + ) % & + $ # # + % . % # $ + : . # % $ $ % + % + & ) / % + . % + * D / C . # # / E # & # 5 +# # * < % & % & ) / + $ * =1 *$ + = 1 & " % : + % . $ % : + + - ) / + + + . % D ; + # 2 # - # # / # ? # < # * # & # # * # * F # G # # 5 # ( # = . $ + & + ) / + + $ & # % + ; +0# # <# # ; 7# +0 * : # ( % <# & # / % % + )
: + % # % + - -> ) / + - :: + : + 9 % . , # # %% % $ % H + I : # , $ + % % + . # %- % ;2 2 ) + , J I" + )
Introduction générale 1
Chapitre 1 : Etude bibliographique……….3
I.
Les Gaz à Effet de Serre (GES) ... 3
1. 1.
Définition des GES ... 3
1. 2.
Sources d'émission des GES ... 3
1. 3.
Effets du réchauffement climatique ... 6
1. 4.
Mesures de lutte contre le réchauffement climatique: Conventions... 8
II.
Techniques de piégeage du CO
2utilisées en industrie... 9
2. 1.
Le piégeage postcombustion du CO
2... 10
2. 2.
Le piégeage précombustion du CO
2... 10
2. 3.
Le piégeage par oxycombustion du CO
2... 11
III.
Généralités sur les phénomènes d’adsorption et de désorption ..
……….12
3. 1.
Phénomène d’adsorption... 12
3. 2.
Phénomène de désorption ... 14
IV.
Etat de l’art de l’adsorption du CO
2sur adsorbants solides ... 15
4. 1.
Les oxydes de métaux ... 15
4. 1. 1. Sites basiques des oxydes de métaux alcalino-terreux et adsorption du CO2……… ...16
4. 1. 2. Influence des caractéristiques des oxydes de métaux pour l’adsorption du CO2... 20
4. 2.
Les zéolithes... 21
4. 2. 1. Généralités ... 21
Sommaire
4. 3.
Les zéolithes de type faujasite (FAU)... 23
4. 3. 1. Les sites cationiques ... 25
4. 3. 2. Les sites protoniques... 26
4. 4.
Adsorption du CO
2sur les zéolithes ... 27
4. 5.
Influence des caractéristiques structurales des zéolithes sur
l’adsorption du CO
2... 29
4. 5. 1. Influence des propriétés basiques des zéolithes et du cation échangeable .. 29
4. 5. 2. Influence de la taille des pores... 31
4. 5. 3. Influence du rapport Si/Al ... 31
4. 6.
Autres facteurs influençant l’adsorption du CO
2sur les zéolithes ... 32
4. 6. 1. Influence des caractéristiques de l’adsorbat ... 32
4. 6. 2. Influence de la formation des carbonates... 32
4. 6. 3. Influence de la présence d’eau ... 33
4. 6. 4. Influence de la pression et de la température... 34
4. 7.
Les mélanges mixtes oxydes/zéolithes ... 35
Chapitre 2 : Partie expérimentale.…...37
I.
Préparations des adsorbants ... 37
1. 1.
Les échanges cationiques ... 37
1. 2.
Les mélanges mécaniques... 37
1. 3.
Les imprégnations d’acétates ... 37
1. 4.
Procédures de calcination et d’activation des adsorbants... 38
II.
Techniques de caractérisations... 40
2. 1.
Analyses élémentaires... 40
2. 3.
Etude texturale par physisorption d’azote... 41
2. 4.
Diffraction de rayons X... 43
2. 5.
Microscopie Electronique à Transmission... 44
2. 6.
Spectroscopie infrarouge à transformée de Fourier ... 45
III.
Expériences d’adsorption et de désorption du dioxyde de
carbone ... 46
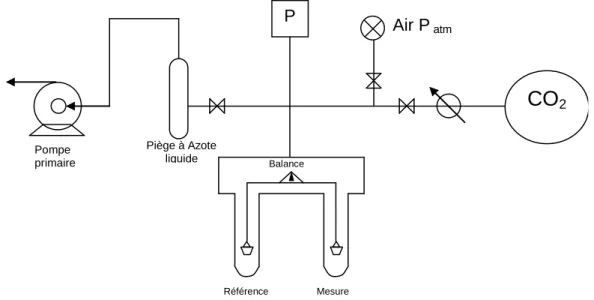
3. 3.
Adsorption en mode statique : technique microgravimétrique... 46
3. 4.
Etude de la désorption programmée en température ... 49
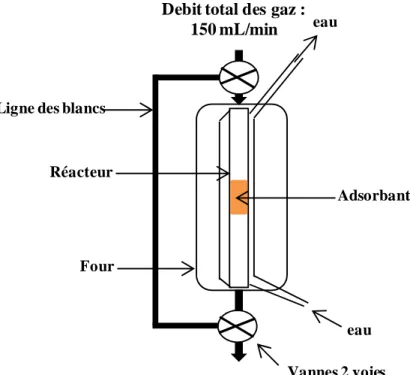
3. 5.
Adsorption / désorption en réacteur dynamique à lit fixe... 50
Chapitre 3 : Résultats et discussions………..56
I.
Caractérisations ... 56
1. 1.
Analyses élémentaires... 56
1. 2.
Apports de la microscopie électronique à transmission... 57
1. 3.
Etude de la structure cristalline des adsorbants ... 59
1. 4.
Etude texturale par physisorption de diazote ... 62
1. 5.
Etude de la déshydration optimale des adsorbants ... 69
1. 6.
Analyses microgravimétriques... 74
1. 6. 1. Etude des capacités d’adsorption du dioxyde de carbone... 74
1. 6. 2. Détermination des chaleurs d’adsorption... 83
II.
Etude des sites d’adsorption du CO
2... 90
2. 1.
Thermodésorption du dioxyde de carbone... 90
2. 1. 1. Sur les zéolithes échangées par des cations bivalents... 93
Sommaire
2. 1. 3. Sur les zéolithes imprégnées ... 104
2. 1. 4. Conclusions... 111
2. 2.
Identification des espèces de CO
2absorbées ... 113
2. 2. 1. Zéolithes échangées par des cations bivalents ... 113
2. 2. 2. Mélanges mécaniques ... 117
2. 2. 3. Effet de la nature de l’oxyde dans les zéolithes imprégnées ... 120
2. 2. 4. Conclusions... 123
III.
Piégeage du CO
2en dynamique à lit fixe ... 124
3. 1.
Adsorption / désorption du dioxyde de carbone ... 124
3. 1. 1. Adsorption du CO2... 124
3. 1. 2. Désorption... 129
3. 2.
Adsorption / désorption du CO
2en présence d’humidité ... 134
3. 2. 1. Adsorption : Influence de l’eau... 134
3. 2. 2. Désorption... 139
3. 2. 3. Influence de l’oxyde dans les mélanges mécaniques... 142
3. 3.
Etude du piégeage du CO
2en mélange gazeux ... 146
3. 3. 1. Adsorption du mélange binaire dioxyde de carbone / méthane : diamètres moléculaires voisins... 146
3. 3. 2. Effet du méthane sur les sites d’adsorption du CO2... 150
3. 3. 3. Adsorption du mélange binaire dioxyde de carbone / méthane en présence d’eau ... 152
3. 3. 4. Influence du mélange méthane / vapeur d’eau sur les sites d’adsorption du CO2... 154
3. 3. 5. Adsorption du mélange binaire dioxyde de carbone / propane : même masse molaire ... 157
3. 3. 7. Adsorption du mélange binaire dioxyde de carbone / propane en présence d’humidité... 164
3. 3. 8. Influence du mélange propane / vapeur d’eau sur les sites d’adsorption du CO2... 167
Chapitre 4 : Conclusions générales et perspectives
……….
