BRET MACHA
Synthesis, Characterization, and Coordination of a
Boratabenzene-Phosphine Ligand with Group 10
Transition Metals
Mémoire présenté
à la Faculté des études supérieures et postdoctorales de l’Université Laval dans le cadre du programme de maîtrise en chimie
pour l’obtention du grade de Maîtrise ès sciences (M.Sc.)
DÉPARTEMENT DE CHIMIE FACULTÉ DES SCIENCES ET DE GÉNIE
UNIVERSITÉ LAVAL QUÉBEC
2012
Résumé
Les ligands à base de borabenzène/boratabenzène, uniques en leur genre, demeurent l’un des intérêts centraux du groupe Fontaine en raison de leur aromaticité et de leur stabilité en tant que ligands acides de Lewis. La synthèse d’une phosphine de type L possédant un groupe boratabenzène (di-tert-butylphosphidoboratabenzène) a été faite en deux étapes. D’abord, l’espèce neutre borabenzène-di-tert-butylchlorophosphine est préparée à partir de la di-tert-butylchlorophosphine et de la 1-chloro-2-(triméthylsilyl)boracyclohexa-2,5-diène. La réduction de l’intermédiaire neutre avec du potassium génère le sel de potassium équivalent (di-tert-butylphosphidoboratabenzène de potassium). Des complexes de coordination de métaux du groupe 10 présentant des interactions bis-di-tert-butylphosphidoboratabenzène ont été préparés. Les données cristallographiques des complexes de nickel et de platine correspondants montrent qu’une pléiade de modes de liaisons peuvent être atteints avec le ligand obtenu. La voie réactionelle, ainsi que les calculs de théorie de la functionelle de la densité, permettront de démontrer les qualités de ce ligand en tant qu’excellent donneur d’électrons. Le but ultime demeure non seulement l’étude des interactions se déroulant dans la sphère de coordination d’un complexe montrant un groupement borabenzène/boratabenzène, mais aussi l’étude de leur capacités à activer des petites molécules à base d’éléments du groupe principal.
Abstract
Borabenzene/boratabenzene transition metal systems remain of active interest within the Fontaine group due to a unique combination of aromaticity and stability within a Lewis acidic ligand framework while in a complexed state. Augmentation to a classic L-type phosphine possessing a boratabenzene element has been achieved (tert-butylphosphidoboratabenzene). This ligand was successfully synthesized by reaction of
di-tert-butylchlorophosphine with 1-chloro-2-(trimethylsilyl)boracyclohexa-2,5-diene which
yielded the neutral borabenzene-di-tert-butylchlorophosphine species. Reduction of this borabenzene-di-tert-butylchlorophosphine species with potassium then yielded the target ligand coordinated to potassium (potassium di-tert-butylphosphidoboratabenzene). Utilizing this new phosphine, reactions were carried out where the species was successfully coordinated within three group 10 metal complexes. Crystallographic data from these nickel and platinum bis-phosphine complexes have shown that a variety of binding modes for this ligand with metal centers can be achieved. The synthetic process along with density functional theory calculations will be discussed. The end goal is synthesis of new transition metal complexes displaying prominent borabenzene/boratabenzene moieties and investigating their interactions within the coordination sphere of metal centers as well as their ability to activate small molecules composed of main group elements.
Table of Contents
Résumé...………..………..………..………...ii Abstract...……….………..………..………...iii Table of Contents………..……….…………..………....iv List of Tables………...…...….………...vi List of Figures………....…….……….vii List of Schemes………...………....ix List of Abbreviations………..…...………....xi List of Compounds………...……...………xiii Preface………..………...xiv Chapter 1 – Introduction………....………..1 1.1 General………....……….1 1.2 Borabenzene-Boratabenzene………...………1 1.2.1 Structure………....………11.2.2 Synthesis of Borabenzene/Boratabenzene Species………....…………...6
1.2.3 Binding Modes of Borabenzene/Boratabenzene to Transition Metals…...……..8
1.3 Phosphine Background………...…...12
1.3.1 General Statements………...…..12
1.3.2 Tolman Electronic Parameter………...……..12
1.3.3 Tolman Angle and Solid Angle………...……...14
1.4 Group 10 Metal Complexes………...…....16
1.4.1 Electronic Structure………...…….16
1.4.2 Oxidation States and Common Geometries………....…....16
1.4.3 Applications to Carbon Dioxide Coordination and Activation…………...…...17
1.5 Zwitterionic Transition Metal Species………....…...19
1.5.1 Overview………....…….19
1.5.2 Development of Borate Based Anionic Ancillary Ligand Structures…...….20
1.5.3 Group 10 Zwitterionic Complexes………...…..22
1.6 Project Outline………...…....23
1.6.1 Fundamentals………...…...23
1.6.2 Making Nickel a Viable Alternative………...…....24
Chapter 2 – Experimental Techniques………...………....…...25
2.1 Nuclear Magnetic Resonance Spectroscopy………....…..25
2.2 X-Ray Diffraction………...……...27
2.3 Density Functional Theory………...…….29
2.4 Inert Atmosphere Manipulations………...…...29
2.4.1 Schlenk Techniques………...…….30
2.4.2 Glove Box Techniques………...31
Chapter 3 – Synthesis and Characterization of Potassium Di-tert-butyl-phosphidoboratabenzene (K+(DTBPB)-)………....….…..32
3.1 Obscure Binding Modes for Boratabenzene………...…...32
3.2 DFT Calculations of Phosphine Donation………...…..35
3.3 Synthesis of Potassium Di-tert-butylphosphidoboratabenzene...38
3.3.2 Reactivity Studies for Creation of Di-tert-butylchlorophosphidoborabenzene
(DTBCPB) ………...………...40
3.3.3 Reduction of Di-tert-butylchlorophosphidoborabenzene (DTBCPB) …....…...46
3.4 Crystal Structures of Di-tert-butylphosphidoboratabenzene Ligand………....….48
Chapter 4 – Ligand Reactions with Group 10 Metals………...53
4.1 Literature Precedent for Metal Reactions………...………...53
4.2 Synthetic Route and Characterization………...………....53
4.3 Reactions with NiX2 Precursors………...……….55
4.4 Reactions with PtX2 Precursors………...…………..66
4.5 Test Reactivity for Complexes………...………...76
Chapter 5 – Conclusions...………...……...79
5.1 Immediate Conclusions………...…..79
5.2 Future Work………...80
Chapter 6 – Experimental Results………...………..82
6.1 General………....………...82
6.2 Chapter 3 Species………...………...83
6.2.1 General Considerations………....………...83
6.2.2 Synthesis of the Boracycle Starting Material………...…………..83
6.2.3 Synthesis of K+(DTBPB)-………...…………85
6.3 Chapter 4 Species………...……...87
6.3.1 General Considerations………...87
6.3.2 Preparation of Group 10 Metal Reagents……….…...87
6.3.3 Synthesis of Bis-Phosphine Group 10 Metal Products………....…...88
6.3.4 Bis-Phosphine Metal Complex Activation Testing……….…...91
6.3.5 Products Synthesized in Chapter 5.2 – Future Work………...93
