High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance
Texte intégral
Figure
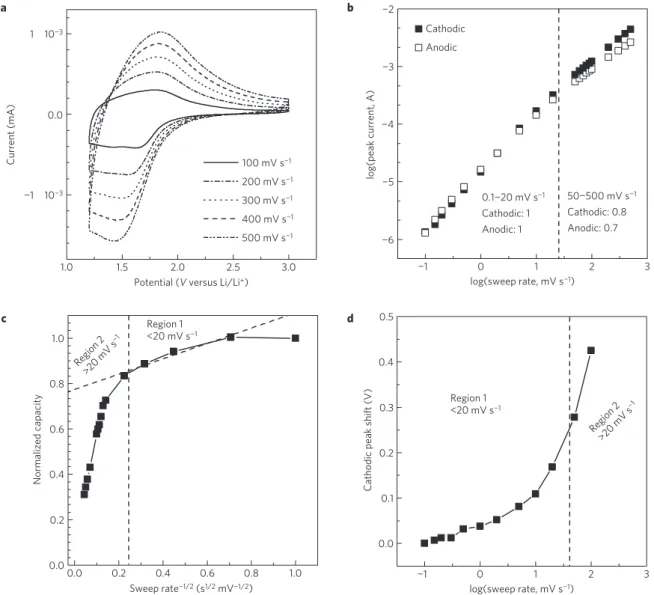


Documents relatifs
The Battery Energy Storage (BES) offers significant potential benefits at generation, transmission, distribution, and consumption levels of power systems. More specifically,
Feed Feed Fodder crops Excreta Waste Household Maize- sweet potatoes Maize- beans Maize- perennials Maize- beans Cattle Manure storage Livestock Food crops Feed Food Excreta
Second, a strong correlation was detected between the Capon profiles at different polarization channels with the LVIS waveform and the distribution of the SFL data, opening prospects
Progression & Tumor Resistance, Innovative Therapies 38 The Splicing Factor SRSF2 is an Early Component of the DNA Damage Response in Lung Cancer Cells.. KHALIFE Manal ;
If we classify the production process by the sign of the hyperon polarization, as shown in Figure 6, it is apparent that the spin of the hyperon is in the same direction as
well as the institutional status of the HEI: booths for public universities and small professional écoles usually have fewer hosts, sometimes as few as one or
- les caractérisations rhéologiques de différents bétons autoplaçants réalisées à l’aide de la sonde Viscoprobe implantée dans un malaxeur planétaire et
resistance, enabling a long-term high rate operation of the battery. Remarkably, such a low internal resistance revealed in our SSE-based hybrid-electrolyte Li-S. batteries is one
