T
T
H
H
È
È
S
S
E
E
En vue de l'obtention duD
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l’Université Toulouse III – Paul Sabatier Discipline ou spécialité : Cancérologie
Présentée et soutenue par Aline Meulle Le 10 octobre 2008
Titre : Place des adipocytes dans la réponse tissulaire aux radiations
ionisantes
JURY
Dr Yannick Saintigny, Chargé de recherche, CEA Fontenay aux Roses Dr Fréderic Bost, Chargé de recherche, Nice
Dr Marc Benderitter,Directeur de recherche , IRSN Pr Catherine MULLER, Professeur Université Toulouse III
Pr Philippe Valet, Professeur Université Toulouse III
Ecole doctorale : Ecole doctorale Biologie Santé Biotechnologie Unité de recherche : IPBS CNRS UMR5089
Directeur(s) de Thèse : Pr Catherine Muller
« Il faut toujours viser la lune, car même en cas d’échec, on atterrit dans les étoiles »
Oscar Wide
MERCI
Mes premiers remerciements sont pour le Dr Yannick Saintigny et le Dr Frédéric Bost pour avoir accepté de juger mon travail et de faire partie du Jury. Je voudrais aussi remercier le Dr Marc Benderitter d’avoir accepté de prendre la présidence du jury. J’espère que vous avez apprécié ce séjour toulousain.
Je voudrais remercier Denis Biard pour nous avoir fourni la séquence du shRNA codant pour la DNA‐PKcs, ainsi que le Dr Guilaine Escourrou du service d’anatomo‐pathologie de Rangueil pour son aide.
J’ai effectué ma thèse dans l’équipe du Pr Bernard Salles. Merci Bernard de m’avoir accueillie au sein de ton équipe durant ces années, d’avoir toujours eu la porte de ton bureau ouverte pour mes questions scientifiques et celles un peu moins scientifiques. Mais merci surtout pour m’avoir initiée aux joies du pulse field… Je garderai un très bon souvenir de mon passage dans ton équipe, mais je me souviendrais plus de toi comme un chef d’équipe qui saute à l’élastique un soir de juillet… Durant ces trois années de thèse, j’ai été co‐encadrée par Philippe Valet et Catherine Muller. Je voudrais vous remercier de m’avoir fait confiance en me confiant ce sujet. Si j’ai chevauché la route du Far‐West des adipocytes, c’est que je savais où je mettais les pieds avec vous deux. Philippe, j’aimerais te remercier d’avoir plongé dans le monde de la réparation pour m’aider dans mon travail, et de toujours avoir été enthousiasmé par mes résultats, ce qui me motivait un peu plus à chaque fois. Cathie, par ces quelques mots, j’aimerais te remercier pour tout ce que tu as pu me donner sans compter durant ces quelques années, merci d’avoir été présente à chaque fois que j’en avais besoin. Si aujourd’hui je me sens prête à partir pour de nouvelles aventures, c’est grâce à tout ce que tu as pu transmettre. Et bonne chance avec tous ces « sudistes »…
Je remercie aussi l’équipe de Bernard Salles ou les « garçons ». Je voudrais remercier Patrick Calsou pour ses conseils de réparateur et ses critiques constructives envers mes résultats, Philippe Frit pour toutes ses discussions scientifiques et surtout politiques, ainsi que Denis Gomez. J’aimerais surtout remercier les filles des « garçons » : Stéphanie Dauvillier pour ces discussions autour d’un café et pour nous aider dans nos expériences d’IF (surtout n’oublie de régler la clime du confocal…), mon binôme Christine Delteil pour tous ces moments de corvées passés en salle de culture qui furent agréables grâce à ta bonne humeur, Oriane Bombarde depuis que tu es arrivée, tu as su faire ta place, si bien que tu es presque une O’Hara, Cindy Iglésias pour tes conseils surnaturels et Elisa Boutet. J’ai aussi une pensée pour toutes les personnes qui sont passées dans cette équipe : Jérôme Drouet, Karine Jaquet, Céline Boby….
Quand je suis arrivée à l’IPBS, je suis arrivée avec Sébastien Britton. On a passé plus de 4 ans ensemble dans cette équipe et on a partagé de bons moments et de moins bons.
Maintenant c’est à toi de faire ton show, alors bon courage et bonne chance pour cette dernière année de thèse et pour toutes celles qui vont suivre. Je remercie aussi mon équipe qui se situe là haut sur la colline de Rangueil. Merci à Danièle Daviaud pour ses précieux conseils sur les 3T3F442A et son aide pour me simplifier la vie, Camille Attané pour son gel retard, car il ne faut pas oublier que c’est toi qui as commencé ce sujet, Charlotte Guigné pour m’avoir fait quelques manips et surtout pour ton sourire, et Sophie Le Gonidec, je suis sure qu’un jour tu arriveras à implanter des SUM‐159‐PT dans des souris. C’était toujours un plaisir pour moi de venir vous voir.
Durant cette thèse, j’ai été entourée par les O’Hara que je remercie de tout mon cœur, car elles ont mis un grain de folie à cette thèse :
Quand je suis arrivée, Jenny Paupert et Fanny Bouquet m’attendaient pour former les sardines, ce trio a été une vraie histoire d’amitié. Jenny, tu m’as appris tout ce que tu savais de la paillasse et tu m’as transmis ton virus de la recherche, et en plus tu m’as donnée ton amitié. C’est en travaillant à tes côtés que j’ai compris que je voulais faire une thèse, donc ma thèse est un peu la tienne. Fanny, même si on n’était pas toujours d’accord, ton soutien et ton aide ont beaucoup comptés pour moi durant ces années, bonne chance pour ta nouvelle vie de new‐yorkaise. Un jour, on reformera notre trio (mais pas à NY, désolé Fanny….). Puis notre trio a été agrandi par l’arrivée de Marielle Ousset et Béatrice Dirat. Marielle (ou dois‐je dire ma syndicaliste préférée), ton amitié à été importante pour moi car tu as toujours été là pour écouter mes petits états d’âme scientifiques ou non (« j’en ai marre du pulse field !!!! »), et pour me donner des conseils, alors surtout ne change pas ! Béa, grâce à toi j’ai rencontré une vraie toulousaine (avec accent et expressions !!!!), et je suis contente de cette rencontre, merci pour ces discussions qui souvent se terminaient par un fou rire. Puis j’ai eu la chance de voir mes 2 mains se transformer en 4 mains grâce à l’arrivée de Sandrine Imbert. Ma petite Sandrine, ce fut un réel plaisir de travailler avec toi, tu as des mains en or pour la PCR, et un jour tu seras la reine des clono…. Tu sais j’ai une petite place dans ma valise, je peux te prendre pour venir en post‐doc avec moi….. Puis on a vu débarquer une pharmacienne parisienne, Ludivine Bochet, qui s’est très vite intégrée et qui est devenue une vrai toulousaine. On n’a pas beaucoup travaillé ensemble, mais j’espère t’avoir aidée. Tu as apporté à cette équipe une nouvelle fraîcheur grâce à ta bonne humeur. Bonne chance pour cette aventure qu’on appelle la thèse.
Le côté « hypoxie » de l’équipe s’est agrandi grâce à l’arrivée de Frédérique Fallone et celle récente de Catherine Botanch, que je connais depuis peu mais que j’apprécie surtout pour ces fous rires. Pendant ces années, plusieurs personnes ont passés quelques mois avec nous et en particulier Murielle Serres qui a eu la lourde de tâche de travailler sur un sujet pas facile avec moi mais qui est toujours restée motivée, ainsi que Anne, Olivier, Pierre‐Yves, Audrey, Amélie... J’ai eu la chance d’organiser des réunions entre thésards à l’institut avec Jean‐Michel Escoffre et Gaëlle Huet. Ce fut une jolie aventure grâce à vous deux.