Bibliography………...………...………...95
Appendix A – DFT Calculations………..……...……….100
Appendix B – Crystallographic Data………...……...104
List of Tables
Table 1-1. Stretching frequencies of various carbonyl complexes…....………..13
Table 1-2. TEP data for common phosphines………...………..14
Table 1-3. Chart referencing Tolman angles to the solid angle values………...…....15
Table 1-4. Table of metal carbon dioxide captures………...……..18
Table 2-1. Nuclei of interest with basic information………...……....25
Table 2-2. NMR resonance frequencies of common nuclei………...……….26
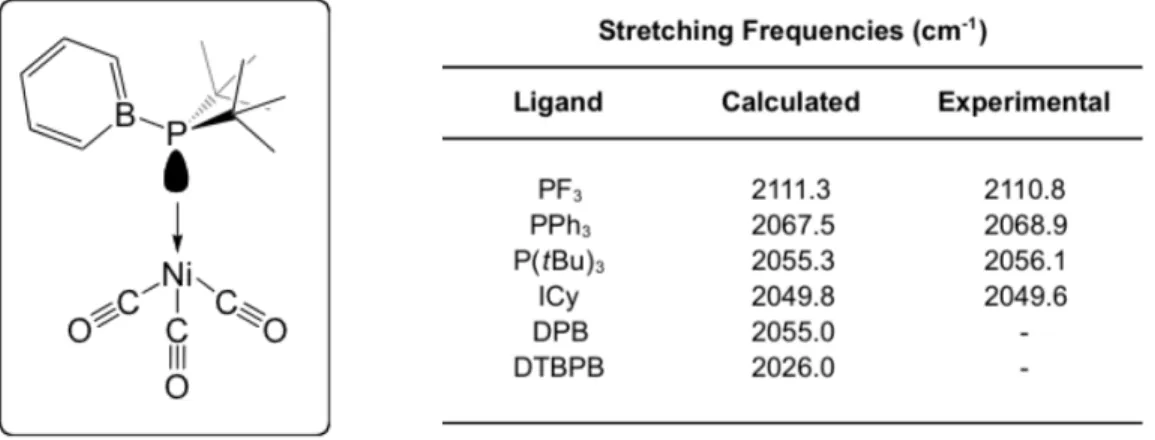
Table 3-1. Gusev’s plot of experimental versus calculated νCO stretching frequencies along with a fitted linear correction coefficient…………...36
Table 3-2 A few selected νCO stretching frequencies of phosphine and carbene ligands compared to the DFT results for DTBPB………...…...37
Table 3-3. Boratabenzene species synthesized by exchange mechanisms with borabenzene-trimethylphosphine………...42
Table 3-4. Selected bond distances and bond angles for K+(DTBPB)-………...……49
Table 3-5. Selected bond distances, bond angles, and torsion angles for K+(DTBPB) -(18-crown-6) compared to data for K+(DPB)-(18-crown-6) and K+(DAB) -(18-crown-6)………...……51
Table 4-1. MX2 species used in bis-phosphine reactions………...…………...54
Table 4-2. NMR assignment for DPB boratabenzene protons………...……...58
Table 4-3. Selected bond distances bond angles for Ni2(DTBPB)2…………...…….62
Table 4-4. Calculated bond distances for the DFT structures Ni(DTBPB)2 and Ni2(DTBPB)2 relative to X-ray structure reported for Ni2(DTBPB)2…....66
Table 4-5. Selected bond distances and bond angles for Pt(DTBPB)2…………...68
Table 4-6. Structural information for [(tBu3P)Pt{η3-BCC-VBPh}] and [(tBu3P)2Ni{η3 -BCC-VBPh}]...70
Table 4-7. Structural information for [Pd(TXPB)] and [Ni(TXPB)]...72
Table 4-8. Calculated bond distances and bond angles for the DFT structures Pt(DTBPB)2 and Pt2(DTBPB)2 relative to X-ray structure reported for Pt(DTBPB)2………....…...73
Table 4-9. Selected bond distances and bond angles for Pt(COD)(DTBPB)2…....….75
Table A-1. DFT Data for Ni(DTBPB)2………...…...100
Table A-2. DFT Data for Ni2(DTBPB)2………...101
Table A-3. DFT Data for Pt(DTBPB)2………...…....102
Table A-4. DFT Data for Pt2(DTBPB)2………...103
Table B-1. Crystal data and structure refinement for K+(DTBPB)-…………...……104
Table B-2. Crystal data and structure refinement for K+(DTBPB)-(18-crown-6)...105
Table B-3. Crystal data and structure refinement for Ni2(DTBPB)2…………...106
Table B-4. Crystal data and structure refinement for Pt(DTBPB)2………....…107
List of Figures
Figure 1-1. Varying electronic structures of borabenzene, benzene, and pyridine…...2
Figure 1-2. Representation of a classic Lewis base stabilizing the aromatic system of a borabenzene ring...………...……3
Figure 1-3. Structural representations and nomenclature of dative versus anionic Lewis base interactions with borabenzene………...…4
Figure 1-4. Representations of lone pair orbital overlaps of common anionic (X) Lewis basic substituents on boratabenzene systems ………...……5
Figure 1-5. Charge representations of the second period heterocycles…………...…....6
Figure 1-6. ORTEP structure of the first neutral Lewis base-borabenzene adduct, borabenzene-pyridine………....…....8
Figure 1-7. Comparison between a classic three-legged piano stool complex and a borabenzene facsimile………....…...8
Figure 1-8. Diagram of degree of slip distortion in boratabenzene complexes……...9
Figure 1-9. Exerts from group 8 and 9 borabenzene/boratabenzene transition metal complexes………...…....10
Figure 1-10. Exerts from group 10 boratabenzene transition metal complexes……...10
Figure 1-11. Exerts from group 5 and 6 borabenzene/boratabenzene transition metal complexes………...…....11
Figure 1-12. Exerts from group 3 and 4 borabenzene/boratabenzene transition metal complexes………...11
Figure 1-13. Diagram of boratabenzene-phosphine ligand complexes………...12
Figure 1-14. Scaffold of the complex used to test IR stretching frequencies for TEP characterization………...14
Figure 1-15. Schematics for Tolman angle and solid angle steric parameters……...….15
Figure 1-16. Representations of group 10 metal complexes in their M(IV) oxidation state………...…..17
Figure 1-17. ORTEP structure of (carbon dioxide)-bis(tricyclohexylphosphine) nickel………...18
Figure 1-18. Basic mock for a zwitterionic platinum group metal complex…….…...19
Figure 1-19. Cationic rhodium complex versus the unexpected zwitterionic rhodium borate species………....………...21
Figure 1-20. Diagram of tridentate phosphine-borate zwitterions………....……...…...22
Figure 2-1. Geometric representation of Bragg’s Law………...…..27
Figure 3-1. Representations of obscure binding modes for borabenzene/boratabenzene………...32
Figure 3-2. Diagram of boratabenzene-phosphine ligand complexes reported by Fu………...33
Figure 3-3. Crystal structure comparisons between DPB and DAB………....….34
Figure 3-4. Structural comparison between DPB and DTBPB………....…….35
Figure 3-5. Mock of DTBPB coordinated to LNi(CO)3………...37
Figure 3-6. DFT images for HOMO and HOMO-1 orbitals for DTBPB and DPB………...37
Figure 3-7. Comparisons between the 31P NMR spectra of borabenzene-trimethylphosphine and borabenzene-di-tert-butylchlorophosphine…...45
Figure 3-8. The conversion of 1-chloro-2-(trimethylsilyl)boracyclohexa-2,5-diene to
di-tert-butylchlorophosphidoborabenzene………...……..46
Figure 3-9. ORTEP structural representation of K+(DTBPB)-………...……..49
Figure 3-10. Full diagram of the structure showing the three coordinate nature of the K+(DTBPB)- ligand to potassium………....……...50
Figure 3-11. ORTEP structural representation of K+(DTBPB)- coordinated to 18-crown-6………...……...…51
Figure 4-1. 1H NMR spectrum for what is believed to be H-P(tBu) 2(BC5H5)……...56
Figure 4-2. 31P NMR spectrum for the reaction of K+(DTBPB)- and NiCl 2(DME)...57
Figure 4-3. 1H NMR spectrum of K+(DTBPB)-(18-crown-6) with NiBr 2(PPh3)2…....57
Figure 4-4. Extracted crude from the NiBr2(PPh3)2 reaction with known isolated boratabenzene products, side products, and reagents………...58
Figure 4-5. [M]DPB proton assignment scheme………...58
Figure 4-6. Proposed structures of the possible Ni(DTBPB)2 species………...59
Figure 4-7. 1H NMR data for green and red fractions of reaction with NiBr2(PPh3)2………...59
Figure 4-8. 31P NMR data for the red fraction from the reaction for Ni2(DTBPB)2...61
Figure 4-9. ORTEP structural representation of Ni2(DTBPB)2………...61
Figure 4-10. 1H NMR spectrum for reaction with 1:1 ratio of K+(DTBPB)- with NiBr2(PPh3)2………...63