168
Page 1
Cette thèse effectuée au sein de l’Institut de Chimie des Milieux et des Matériaux de Poitiers (IC2MP) dans l’équipe des zéolithes et solides apparentés s’inscrit dans le cadre actuel de la protection de l’environnement et la réduction des composés à effet de serre.
Les changements climatiques sont attribués à un certain nombre de molécules gazeuses, appelées gaz à effet de serre (GES), qui par leurs propriétés physiques contribuent au réchauffement de la Terre. Parmi eux, le dioxyde de carbone (CO2) est celui qui présente une
augmentation importante des concentrations dans l’atmosphère, en grande partie due aux activités humaines. En effet, de nos jours, le dioxyde de carbone représente environ 70% des émissions des GES.
La Convention de Rio (1992) et le Protocole de Kyoto (1997) expriment la prise de conscience et l'engagement des institutions internationales et des gouvernements à prendre en charge cette problématique. Rappelons qu'à Kyoto, l'Union Européenne s'est engagée à réduire de 8% les émissions de dioxyde de carbone entre 2008 et 2012. Par conséquent, des réglementations de plus en plus strictes des émissions de dioxyde de carbone dans les secteurs industriels ont été mises en place et la recherche de techniques plus efficaces visant à réduire au maximum les émissions de ce gaz dans l’atmosphère s’est avérée importante.
L’amélioration des procédés de réduction des émissions passe par la recherche d’adsorbants qui présentent de grandes capacités d’adsorption et une forte sélectivité du CO2
en présence d’impuretés, des faibles conditions améliorées de régénération, une bonne stabilité dans les procédés d’adsorption / désorption et un faible coût de production.
A l’heure actuelle, les solides à structure poreuse tridimensionnelle (zéolithes) font partis des adsorbants les plus utilisés. Les zéolithes de type faujasite X semblent intéressantes pour le piégeage du dioxyde de carbone, à cause de leur grand volume microporeux. Nous nous sommes donc intéressés essentiellement à ce type de matériaux.
L’objectif global de cette étude est d’augmenter les quantités de CO2 adsorbées à basse
température et d’optimiser la régénération de l’adsorbant dans un domaine de température peu élevée (40 – 300°C).
Ce manuscrit sera donc divisé en quatre chapitres.
Le premier chapitre est consacré à l’étude bibliographique. Dans ce chapitre, les gaz à effet de serre (en particulier le dioxyde de carbone), les effets dus à l’augmentation des teneurs en CO2 dans l’atmosphère, les mesures de lutte contre le réchauffement climatique
Introduction générale
Page 2
dans l’atmosphère seront détaillés. Les phénomènes d’adsorption et de désorption, les différents adsorbants solides (zéolithes, oxydes tels que MgO et CaO…) et les paramètres influençant l’adsorption du dioxyde de carbone seront également présentés.
Le second chapitre est consacré à la partie expérimentale. Dans ce dernier, les méthodes de préparation des échantillons ainsi que les techniques de caractérisation et l’appareillage utilisés pour les expériences d’adsorption et de désorption du CO2 seront décrits.
Le troisième chapitre présente les résultats et les discussions concernant les caractérisations, les études d’adsorption et de désorption du dioxyde de carbone. Les capacités d’adsorption du CO2 à saturation sont comparées entre les adsorbants étudiés. L’influence de
l’eau, du méthane et du propane sur les capacités et les sites d’adsorption du CO2 à basse
température a été également étudiée.
Enfin, le quatrième chapitre résume les principales conclusions qui ont été esquissées en s’appuyant sur les résultats expérimentaux. Les perspectives envisagées seront également présentées.
Chapitre 1 : Etude bibliographique
Page 3
I.
Les Gaz à Effet de Serre (GES)
1. 1.
Définition des GES
L'atmosphère terrestre est constituée pour l'essentiel de diazote, de dioxygène et d’autres composés en faibles quantités appelés « gaz traces » (< 1% volumique). Parmi eux se trouvent notamment le dioxyde de carbone et la vapeur d'eau, qui possèdent la propriété particulière de « piéger » l'énergie solaire en adsorbant une partie des rayonnements infrarouges réémis par la Terre. Ces derniers sont appelés gaz à effet de serre (GES) car leur accumulation en une couche concentrique (située à une altitude inférieure à 16 km) provoque un « effet de serre » naturel permettant une température moyenne terrestre de +15°C, au lieu des -18°C qui résulteraient de l'absence de cette couche [1]. L'effet de serre naturel est donc indispensable à la vie sur Terre.
Les principaux gaz à effet de serre naturel sont : la vapeur d'eau (H2O), le dioxyde de
carbone (CO2), le méthane (CH4), l’oxyde de diazote (N2O) et l'ozone (O3). Cependant, il
existe d'autres gaz, issus de l'activité humaine (émissions anthropiques), ou l'on retrouve ceux cités précédemment mais également les gaz fluorés, notamment les chlorofluorocarbures (CFC), les hydrofluorocarbures (HFC), le perfluorométhane (CF4) et les hexafluorures de
soufre (SF6). Par ailleurs, les GES issus des activités humaines sont produits dans plusieurs
secteurs d'activités avec des pourcentages d’émissions différents.
1. 2.
Sources d'émission des GES
Les émissions des GES sont à la fois d'origine naturelle (essentielle pour la vie sur terre) et anthropique. La figure 1 montre les émissions mondiales des gaz à effet de serre en 2004 par secteur d'activité. D'après le Groupe d'experts Intergouvernemental sur l'Evolution du Climat (GIEC) [2], les domaines d'activité dont la production est la plus importante sont : la production d'énergie (25,9%), suivi du secteur industriel (19,4%) et des déforestations (17,4%). Le secteur des transports, quant à lui, contribue à hauteur de 13,1%. Au contraire, le domaine des déchets et eaux usées (2,8%) ainsi que celui des bâtiments résidentiels et commerciaux (7,9%) sont les secteurs d'activité à faibles émissions au niveau mondial.
Page 4
Figure 1 : Emissions mondiales de gaz à effet de serre par secteur d'activité [2]. En France, selon le groupe dit « Facteur 4 » [3], les émissions des GES proviennent des transports pour 26 %, suivis de l'industrie (22 %), de l'agriculture (19 %), des bâtiments et habitations (19 %), de la production et de la transformation de l'énergie (13 %) et du traitement des déchets (3 %). Depuis 1990, les émissions ont augmenté de plus de 22 % pour les transports et les bâtiments. En revanche, elles ont diminué de 22 % dans l’industrie, de 10 % dans le secteur agricole, de 9 % dans le secteur de l’énergie et de 8 % pour le traitement des déchets. Cette baisse d’émission de CO2 est due essentiellement à l’évolution structurelle
de l’industrie vers des marchés spécifiques porteurs en termes d’innovation. Enfin, il y a l’influence des modes de développement et des styles de vie selon que les préférences des consommateurs s’orientent vers plus de sobriété en utilisant des machines (chauffages, voitures…) émettant moins de GES.
Le CO2 est toutefois le composé, parmi les GES, dont les émissions mondiales restent
les plus importantes (figure 2). 56,6% de production de ce gaz viennent des combustibles fossiles (charbon, pétrole, gaz), 17,3% des déforestations et 2,8% d'autres secteurs (transports, résidences et commerces etc...). La production mondiale du CH4 (14,3%) et du N2O (7,9%)
parait négligeable devant celle du CO2. Par conséquent, la cause première de la hausse de la
concentration en CO2 dans l’atmosphère est l’utilisation de combustibles fossiles. Le
Chapitre 1 : Etude bibliographique
Page 5
Figure 2 : Emissions mondiales des gaz à effet de serre [2].
D'après le dernier rapport du GIEC en 2007, les émissions mondiales de GES imputables aux activités humaines ont augmenté depuis l’époque préindustrielle, la hausse a été de 70 % entre 1970 et 2004. Les activités humaines engendrent des émissions de quatre GES à longue durée de vie : le CO2, le CH4, le N2O et les hydrocarbures halogénés.