Je remercie aussi toutes les personnes de l’institut qui m’ont soutenu durant cette thèse. Je n’oublie pas nos voisins, l’équipe de Martine Defais pour nous avoir supportés et pour leur roue en chambre froide qui m’a aidée lorsque je me suis transformée en une usine à pulse field, l’équipe d’Olivier Cuvillier pour leur bonne humeur et l’équipe de Justin Teissié pour ces nombreuses discussions.
Les couloirs sont remplis de thésards qui sont prêts à nous aider et certain d’entre eux sont devenus des amis : je voudrais remercier en particulier Flavie Kersanté pour son soutien durant les derniers mois de nos thèses, et pour ces moments de délires nécessaires à notre bien‐être mental en cette fin de thèse ; Chloé Mauroy que je ne connais que depuis quelques mois mais qui est devenue une vrai copine qui m’a soutenue pendant les derniers moments de ma thèse ; Leyre Brizuela‐Madrid pour cette amitié qui tu m’a apportée et qui a une grande importance à mes yeux ; le belle ragazze italiane Guisy Biancha e Guida Locatella pour leur joie de vivre à l’italienne ; Laury Rey et Fanny Lemee (promo 2005 power) pour m’avoir accueillie dans leur équipe lors de mes visites au Typhoon.
Mes derniers remerciements vont aux personnes les plus importantes pour moi : ma famille. Je veux remercier mon oncle et ma tante (Bibi et Bobo pour les intimes), ma mamie, mon papi et mes beaux‐parents Nicole et Guy pour m’avoir soutenue et surtout d’avoir fait des kilomètres pour avoir été là le jour de ma thèse.
Je veux remercier ma sœur et sa petite famille, car même si tu n’as pas toujours compris pourquoi je me donnais autant pour cette thèse, tu m’as toujours soutenue et encouragée. Puis je remercie mes parents pour leur amour, leur soutien et leur présence qui me donnent tous les jours et sachez que la fierté que je vois dans vos yeux est mon moteur.
Pour finir, je veux remercier Stéphane pour le soutien qu’il m’a apporté tous les jours de cette thèse, pour m’avoir supportée et pour avoir continué à m’aimer même quand le stress me rendait chiante. Ta présence m’a permis de mieux vivre ces moments. J’ai donné beaucoup d’énergie pour cette thèse, mais sachez que cela en valait la peine. Merci
PLACE DES ADIPOCYTES DANS LA RÉPONSE TISSULAIRE AUX RADIATIONS IONISANTES
La radiothérapie est un des traitements majeurs utilisés en cancérologie en particulier dans les cancers du sein. Les cellules exposées aux radiations ionisantes (RI) présentent de nombreux dommages à l’ADN. Parmi ces lésions, les dommages les plus cytotoxiques sont les cassures double brin (CDBs) et un déterminant majeur de la radiosensibilité cellulaire est la capacité des cellules à réparer ces cassures. De plus, au cours de l’irradiation de la tumeur, les cellules du microenvironnement, irradiées au même titre que les cellules tumorales, pourrait modifier la réponse des cellules cibles. Ce concept de radiobiologie tissulaire est soutenu par une étude du groupe de Fucks et Kolesnik qui montrent l’apparition d’une radiorésistance de tumeurs xénogreffées lorsque les cellules endothéliales sont résistantes à l’apoptose radio-induite. Parmi les cellules du stroma mammaire, nous avons décidé de nous intéresser aux adipocytes. En effet, ces cellules sont les plus abondantes dans le microenvironnement des tumeurs mammaires et elles sécrètent de nombreux facteurs de croissance et cytokines susceptibles de modifier le comportement de la tumeur. Durant ma thèse, je me suis intéressé dans un premier temps à la réponse des adipocytes à l’exposition aux RI, et plus spécifiquement à la régulation de l’activité de réparation des CDBs au cours de la différenciation adipocytaire et dans un deuxième temps à l’influence des adipocytes sur la radiosensibilité de cellules tumorales mammaires.
Nous avons pu montrer qu’au cours de la différenciation adipocytaire, il existait une régulation positive de l’activité de réparation des CDBs de l’ADN. Cette augmentation de la réparation des CDBs est expliquée par une régulation positive de l’expression de la protéine kinase dépendante de l’ADN (ou DNA-PK) qui joue un rôle de plate-forme dans la réaction de réparation par jonction d’extrémités non homologue (ou JENH). L’activité DNA-PK est spécifiquement augmentée au cours de la différentiation adipocytaire, murine et humaine, ce qui apporte des éléments nouveaux et originaux sur les liens entre différenciation et réparation dans des cellules post-mitotiques à durée de vie prolongée. Nos résultats montrent par ailleurs que les adipocytes matures sont des cellules radiorésistantes par rapport aux pré-adipocytes, leur survie prolongé face à ce stress génotoxique favorisant l’hypothèse de leur participation dans la réponse tissulaire aux RI. Effectivement, les premiers résultats obtenus montrent que dans un système de coculture en 2D, les adipocytes protègent les cellules tumorales mammaires de la mort clonogène, cet effet étant dépendant de facteurs solubles sécrétés par les adipocytes. En conclusion, nos résultats montrent que les adipocytes matures présentent, contrairement à la majorité des cellules différenciées étudiées jusque lors, une augmentation des activités de réparation des CDBs par rapport à leurs progéniteurs pré-adipocytaires. Ces résultats confirment leur participation à la radiorésistance des cancers du sein, ce qui pourrait être particulièrement intéressant à étudier dans un contexte d’obésité, obésité qui est associée à un mauvais pronostic dans les cancers du sein selon des mécanismes incomplètement élucidés.
ROLE OF ADIPOCYTES IN THE TISSUE RESPONSE TO IONIZING RADIATION
Radiotherapy is a major therapeutic strategy in the treatment of many human tumors including breast cancer. Exposure of cells to ionizing radiation (IR) induces DNA damage and among them, DNA double strand breaks (DSBs) are considered to be responsible for the cytotoxic effect of IR. In fact, the ability of cells to repair DNA DSBs is a major determinant of the cellular response to IR. However, cancer is a tissue-based disease in which malignant cells interact dynamically with multiple normal cell types such as fibroblasts, adipocytes, infiltrating immune cells and endothelial cells within the context of extra-cellular matrix. With regards to the response to IR, emerging studies suggest that the irradiated stroma can control the overall fate of epithelial cells such as survival/death. For example, a study by Fucks and Kolesnick group has shown that expression of radiation damage by tumor cells is conditionally linked to radiation-induced apoptosis in the affected tissue microvasculature. According to these recent results, the goal of our project was to investigate if the ability of IR to induce changes in tissue microenvironment through the response of adipocytes is a critical component of the cytotoxic response of breast cancer cells. In fact, adipocytes are highly represented in breast tumor stroma and are able to secrete many growth factors and cytokines. They are therefore excellent candidates to play an important role in tumor behavior in response to IR through heterotypic signaling processes, since they.