Figure 4-11. Dimeric phosphine-alkynyl-borane nickel(0) species published by Stephan along with a POV-ray drawing of species………...65
Figure 4-12. DFT optimized structures for Ni(DTBPB)2 and Ni2(DTBPB)2……...…..65
Figure 4-13. ORTEP structural representation of Pt(DTBPB)2………...67
Figure 4-14. VT-NMR data for Pt(DTBPB)2………...68
Figure 4-15. [ScCl(3,5-Me2C5H3BNMe2)2]2 reported by Herberich………...69
Figure 4-16. [(tBu3P)Pt{η3-BCC-VBPh}] and [(tBu3P)2Ni{η3-BCC-VBPh}] reported by Emslie………...70
Figure 4-17. ORTEP structures for [(tBu3P)Pt{η3-BCC-VBPh}] and [(tBu3P)2Ni{η3 -BCC-VBPh}]...70
Figure 4-18. [Pd(TXPB)] and [Ni(TXPB)] as reported by Emslie...71
Figure 4-19. ORTEP structures for [Pd(TXPB)] and [Ni(TXPB)]...71
Figure 4-20. DFT optimized structures for Pt(DTBPB)2 and Pt2(DTBPB)2...72
Figure 4-21. NMR data for reaction synthesizing Pt(COD)(DTBPB)2………...74
Figure 4-22. ORTEP structural representation of Pt(COD)(DTBPB)2………...74
Figure 4-23. Comparisons of Pt(COD)(DTBPB)2 versus Ni(COD)(C5H5BPh)……...76
Figure 5-1. Comparisons of Ni2(DTBPB)2 and Pt(DTBPB)2………...79
Figure 5-2. Proposed Mes* functionalized di-tert-butylphosphidoboratabenzene…...80
List of Schemes
Scheme 1-1. Flash thermolysis scheme for testing viability of “free”
borabenzene………...2
Scheme 1-2. Three proposed mechanisms for borabenzene exchange reactions……...3
Scheme 1-3. Reaction showcasing the acidity of the 1-methylboracyclohexadiene versus cyclopentadiene………...…...5
Scheme 1-4. Initial synthesis of a boron planar heterocycle………...………..6
Scheme 1-5. Synthesis by Ashe of the phenylboratabenzene adduct………...….7
Scheme 1-6. Innovation by Schmidt utilizing a species which reacts with neutral Lewis bases resulting in a borabenzene-base interaction………...7
Scheme 1-7. Reaction used to synthesize the first borabenzene neutral Lewis base adduct………....……7
Scheme 1-8. Practical application of a “simple salt reaction”………...……9
Scheme 1-9. Practical application of a “boratabenzene ring exchange”…………...…..10
Scheme 1-10. Reaction performed by Aresta capturing carbon dioxide…………...…....17
Scheme 1-11. Catalytic reaction demonstrating the hydroformylation of p-isobutylstyrene………...….21
Scheme 1-12. Reactivity profile for the Rh(COD)BPh4 zwitterion………...22
Scheme 1-13. Reaction scheme for bidentate group 10 zwitterions………...23
Scheme 3-1. Roadmap for creation of neutral borabenzene adducts………....…...38
Scheme 3-2. Simplified reaction for reduction of DTBCPB to M+(DTBPB)-……...….38
Scheme 3-3. Cyclization reaction used to synthesize large quantities of the 1-chloro-2-(trimethylsilyl)boracyclohexadiene precursor………...…...39
Scheme 3-4. Reaction for common neutral borabenzene compounds utilizing Lewis basic reagents………...…...40
Scheme 3-5. Fu nucleophilic method to synthesize boratabenzene adducts………...41
Scheme 3-6. Reaction for K+(DTBPB)- utilizing the exact method used by Fu…...…..41
Scheme 3-7. Reaction testing ability to effectively reduce di-tert-butylchlorophosphine with solid potassium………...……42
Scheme 3-8. Wurtz coupling side reaction or phosphine-halide reductions……....…....43
Scheme 3-9. Testing for synthesis of di-tert-butylchlorophosphidoborabenzene…...…43
Scheme 3-10. Test reaction for the borabenzene exchange of PMe3 with PtBu2Cl...44
Scheme 3-11. Reaction used to effectively generate di-tert-butylchloro-phosphidoborabenzene in situ for subsequent reductions………...46
Scheme 3-12. Reduction for DTBCPB to K+(DTBPB)-………...47
Scheme 3-13. Synthesis of K+(DTBPB)-(18-crown-6)……….…....….48
Scheme 4-1. Reaction by Fu coordinating a reduced phosphido-boratabenzene ligand (DPB) onto a metal center………...53
Scheme 4-2. Basic mock for bis-phosphine reactions on Group 10 metals…………....54
Scheme 4-3. Original reaction for synthesis of Ni(DTBPB)2 from NiCl2…………...55
Scheme 4-4. Reaction of K+(DTBPB)- with NiCl 2(DME)………...56
Scheme 4-5. NMR reaction between K+(DTBPB)-(18-crown-6) and NiBr2(PPh3)2………...57
Scheme 4-6. Synthetic scheme for the synthesis of Ni2(DTBPB)2 ………...60
Scheme 4-8. Reaction for dimeric nickel species reported by Herberich………....…....64
Scheme 4-9. Reaction yielding the bis-phosphine Pt(DTBPB)2 species…...67
Scheme 4-10. Reaction utilizing PtCl2(COD)………...…....73
Scheme 4-11. Test reactions for activation of carbon dioxide………...…...76
Scheme 4-12. Diagram of Tilley Si-H activation reaction………...………...…..77
Scheme 4-13. Test reactions for the activation of a Si-H bond………...77
Scheme 4-14. Test reactions for the activation of a Si-H bond………...77
Scheme 4-15. Test reactions for the activation of a P-H bond………...78
Scheme 4-16. Test reactions with chromium(0) hexacarbonyl (Cr(CO)6)…………...78
Scheme 5-1. Reaction scheme to block ring coordination of the phosphine………...…80
List of Abbreviations
Å Angstrom (bond length) Bcat catecholatoboryl
bp boiling point br broad (spectral) CCD charge coupled device COD 1,5-cyclooctadiene Cp cyclopentadienyl
Cy cyclohexyl
d doublet (spectral) dn electron configuration
DFT Density Functional Theory DME dimethoxyethane DPB diphenylphoshido-boratabenzene anion DTBCPB di-tert-butylchloro-phosphidoborabenzene DTBPB di-tert-butylphosphido-boratabenzene e- electron
E main group element GC gas chromatagraphy
h hour
HMQC Heteronuclear Multiple Quantum Coherence HOMO Highest Occupied
Molecular Orbital
Ι nuclear spin
iPr iso-propyl
IR infrared
J coupling constant (Hz) L neutral (2-electron) donor
ligand
LUMO Lowest Unoccupied Molecular Orbital m- meta M metal, molar Me methyl Mes mesityl, 2,4,6-trimethylphenyl min minute mp melting point MS mass spectroscopy o- ortho OTf trifluoromethane-sulfonate nBu n-butyl NMR Nuclear Magnetic Resonance Nu nucleophile
ORTEP Oak Ridge Thermal Ellipsoid Plot
p- para
Ph phenyl
ppm parts per million
rt room temperature s singlet (spectral)
t triplet
tBu tert-butyl
TEP Tolman Electronic Parameter
THF tetrahydrofuran TMS trimethylsilyl
X halogen, anionic (1 electron) donor ligand Z Lewis acid (2 electron
acceptor) Δ heating δ chemical shift (ppm) η hapticity (π-coordination) θ solid angle (°) κ hapticity (σ-coordination) λ wavelength μ bridging ν frequency π pi (bonding) σ sigma (bonding) ϕ angle (spherical coordinates)
Preface
First and foremost, I would like to thank the professors who have influence my journey throughout my path in chemistry. Without wonderful teachers and guides educating and encouraging young researchers, the world would cease to evolve in culture and technology. Especially helpful in my academic growth are my previous professors at Texas A&M University, especially Drs. John Hogg, François Gabbaï, and John Gladysz.