Cependant, seules les concentrations atmosphériques des trois premiers gaz ont augmenté de manière significative de 1970 à 2004 et en particulier celle du CO2 (figure 3).
Figure 3 : Emissions mondiales des GES depuis 1970 [2].
La concentration atmosphérique mondiale du CO2 est passée de 280 parties par million (ppm) environ à l’époque préindustrielle à 379 ppm en 2005. Le rythme d’accroissement annuel de la concentration du CO2 a été plus rapide au cours des 10 dernières années (1,9 ppm par an en moyenne entre 1995 et 2005) qu’il ne l’a été depuis le début des mesures atmosphériques directes continues (1,4 ppm par an en moyenne entre 1960 et 2005), bien qu’il puisse varier
Page 6 d’une année à l’autre [4]. La concentration du méthane est passée d’environ 715 parties par milliard (ppb) à l’époque préindustrielle à 1732 ppb au début des années 1990, pour atteindre 1774 ppb en 2005. Le taux de croissance a fléchi depuis le début des années 1990, en cohérence avec les émissions totales (somme des sources anthropiques et naturelles) qui sont restées pratiquement constantes au cours de cette période [4]. La concentration atmosphérique globale de N2O est passée de 270 ppb à l’époque préindustrielle à 319 ppb en 2005. Celle de nombreux hydrocarbures halogénés (dont les hydrofluorocarbones) a augmenté, essentiellement sous l’effet des activités humaines, alors qu’elle était proche de zéro à l’ère préindustrielle.
D'après les scientifiques du GIEC, l'augmentation des GES dans l'atmosphère est à l'origine du réchauffement de la planète. En effet, plus la concentration des GES dans l'atmosphère est grande et plus le rayonnement infrarouge (donc la chaleur) piégé par la Terre sera important. Par conséquent, les GES accentueraient la quantité d'énergie piégée par l'atmosphère et entrainerait donc une augmentation des températures. Ils estiment également que le réchauffement climatique de 1970 à 2004 a eu des effets sur l'environnement et qu'on pourrait s'attendre à des événements climatiques particuliers si la température mondiale continuait à augmenter.
1. 3.
Effets du réchauffement climatique
Les effets du réchauffement climatique sont une certitude, puisqu’on observe une augmentation des températures moyennes globales terrestres et maritimes. Une fonte généralisée des neiges et de la glace est également observée induisant une augmentation du niveau moyen des mers. On remarque également que de nombreux écosystèmes sur tous les continents et les océans sont affectés [4]. En se basant sur les observations actuelles et grâce aux modèles du réchauffement climatique de 1980 à 1999, le GIEC estime qu’une augmentation continue des températures mondiales causerait des problèmes au niveau des eaux, de la santé, de l'alimentation, etc… (tableau 1).
Tableau 1 : Exemples d'incidences associées à l'élévation de la température moyenne à la surface du globe [4].
Chapitre 1 : Etude bibliographique
Page 7
Selon les conclusions du dernier rapport de 2007 [2], le dioxyde du carbone représente à lui seul plus de trois quart des émissions des GES. C’est pourquoi depuis plusieurs années, un intérêt particulier est porté sur ce gaz à effet de serre.
Le GIEC a alarmé la communauté internationale (CI) sur le phénomène du réchauffement climatique et ses conséquences sur la planète. Face à cela, une prise de conscience générale à été observée et plusieurs mesures ont été prises pour lutter contre le réchauffement de la planète.
Page 8
1. 4.
Mesures de lutte contre le réchauffement climatique:
Conventions
En signant des conventions, la communauté internationale a pu se mettre en accord sur la nécessité de fixer un objectif visant à stabiliser les concentrations des GES, dans le but de stabiliser le climat en signant des conventions. Par exemple, le Protocole de Kyoto est un programme international de lutte contre le changement climatique par la réduction des GES issus des activités humaines. Il a été négocié en novembre 1997 et les pays industrialisés ayant ratifié le protocole (dont la France) se sont engagés à réduire leurs émissions de GES respectives, avec une réduction globale de 5,2 % des émissions d'ici 2012 par rapport aux émissions de 1990 [5]. Une stratégie de réduction de six gaz à effet de serre a été établie. Le CO2, le CH4, le N2O, les HydroFluoroCarbures (HFC), les PerFluoroCarbures (PFC) et
l’Hexafluorure de soufre (SF6). Pour les pays membres de l'Union Européenne, l'objectif de
réduction est fixé à 8 % par rapport aux niveaux de 1990 au cours de la première "période d'engagement" (2008 - 2012). Cet objectif est partagé entre les 15 états membres. Les pays en voie de développement n'ont pour le moment qu'une obligation d'inventaire de leurs émissions [5].
Le Protocole de Kyoto prendra fin début 2013. Un traité international sur le réchauffement climatique devait voir jour lors du Sommet de Copenhague sur le climat en décembre 2009 pour prendre le relais de Kyoto, à travers un accord encore plus ambitieux. Mais contrairement aux décisions attendues, aucun accord chiffré entre les pays industrialisés n'a eu lieu.
Par ailleurs, réduire les émissions des GES dans l'atmosphère, en particulier le CO2,
entraîne l'adoption ou le développement des procédés de piégeage ou de stockage relatifs à différentes industries (énergie, pétrole, transports, bâtiment, agriculture etc...). Cependant, de nombreuses techniques de piégeage et de stockage du CO2 existent déjà en industrie.
Chapitre 1 : Etude bibliographique
Page 9
II.
Techniques de piégeage du CO
2utilisées en industrie
Dans le domaine industriel, le piégeage et le stockage du CO2 (PSC) est un processus consistant à séparer le CO2 de ses sources industrielles et énergétiques, à le transporter dans un lieu de stockage et à l'isoler de l'atmosphère sur le long terme (environ 200 ans). Le PSC est considéré comme une option parmi les mesures d'atténuation visant à stabiliser la concentration de gaz à effet de serre dans l'atmosphère [6].
Quatre procédés sont aujourd'hui utilisés pour piéger le CO2 au niveau de l’étape de
séparation. Ces différents procédés sont (figure 4) : - l’absorption
- l’adsorption
- la séparation cryogénique - les membranes.
Figure 4 : Techniques de séparation du CO2 des effluents gazeux [7].
Le piégeage du CO2 est d'ores et déjà une technologie industrielle utilisée aujourd'hui pour le
traitement du gaz naturel. Elle est pratiquée couramment dans la fabrication d'engrais, dans l'industrie agroalimentaire et dans le secteur de l'énergie (industrie pétrolière et gazière). Le principal problème est en général la faible concentration en dioxyde de carbone. Selon l'industrie concernée, cette teneur peut aller de quelques pour cent à 20 %. D'autres gaz, tels que l'oxygène, la vapeur d'eau ou l'azote, se retrouvent aussi dans ces effluents. Il serait
Page 10
impensable de vouloir tous les compresser pour les stocker, pour des raisons de coût énergétique et de place.
Des méthodes de séparation sont donc nécessaires pour pouvoir capter exclusivement le CO2. Un grand nombre de procédés industriels de piégeage existent sur le marché, chacun
ayant un domaine d'application spécifique en fonction de la nature des fumées à traiter (composition, température, pression). Ils relèvent tous de trois catégories principales : le piégeage postcombustion, le piégeage précombustion et le piégeage par oxycombustion [6,8].
2. 1.