As a first step, to elucidate the potential role of adipocytes in the response of the tumor to IR, we investigated their own ability to repair IR-induced DNA DSBs. We have demonstrated that the ability to repair DSBs is increased during adipocyte differentiation. During the first 24 h following the commitment into adipogenesis, we showed an increase in expression and activity of the catalytic sub-unit of the DNA-PK complex, DNA-PKcs. DNA-PKcs is a key factor of the DSBs repair by the non-homologous end joining pathway (NHEJ). Finally, similar positive regulation of DNA-PKcs expression and activity was observed during differentiation of primary culture of pre-adipocytes isolated from human sub-cutaneous adipose tissue. This efficient response to DNA damage might contribute to the life-long maintenance of adipocytes. Moreover, our results showed that mature adipocytes are radioresistants cells compared to their immediate precursor pre-adipocytes cells. This ability to resist to genotoxic stress supports the idea that adipocytes might participate to radioresistance at a tissue level. In fact we demonstrated, in a 2D co-culture model that adipocytes protect breast cancer cell from clonogenic cell death in a manner that is dependent of secreted soluble factors.
In conclusion, our results show that matures adipocytes cells, in opposition to the majority of terminally differentiated cells, increases their ability to repair DSBs during differentiation. These results confirm the participation of mature adipocytes to the radioresistance of breast cancer cells. Accordingly, it would be of interest to evaluate the paracrine role of adipocytes within the context of obesity that has been recently established a negative prognostic factor even for localized breast tumors.
Abréviations
ACC Acétyl-CoA carboxylase
APE1 Apurinic/apyrimidinic endonuclease 1 ATGL Adipocyte Triacyglycéride Lipase ATM Ataxia Telangectasia Mutated ATR Ataxia Telangectasia Related
B-NHEJ Backup Non Homologous End Joining (NHEJ alterne) BER Base Excision Repair
CAF Cancer associated Fibroblaste CDB Cassure Double Brin
CS Cockayne Syndrome
CSB Cassure Simple Brin
D-NHEJ DNA-PK dependent Non Homologous End Joining (NHEJ classique) DAR Differentiation-Associated Repair
DG Digkycéride
DNA-PK DNA Dependent Kinase EGF Epidermal Growth Factor
EGFR Epidermal Growth Factor Receptor ERO Espèce Réactive de l’Oxygène FAS Fatty Acid Synthetase
GGR Genome Global repair GLUT GLUcose Transporteur HAT Histone Acétyltransférase IL-1 Interleukine 1
IL-6 Interleukine 6 IL-11 Interleukine 11
LHS Lipase Hormono-Sensible LMG Lipase des Monoglycérides LPL Lipoprotéine Lipase MMP Métalloprotéase MMR Mismatch Repair
MP Membrane Plasmique
MRN Complexe MRN : Mre11 – Rad 50 – Nbs 1 MSI Phénotype d’Instabilité des Microsatellites NER Nucleotide Excision repair
NHEJ Non Homologous End Joining RI Radiation Ionisante
SVF Fraction Stroma Vasculaire
TA Tissu Adipeux
TCR Transcription-Coupled repair
TG Tryglycérides
TTD Forme photosensible de la Trichothiodystrophy
UV Ultra-Violet
SOMMAIRE
INTRODUCTION...……….………...5
INTRODUCTION GENERALE... 6
I-LES DIFFERENTES VOIES DE REPARATION DES LESIONS DE L’ADN... 7
A. Les cassures doubles brins...7
1. Origine des cassures doubles brins ... 7
1.1. Origine endogène... 7
1.2. Origine exogène... 9
a. Agents physiques : les radiations ionisantes... 9
b. Agents Chimiques ... 10
2. Signalisation et réparation des dommages... 11
2.1. Signalisation des dommages... 11
2.2. Réparation des CDBs ... 13
a. La recombinaison homologue... 13
b. Le Single-Strand Annealing ... 14
c. La réparation par jonction d’extrémité non homologue ... 14
a. Le mécanisme de la NHEJ... 14
b. Les acteurs majeurs de la NHEJ... 15
1. L’hétérodimère Ku ... 15
2. La DNA-PKcs ... 16
3. Artémis... 18
4. Le complexe XRCC4/Ligase IV ... 18
5. XLF/Cernunnos... 19
c. La voie alternative de la NHEJ : la B-NHEJ ... 19
B. Les autres systèmes de réparation...21
1. Nucleotide Excision Repair ou NER... 21
1.1. Global Genome repair ou GGR... 22
1.2. Transcription-Coupled Repair ou TCR ... 23
2. Base Excision Repair ou BER... 24
3. Mismatch repair ou MMR ... 25
3.1. La voie de réparation du MMR ... 25
3.2. Les conséquences d’un défaut de MMR... 26
3.3. Les protéines du MMR dans la réponse aux dommages à l’ADN ... 27
II-REGULATION DE LA REPARATION DE L’ADN DANS LES CELLULES DIFFERENCIEES... 27
B.Régulation des voies de réparation de l’ADN lors de la différenciation....29
1. Régulation du NER durant la différenciation cellulaire... 29
1.1 Le DAR ... 30
1.2 Le DAR est-il présent dans toutes les cellules différenciées ?... 31
a. Les neurones ... 31
b. Les cellules musculaires ... 31
c. Les adipocytes ... 31
d. Les cellules hématopoïétiques... 32
e. Les kératinocytes ... 32
f. Les hépatocytes ... 33
g. Les spermatozoïdes ... 33
h. En conclusion… ... 33
2. Régulation du BER durant la différenciation cellulaire ... 34
2.1. Existe-t-il une réparation du BER couplée à la transcription ? ... 34
2.2. La régulation du BER au cours de la différenciation cellulaire... 35
2.3. Existe-t-il une activité du BER spécifique à la différenciation ?... 36
3. Régulation du MMR durant la différenciation cellulaire ... 36
C.Régulation des voies de réparation des CDB de l’ADN lors de la différenciation...37
1. Régulation de la réparation des CDBs au cours du cycle cellulaire... 37
1.1. Régulation entre RH et NHEJ durant le cycle cellulaire... 37
1.2. Régulation entre la D-NHEJ et la B-NHEJ ... 39
2. Régulation de la réparation des CDBs au cours de la différenciation cellulaire... 39
2.1. La réparation des CDBs dans les tissus à renouvellement constant ... 39
a. Les cellules hématopoïétiques ... 39
b. Les cellules germinales mâles ... 41
c. les cellules intestinales... 41
d. Les ostéoblastes ... 42
e. Les kératinocytes ... 42
f. Ce qu’il faut retenir de la régulation de la réparation des CDBs dans les tissus à renouvellement constant !... 43
2.1. La réparation des CDBs des cellules différenciées à durée de vie longue ou prolongée ... 44
a. Les neurones ... 44
b. Les cellules musculaires ... 46
c. Ce qu’il faut retenir de la régulation de la réparation des CDBs dans les tissus à durée de vie prolongé ! ... 47
III.LA REPARATION DES ADIPOCYTES... 48
A. Qu’est-ce qu’un adipocyte ?...48
1. Les fonctions métaboliques du TA ... 48
1.1. Le stockage des lipides : la lipogenèse... 49
a. Captage d’acide gras... 49
b. La lipogenèse de novo ... 49
1.2. La libération des lipides : la lipolyse ... 50
2. Le développement du TA ... 50
2.1. La différenciation adipocytaire ... 50
a. La détermination adipocytaire ... 50
b. Le processus de différenciation adipocytaire ... 51
c. Contrôle de la différenciation ... 52
a. Les facteurs de transcription... 52
b. Régulation par les hormones ... 53
2.2. Le développement pathologique du TA : L’obésité ... 53
3. Activité sécrétoire du TA... 54
3.1. Les facteurs lipidiques ... 54
3.2. Les facteurs protéiques ... 55
B. Les adipocytes et les génotoxiques...57
1. Est-ce qu’un adipocyte possède des voies de mort ? ... 58
2. La réponse des adipocytes aux agents génotoxiques ... 59
3. La réparation de l’ADN d’un adipocyte... 60
IV. LES ADIPOCYTES COMME ACTEURS DE LA REPONSE A LA RADIOTHERAPIE... 61
A. La radiothérapie...62
1. Le mode d’action de la radiothérapie... 62
2. La mort radio-induite... 63