Most importantly within this preface, I would like to extend a tremendous amount of gratitude and thanks to my professor, mentor, and friend Frédéric-Georges Fontaine. In a world of often overbearing personalities, his guidance and care for the well-being and development of his students can truly never be underestimated. I would specifically like to thank him for his faith in accepting me into the group and entrusting me with this wonderful project.
For technical help I graciously would like to thank all of the members of the chemistry department staff especially Pierre Audet for his aid in NMR spectroscopy. I would also specifically like to thank Chris Garon and Christian Tessier for their help interpreting data from X-ray crystallography. Thanks are also due to Josée Boudreau and Laurent Maron for work with DFT calculations.
Finally, I would like to thank all the members (past and present) of the Fontaine group with whom I have had the utmost pleasure in working with. I would especially like to thank Guillaume Bélanger-Chabot, Stephanie Barnes, Marc-André Légaré, Philippe Rioux, and Viridiana Perez for sharing in the love/hate relationship of borabenzene chemistry. My time in Canada was short, but the memories were profound! I wish you all the best and
To students who trust that seamanship acquired in placid, charted waters will contribute to their handling of craft on troubled seas.
W. Mansfield Clark
Chapter 1 – Introduction
1.1 General
As a practical matter for fundamental understanding of the chemistry discussed in this thesis, the following chapter is meant to clarify underlying concepts used within this research project. A proper background and detailed history of the development and synthesis of borabenzene ligand substituents will be provided. Also, discussion of pertinent information concerning phosphine applications in organometallic chemistry, group 10 organometallic complexes, and zwitterionic complexes will also be included. Ultimately this chapter will introduce concepts that should be kept in mind as discussion progresses in regard to applications and uses for this chemistry project as a means for research knowledge and understanding, as well as a practical scheme for new industrial catalyst design.
The last section of the introduction will attempt to condense the knowledge of the previous chapters into a focused view of the project goal and knowledge that was initially sought when synthesis began. Although discussion of experimental results will be provided with literature backgrounds in the appropriate chapter, this section should be thought of as a means to understand where the project’s completion intends to head in the future and why the initial project goal is still attainable and desired.
1.2 Borabenzene-Boratabenzene
1.2.1 Structure
Borabenzene, as the name suggests, is a 6 π-electron aromatic analog of the classic benzene ring with boron substituted for one of the carbon atoms. Structurally the system is analogous to both benzene and pyridine with all six atomic members of the ring structurally planar. Boron remains σ-bonded to the two adjacent carbons with its last valence electron involved in the aromatic π-system delocalized over boron and the five carbons as illustrated
in Figure 1.1. With all three valence electrons of boron involved in bonding interactions with the neighboring carbons, an unoccupied sp2 orbital emerges within the plane of the
ring resulting in boron’s electron deficient nature. This unoccupied low energy orbital on boron allows for a coordination site which can interact with various compounds possessing a lone pair of electrons [1].
Figure 1-1. The electronic structures of (a) borabenzene, (b) benzene, and (c) pyridine
The exchange of a boron atom within the normally homoaromatic benzene framework of carbon atoms imparts drastically altered stability and electronic properties to the superstructure of the molecular system. With two fewer valence electrons versus the benzene analog, borabenzene (while structurally stable and aromatic by density functional theory calculations) is always experimentally witnessed stabilized by an exocyclic pair of electrons supplied by a Lewis base (see Figure 1-2). Borabenzene species not stabilized by an exocyclic adduct are thought to be extremely reactive and unstable [1]. Some flash thermolysis studies however (see Scheme 1.1), were performed in which a product was condensed onto a 10K cold window and was characterized as a possible borabenzene-N2
product by infrared spectroscopy, however, this adduct can still be classified as a formal Lewis base adduct [2]. As to the date of this thesis, all practical attempts to synthesize a “free” non-Lewis base coordinated borabenzene species have met with failure.
Scheme 1-1. Flash thermolysis scheme showing the detectable molecules synthesized from
Figure 1-2. Representation of a classic Lewis base stabilizing the aromatic system on the
borabenzene ring via lone pair of electrons occupying the vacated sp2 orbital
While stabilized by neutral Lewis bases, various borabenzene compounds are known to readily undergo exchange reactions either with other stronger Lewis bases or less bulky Lewis base adducts. The tendencies tend to predict the borabenzene Lewis acid/Lewis base systems to increase in stability with the strength of the Lewis base involved in the interaction [1, 3]. As can be seen below in Scheme 1-2, three different mechanisms were tested by Fu for the exchange reaction. Mechanistically, the exchange reaction was confirmed to proceed via an associative manner in which the borabenzene adduct interacts with a stronger Lewis base resulting in a four-coordinate boron intermediate prior to the release of the weaker Lewis base [4].
Scheme 1-2. Diagram showing three proposed mechanisms for borabenzene exchange
reactions. The top two mechanistic pathways (Grey) were disproved while the associative mechanism (Black) was experimentally confirmed [4]
boron on borabenzene, the overall charge of the borabenzene complex remains neutral. However, depending upon the nature of the bond between the boron and interacting group outside of the conjugated ring system, the boron atom can be witnessed to adopt a formally anionic charged state (see Figure 1-3). In borabenzene compounds where the stabilizing fragment bound to boron forms a covalent interaction rather than Lewis base donation, the ring system is witnessed to become formally anionic. This anionic charge is observed to be centered predominantly on the two carbons flanking the boron atom within the ring, which allows the boron atom to remain electrophilic in nature. These anionically charged species are referred to as boratabenzene species and are witnessed to behave drastically different than their neutral analogs [1].
Figure 1-3. Representations of (a) dative versus (b) anionic Lewis base interactions with
borabenzene along with subsequent charge formations and nomenclature [1]
The charge from the covalent X fragment on the boron allows for the electron density to shift towards the five carbons on the ring and not occupy the π-system on boron as completely as is observed in the neutral form. This anionic charge dramatically changes the behavior of these ring systems when coordinated onto transition metals and has been shown to exhibit behavior more analogous to the common cyclopentadiene ligand family. In 1996, Herberich published a study finding that 1-methylboracyclohexadiene is even more acidic than cyclopentadiene in solution (see Scheme 1-3) [5]. The anionic form of borabenzene is thought to be more stable than the neutral form and will also cause the ligand to lose its ability to exchange with other Lewis bases [1].
Scheme 3. Reaction showcasing the increased acidity of the
1-methylboracyclohexadiene versus cyclopentadiene [5]
In boratabenzene systems, the nature of the exocyclic interaction with boron can vary dramatically depending upon the covalent species possessing a strong Lewis base lone pair of electrons as well as overall size of the atom involved in the interaction.
Figure 1-4. Representations of lone pair orbital overlaps of anionic (X) Lewis base
adducts on boratabenzene systems: (a) amido interactions (b) phosphido interactions [6] Due to the partially vacated π-system over boron resultant of the shift in electron density away from the negatively charged atom, the lone pair of electrons from an exocyclic Lewis base can readily stabilize into a π-interaction with boron [5-7]. This interaction is very common when the exocyclic fragment lies within the same period as boron (nitrogen being the most notable as seen in Figure 1-4).
Interestingly enough, just as with the covalent characteristic denoting an anionic charge upon the ring, the same charge formation also happens to covalent interactions in pyridine systems as well. The species can be seen to adopt the predicted positive charge when arranged against the benzene analogs while in their protonated forms (see Figure 1-5).
Figure 1-5. Charge representations of second period aromatic heterocycles: (a)
Hydridoboratabenzene and (c) Pyridinium relative to (b) Benzene
1.2.2 Synthesis of Borabenzene/Boratabenzene Species
Synthesis of boratabenzene complexes were originally reported in the early 1970’s with development of increasingly creative synthetic routes for both functionalization and coordination of substituents both on and off of the ring system. Starting in 1970, Herberich reported the insertion of Br2B-C6H5 into a cobaltocene complex [8]. The complex that was
formed exhibited aromatic character and led to the theory that the boracycle was isoelectric when compared to cyclopentadiene (see Scheme 1-4).