Le piégeage postcombustion du CO
2Le piégeage postcombustion a pour objectif d'extraire le CO2 dilué dans les fumées de
combustion. Il peut s'intégrer sans apporter trop de modifications aux installations existantes. Le procédé le plus couramment utilisé est le piégeage du CO2 par un solvant, en général des
amines. D'autres procédés sont envisagés par cycle calcium et par voie cryogénique [8]. Le premier consiste à capter le CO2 par de la chaux vive pour produire du calcaire. Celui-ci est
chauffé, ce qui libère le CO2 tout en produisant à nouveau de la chaux vive. Le procédé par
voie cryogénique repose sur la solidification du CO2 par givrage pour le séparer. La
séparation du CO2 peut également se faire au contact d'un adsorbant solide ou à travers une
membrane [8]. Le piégeage du CO2 en postcombustion est le procédé le plus utilisé dans
l’industrie.
2. 2.
Le piégeage précombustion du CO
2Pour ce type de procédé, l'objectif est de capter le carbone avant combustion, lors du processus de fabrication du combustible. Il est converti en entrée d'installation en gaz de synthèse, un mélange de monoxyde de carbone (CO) et de dihydrogène. La technique utilisée est soit le vaporeformage de gaz naturel en présence d'eau, soit l'oxydation partielle en présence d'oxygène. Le CO présent dans le mélange réagit avec l'eau au cours de l'étape de conversion pour former du CO2 et du dihydrogène. Le CO2 est alors séparé du dihydrogène,
lequel peut être utilisé pour produire de l'énergie (électricité ou chaleur) sans émission de CO2
Chapitre 1 : Etude bibliographique
Page 11
2. 3.
Le piégeage par oxycombustion du CO
2Cette technologie n'est pas à proprement parler un piégeage du CO2. La question est ici réglée à l’entrée et non à la sortie, il s'agit de produire une fumée concentrée à 90% de CO2 en réalisant une combustion à l'oxygène pur avec un recyclage d'une partie du CO2 en substitution de l'azote de l'air. L'oxycombustion est particulièrement adaptée à une remise à niveau d'une installation existante. Cependant, la séparation de l'oxygène de l'air obtenue principalement par la voie cryogénique est coûteuse et consommatrice d'énergie. À titre indicatif, la consommation d'énergie de l'apport en oxygène pur pour une centrale à charbon d'une puissance de 500 MW fonctionnant 8000 heures par an représenterait 15% de sa production électrique annuelle. Pour éviter le coût de séparation de l'oxygène de l'air, une technologie prometteuse est envisagée : la combustion dite en boucle chimique (chemical looping combustion). Elle consiste à apporter l'oxygène de l'air sur un support métallique, qui en circulant transfère l'oxygène [8].
Toutefois, grâce aux techniques actuelles on piège 85 à 95% du CO2 traité dans les
installations de piégeage [6]. Les techniques d’adsorption utilisant des adsorbants solides, (figure 4) apparaissent comme étant plus efficaces comparées aux autres techniques [9]. Elles permettent une meilleure sélectivité du gaz à traiter et de concentrer des quantités importantes de gaz. Elles traitent également des flux de débits importants grâce au pouvoir adsorbant de certains de ces solides. Dans cette étude bibliographique, nous allons nous intéresser uniquement aux techniques d’adsorption du CO2 utilisant des solides telles que les zéolithes.
Page 12
III.
Généralités sur les phénomènes d’adsorption et de
désorption
3. 1.
Phénomènes d’adsorption
L’adsorption consiste en l’accumulation d’un fluide (adsorbat) à la surface d’un solide (adsorbant). Il existe deux principaux modes d’adsorption :
la physisorption qui repose sur des forces intermoléculaires faibles (liaisons de Van der Walls ou forces électrostatiques) entre la surface d’un solide et les molécules à proximité de cette surface. L’adsorption physique se caractérise par un processus réversible.
la chimisorption qui repose, principalement, sur des interactions de nature covalente c’est-à-dire qui implique la formation d’une liaison entre l’adsorbat et l’adsorbant dont les effets sont souvent irréversibles.
Pour une température donnée et à l’équilibre, l’évolution de la quantité adsorbée par l’adsorbant en fonction de la pression d’adsorbat décrit l’isotherme d’adsorption. Celles-ci peuvent avoir plusieurs allures et dépendent du couple adsorbat/adsorbant. Un classement en six catégories a été réalisé (figure 5) par IUPAC (International Union of Pure and Applied Chemistry)[10] :
Chapitre 1 : Etude bibliographique
Page 13
•
Isotherme de type I
Elle est caractéristique des matériaux microporeux très hydrophiles. Cette courbe est théoriquement réversible et concave par rapport à l’axe des abscisses. Elle présente une capacité d’adsorption élevée dans le domaine des faibles pressions relatives (P/P0), suivi d’un
plateau sur un grand domaine de pression relative. Cette allure est caractéristique des matériaux possédant de petits pores avec une distribution très étroite et des matériaux à grands micropores tels que les zéolithes présentant beaucoup de défauts.
•
Isotherme de type II
Elle est caractéristique d’une adsorption très progressive sur des solides hydrophiles non poreux ou macroporeux, mais dont l’affinité avec l’eau est moins importante que dans le cas précédent. L’isotherme est généralement réversible. Elle a une forme concave par rapport à l’axe des abscisses, suivie d’une droite à faible pente et change de courbure pour les grandes valeurs de P/P0. Cette forme indique une adsorption monocouche puis multicouche sur une
surface ouverte.
•
Isotherme de type III
Elle est réversible et convexe par rapport à l’axe des abscisses sur tout le domaine de pression. Cette isotherme caractérise des interactions adsorbant-adsorbat très faibles. Il est très rare de rencontrer ce genre de courbe dans la pratique. Elle est également caractéristique des matériaux hydrophobes comme le type V.
•
Isotherme de type IV
Elle est caractéristique d’une adsorption dans les matériaux mésoporeux. Elle a la même allure que l’isotherme de type II mais pour des pressions plus élevées, on observe une augmentation de la quantité adsorbée liée à la condensation capillaire dans les mésopores. Une fois le remplissage des mésopores achevé, l’isotherme se termine par un palier de saturation de longueur variable. De plus, la désorption n’est pas réversible et se caractérise généralement par une hystérèse.
Page 14
•
Isotherme de type V
Elle est très rare et indique une très faible interaction adsorbant-adsorbat, comme le type III, avec une hystérèse liée à la présence de mésopores.
•
Isotherme de type VI
Elle est caractérisée par la présence de marches qui traduisent un processus d’adsorption par couches successive.
3. 2.
Phénomène de désorption
La désorption est l’inverse du phénomène d’adsorption. Il s’agit ici d’un phénomène par lequel un fluide est libéré de la surface d’un solide (adsorbant) ou des pores par apport d’énergie extérieure sous forme de chaleur [11,12], ou par diminution de la pression du milieu [12]. Le plus souvent, la désorption est réalisée dans le but de régénérer l’adsorbant. Par ailleurs, lors des traitements de régénération, l’augmentation de la température peut avoir des effets néfastes sur les adsorbants (charbon actif, 13X…).
En industrie, la régénération des adsorbants solides (charbon actif, zéolithes…) après saturation est une opération indispensable compte tenu des coûts élevés des adsorbants utilisés. Plusieurs procédés, dont la régénération thermique (TSA : Temperature Swing Adsorption) sont souvent utilisés [13]. Cependant, certains solides (tel que le charbon actif) sont difficilement régénérables, du fait de leur sensibilité à haute température. Par conséquent, seules les espèces adsorbées faiblement liées seront désorbées, d’où une régénération partielle de l’adsorbant. Dans le cas des zéolithes (exemple : 13 X), la régénération de l’adsorbant à haute température est meilleure que dans le cas du charbon actif, due à leurs structures poreuse bien cristallisée [13,14]. Cependant, l’application d’une température élevée (> 500°C) lors des traitements de régénération sur ce type d’adsorbants (zéolithes) peut détruire certains sites d’adsorption ou causer l’effondrement de la structure. Par conséquent, cela entraîne une perte de capacité d’adsorption [13,14]. Selon la température de désorption utilisée (cycles d’adsorption/désorption), les quantités des espèces adsorbées peuvent diminuer à cause l’adsorption irréversible des espèces (formation des carbonates par exemple).