2.1. L’apoptose ... 63
2.2. La nécrose... 64
2.3. La mort post-mitotique ou la catastrophe mitotique ... 65
B. Le microenvironnement de la tumeur peut-il avoir un rôle dans la réponse à la radiothérapie ?...66
1. Le microenvironnement stimule la progression tumorale... 66
2. Influence du microenvironnement sur la réponse aux RI ? ... 67
C.L’adipocyte : la cellule oubliée du microenvironnement...70
1. L’adipocyte influence-t-il la cellule tumorale ?... 70
1.1. Le rôle des adipocytes dans la progression tumorale ... 70
1.2. Quels sont les modes d’action des adipocytes sur la progression tumorale ... 71
b. Les adipokines anti-oncogéniques... 73
1.3. Le dialogue entre adipocytes et cellules tumorales semblent être dans les deux sens afin de stimuler la progression tumorale... 74
1.4. L’influence des adipocytes sur d’autres types de cellules cancéreuses... 76
2. Données épidémiologiques : l’obésité est un facteur de risque d’apparition du cancer 77 V.OBJECTIF... 80
RESULTATS………...81
PARTIE I : REPONSE DES ADIPOCYTES AUX RI... 82
A.Régulation positive de l’activité de réparation des cassures doubles brins durant l’adipogenèse, implication de la protéine kinase dépendante de l’ADN...82
B. Résultats complémentaires...83
1. Régulation de la DNA-PKcs durant l’adipogenèse ... 83
2. La régulation de la voie alternative de la NHEJ durant l’adipogénèse... 84
3. Les adipocytes sont-ils radiorésistants ? ... 84
4. Matériels et méthodes des résultats complémentaires ... 85
C.Discussion de la partie I...87
PARTIEII :INFLUENCE DES ADIPOCYTES SUR LA REPONSE A LA RADIOTHERAPIE... 89
A. Les adipocytes favorisent la prolifération des cellules tumorales...90
B. Les adipocytes protègent de la mort radio-induite les cellules cancéreuses mammaires 90 C. Les adipocytes induisent une hyperphosphorylation de la protéine Chk1 dans les SUM-159-PT irradiées....92
D. L’interleukine-6, un candidat potentiel...93
E. Matériels et méthodes...94
F. Discussion de la partie II...96
CONCLUSIONS ET PERSPECTIVES………..100
REFERENCES BIBLIOGRAPHIQUES………104
INTRODUCTION GENERALE
Comprendre les mécanismes contrôlant la carcinogenèse et la progression tumorale est un objectif important de la recherche. Des études récentes ont montré que la tumeur devait être considérée comme un tissu où les cellules tumorales interagissent avec les cellules saines environnantes, et que cette interaction pouvait être délétère. En effet, différentes études ont montrées que l’environnement proche de la tumeur peut stimuler la progression tumorale et la carcinogenèse mais aussi avoir une influence sur la réponse à la thérapeutique. Cet environnement est composé de divers types cellulaires dont les cellules endothéliales, les macrophages, les fibroblastes, les adipocytes…. Parmi les différents types cellulaires, peu d’études se sont intéressées au rôle des adipocytes qui sont susceptibles de modifier le comportement de la tumeur. Pourtant les caractéristiques de ces cellules en font des candidats potentiels. En effet, les adipocytes sont les composants majeurs du tissu adipeux, tissu très abondant dans l’organisme, et présents dans de nombreux stromas tumoraux dont le stroma mammaire. De plus, ces cellules ont une activité sécrétoire importante, ils vont sécréter de nombreux facteurs qui sont connus pour avoir un rôle sur la tumeur tels que le TNFα l’interleukine 6 ou encore différentes métalloprotéinases. Au cours de ma thèse, nous avons observé l’influence des adipocytes sur le cancer du sein et plus précisément sur la réponse à la radiothérapie, traitement très utilisé dans ce type de cancer.
Dans un premier temps, le comportement des adipocytes face à l’exposition aux radiations ionisantes (RI) a été étudié, car il n’existe pas de données dans la littérature sur leur sensibilité à ce génotoxique. Les RI sont cytotoxiques car elles endommagent la molécule d’ADN en créant des cassures doubles brins. La réparation de ces cassures est un élément déterminant de la réponse aux RI. Les adipocytes sont des cellules matures différenciées qui ne possèdent pas ou peu de renouvellement. Quelques études ont montré que les cellules différenciées à faible renouvellement adaptaient leur activité de réparation durant le processus de différenciation. La première partie de ma thèse a été d’observer le comportement des adipocytes après exposition aux RI et plus précisément d’observer si l’activité de réparation des cassures doubles brins était régulée au cours de la différenciation adipocytaire.
La seconde partie de cet exposé présentera l’influence des adipocytes sur la réponse à la radiothérapie des cellules cancéreuses du sein. Des études ont montré que les cellules endothéliales pouvaient modifier la réponse des cellules cancéreuses à la radiothérapie. Nous avons donc étudié si ce concept pouvait être étendu aux adipocytes.
I- Les différentes voies de réparation des lésions de l’ADN
La réponse cellulaire aux agents génotoxiques est en grande partie conditionnée par la capacité des cellules à réparer ces lésions. Nous allons donc voir les différents systèmes de réparation en insistant les cassures doubles brins (CDBs) car elles représentent les lésions les plus cytotoxique induite par les radiations ionisantes (RI).
A. Les cassures doubles brins
1. Origine des cassures doubles brins
1.1. Origine endogène
Les CDBs sont des lésions très toxiques pour la cellule dont l’origine peut être multiple. Outres toutes les sources exogènes connues (en particulier les radiations ionisantes ou RI), les CDBs peuvent avoir une origine endogène qui est contrôlée ou non par la cellule. Une étude a montré que le taux de CDBs spontanées lors d’un cycle cellulaire est de 50, soit une cassure toutes les 1.108 paires de bases (Vilenchik and Knudson 2003). Ce taux de CDBs endogènes peut
sembler élever car il correspond au nombre de CDBs formées après irradiation à 1.5 Gy, on peut donc dire que le métabolisme cellulaire est une des sources endogènes la plus importante de CDBs. Les organismes aérobies, tel que l’homme, ont un métabolisme qui est dépendant de l’oxygène. En effet, ce dernier est impliqué dans de nombreuses réactions chimiques qui sont génératrices de chaleur et d’énergie. Toutefois, 1 % de l’oxygène respiré échappe aux chaînes de production et va se réduire de façon incomplète pour se transformer en espèces réactives de l’oxygène (ERO). Par leur nature radicalaire, les ERO représentent une menace potentielle pour l’ADN car elles peuvent conduire à des lésions oxydatives de bases ou de sucres, à l’apparition de sites abasiques, conduisant secondairement à l’apparition de CSBs et de CDBs (Su 2006). Le stress oxydatif est donc une source endogène importante de dommages de l’ADN. Une autre source endogène importante de génération des CDBs est la réplication. Lorsque la machinerie de réplication rencontre une lésion simple brin d’origine endogène ou exogène, la progression de la fourche de réplication peut se trouver bloquée, ce qui peut induire une cassure sur le brin porteur du dommage et peut se traduire par une rupture interne de l’une des chromatide sœur (Kogoma 1997). Par ailleurs dans certains tissus et en particulier le tissu lymphoïde, la réexpression de certaines endonucléases telles que les protéines RAG peut être aussi à l’origine de CDBs (Gladdy et al. 2003).