Scheme 1-4. Initial synthesis of a planar boron heterocycle [8]
Later in 1971, Ashe reported an isolated boratabenzene-phenyl adduct complexed to lithium [9]. These two discoveries marked the start of research into borabenzene chemistry; however, synthetic routes to readily access the neutral borabenzene species were not developed until later in the mid 1980’s [10-12]. Ashe’s scheme for the creation of lithium phenylboratabenzene also showcases a key innovation in synthesis of this class of structures. As can be seen, the synthetic route utilizes a stannacycle precursor which undergoes transmetallation with a boron dihalide reagent to yield a parent boracycle. This boracycle reagent can then simply undergo deprotonation to aromatize the ring into a boratabenzene product (see Scheme 1-5).
Scheme 1-5. Synthesis by Ashe of the aforementioned phenylboratabenzene adduct [9]
The first neutral borabenzene molecule to be successfully synthesized and characterized was a borabenzene-pyridine species developed through a process discovered by Schmid and coworkers in 1985 (see Scheme 1-6) [10, 11]. A key step in the development for synthetic routes to neutral borabenzene adducts was achieved by modifying the stannacycle precursor with a TMS group which could then be transmetalated with a boron dihalide species to give a stable boracycle reagent. This methoxyboracyclotrimethylsilyl species would become a key intermediate in readily accessing the up to this point neutral species in predictable and reliable fashions (see Scheme 1-7). The key step in this reaction scheme is the isomerization of the boracycle into the active intermediate which reacts with species possessing lone pairs of electrons. This type of reaction yields a neutral borabenzene compound while also irreversibly releasing TMS-OMe [10] or TMS-Cl [13] as a byproduct.
Scheme 1-6. Reaction by Schmidt utilizing a modified version of the reagent reported by
Ashe in reactions with neutral Lewis bases to displace TMS-OMe [10, 11]
Scheme 1-7. Reaction used to synthesize the first borabenzene neutral Lewis base adduct
(borabenzene-pyridine) [10, 11]
The classic Lewis-pair of borabenzene and pyridine experimentally display how the dramatic stabilizing effect of strong Lewis bases effectively occupy the empty sp2 orbital in
π-systems is completely intact with little π-overlap in the B-N bond displayed by a rather large torsion angle of 43.3° appearing between the two planes of the rings [10, 11].
Figure 1-6. ORTEP diagram of borabenzene-pyridine showing an angle between the ring
planes of 43.3° [10]
1.2.3 Binding Modes of Borabenzene/Boratabenzene to Transition Metals
When examining the known binding modes for borabenzene and boratabenzene metal complexes a pattern quickly emerges in which the most common binding mode of these species with a metal center is through the π-electron system. This aromatic electron coordination to a metal center is typically thought of as analogous to the classic arene ligand motif and borabenzene complexes traditionally exhibit many common parameters with these arene classes of transition metal complexes. This often results in formation of sandwich or half-sandwich complexes (see Figure 1-7) [1, 12].
Figure 1-7. Comparison between a (a) classic three-legged piano stool complex and a (b)
borabenzene facsimile
The charged boratabenzene version of this species however, loses the six-membered aromaticity of the ring and is often observed to coordinate to metal complexes via a “slip-distorted” coordination mode in which the ring does not sit on the true center of the metal, but rather “slips” away from the boron atom. This means that a cationic metal center will shift the center of coordination towards the five carbons and not center itself directly on the ring. In severe cases the aromaticity of the ring can be broken so that the ring will actually be hinged between the first/fifth carbon and the boron atom (see Figure 1-8). This sort of
slipped coordination is experimentally observed to increase in decentralization relative to the cationic charge of the metal coordinated to the ring [5, 7, 14, 15].
Figure 1-8. Diagram for the geometry of slip distorted boratabenzene complexes resulting
in a buckled ring structure between the C1-B-C5 carbons [15]
Because of this typical π-system coordination mode, many of the first crystallographically characterized borabenzene complexes were sandwich complexes and three-legged piano stool conformations. These complexes were typically synthesized from reactions with transition metal halides and alkali metal boratabenzenes of the form M+(C
5H5BR)- [12].
These reactions are usually witnessed to proceed cleanly affording the boratabenzene complex while displacing a simple salt as a byproduct as can bee seen in Scheme 1-8.
Scheme 1-8. Practical application of a “simple salt reaction” used to synthesize
borabenzene sandwich complexes [16]
Borabenzene/boratabenzene transition metal complexes can be crudely divided into three classes of complexes based on the types of transition metals to which they are coordinated: complexes of group 3 and 4 transition metals, group 5, 6, and 7 transition metal complexes, and group 8, 9, and 10 transition metal complexes. As was previously discussed, many of
the first borabenzene/boratabenzene complexes were derived from a cobaltocene or ferrocene framework. This is very common for the group 8 and 9 complexes as can be seen below in Figure 9. A few examples of group 10 complexes are also reported in Figure 1-10 but are far less common than the group 8 and 9 adducts.
Figure 1-9. Examples of group 8 and 9 boratabenzene metal complexes†
Figure 1-10. Examples of group 10 boratabenzene complexes‡
The second major class of borabenzene/boratabenzene complexes are taken from interactions with group 5, 6, and 7 transition metals (see Figure 1-11). They usually feature three-legged piano stool like complexes comprised of boratabenzene-triscarbonyl transition metal interactions. These species are formed from exchange of alkali or transition metal boratabenzene species with metal-carbonyl precursors [12] and are well documented for groups 5, 6, and 7 transition metals (see Scheme 1-9).
Scheme 1-9. Practical application of a “boratabenzene ring exchange” reaction [22]
†
(a) Co[17] (b) Ru[18, 19] (c) Os[20]
‡
Figure 1-11. Examples of group 5 and 6 borabenzene/boratabenzene complexes†
The third and final class of borabenzene/boratabenzene transition metal complexes are derived from interactions with group 3 and 4 transition metals (see Figure 1-12), and were developed primarily through olefin polymerization catalysis studies [1, 15, 25].
Figure 1-12. Examples of group 3 and 4 boratabenzene complexes‡
In 1996, Fu published crystallographic data displaying several transition metal species coordinated to diphenylphosphidoboratabenzene (DPB) [28]. These structures all displayed the ligand coordinated through the phosphine substituent rather than through the π-system as traditionally witnessed in every other circumstance up until that point (see Figure 1-13). His published results show that a phosphidoboratabenzene system developed to mimic PPh3 can coordinate to zirconium, rhodium, and iron through the lone pair of
electrons on phosphorus [1, 28]. A more detailed analysis of obscure binding modes of borabenzene and boratabenzene moieties with transition metals will be discussed in Chapter 3.1.
Figure 1-13. Generic diagram of boratabenzene-phosphine ligand reported by Fu [28]
The DPB ligand was also seen to experimentally be much more electron donating and strongly coordinating than triphenylphosphine. Since this property specifically applies to my project, synthesis will be discussed in chapter 3.3 along with the crystal structures of his reported ligand with the aforementioned transition metals (Zr, Fe, and Rh).
1.3 Phosphine Background
1.3.1 General Statements
Phosphines (PR3) constitute one of the best classes of L-type ligands for chemists to
synthesize a combination of both steric and electronic effects within the coordination sphere of an organometallic complex [29, 30]. Historically, use of organometallic phosphine species has led to many fruitful developments in catalysis via tuning of electron donating properties towards the metal center [29]. Phosphines are also variable in their π-acidic character, which makes them able to be strong lone electron pair donors as well as mildly weak π-acceptors (the exception is PF3, which is known to accept π-electron density
exceptionally well).
1.3.2 Tolman Electronic Parameter
Since carbonyl ligands are known to absorb electron density from a metal center in an organometallic complex, a wide range of information can be accrued from comparisons of various organometallic carbonyl complexes. When examining IR frequencies of the νCO
carbonyl stretching mode in HOMO-ligand carbonyl transition metal complexes (see Table 1-1), a standard can be formulated for comparison against various electron donating ligands coordinated to the same metal-carbonyl system [31-33]. Exchanging one carbonyl for
another dative ligand will shift the amount of electron density on the metal center and alter the amount of electron density occupying the π*-orbitals on the remaining carbonyl fragments. This will inherently yield useful information about the effectiveness of a ligand to donate electron density and overlap with the metal’s d-orbitals [31-33].