Chapitre 1 : Etude bibliographique
Page 15
IV.
Etat de l’art de l’adsorption du CO
2sur adsorbants
solides
Dans la littérature, les adsorbants solides les plus utilisés dans les procédés de piégeage du CO2 sont :
- le charbon actif [13-16],
- les solides inorganiques hybrides (MCM-41, MCM-48…) [13,15,17,18], - les hydrotalcites (Layered double hydroxides : LDHs) [13,19-21], - les MOFs (Metal-Organic Frameworks) [15,16,22-24],
- les oxydes des métaux à caractère basique (CaO, MgO…) [13,25-27], - les zéolithes (A, Y, X…) [13,15-35].
De nombreuses recherches dans la littérature ont montré que les zéolithes présentaient de bonnes capacités d’adsorption du CO2 à faible température et pression [36,37]. De plus, les
oxydes basiques (tels que MgO, CaO etc…) ont des sites de force basique supérieure à ceux des zéolithes [13,26,27]. L’étude bibliographique de l’adsorption du CO2 sera donc axée
principalement sur ces adsorbants.
4. 1.
Les oxydes de métaux
L’adsorption du CO2 sur les oxydes de métaux a été largement étudiée [26,38]. Les
oxydes des métaux à caractère basique sont ceux qui sont formés à partir des éléments des groupes IA et IIA de la classification périodique (figure 6). Les oxydes basiques qui sont considérés comme étant les meilleurs adsorbants pour le CO2 sont l’oxyde de magnésium
(MgO) et l’oxyde de calcium (CaO). Leur faible coût et leur résistance à haute température, et surtout leur caractère basique supérieur à ceux de la colonne IA en font des candidats de choix [13].
En reportant dans un Tableau Périodique le comportement de l'oxyde (figure 6), on observe que les oxydes basiques correspondent aux combinaisons de l'oxygène avec les métaux peu électronégatifs (métaux alcalins et alcalino-terreux), c'est-à-dire des oxydes ioniques. Au contraire, les oxydes acides résultent de la formation de liaisons covalentes avec les non-métaux. Les oxydes amphotères se situent à la frontière des deux groupes. D’autre part, notons que le CO2 est considéré comme une molécule acide (oxyde acide) et s’adsorbe
Page 16
Figure 6 : Evolution du caractère acido-basique des métaux et gaz [39].
Par conséquent, seuls les oxydes à caractère basique seront à priori des bons adsorbants pour le dioxyde de carbone. Dans la littérature, ce dernier est souvent utilisé comme molécule sonde afin d’identifier et de caractériser les sites ou les propriétés basiques des adsorbants [13,38,40]. En outre afin de pouvoir piéger (adsorber) le CO2, les oxydes doivent posséder
dans leurs structures des sites basiques.
4. 1. 1.
Sites basiques des oxydes de métaux alcalino-terreux
et adsorption du CO
2Le caractère basique des oxydes des métaux alcalino-terreux est attribué principalement à trois types de sites d’adsorption. Les sites O2- (oxygène anionique), les sites HO- (groupement hydroxyle) et la paire Mn+ O2- (paire métal/oxygène) [26,38,40,41].
Les sites O2- sont considérés comme des sites basiques forts et présentent deux types de coordination (figure 7) :
- O2- de coordination 4 (sites sur les arrêtes), où la formation des espèces de carbonates monodentés est favorable.
- O2- de coordination 3 (sites sur les coins), où la formation des espèces de carbonates bidentés est favorisée. Par ailleurs, ces espèces peuvent également se former, ainsi que les carbonates polydentés, sur la paire Mn+ O2-. Mais cela dépendra du nombre de cation métallique qui vont interagir avec les anions oxygènes [38].
Chapitre 1 : Etude bibliographique
Page 17
Figure 7 : Schéma d’un monocristal de MgO. La coordination des ions Mg2+ situés sur les plans des faces, des arrêtes et des coins sont indiquées [41].
Les sites OH- quant à eux sont considérés comme des sites basiques faibles et favorisent la formation des espèces hydrogénocarbonates (HO-CO2-) à la surface des oxydes [38,41]. La
présence de ces sites basiques sur ces oxydes (exemple MgO et CaO) est due au fait que la surface des oxydes est naturellement hydroxylée et parfois carbonatée [40]. Des études théoriques et expérimentales récentes sur MgO ont montré que ces hydroxyles peuvent rester à la surface jusqu'à 900°C [40,42]. La paire Mn+ O2- est considérée comme un site de force basique intermédiaire, où la formation de carbonates bidentés est la plus favorable par rapport aux sites O2- de coordination 3 [38,40].
Pour identifier ces différentes espèces et déterminer la force de ces sites basiques, la microscopie infrarouge (FTIR) et la désorption programmée en température (TPD) sont les techniques d’analyses les plus utilisées [26,38,40,41,43].
Les études réalisées par C. Pighini et al. [40] et J. C. Lavalley [38], sur MgO ont mis en évidence par spectroscopie infrarouge les bandes d’adsorption du CO2 (entre 1200 – 1700 cm -1
) correspondant aux différentes espèces carbonyles formées à la surface. Ainsi, les bandes d’adsorption correspondant aux carbonates monodentés ont été observée entre 1500 – 1490 cm-1 (vibrations symétriques υsO-C-O) et entre 1440 – 1400 cm-1 (vibrations asymétriques
υasO-C-O). Celles des carbonates bidentés ont été observées entre 1360 – 1340 cm-1
(vibrations symétriques υsO-C-O) et entre 1600 – 1540 cm-1 (vibrations asymétriques υas
O-C-O). Les bandes d’adsorption des bicarbonates ont été, quand à elles, observées entre 1260 – 1240 cm-1 (δOH), entre 1470 – 1450 cm-1 et entre 1650 – 1630 cm-1 correspondant
respectivement aux élongations de la liaison C-OH, aux vibrations symétriques et asymétriques (O-C-O).
Page 18
Les travaux effectués par V.K. Díez et al. [43] en thermodésorption du CO2 sur MgO
montrent trois types d’espèces désorbant à différentes températures (figure 8). Selon eux, les sites basiques faibles ont une température de pic de désorption maximale (Tm) à 117°C et sont
assignés aux bicarbonates formés à partir des groupements hydroxyles. Les sites basiques moyens ont un Tm à 167°C et sont assignées aux carbonates bidentés formés sur la paire Mn+
O2-. Quant aux sites basiques forts, leur Tm est à 273°C. Ces sites sont attribués aux
carbonates monodentés formés sur les sites O2-.
Figure 8 : Profils TPD du CO2 obtenus sur MgO calciné à différentes températures [43].
Les travaux effectués par Di Cosimo et al. [26] ont également donné trois différentes Tm. Ces
températures (100, 200 et 300°C) ont été respectivement attribuées, comme dans les travaux de V. K. Diez, aux bicarbonates, carbonates bidentés et aux carbonates monodentés. En revanche, ceux de M. Manfred et al. [44] réalisés sur différents oxydes montrent que la surface des sites basiques forts sur SrO (757°C) est supérieure à celle de CaO (547°C) et à celle de MgO (347°C). Donc, la force des sites basiques (énergie du CO2 chimisorbé) de CaO
est supérieure à celle de MgO. Cette différence de force entre MgO et CaO peut être attribuée à la basicité de CaO qui est 25 à 30% supérieure à celle observée sur MgO [45].
Le caractère acide du CO2 facilite son adsorption sur les sites de quelques oxydes
métalliques. Plus précisément, ceux qui sont de nature ionique et qui présentent des sites fortement basiques [36]. Ce groupe de solides inclut les oxydes des métaux alcalins (Na2O,
Chapitre 1 : Etude bibliographique
Page 19
utilisés pour le piégeage du CO2 dans le milieu industriel. Le CO2 peut être adsorbé sur ces
solides en formant à la surface des nombreuses espèces carbonates mono ou polydentés selon la réaction suivante :
MO(s) + CO2 (g) ↔ MCO3 (s)
Où M peut être par exemple Mg, Ca, Sr, Ba.