Aussi cytotoxique que puisse être une CDB, dans certain cas elle est programmée au cours de processus tels que la recombinaison V(D)J ou la commutation isotypique. La recombinaison V(D)J est un mécanisme de recombinaison de l’ADN qui permet de créer une grande variété
d’immunoglobulines (Ig) et de récepteurs des cellules T (TCR) différents, nécessaire à la reconnaissance de l’immense diversité d’antigènes étrangers. Les chaînes polypeptidiques lourdes et légères des Ig et des TCR sont constituées d’une région variable responsable des interactions avec l’antigène et d’une région constante. La région variable des chaînes lourdes est composée de la recombinaison quasi-aléatoire d’un des 200 segments variables (V) avec un des 12 segments de jonction (J) et un des 4 segments de diversité (D), par contre la région variable des chaînes légères possède de nombreux segments V et J mais aucun D. Ce mécanisme de recombinaison aléatoire est initié par les protéines RAG 1 et RAG 2 (Recombination Activating Gene) qui vont reconnaître des séquences spécifiques appelées séquences RSS pour Recombination Signal Sequence, et cliver l’ADN. Ces séquences sont situées de part et d’autres des segments V, D et J et au locus des IgG et des TCR, et sont respectivement clivées dans les cellules pré-B (moelle osseuse) et pré-T (Thymus). La recombinaison V(D)J est destinée a produire des récepteurs antigéniques, elle est présente exclusivement dans les cellules de la lignée lymphoïde (Oettinger et al. 1990). La recombinaison V(D)J se fait en deux étapes principales :
- l’étape du clivage double brin qui nécessite l’intervention du complexe RAG1/RAG2/HMG1. Ce complexe synaptique va se lier au niveau des séquences RSS et favoriser leur clivage par l’activité endonucléase des RAG. Pour cela, RAG va d’abord créer une CSB avec une extrémité 3’-OH et 5’-phosphate. Il va ensuite activer le groupement 3’-OH et catalyser une réaction de trans-estérification qui va libérer deux types d’extrémités, une extrémité codante en épingle à cheveux « hairpin » et une extrémité signal terminée par un bout franc phosphorylé en 5’. Suite à la création de deux CDBs, la deuxième étape intervient.
- l’étape de réparation des CDBs est une étape de jonction qui fait intervenir l’ensemble des facteurs de la NHEJ (Non Homologous End Joining pathways). La NHEJ est la voie de réparation majoritaire des CDBs (voir paragraphe I.A.3.). La protéine Artémis va maturer l’extrémité codante en hairpins grâce à son activité nucléase, ce qui va ouvrir l’ADN, et la TdT va permettre l’ajout de nucléotides en une extension aléatoire favorisant d’autant plus la diversité jonctionnelle.
La commutation isotypique participe aussi à la réponse immunitaire puisqu’elle permet la substitution de la chaîne lourde IgM par celle d’un autre isotype (IgG, IgA ou IgE) sans modifier la spécificité de l’anticorps mais en lui fournissant de nouvelles fonctions. Cette commutation isotypique se produit de la même façon que la recombinaison V(D)J. Des CDBs sont crées puis réparées pour permettre le changement du gène de la chaîne lourde. La voie de réparation de ces CDBs la plus probable semble être la NHEJ puisque Artémis et la DNA-PKcs semblent nécessaires à la commutation isotypique (Franco et al. 2008). Mais cela ne semble pas si simple, puisque de nombreux travaux montrent que la DNA-PKcs ne participe pas à la commutation
isotypique car le niveau des différentes classes d’immunoglobulines est le même quelque soit le statut en DNA-PKcs (Bosma et al. 2002; Cook et al. 2007; Kiefer et al. 2007).
1.2. Origine exogène
Les agents exogènes pouvant conduire à la formation de CDBs sont classés en deux catégories : les agents physiques, principalement représentés par les RI et les agents chimiques parmi lesquels on va retrouver de nombreux médicaments anti-tumoraux.
a. Agents physiques : les radiations ionisantes
Les RI ont plusieurs origines, cosmique, nucléaire mais aussi interne due à l’air respiré et aux aliments ingérés. Une des origines les plus connues et les plus étudiées est la radiothérapie, c'est-à-dire l’utilisation de l’effet cytotoxique des RI dans le cadre d’un traitement antitumoral.
Si les RI sont aussi cytotoxiques pour les cellules, c’est qu’ils sont capables d’interagir avec la matière vivante et inorganique engendrant des dommages sur tous les types macromoléculaires de la cellule (carbohydrates, lipides, protéines et acides nucléiques). En effet, les RI peuvent induire des dommages dans différents compartiments cellulaires tel que la membrane plasmique, mais la cible préférentielle des RI est la molécule d’ADN (Albanese and Dainiak 2003). Les dommages radio-induits de l’ADN sont de natures très variables et peuvent induire des lésions sur un ou deux brins simultanément (Goodhead 1994). (Figure 1). Les lésions induites par les RI sont les modifications de bases avec plus de 70 types de lésions oxydatives différentes (2000 lésions pour 1Gy), l’oxydation des sucres, les pontages covalents entre protéines et ADN, ainsi que les CSBs (1000 lésions pour 1Gy). Mais les lésions les plus dangereuses causées par les RI sont les CDBs (40 lésions pour 1Gy).
Si les RI induisent autant de dommages à l’ADN, cela est du majoritairement à l’action radicalaire du rayonnement. Les RI en interagissant avec des molécules d’eau vont produire des ERO par le processus de la « radiolyse de l’eau », c'est-à-dire quand l’énergie du rayon est transférée à la molécule d’eau qui conduit à son ionisation (Feldberg and Carew 1981). La molécule d’eau devient instable et va se fragmenter en espèces chimiques radicalaires extrêmement réactives : un ion H+ très réducteur et un radical hydroxyle OH- très oxydant.
L’électron éjecté va pouvoir réagir soit avec une autre molécule d’eau, soit si ce processus est réalisé à proximité de l’ADN, réagir avec les atomes de l’ADN. Cela va induire une altération de sa structure par oxydation ou réduction conduisant à un très large spectre de lésions. Ces lésions dues au mécanisme indirect des RI peuvent générer des CDBs qui peuvent être soit dues à la nature aléatoire d’apparition des CSBs soit dues à la rencontre d’une CSB avec un système de réparation. En effet, la formation de 2 CSBs à moins de 20 paires de bases l’une de l’autre
O O O O P OH O O O O O O O O O A T G C Particule ionisante Création de site abasique Création de CSB Dégradation de base Protéine Pontage ADN- Protéine O P OH O P OH O P OH
Figure 1 : Mécanisme d'action des RI sur l'ADN
O O O base C1' C2' C3' C4' C5' Ncs, Cali Ncs, Cali, Bléo Ncs, Cali
Figure 2 : Sites majoritaires d'oxydation nucléotidique par las agents radiomimétiques sur la particule d'ADN. Bléo : bléomicine ; Ncs : Néocarsinostatine ; Cali : Calichéamicine
pourrait engendrer une CDB. La réparation par le système du BER (réparation par excision de base) des dommages dus aux RI peut aussi générer des CDBs, si cette machinerie de réparation rencontre une CSB ou une autre machinerie de réparation. Même si la radiolyse de l’eau est la voie majoritaire d’apparition des CDBs par les RI, les rayons peuvent générer des CDBs directement. Une particule irradiée pourrait traverser une même région de l’ADN créant une CSB puis une CDBs, ou une radiolésion sur un brin pourrait générer un radical libre transféré par réaction en chaîne sur le brin complémentaire.