Table 1-1. Stretching frequencies of various organometallic carbonyl complexes [31, 33]
One effective organometallic scaffold for testing phosphine electron donation has traditionally been infrared spectroscopy of a LNi(CO)3 framework (see Figure 1-14). The
tris(carbonyl)nickel νCO stretching frequency, designated as the Tolman Electronic Property
(TEP), was one of the first reported manners for comparison of electron donating ligands (see Table 1-2) [34] and the residual data from years of ligand testing provides concrete evidence for comparing and evaluating new ligand designs [35]. In the context of this thesis, the Tolman electronic property proves especially effective due to the fact that all research reported was conducted on group 10 organometallic complexes.
Figure 1-14. (Left) Scaffold of the complex used to test IR stretching frequencies for TEP
characterization [34] Table 1-2. (Right) TEP data for common phosphines [29, 34, 35]
1.3.3 Tolman Angle and Solid Angle
With advancement in the field of ligand design and creation of “super” ligands specific to cross-coupling catalytic systems, many useful characterization properties must be developed and implemented in order to find ways of quantifying structural characteristics of organometallic complexes. These factors become evermore relevant as recent phosphine development witnesses new multidentate bridging ligands engineered to effectively occupy more than one half of a metal’s coordination sphere [36].
Along with the Tolman Electronic Property, phosphines are also classified by their ability to impart steric hindrance upon a metal coordination center (see Figure 1-15). Determining a value for this property can be difficult due to the wide range of organometallic complexes and ligand systems in consideration, but two main factors are typically used: the Tolman angle [34, 35, 37] and the solid angle [38-43]. Both of these properties prove effective in evaluating the ability of a phosphine to facilitate processes such as reductive elimination in cross-coupling catalytic applications.
Figure 1-15. Schematics for determination of (a) Tolman angle and (b) solid angle steric
parameters [29, 44]
The definition the Tolman angle or cone angle stems from an imaginary cone of three-dimensional space within a metal’s coordination sphere occupied by a phosphine ligand. This property has typically proved effective in evaluating broad steric trends in organometallic chemistry up until recently, when computer modeling and energy optimization calculations became commonplace.
One other major problem with this definition is that phosphines can dramatically vary in their Tolman angle when coordinated to different transition metals. A more accurate projection of steric bulk has proven to be the solid angle (θ) property. As can be seen in Table 1-3 these steric values can differ significantly. This property is defined as the normalized two-dimensional area occupied by the phosphine from a projection of light emitted at the metal’s center onto a sphere encompassing the entire complex [29-30, 34].
1.4 Group 10 Metal Complexes
1.4.1 Electronic Structure
The metals of group 10, (nickel, palladium, and platinum), are all known to be exceedingly electrodense due to their position on the far right side of the transition metal portion of the periodic table. This high electrodensity (in an M(0) oxidation state) is also explained in part because they contain a full subshell of d-electrons (d10) while witnessed in an
organometallic complexed state. Complexes of these group 10 metals in the M(0) oxidation state usually contain variants of strong π-acidic ligands in order to aid in electron stabilization of the metals d-electrons. These group 10 metal complexes will also become even more electrodense in charged states meaning that with increasingly high oxidation states they also become increasingly difficult to stabilize while in a complexed form [45-49].
1.4.2 Oxidation States and Common Geometries
Typical oxidation states for group 10 metal complexes range from M(0) to M(IV) (platinum can attain a Pt(VI) oxidation state) with the most common oxidation state for all three elemental members of the group being M(II). Group 10 organometallic complexes in the second oxidation state are predominantly witnessed in a 16e- “square-planar” geometry due
to an increase in energy of the dx2-dy2 orbital preventing electron occupation [45-49].
Nickel, as the exception of the group 10 transition metals, is extremely rare in the M(IV) oxidation state and has never been reported within Ni(IV)-C bond framework. Palladium and platinum commonly exist in the M(IV) oxidation state, but this form is usually reserved
for metal-halide species and are rarely attained as a stable M(IV)-C organometallic species (some examples are listed below in Figure 1-16) [45-49].
Figure 1-16. Three representations of group 10 metal complexes in their M(IV) oxidation
state
As a general rule, most transitions between oxidation states for group 10 metal complexes occur through the well-documented oxidative addition/reductive elimination pathways. This set of transitions usually ensures that the oxidation states commonly jostle from M(0) to M(II) to M(IV) in catalytic applications. In organometallic M-C bond chemistry M(0) species are very prone to oxidative additions while the likelihood for reductive eliminations is reversely proportional (dramatically increases with oxidation states) [45-49].
1.4.3 Applications to Carbon Dioxide Coordination and Activation
In 1975, Aresta published carbon dioxide capture by a tris(tricyclohexylphosphine)nickel complex (see Scheme 1-10) [50]. Although reports of carbon dioxide capture by an organometallic species had appeared in literature reports [50-54], this was the first reported crystal structure of such an interaction [50].
Scheme 1-10. Reaction performed by Aresta capturing carbon dioxide [50]
The (CO2)-bis(tricyclohexylphosphine)nickel complex would eventually become known as
Aresta’s complex and would become a key structure for evidence of the ability of organometallic complexes in being able to possibly functionalize the greenhouse gas [55].
Figure 1-17. Reported X-ray structure of carbon dioxide captured nickel complex (carbon
dioxide)-bis(tricyclohexylphosphine)nickel [50]
As can be seen above the feature of Aresta’s complex that differentiates it from average organometallic complexes is the ability for carbon dioxide capture in a “side-on” manner (see Figure 1-17), which is drastically different than previously reported captures which appear “end-on” [50-54]. This allows for a bond from the nickel center to both carbon and oxygen in a triangular manner (one carbon-oxygen bond still remains). This type of organometallic carbon dioxide bonding is still uncommon as most other complexes contain only M-O or M-C bonds, but not both (see Table 1-4).
Table 1-4. Table of metal carbon dioxide captures [56-74]†
†
The possibility to create a catalytic cycle could theoretically be accomplished from an organometallic species containing both M-O and M-C bonds on the same metal center. In 2008, catalytic cross-coupling between organozinc reagents and CO2 was reported by Dong
[75], which was described as an extension of Negishi coupling in which CO2 is considered
to be the electrophilic partner within the cycle. Aside from this publication though, few catalytic applications have been subsequently reported.
1.5 Zwitterionic Transition Metal Species
1.5.1 Overview
Zwitterionic organometallic complexes have recently become a desirable addition to the overall structure of an organometallic catalyst [76]. By definition, these species are complexes that feature both positive and negative charged sub-portions of their structure, however the distance between these charged sub-portions remain wide enough to limit ionic pairing interactions, but allow for the overall complex to remain fundamentally neutral in nature. Structural elements within the complex are traditionally used to force greater separation distances between the cationic/anionic charges than normally observed. This feat is usually accomplished by utilization of sterically hindering ligand species that will allow for anionic charged moieties to be sequestered away from the cationic metal center. This ligand design results in the overall distance between the cationic metal center and the anionic ligand around 3.7Å (for the borate zwitterion seen in Figure 1-18) [76].
The sequestering of charges within the framework of the organometallic complex renders several benefits for the applications of these complexes versus non-zwitterionic species. Cationic platinum group metal complexes have been shown to promote many industrial catalytic reactions. In the case of catalytic asymmetric hydrogenation, a cationic platinum group catalyst was the developmental platform for exploring the chemistry [78-80]. However, these reactions performed by cationic platinum group metal complexes are traditionally conducted in very narrow solvent conditions. These species are usually poorly soluble in solvents with low polarity but while in highly coordinating solvents, the species are witnessed to lose much of their reactivity due to solvent coordination interference [81-83]. Also, due to the presence of a strongly bound counter ion, certain interferences from ion-pairing can be witnessed to degrade the overall catalytic performance [84, 85]. Zwitterionic complexes have a wider solvent solubility profile due to the organic functionalization of the species imparted by the negative ligand substituents as well as minimal ion-pairing interactions due to the structural coordination of the negative charge to the cationic metal center [76]. Also, studies among similar ligand metal frameworks contrasting zwitterionic and non-zwitterionic species have shown significant evidence for the zwitterionic species to promote increased activity and selectivity in common catalytic applications [86, 87].