Bonenfant et al. [45] a classé les oxydes en trois catégories selon leur basicité et l’affinité du CO2 avec leurs surfaces. Ainsi, l’affinité du CO2 avec la surface des oxydes
basiques (CaO et MgO) est supérieure à celle des oxydes amphotères (Al2O3, Cr2O3, TiO2,
MnO, oxydes de fer) et des oxydes acides (SiO2).
Dans la littérature, les capacités maximales du CO2 chimisorbé obtenues sur CaO à 1
bar sont de 4,5 mmol.g-1 et de 2,3 mmol.g-1 à 500 et 650°C respectivement [21,36]. Par ailleurs, les travaux de Barker [46] ont démontré que les capacités d’adsorption du CO2 sur
CaO dépendent de la taille des particules de poudre CaO et de la surface développée. D’après les expériences qu’il a réalisé, les capacités d’adsorption du CO2 obtenues au bout de 24 h sur
poudre de CaO avec une taille de particules de 10 µm étaient de 13,4 mmol.g-1 à 866°C. Par contre, celles obtenues à 577°C avec des tailles de particule de 10 nm étaient de 16,6 mmol.g
-1
. D’autre part, dans le cas où les nanoparticules de CaO étaient compressées, les capacités d’adsorption du CO2 diminuaient à 8,9 mmol.g-1 [36,46]. Par ailleurs, le CO2 est
principalement chimisorbé sur CaO. Les espèces formées à la surface sont sous forme de carbonates, donc fortement liées [36].
Les capacités maximales d’adsorption du CO2 obtenues sur MgO à 0,2 bar sont de 0,64
et 0,13 mmol.g-1 à 25 et 500°C respectivement [21]. Comme pour CaO, les quantités de CO2
adsorbées diminuent quand la température augmente, traduisant l’exothermicité de l’adsorption. Cependant, contrairement à CaO, les travaux de Gregg et al. [47] montrent qu’à basse température (-100°C), il y a une physisorption et une chimisorption du CO2 sur MgO.
Par contre à haute température (500°C), seule la chimisorption du CO2 est logiquement
observée. D’autre part, Sunho et al. [36] montrent que les capacités de CO2 adsorbées sur
MgO et CaO peuvent augmenter en présence d’eau (H2O). Whateley [48] explique que cette
augmentation (sur MgO) est liée au fait que l’eau peut interagir avec les espèces bidentées pour produire à la surface de l’oxyde, des espèces mondentées et des groupements hydroxyles (hydrogénocarbonates).
Page 20
Les travaux de Lee et al. [49] ont montré que la vitesse de formation des carbonates sur CaO était favorisée par une augmentation de la température. Par conséquent, les températures de régénération de CaO sont importantes. Les quantités de CO2 désorbées sont de 1,43 ; 2,29 ; 3,50 et 5,09 mmol g-1 de CaO quand les températures de désorption atteignent600°C, 650°C, 700°C et 750°C respectivement. Contrairement à CaO, les températures appliquées sur MgO pour les traitements de régénération sont inférieures. Les travaux Beruto et al. [50] indiquent que la désorption totale du CO2 adsorbé sur MgO sous vide à 700°C se fait en chauffant pendant 1h. Or, concernant CaO, cette désorption se fait au bout de 4h de chauffage dans les mêmes conditions. Margandan et al. [51] indiquent également que la régénération du MgO peut se faire entre 400 et 800°C, contrairement à CaO ou les températures peuvent dépasser les 900°C.
En se basant sur ses informations, l’oxyde de magnésium et l’oxyde de calcium semblent être des bons adsorbants pour le dioxyde de carbone, du moins à basse température, à cause de leur basicité. Toutefois, MgO semble avoir des interactions avec CO2 plus faibles que CaO, ce qui permet d’appliquer des températures peu élevées (400 – 900°C) lors des traitements de régénération.
4. 1. 2.
Influence des caractéristiques des oxydes de métaux
pour l’adsorption du CO
2Les oxydes de métaux (tels que MgO, CaO, BaO, etc.. .) sont des solides qui peuvent être monocristallins et présenter des surfaces parfaitement définies. Ils présentent dans leurs environnements (surface) des sites très réactifs sur les arrêtes et les coins vis-à-vis des molécules acides ou basiques, due à leur structure cristalline simple [52].
Par exemple, au cœur de l’oxyde tous les ions Mg2+ et O2- ont une coordinence de six. Par contre, les cations (Mg2+) présentent une coordination de cinq (situé au niveau des arrêtes) et sont plus abondants sur la face (001). Donc ils sont favorables pour compléter leur sphère de coordination à l’adsorption d’une molécule en phase gaz [41]. De plus, sur ce type d’oxydes la face la plus exposée est la face (001) [52].
Par contre, les anions (O2-) sont plus abondants sur les faces (100). Ceux présentant une coordination de cinq sont situés au niveau des coins, mais sont énergétiquement plus stables que les cations qui ont la même coordinence [40]. Dans ce cas, les sites O2- situés au niveau de la face (100) ne sont pas favorables à l’adsorption du CO2, seuls les sites se trouvant sur les faces les plus exposées (001) seront favorables à l’adsorption du CO2.
Chapitre 1 : Etude bibliographique
Page 21 Des solides, à réseau poreux tridimensionnel, tels que les zéolithes ont également fait l’objet d’études sur l’adsorption du CO2 [45].
4. 2.
Les zéolithes
L’histoire des zéolithes débute en 1756 lorsque le baron Crönstedt, minéralogiste suédois, découvre la stilbite [53]. Il s’agit d’une nouvelle famille de minéraux, les aluminosilicates hydratés, qu’il nommera zéolithes, du grec zeô, bouillir et lithos, pierre. Les zéolithes étant présentes en très petite quantité sur Terre, de nombreux travaux de synthèse ont été entrepris.
De nos jours, il existe un peu plus de 170 structures zéolithiques répertoriées, seulement 130 peuvent être synthétisées (plus de quatre millions de tonnes sont produites par an) et 40 seulement sont d’origine naturelle. Les ouvrages de Breck [54], Rabo [55], Jacobs [56], Guisnet et Gilson [57] ainsi que de nombreux articles regroupés dans des actes de congrès internationaux sur les zéolithes [58-60] décrivent les structures des zéolithes, leurs caractéristiques physico-chimiques et leurs propriétés en adsorption ou en catalyse.
4. 2. 1.
Généralités
Les zéolithes sont composées de canaux et de cavités interconnectés (figure 9) qui après l’évacuation de l’eau de constitution, possèdent un volume poreux accessible à un grand nombre de molécules. Cette propriété remarquable a ouvert la voie à de nombreux champs d’application, tels que le tamisage moléculaire, le séchage des fluides, la séparation des mélanges gazeux, l’élimination des composés soufrés du pétrole et la séparation de l’air en ses différents constituants [54,57,59]. De plus, la présence de cations extra-réseau interchangeables entraîne une large utilisation des zéolithes dans les procédés d’adoucissement des eaux, le remplacement des polyphosphates dans les lessives ou la purification des eaux usées par élimination de l’ammoniac [58,61]. L’échange de cations alcalins ou alcalino-terreux par des protons offre également de nombreuses applications dans le domaine de l’industrie comme adsorbant en catalyse [55,58].