b. Agents Chimiques
Les agents chimiques induisant des CDBs peuvent être classés en plusieurs catégories : - les agents radiomimétiques : les molécules de cette catégorie sont qualifiées de radiomimétiques car ils induisent des CDBs de la même façon que les RI, en attaquant la molécule d’ADN. La spécificité de ces agents est d’avoir un ratio CSB/CDB plus petit que pour les RI, c'est-à-dire qu’elles génèrent plus de CDBs. Les CDBs chimio-induites résultent de l’attaque radicalaire, par déshydrogénation sur une position bien spécifique du désoxyribose, sur l’un des deux brins d’ADN qui peut conduire à la perte de la base associée, du sucre oxydé ou d’un fragment associé à la base (Figure 2). Il existe différents agents qui sont étudiés pour leur capacité anti-tumorale :
• la calichéamicine (cali) : c’est le plus puissant agent inducteur de CDBs (30 % de plus de CDBs que les RI). Les CDBs sont obtenues de façon directe par 2 CSBs adjacentes générées simultanément sur les 2 brins. La cali va agir, par une attaque nucléophile sur les atomes de carbone en C3 et C6 de la molécule d’ADN dans le sillon mineur et sur les 2 brins. La cali est capable de cliver des régions polypyrimidiniques/polypuriniques de 4 paires de bases contenant au moins 3 pyrimidines dont 2 cytosines, le site majoritaire est 5’-tcct-3’ (Povirk 1996). La cali a été testée en clinique comme agent anti-tumoral mais semble trop toxique.
• la bléomicine (bléo) : c’est un antibiotique glycopeptidique naturel, basique et hydrosoluble. La bléo va entrer dans la cellule sous une forme inactive complexée avec des ions cuivre. L’ion cuivre est alors remplacé par chélation d’un ion fer générant un nouveau complexe, Bleo/Fer[II]. Un complexe ternaire va être formé avec l’oxygène moléculaire (un hydroperoxide ou de l’oxygène singulet) puis il va subir une réduction d’électron pour former un complexe actif. L’action de la Bléo passe majoritairement par la formation de sites AP et de CSDs. La bléo forme 10 à 20 fois plus de CSBs que de CDBs, mais la quantité de CDBs est 10 % plus importante que les RI. La bléo génère des CDBs franches, dues principalement à l’apparition aléatoire de 2 CSBs sur une région restreinte, ou dues à l’apparition d’une CSB qui va créer une structure de l’ADN favorisant une deuxième attaque par la bléo (Povirk 1996). Cet agent a été utilisé dans le
traitement des lymphomes en association avec d’autres médicaments (traitement appelé CHOP-Bléo en clinique).
• la Néocarcinostatine (Ncs) : c’est une pro-drogue nécessitant une activation réductrice par ces co-facteurs (sulfhydryles, glutathions). Après activation, la Ncs va s’intercaller parallèlement aux bases dans le sillon mineur de l’ADN et intéragir avec les phosphates de la molécule d’ADN. Les clivages simples sont les lésions majoritaires de cet agent, et se produisent de façon spécifique sur les bases désoxythymidates (t) et désoxyadénylates (a). La Ncs produit 20 % de CDBs, ce qui représente 20 % de CDBs en plus que les RI (Linenberger et al. 2001). Un quart des CDBs sont dues à une action directe de la Ncs, une molécule peut attaquer simultanément les 2 brins d’ADN. Les séquences majeures comme sources potentielles de CDB sont les séquences 5’-agc/t-3’ et 5’-tga/t :c-3’ et leurs complémentaires (Povirk 1996).
- les inhibiteurs de topoisomérase : les topoisomérases sont des enzymes qui modifient la topologie de l’ADN en provoquant des cassures de l’ADN. Ces coupures permettent de maintenir la structure de l’ADN en évitant les phénomènes de surtension qu’occasionnent la réplication, la recombinaison et la transcription. Les topoisomérases I et II vont agir en plusieurs étapes : la formation d’un complexe clivable enzyme-ADN suivie d’une religation de l’ADN. Les inhibiteurs des topoisomérases se lient au complexe topoisomérase-ADN au stade de clivage et empêchent l’étape de religation. Cela rend définitive la CDB lorsque qu’il s’agit d’un inhibiteur des topoisomérases II (Etoposide, Ténoposide). Pour les inhibiteurs des topoisomérases I (Irinotécan, Topotécan), la CSB peut être convertie en une CDB à la suite de la rencontre avec une ADN polymérase ou une ARN polymérase (Nitiss and Wang 1996).
- les agents alkylants bifonctionnels : ces agents sont utilisés en clinique pour leur capacité à générer des pontages covalents interbrins. Le mécanisme de réparation de ces lésions est mal connu, mais il semblerait que la formation d’une CDB soit nécessaire (Lawley and Phillips 1996).
2. Signalisation et réparation des dommages 2.1. Signalisation des dommages
La réponse cellulaire aux CDBs (Figure 3) passe par l’activation rapide de différentes cascades de signalisation toutes initiées par les protéines kinases ATM (Ataxia Telangectasia Mutated) et ATR (AT Related). Ces protéines font parties de la famille des analogues des phosphatidyl-inositol-3 kinase (PI3K), la famille des phosphatidyl-inositol-3 kinase like kinase (PI3KK), comme la protéine DNA-PKcs sous-unité catalytique du complexe DNA-PK (Protéine Kinase dépendante de l’ADN), et elles pourraient aussi jouer un rôle dans la détection du dommage (Abraham 2004). Dans un premier temps, la réponse aux CDBs se fait par l’activation
Figure 3 : Représentation schématique de la réponse aux cassures doubles brins (d’après Bakkenist and
Kastan. 2004). Après création de la CDB, les protéines détectrices (complexe MRN) vont détecter la cassure, puis ATM est recrutée rapidement. ATM va induire le signal en activant différents effecteurs qui vont avoir des effets biologiques tels que l’arrêt du cycle cellulaire, la réparation de l’ADN et dans certains cas la mort cellulaire.
RH : Recombinaison Homologue ; RNH : Recombinaison Non Homologue ou NHEJ
d’ATM qui va permettre la phosphorylation de nombreux substrats, dont les plus connus sont p53, NBS1, Chk1, Chk2 ce qui aura pour but l’activation des contrôles de restriction du cycle cellulaire, l’initiation de la réparation de l’ADN et la mort cellulaire.