1.5.2 Development of Borate Based Anionic Ancillary Ligand Structures
For the purpose of this thesis consideration of borate based anionic fragments of zwitterionic complexes will only be discussed. These borate ligand frameworks were among the first and most thoroughly explored zwitterion classes to appear in literature. This allows for the most accurate catalyst performance comparisons to be discussed when reviewing the literature. In the early 1970’s Osborn reported decreased catalytic activity of a cationic rhodium species when ion paired with a [BPh4]- counter ion versus [PF6]- and
[ClO4]- counter-ion catalyst species [88, 89]. Explanation of this discovery was provided
when elucidation of the structure of a similar complex [90] hinted that the parent [Rh(nbd)(PPh3)2]+BPh4- species was rearranging to form the η6-coordinated borate species
as shown below in Figure 1-19 [91]. Upon confirmation of the structure of the catalyst, Schrock and Osborn realized that the complex could formally be classified as zwitterionic
in nature due to the inability to place the anionic charge on the coordinating ring through resonance structures of the complex [91]. The first crystallographically characterized structure of rhodium complexes of this type yielded data showing that the negative borate charge was located approximately 3.7Å away from the positive Rh center [90]. This data confirms that little direct ion pairing interactions are witnessed in these zwitterionic species.
Figure 1-19. (a) Typical cationic rhodium complex versus the (b) unexpected zwitterionic
rhodium borate species synthesized by Osborn [88]
Scheme 1-11. Initial catalytic reaction demonstrating the increased activity for zwitterionic
catalysts in hydroformylation of p-isobutylstyrene [86]
Research for reactivity of these borate zwitterionic complexes was limited to dehydrogenation probing until the early 1990’s when a host of potential catalytic applications were tested with a [Rh(COD)]+BPh
4- species. Alper and co-workers made the
initial discovery of this rhodium catalyst being able to perform difficult hydroformylation reactions on vinylarenes and vinyl ethers to branched aldehydes at near quantitative yields and with exceptional regioselectivity (see Scheme 1-11) [86]. Through a host of subsequent studies with this species Alper and co-workers along with efforts from other groups (see Scheme 1-12) were able to show the ability for this species to cleave as well as initiate many variants of E-H as well as E-C bonds (E = main group element) [76, 86, 92-99].
Scheme 1-12. Reactivity profile for the Rh(COD)BPh4 zwitterion [76, 86, 92-99]
1.5.3 Group 10 Zwitterionic Complexes
Focusing attention specifically on a few group 10 metal borate zwitterionic species for a quick review yields interesting chemistry in catalytic applications. Utilizing a κ3-[P
3BR]
-[M]+L
x type ligand system developed by Tilley in the late 1990’s [100], a platinum
zwitterion was synthesized and tested for catalytic applications based upon similar success found with Tilley’s iridium species (see Figure 1-20) [101].
Figure 1-20. Diagram of (a) the first tridentate phosphine borate zwitterion and (b) the
The nature of this tridentate zwitterion framework placed the charge at a distance of approximately 3.6Å which is relatively close to the distance of 3.7Å first reported by Haines for the bond distance of [Rh(P(OMe)3)2]+[BPh4]- [90, 100]. However, in this case,
due to the three phosphine fragments binding to the metal center, the negative charge is “locked” into a generic spatial area around the metal center preventing free rotation around the metal coordination as observed in the tetraphenylborate zwitterions. This structure displayed promising reactivity in the area of hydrosilylation catalysis and was further developed by Peters to yield a new form of zwitterion utilizing a [κ2-P2BR2]-[M]+Lx
framework [102]. These zwitterionic species have been shown to activate increasingly difficult E-H bonds including the C-H bonds on benzene (see Scheme 1-13) [103].
Scheme 1-13. Typical reaction scheme for bidentate group 10 zwitterions, which have
been shown to activate the C-H bond in benzene [103]
1.6 Project Outline
1.6.1 Fundamentals
With the publication by Aresta in 1975, a precedent was set that nickel could bind carbon dioxide in a side-on manner, which could possibly allow for cleavage or functionalization of one of the C-O bonds. Through active research on “Aresta’s complex” chemists have been unable to perform any catalytic chemistry with the bound carbon dioxide complex until recently by the aforementioned research by Dong [75]. It would seem that the novelty of this interaction has been slowly waxing with the problematic increase of carbon dioxide levels in the atmosphere inspiring many chemists to review important breakthroughs in carbon dioxide functionalization.
has been building amongst the chemistry society to push for development of nickel as a viable alternative to the heavier analogues of group 10. Researching manners in which the applicability of nickel could be promoted to perform many “mundane” catalytic cycles typically reserved for the platinum group metals would also have potential to greatly reduce cost in industrial applications from catalyst synthesis to waste disposal.
1.6.2 Making Nickel a Viable Alternative
Nickel, due to its electrodense nature is rarely witnessed in the M(IV) oxidation state. This property is one of the first significant drawbacks for its use in facilitating known platinum group catalysis as many of the platinum group metals use this transition from a M(II) oxidative addition to a M(IV) species to facilitate a plethora of known coupling reactions. The question arises that if a ligand system was created which could donate enough electron density to a nickel(II) center, could oxidative addition take place yielding a nickel(IV) organometallic intermediate species? Could this process be applied to utilizing a nickel(II) species to try to capture carbon dioxide? These questions were paramount at the commencement of this project and inspiration struck when Fu was able to show through his research that a boratabenzene-phosphine (while shown to be extremely strongly donating by density functional theory calculations), is witnessed to bind through the lone pair of electrons on the phosphorus atom rather than through the π-system of borabenzene. In this sort of ligand framework a combination of strong phosphine donation into a nickel center as well as inherent zwitterionic character from the close proximity of the anionic boratabenzene ring was thought to be very desirable in advancing the known organometallic chemistry of nickel to more niche applications to industrial synthesis.
Chapter 2 – Experimental Techniques
2.1 Nuclear Magnetic Resonance Spectroscopy
Do to the wide variety of nuclei (both main group elements as well as transition metals) involved in organometallic synthetic processes, the possibility to monitor and extract useful data from intramolecular spin interactions exists. By studying chemical shifts of nuclei as well as heteronuclear spin coupling, nuclear magnetic resonance spectroscopy provides a valuable tool for discerning information regarding organometallic complex properties, such as electron shielding, coordination modes, and structural geometry. This project possesses many useful NMR active nuclei for obtaining complex and ligand data, most notably 1H,
11B, and 31P nuclei (see Table 2-1). When complexed with platinum, the possibility also
existed to examine the 195Pt NMR data to derive general strength of donating ability of this
phosphine to a metal center [104].
Table 2-1. Nuclei of interest with basic information
Phosphorus-31 is a valuable NMR active nucleus for this research project due to the 100% abundance of the nuclei as well as its high receptivity. The nucleus has a spin (I) of 1/2 which makes its ability to couple with other nuclei valuable in determining both coordination modes of ligands and coordination geometries. Also of extreme value to this project is the ability of the 31P nucleus to couple with 195Pt resulting in observable 195Pt
“satellites” which arise from coupling with the 33.8% abundant, I = 1/2, 195Pt nuclei [105,
Boron-11 is another key nuclei for useful data pertaining to the project. The quadrupolar nature of the spin (I) 3/2 nuclei typically results in severe broadening of observed spectra, nevertheless, the nuclei provides yet another way to monitor resonance shift and derive information for justifying the geometry as well as electron shielding of the borabenzene/boratabenzene products [107, 108].
Adding to the standard characterization NMR nuclei monitored during synthesis (1H, 13C),
several other NMR experiments were conducted to elucidate useful data pertaining to these chemical species. Variable temperature (VT) NMR data was taken in order to attempt freeze coordination of the ligand and derive useful data pertaining to the coordination mode of the ligand in solution versus room temperature. Simple 2D experiments (HMQC) were undertaken to allocate proton positions on the borabenzene rings to signal resonances. Also
31P selective decoupling experiments were undertaken to identify coordination of the
phosphine to various other NMR active nuclei [104].