Les zéolithes sont des aluminosilicates parfaitement cristallisés de formule générale Mx/n(AlO2)x(SiO2)y où n est la valence du cation extra-réseau M, x+y le nombre total de
tétraèdres SiO4 et AlO4- par maille élémentaire et y/x le rapport atomique Si/Al [62]. Plus ce
rapport est élevé, plus la zéolithe est hydrophobe. Les zéolithes sont basées sur un arrangement tridimensionnel de tétraèdres TO4 (SiO4 ou AlO4-) liés par leurs atomes
Page 22
d’oxygène (enchaînement T-O-T) pour former des sous-unités et finalement de larges réseaux constitués de blocs identiques (les mailles élémentaires). Les atomes de silicium et d’aluminium étant respectivement tétravalents et trivalents, la présence d’aluminium dans les tétraèdres induit des charges négatives dans la charpente, compensées par des cations alcalins ou alcalino-terreux. Il en résulte une structure très aérée constituée d’un réseau très régulier de canaux et cages avec des ouvertures inférieures à 10 Å (micropores). Pour l’essentiel, les processus intervenant en adsorption, catalyse et échange d’ions se produisent dans ces pores de taille très voisine de celle des molécules organiques ou gazeuses [62].
4. 2. 2.
Les différentes familles des zéolithes
En catalyse et en adsorption, quatre grandes familles de zéolithes, définies par le nombre d’atomes T dans les ouvertures de pores les plus larges, sont utilisées :
- les zéolithes à petits pores avec des ouvertures de pores à 8 atomes T, qui ont des diamètres libres de 3 à 4,5 Å.
- les zéolithes de taille de pore intermédiaire avec des ouvertures à 10 atomes T, qui ont des diamètres libres de 4,5 à 6 Å.
- les zéolithes à larges pores avec des ouvertures à 12 atomes T, qui ont des diamètres de pores libres de 6 à 8 Å.
- les zéolithes à pores ultra-larges comme la cloverite (20 T, 6 x 13,2 Å), VPI-5 (18 T, 12,7 Å), AlPO4-8 (14 T, 7,9 x 8,7 Å).
Cet assemblage donne lieu à diverses structures, conjointes par les canaux, où les molécules d'une taille appropriée tels que les gaz comme le CO2 (diamètre : 3,3 Å), l'eau et des cations
métalliques échangeables (Li+, Na+, K+, Ca2+, Ba2+, Mg2+, etc…) peuvent pénétrer et compenser les charges négatives excédentaires créées par la substitution des tétraèdres SiO4
Chapitre 1 : Etude bibliographique
Page 23
FAU (7,4 Å) BEA (5,6 x 5,6 Å) FER (4,2 x 5,4 Å)
LTA (4,1 Å) MOR (6,5 x 7,0 Å) MFI (5,1 x 5,4 Å)
Figure 9 : Structures de quelques zéolithes, montrant la structure des canaux et de cavités de chaque type de zéolithe [63].
Parmi les différentes familles de zéolithes connues, les zéolithes de type faujasite sont les plus utilisées dans les procédés d’adsorption du CO2 en milieu industriel, due en
particulier à leurs tailles de pores qui permet l’accès à un certain nombre de molécules gazeuses [31,43,45].
4. 3.
Les zéolithes de type faujasite (FAU)
La structure faujasite (FAU) est constituée de 24 tétraèdres TO4 reliés de manière à
former 8 cycles à 6 tétraèdres et 6 cycles à 4 tétraèdres, qui forment un cube-octaèdre encore appelé cage sodalite ou cage β. La structure peut se décrire comme un assemblage de cube-octaèdres reliés les uns aux autres par des prismes hexagonaux (figure 10). Cet assemblage fait apparaître une grande cavité polyédrique à 26 faces appelées supercage (ou cage α) qui constitue l’unité de base de la microporosité de cette zéolithe. Les supercages sont assimilées à des pseudo-sphères de 13 Å de diamètre et d’environ 850 Å3 de volume. Elles communiquent entre elles par l’intermédiaire d’ouvertures à 12 atomes d’oxygène (7,4 Å de diamètre), ce qui permet l’accès à de nombreuses molécules. La cage β a un diamètre de 7 Å
Page 24
et un volume de 160 Å3 environ. Elle est connectée à la supercage par des ouvertures à 6 atomes d’oxygène de 3 Å de diamètre environ. Elle est donc inaccessible au CO2.
Cage sodalite (ou cage β ) Prisme hexagonal Supercage (ou cage α ) 7.4 Å 13 Å
Figure 10 : Représentation de la structure faujasite.
La maille élémentaire de la faujasite comprend 8 cages α et 16 prismes hexagonaux, formant une structure cubique à faces centrées. Son paramètre de maille varie de 24,2 à 24,8 Å selon le nombre d’aluminium de la charpente et les cations de compensation(nature et nombre) [64].
On distingue deux types de zéolithes faujasite (formule de maille NaxAlxSi192-xO384
avec x le nombre d’aluminium et de sodium dans la structure) selon la composition et le rapport Si/Al :
La faujasite X dont le rapport Si/Al est compris entre 1 et 1,5 [65]. La faujasite Y dont le rapport Si/Al est supérieur à 1,5 [66].
Ces zéolithes sont synthétisées par voie hydrothermale. Cette technique ne permet pas d’obtenir un rapport Si/Al supérieur à trois. La valeur des rapports Si/Al de la zéolithe obtenue dépend principalement de la proportion de chacun des constituants de l’hydrogel de depart [64].
Dans la structure des zéolithes de type FAU, les sites cationiques et protoniques responsables de la basicité et/ou de l’acidité (zéolithe protonique) de la structure sont localisés.
Chapitre 1 : Etude bibliographique
Page 25
4. 3. 1.
Les sites cationiques
Dans la structure des zéolithes de type faujasite NaX, les cations de compensation sont des cations sodiques. Ces cations peuvent être échangés très facilement et de manière réversible [67]. L’échange peut être total ou partiel et les cations échangés sont généralement les suivants : Li+, Na+, K+, Rb+, Cs+, NH4+, H+, Ca2+, Mg2+, Ba2+. L’échange cationique
Na+/NH4+ permet d’obtenir la zéolithe (NH4)FAU qui donne par décomposition thermique la
forme protonée HFAU.
Les positions moyennes du cation au sein du réseau ont été déterminées par des études en diffraction de rayons X [68] et de neutrons [69]. Les cations sont mobiles au sein de la zéolithe et leur distribution dépend de certains facteurs comme le degré d’hydratation, la température, la présence des molécules adsorbées ou les échanges cationiques.
C3v
III II
II’ I’ I
Figure 11 : représentation des sites cationiques Na+ (○) dans la structure de la faujasite. A l’exception du site III, localisé au niveau des fenêtres de la supercage (figure 11), les sites cationiques se trouvent sur les axes cristallographiques ternaires de la faujasite :
•Le site I se trouve au centre du prisme hexagonal (16 positions par maille).
•Le site II se trouve dans la supercage centré sur les fenêtres hexagonales du bloc sodalite (32 positions par maille).
Les sites I’ et II’ se situent dans les cavités sodalites. Ils occupent les positions symétriques respectivement des sites I et II par rapport aux faces hexagonales du bloc sodalite. La présence des sites III est spécifique à la structure des zéolithes X. En revanche, le taux
Page 26
d’occupation de chaque site dépend du nombre de cation extra réseau et donc du rapport Si/Al et du type de cation [68,70].
4. 3. 2.
Les sites protoniques
Les groupements hydroxyles formés par les protons liés aux atomes d’oxygène du réseau sont responsables de l’acidité de Brönsted des zéolithes faujasites. Deux types de groupements hydroxyles sont dénombrés en fonction de leur localisation et de leur accessibilité aux molécules adsorbées [71]. Les différents types d’atomes d’oxygène sont représentés sur la figure 12.
1 2
3 4
Figure 12 : représentation des différents types d’atomes d’oxygène (●) dans la structure de la faujasite.
oLes groupements hydroxyles relatifs aux atomes d’oxygène de type O1 et O4 pointant
vers l’extérieur de la supercavité.
oLes groupements hydroxyles relatifs aux atomes d’oxygène de type O2 et O3 pointant
vers l’extérieur de cavité sodalite.