La présence de CDBs augmente l’activité kinase d’ATM et cela passe par l’autophosphorylation de la serine 1981. Cette autophosphorylation permet la conversion de la forme dimérique inactive en une forme monomérique active d’ATM (Bakkenist and Kastan 2003). Plusieurs études ont essayé de comprendre comment et par quelles protéines, les CDBs permettent cette activation. Le complexe MRN, composé de trois protéines MRE11, Rad50 et Nbs1, est le plus connu pour activer ATM. En effet en 2004, Lee and Paull ont montré que MRN active directement ATM (Lee and Paull 2004). En réponse aux CDBs, le complexe MRN va se fixer rapidement au niveau de la cassure et permettre le recrutement de la protéine ATM grâce à l’interaction entre ATM et Nbs1 (Williams et al. 2007). Le complexe MRN stimule l’activité d’ATM (la phosphorylation de Chk2 est augmentée de 15 fois en présence de MRN), car il permet un changement de conformation de la protéine qui va stabiliser son interaction avec ces substrats (Lee and Paull 2004). Mais d’autres études montrent que les modifications de la structure de la chromatine pourraient avoir un rôle dans l’activation d’ATM (Bakkenist and Kastan 2003). Le remodelage de la chromatine passe par les modifications post-traductionnelles des histones comme des méthylations, des phosphorylations ou des acétylations. Les histones acétyltransférases (HAT) ont la capacité d’acétyler les protéines, et plusieurs études montrent qu’ATM pourrait être acétylée rapidement après l’apparition des dommages. L’étude de Sun et al, indique qu’ATM est acétylée en réponse aux dommages et que cela est nécessaire à son activation, ce mécanisme serait dépendant de la HAT, Tip60 (Sun et al. 2005). De plus, une autre étude montre que hMOH, une HAT responsable de l’acétylation de l’histone H4, interagit avec ATM et que l’autophosphorylation d’ATM serait dépendante de l’activité acétylase de hMOF (Gupta et al. 2005). Au vue de ces données, l’activation d’ATM semble dépendante de plusieurs événements, son autophosphorylation mais aussi du contexte chromatinien et de l’acétylation par différentes HAT.
Une fois activée, ATM va induire différentes cascades de signalisation qui vont conduire à différentes réponses, tel que l’arrêt du cycle cellulaire en G1/S et G2/M, la réparation de l’ADN mais aussi la mort cellulaire. Le blocage du cycle cellulaire va dépendre de l’activation de différentes protéines dont les plus connues sont p53 et Chk2 (pour l’arrêt en G1) et, Chk1 et Chk2 (pour l’arrêt en G2/M) (Figure 4). Cet arrêt va permettre la réparation de l’ADN endommagé. Par contre, si la cellule est en phase S au moment de l’apparition des dommages, alors la réplication est ralentie afin de permettre aux machineries de réparation de se mettre en place et de réparer l’ADN sans rencontrer d’ADN polymérases. Parmi les événements impliqués dans la signalisation
Figure 4 : Signalisation de l’activation des points de contrôle du cycle cellulaire en réponse aux RI
(Jeggo and Lobrich. 2007)
Figure 5 : Les différentes voies de réparation des CDBs après détection de la cassure (Jeggo and
Lobrich. 2007)
NHEJ RH
ATM
Artémis
induit par une CDB, l’environnement chromatinien est modifié tel que l’histone H2AX, un variant de l’histone H2A, qui est phosphorylé rapîdement par ATM ou la DNA-PKcs. Cette phosphorylation va s’étendre sur plusieurs mégabases autour de la cassure (Rogakou et al. 1998; Rogakou et al. 1999). Au niveau de la cassure, γH2AX va permettre le remodelage de la chromatine, et va faciliter l’interaction au niveau de la cassure des substrats d’ATM tel que 53BP1, BRCA1 ou le complexe MRN et permettre le recrutement de certains facteurs de réparations (Downs et al. 2004; Fernandez-Capetillo et al. 2004). Une fois phosphorylé, γH2AX va se situer autour de la cassure ce qui va permettre en immunofluorescence de détecter des foyers utilisés comme marqueurs des CDBs.
2.2. Réparation des CDBs
Pour réparer les CDBs, la cellule dispose de différents mécanismes (Figure 5). Quand la chromatide sœur est présente, la cellule va préférentiellement l’utiliser comme matrice afin de réparer le brin endommagé, c’est le mécanisme de recombinaison homologue (RH). Un autre mécanisme appelé jonction d’extrémité non homologue (NHEJ), existe et va permettre la ligation « directe » des brins d’ADN. Un troisième type de réparation est présent, la voie du «Single-Strand Annealing » (SSA) qui s’utilise entre deux séquences répétées directes aux extrémités de la cassure, c’est une voie transitionnelle entre la NHEJ et la RH.
a. La recombinaison homologue
La RH est un processus basé sur un échange d’information génétique grâce à un événement de recombinaison entre deux séquences d’ADN dont l’une est porteuse de dommages, et nécessitant une longue séquence d’homologie (Figure 6). Pour cela, une fois la cassure détectée, l’activité exonucléase 5’-3’ du complexe MRN va engendrer une longue extrémité simple brin 3’ sortant de plus de 100 paires de bases. Cette extrémité va servir de substrat pour recruter la machinerie de réparation. Dans un premier temps, la protéine RPA se fixe sur l’ADN simple brin afin de le protéger de la dégradation et de faciliter la formation du filament Rad51 en position 3’. Rad51, aidé de Rad52 et de Rad54, va initier la recherche de séquence d’homologie au niveau de la chromatide sœur qui va servir de matrice. Une fois la séquence trouvée, une jonction d’HOLLYDAY va être formée. Une des extrémités libre va envahir l’hélice d’ADN intacte et permettre ainsi l’échange de brins. Puis l’extrémité libre 3’ va s’hybrider au niveau de la matrice et une ADN polymérase va permettre la synthèse du brin. Une fois la polymérisation du nouveau brin finie, la jonction de HOLLIDAY se referme et le brin nouvellement synthétisé est ligaturé par l’ADN ligase I (Hoeijmakers 2001; Valerie and Povirk 2003). BRCA1 et BRCA2, activées par ATM, jouent aussi un rôle dans la RH. En effet, ils interagissent avec Rad51 et
semblent stimuler la RH car BRAC2 aide au recrutement de Rad51 sur l’ADN et BRCA1 pourrait servir de coordinateur à la RH (Valerie and Povirk 2003). La RH est un processus très conservatif, il est qualifié d’ « error-free », car il permet la restauration de la séquence sans perte d’information génétique.
b. Le Single-Strand Annealing
Le SSA est un processus non conservatif, car son utilisation entraine des délétions, des insertions et des translocations. Ce mécanisme de réparation est basé sur l’hybridation de deux séquences répétées directes aux extrémités 3’ de la cassure et de la délétion des nucléotides entre l’extrémité 3’ et la séquence répétée (Figure 7). Le SSA peut être considéré comme une variante de la RH, car l’initiation du processus est identique aux deux voies. En effet, après détection de la cassure, une endonucléase, comme le complexe MRN, va générer une extrémité simple brin 3’ sortant qui sera protégée par RPA. Ces extrémités sont générées jusqu’à l’exposition de séquences répétées directes qui sont capables de s’apparier par complémentarité de bases. Le complexe XRCC1/XPF (une nucléase impliquée dans le NER) va permettre l’incision du brin non homologue, puis une ADN polymérase va synthétiser les nucléotides manquants et les brins vont pouvoir être religués (Valerie and Povirk 2003). Contrairement à la RH, Rad51 n’est pas requis de même que l’utilisation d’un brin complémentaire servant de matrice. Par contre la protéine Rad 52 semble jouer un rôle important dans la liaison initiale aux extrémités de la cassure et potentialise la recherche des séquences homologues entre les extrémités (Van Dyck et al. 2001). Le SSA semble être une voie peu utilisée chez les cellules eucaryotes et pourrait réparer le génome au niveau des séquences répétitives.
c. La réparation par jonction d’extrémité non homologue
La voie de la NHEJ utilise plusieurs enzymes qui reconnaissent les extrémités de l’ADN et les maintiennent proches grâce à un complexe synaptique afin de permettre la religature des deux brins. A la différence de la RH, la NHEJ n’a pas besoin d’homologie pour effectuer la ligation des deux extrémités, ce qui lui permet d’être présente tout au long du cycle cellulaire et fait d’elle la voie majoritaire de la réponse aux CDBs.