Table 2-2. NMR resonance frequencies of common nuclei
NMR experimental and characterization data was collected with samples either being placed in Teflon stoppered “J-Young” NMR tubes or NMR tubes sealed with septa to allow for injection of reagents to samples of complexes. Of vital importance for my reactions with carbon dioxide was the ability of the J-Young tubes to be evacuated and refilled with atmospheres of reagent gas. All spectra were recorded on a Varian 400 MHz NMR instrument or a Bruker 300 MHz NMR instrument. Table 2-2 (as can be seen above) describes the individual frequency that each isotope was recorded on each respective instrument.
2.2 X-Ray Diffraction
Of all characterization techniques at the disposal of the organometallic chemist, the most vital is undoubtedly X-ray diffraction for its ability to ascertain solid state data pertaining to the coordination mode and geometry of metal-ligand interactions. This characterization method places a sample of crystalline material within the path of X-ray radiation. The high-energy nature of this radiation allows for penetration into subsurface layers of the crystal lattice and subsequent diffraction of this energy by the electron dense molecular atoms and bonds, which appear as “reflections” upon a CCD (charge-coupled device) detector window. Utilizing the structured nature and precise alignment of molecules while in a crystal lattice, analysis of diffracted or scattered X-ray reflections can be used to determine the relative positions of the atoms of a molecule and assemble a three dimensional picture of the atoms in space.
Data for single crystal X-Ray Diffraction (XRD) is collected by mounting of crystalline samples of material onto a specialized goniometer point designed to be able to rotate the sample for collection of data from all possible object facets of the crystalline surface. The goniometer mounting head is composed of a non-metallic amorphous polymeric substance designed to not interfere with the collected data. Also, as all diffracted compounds reported within this thesis are incredibly air sensitive materials, the crystalline samples were adhered to the goniometer head by freezing of the sample within a small pool of paratone oil, which due to its high viscosity will sufficiently coat the surface of the crystal and protect it from rapid decomposition due to exposure of the material to air.
With single crystal X-ray diffraction, one important principle that allows for success of this method is the elastic nature of the reflection of the incident X-ray radiation as well as the complete uniformity of the frequency of reflected radiation. The fundamental principle that makes this possible is a simple geometrical law known as Bragg’s law (illustrated in Figure 2-1), which correlates spacing between “planes” of lattices (d), incident angle of radiation penetration (θ), and wavelength (λ) to allow for determination of structure. While most scattered X-ray radiation is cancelled out due to deconstructive interference, a predictable scattering pattern resultant of constructive interference can be derived. Essentially, Bragg’s law states that through constructive interference from reflected X-ray radiation off of uniformly spaced lattices, the wavelength will be reducible by a constant, effectively appearing as a direct reflection. X-ray radiation possesses an average wavelength (λ), which can closely correlate to the average spacing between planes of atomic lattice unit cells (1 – 100 Å) within a sample of crystalline material [109-111].
X-ray diffraction has developed into a key tool for organometallic chemists to use when analyzing bond character of various heteromolecular and homomolecular systems with transition metals species. The vast data available from analysis of similar bond systems often contributes to great knowledge of the nature of bonding interactions due to precedence from previously reported structures. Utilizing the calculated bond distance from various points of electron density and calculated atomic centers, the bond distance and hapticity of interactions between main-group elements and transition metals can be quantified into terms composing various known ratios of molecular orbital contributions. Calculations of intramolecular bond angles as well as torsion angles can also greatly aid in designation of bond order as well as aromaticity among many ligand-metal coordination modes.
Several limitations to this characterization technique are relevant to discuss at this time. Most notably, as X-ray radiation is chosen for this characterization technique due to its average wavelength laying relative close to the size of a crystal’s repeating lattice or unit cell, with larger unit cells, the resolution of the reflections becomes less specific and can not be determined as accurately as unit cells of smaller dimensions. Crystalline cells are
always subject to some sort of disorder as defined by the second law of thermodynamics, resulting in a calculated disorder value always representative of the accuracy of the data corresponding to the calculated reflection correlation. Molecules also vary greatly in their tendency to be “crystalline” or form structured crystalline materials and can prove quite difficult to characterize and remain stable long enough for a uniform lattice to develop before product decomposition.
2.3 Density Functional Theory
During the course of this project, DFT calculations were conducted in order to approximate the viability and stability of the boratabenzene-phosphine ligand as well as the energy differences arising from dimeric and monomeric versions of bis-phosphine group 10 transition metal frameworks. DFT calculations (Gaussian 03 MPW1PW91) modeling the boratabenzene-phosphine (DPB) scaffold were conducted by Josée Boudreau (Université Laval) in order to discern varying electron donation between LNi(CO)3 frameworks. DFT
calculations (Gaussian 98 B3PW91) determining the overall energy stability between monomeric as well as dimeric bis-phosphine group 10 transition metal complexes were conducted by Laurent Maron (UPS Toulouse).
2.4 Inert Atmosphere Manipulations
Due to the high affinity of most transition metals with water and oxygen, synthesis and characterization of these organometallic complexes was only possible while utilizing maximum caution against exposure of these reagents/products with air (O2) and moisture
(H2O). All manipulations were carried out either in specialized glove boxes fitted for
storage of compounds under nitrogen atmospheres or in standard nitrogen/vacuum bimanifold Schlenk lines. Solvents used for reactions were dried and degassed by collection after several days under refluxing conditions with added “ketyl” (sodium/benzophenone) or calcium hydride. NMR solvents were dried under sodium/potassium amalgam and vacuum transferred via Schlenk line.
![Figure 1-8. Diagram for the geometry of slip distorted boratabenzene complexes resulting in a buckled ring structure between the C 1 -B-C 5 carbons [15]](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/24.918.206.728.207.477/figure-diagram-geometry-distorted-boratabenzene-complexes-resulting-structure.webp)
![Table 1-3. Chart referencing reported Tolman angles to solid angle values [29, 38-44] †](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/30.918.255.712.584.941/table-chart-referencing-reported-tolman-angles-solid-values.webp)
![Figure 1-17. Reported X-ray structure of carbon dioxide captured nickel complex (carbon dioxide)-bis(tricyclohexylphosphine)nickel [50]](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/33.918.285.677.124.295/figure-reported-structure-dioxide-captured-complex-dioxide-tricyclohexylphosphine.webp)
![Figure 1-20. Diagram of (a) the first tridentate phosphine borate zwitterion and (b) the first group 10 tridentate borate zwitterion [100, 101]](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/37.918.161.807.102.537/figure-diagram-tridentate-phosphine-borate-zwitterion-tridentate-zwitterion.webp)
![Figure 3-1. Representations of obscure binding modes for borabenzene/boratabenzene ligands: (a) Fontaine 2009 [113] (b) Fu 1996 [28]](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/47.918.290.677.554.737/figure-representations-obscure-binding-borabenzene-boratabenzene-ligands-fontaine.webp)
![Figure 3-2. Diagram displaying the metal DPB ligand complexes reported by Fu [28]](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/48.918.157.808.250.655/figure-diagram-displaying-metal-dpb-ligand-complexes-reported.webp)
![Figure 3-3. Crystal structure comparisons of DPB (Left) and DAB (Right) exhibit the differing geometry around the respective phosphorus and nitrogen centers [6]](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/49.918.261.715.267.486/crystal-structure-comparisons-differing-geometry-respective-phosphorus-nitrogen.webp)
![Table 3-1. Gusev’s plot of experimental versus calculated ν CO stretching frequencies along with a fitted linear correction coefficient [117] †](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/51.918.281.689.102.405/table-gusev-experimental-calculated-stretching-frequencies-correction-coefficient.webp)
![Table 3-3. Diagram showing all the boratabenzene species synthesized by Fu in exchange mechanisms with borabenzene-trimethylphosphine [4]](https://thumb-eu.123doks.com/thumbv2/123doknet/7551651.229179/57.918.269.699.107.403/diagram-showing-boratabenzene-synthesized-exchange-mechanisms-borabenzene-trimethylphosphine.webp)