L’affinité relative du proton pour les différents types d’atomes d’oxygène obéit à la séquence O1 > O2 > O3 en fonction des échanges progressifs protons/cations. Il y a en moyenne 3 à 4
protons par supercage dans le cas des zéolithes non désaluminées. Ces protons sont directement accessibles aux molécules (surtout organiques) adsorbées dans la supercage et sont considérés comme les sites de Brönsted les plus acides.
Cependant dans le cas de l’adsorption du CO2, seuls les sites cationiques (responsables
Chapitre 1 : Etude bibliographique
Page 27
augmentation des sites basiques, par insertion d’oxydes basiques, sur ce type de matériau augmenterait les quantités de CO2 piégées [72].
4. 4.
Adsorption du CO
2sur les zéolithes
Le CO2 fait généralement l'objet d'une adsorption physique (physisorption) et d’une
adsorption chimique (chimisorption) dans la porosité des zéolithes [36]. Les zéolithes sont reconnues pour être des puissants adsorbants du CO2 [37]. Les zéolithes les plus connues qui
ont de bonnes capacités d'adsorption du CO2 à haute pression et basse température sont :
H-ZSM-5, M-H-ZSM-5, 13X et NaX [73-75]. De même, Peter et al. [76] ont étudié l’adsorption du CO2 sur des zéolithes synthétiques, 13X, NaY, HZSM-5-30, HiSiv 3000 et HY-5 (figure 13).
Selon eux, les capacités d’adsorption du CO2 varient de 1,2 à 4,5 mmol.g-1 à 298 K et 1 bar.
De plus, la zéolithe 13X serait le meilleur adsorbant [36,37,76].
En comparaison d’autres adsorbants solides tels que les oxydes ou les hydrotalcites, les zéolithes ont des propriétés régénératrices intéressantes. Elles retrouvent leurs capacités d’adsorption après plusieurs cycles d’adsorption / désorption sans dégradation importante du matériau [36]. Le plus souvent, la régénération des zéolithes peut être faite par variation de pression (PSA : pressure swing adsorption) ou de température (TSA : temperature swing adsorption).
Page 28
Ainsi, Aguila-Armenta [77] compare les isothermes d’adsorption du CO2 obtenues sur une
zéolithe naturelle ZAPS (IRI) avant et après régénération (figure 14).
Figure 14 : Isothermes d’adsorption du CO2 de la zéolithe naturelle ZAPS. Zéolithe
fraiche (●), après régénération (○) [77].
Les résultats obtenus indiquent une perte de capacité inférieure à 0,5 mmol.g-1 (CO2 adsorbé
de manière irréversible) à 298 K en PSA. Cette quantité serait liée au CO2 chimisorbé. Ils
indiquent également que le dioxyde de carbone chimisorbé désorberait totalement quand la température augmente à 393 K. Ruthven et al. [14] indiquent, en mesurant les quantités désorbées à 308 et 573 K sur une zéolithe de type X, que les quantités de CO2 irréversibles
sont négligeables (environ 0,1 mmol.g-1). De même, Peter [76] indique que les capacités d’adsorption du CO2 sur la zéolithe 13X seraient les mêmes (même isotherme d’adsorption)
en la régénérant à 473 K pendant 12 h.
Toutefois, les capacités d’adsorption du CO2 sur les zéolithes peuvent être influencées
Chapitre 1 : Etude bibliographique
Page 29
4. 5.
Influence
des
caractéristiques
structurales
des
zéolithes sur l’adsorption du CO
2L'adsorption du CO2 sur zéolithe est influencée par divers paramètres liés aux
caractéristiques structurales, à savoir [37] :
- les propriétés basiques, - l’échange cationique, - La taille des pores, - Le rapport Si/Al.
4. 5. 1.
Influence des propriétés basiques des zéolithes et du
cation échangeable
Les propriétés basiques des zéolithes viennent des cations qui permettent de capturer fortement des molécules acides en modifiant la densité électronique des atomes d’oxygène de la structure [78,79]. La force de ces sites augmente avec l’électropositivité des cations échangeables [80]. Certains travaux dans la littérature [81,82] ont montré que la force basique des zéolithes cationiques (faujasite Y et X) contenant les cations du groupe 1A augmente comme suit : Li+ < Na+ < K+ < Rb+ < Cs+.
Un autre exemple est la diminution de la basicité des atomes d’oxygènes des zéolithes NaX et NaY lors de la substitution de Na+ par le cation Ba2+ [78]. Ce phénomène pourrait être causé par une diminution partielle de la charge négative des atomes d'oxygènes adjacents aux cations Ba2+.
L’adsorption des gaz par des zéolithes est aussi influencée par la force de polarisation et la distribution des cations échangeables. La taille et le nombre des cations influencent le champ électrique local et la polarisation des molécules adsorbées sur la zéolithe [83]. En général, la force de polarisation est inversement proportionnelle aux rayons ioniques [84]. Khelifa et al. [31] ont montré que l’énergie d’interaction entre le CO2 et les sites cationiques
de la zéolithe M-ZSM-5 (M= Li+, Na+, K+, Rb+, Cs+) diminue avec une augmentation de la taille des cations. Par conséquent, plus le cation est petit, plus il a des chances d’interagir fortement avec le CO2. De même, Krista et al. [61] indiquent une augmentation des capacités
Page 30
Figure 15 : Isothermes d’adsorption du CO2 obtenues sur des zéolithes X échangées à
298 K [61].
Khelifa et al. [31,85] ont observé une diminution de l'affinité d’adsorption du CO2, sur une
zéolithe de type X, lorsque les cations Na+ sont échangés par les cations M2+ (Mg2+, Sr2+, Zn2+, Cu2+). Cependant, il existe une augmentation de l'affinité d'adsorption du CO2 lorsque le
degré d’échange de Na+ augmente. Coughlan [86] et Khelifa [87] ont également montré une diminution de l'affinité d’adsorption après une introduction de cations trivalents (Fe3+, Y3+, Cr3+ et Co3+) dans les zéolithes de type X, Y et A.
Dans le cas de la zéolithe NaX, les ions sodium qui sont accessibles aux molécules de CO2, sont localisés dans deux sites : les sites II qui sont fortement liés à la structure
zéolithique et les sites III qui sont moins liés et responsables de l'hétérogénéité énergétique de l’adsorption du CO2 [73]. Khelifa [31,85] a également montré que cet effet intervenait
seulement quand le taux d’échange était supérieur à 40 - 50%. Ainsi, le nombre de cations qui sont en mesure d'interagir avec les adsorbats a également une importance dans le processus d'adsorption.
Par conséquent, les sites cationiques des zéolithes sont des sites d’adsorption du CO2
![Figure 8 : Profils TPD du CO 2 obtenus sur MgO calciné à différentes températures [43]](https://thumb-eu.123doks.com/thumbv2/123doknet/7934125.265784/30.892.285.609.352.691/figure-profils-tpd-obtenus-mgo-calciné-différentes-températures.webp)
![Figure 9 : Structures de quelques zéolithes, montrant la structure des canaux et de cavités de chaque type de zéolithe [63]](https://thumb-eu.123doks.com/thumbv2/123doknet/7934125.265784/35.892.139.759.114.551/figure-structures-zéolithes-montrant-structure-canaux-cavités-zéolithe.webp)
![Figure 14 : Isothermes d’adsorption du CO 2 de la zéolithe naturelle ZAPS. Zéolithe fraiche ( ● ), après régénération ( ○ ) [77]](https://thumb-eu.123doks.com/thumbv2/123doknet/7934125.265784/40.892.213.680.202.535/figure-isothermes-adsorption-zéolithe-naturelle-zéolithe-fraiche-régénération.webp)