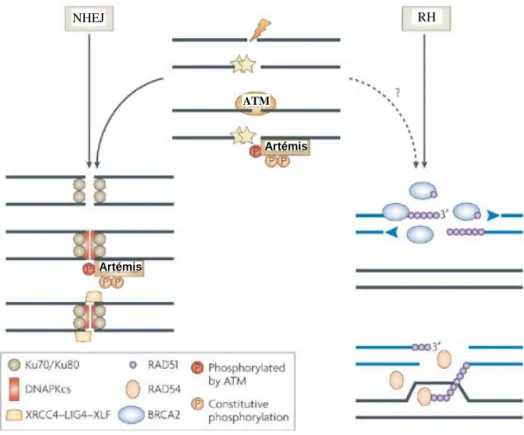
a. Le mécanisme de la NHEJ
Le processus de la NHEJ (Figure 8) est initié par la fixation de l’hétérodimère Ku, composé de deux sous unités Ku70 et Ku80, aux extrémités libres de l’ADN. Une fois fixé, Ku va recruter la sous unité catalytique du complexe, la protéine DNA-PKcs pour former le complexe PK (pour Protéine Kinase dépendante de l’ADN) (Uematsu et al. 2007). Lorsque la
Figure 7 : Schéma représentatif du Single-Strand Annealing d’après (Valerie and Povrick. 2003)
Reconnaissance de la CDB et génération de l’extrémité 3’ simple brin
Recrutement de Rad52
Séquence répétée trouvée
Incision du brin non homologue
Polymérisation Ligation
PK est recrutée sur l’ADN, cela va permettre son activation et l’induction d’un changement de conformation pour mettre en contact les deux DNA-PKcs afin de former une synapse qui va maintenir la proximité des deux brins d’ADN. Puis la DNA-PKcs va s’autophosphoryler, notamment au niveau du cluster 2609-2647, induisant un nouveau changement de conformation qui va libérer les extrémités pour laisser l’accès aux enzymes de maturation si les extrémités ne sont pas compatibles. Il existe différentes enzymes de maturations, Artémis, PNK, APE 1, qui vont prendre en charge différentes extrémités. Par exemple, la polynucléotide kinase (PNK) va ajouter un groupement phosphate en 5’. Dans certains cas, les extrémités présentent un groupement 3’ phosphoglycolate qui doit être enlevé pour permettre la ligature, et cela peut être fait par la PNK, APE1 (apurinic/apyrimidinic endonuclease) ou par l’enzyme de maturation la plus connue Artemis qui permet aussi de cliver les extrémités formant une structure en hairpin (Weterings and Chen 2008). Puis si une étape de polymérisation est nécessaire, les ADN polymérases μ ou λ vont intervenir. Pour finir, un deuxième complexe, composé de la ligase IV et de XRCC4 va être recruté par le complexe Ku/DNA-PK/ADN, et va permettre la ligature des deux brins d’ADN (Costantini et al. 2007). Récemment, un nouveau facteur de la NHEJ a été découvert, c’est la protéine XLF (XRCC4 like Factor) ou Cernunnos, qui serait recruté par Ku au même moment que le complexe XRCC4/Ligase IV, et stimulerait la capacité de ligation de la ligase (Yano et al. 2008). Une fois la cassure réparée, le complexe DNA-PK va permettre la dissociation de la plateforme protéique grâce à une nouvelle phosphorylation de la DNA-PKcs : le complexe XRCC4/Ligase IV et la DNA-PKcs se détache de l’ADN et Ku migre le long du brin d’ADN (Meek et al. 2004; Costantini et al. 2007; Lieber 2008; Weterings and Chen 2008).
b. Les acteurs majeurs de la NHEJ
Les protéines centrales de la NHEJ sont aux nombres de sept : Ku70, Ku80, DNA-PKcs, Artémis, XRCC4, ligase IV et le dernier facteur découvert XLF/Cernunnos.
1. L’hétérodimère Ku
En 1981, Ku a été découvert comme un auto-antigène (Mimori et al. 1981) avant d’être découvert comme protéine de réparation. La purification de Ku a permis de mettre en évidence que cette protéine était composée de deux sous-unités, une de 70 kDa appelée Ku70 et l’autre de 83 kDa nommée Ku80 (Mimori and Hardin 1986). Ces protéines sont conservées et sont très abondantes, environ 40 000 molécules par cellules soit 1 % des protéines totales. L’une des caractéristiques de Ku est sa grande affinité pour les extrémités libres de l’ADN et cela de façon séquence indépendante (Mimori and Hardin 1986; Falzon et al. 1993). En effet, Ku peut se fixer au niveau des CSBs, des CDBs ainsi que sur des structures tige-boucle. Grâce à ses propriétés, Ku
Ku70-Ku80 DNA-PKcs XRCC4 ADN ligase IV P P P XLF/Cernunnos Pas de maturation Artémis PNK, ADN polymérases Reconnaissance du dommage
Activation du complexe DNA-PK : formation de la synapse
Recrutement de la DNA-PKcs
Recrutement du complexe
XRCC4/XLF:Ligase IV ainsi que des enzymes de maturation si nécessaires
Maturation des extrémités
Réparation
Figure 8 : Représentation schématique du mécanisme de la NHEJ P
intervient dans la première étape de la réparation des CDBs en reconnaissant les extrémités libres de l’ADN. Une fois fixé, Ku va permettre le recrutement de la sous-unité catalytique la PKcs, grâce à la séquence C-terminale de Ku80, dont l’assemblage constitue l’holoenzyme DNA-PK qui devient actif (Gottlieb and Jackson 1993). La deuxième caractéristique de Ku est de permettre le recrutement des facteurs de la NHEJ, comme le complexe XRCC4/Ligase IV (Costantini et al. 2007; Weterings and Chen 2008). En résumé, les fonctions de Ku dans la NHEJ sont dans un premier temps de reconnaître les CDBs, puis dans un deuxième temps d’optimiser le recrutement des facteurs de la NHEJ au site de la cassure.
Différentes études ont pu montrer que le rôle de Ku ne se limitait pas seulement à la réparation des CDBs par la NHEJ. Ku est retrouvé au niveau des télomères où il peut se fixer directement à l’ADN télomérique ou via des interactions protéines-protéines. Ku aurait un rôle dans la protection des télomères, en effet la réduction de l’expression de Ku80 par une technique de siRNA ou dans des cellules somatiques humaines Ku80-/+, aurait pour conséquences une
augmentation de la fusion télomérique et une diminution de la longueur des télomères. De plus, il a été montré que Ku interagit avec TRF1 et TRF2, protéines prévenant la fusion télomérique, et régulerait la protéine télomérase, protéine majeure de la régulation des télomères (Jaco et al. 2004; Myung et al. 2004; Fisher and Zakian 2005). D’autres études ont observé un rôle de Ku dans la transcription où il interagirait avec des facteurs de transcription tel que PDX-1 et serait aussi présent au niveau des sites d’élongation de l’ARN polymérase II. De plus les cellules d’hamster déficientes en Ku80 possèdent un taux de transcription 5 fois moins élevé par rapport aux cellules proficientes (Woodard et al. 2001; Mo and Dynan 2002; Lebrun et al. 2005).
Les rôles de Ku ne sont pas limités au noyau, il est présent au niveau du cytoplasme où il aurait un rôle anti-apoptotique en séquestrant la protéine BAX (Sawada et al. 2003). Mais de façon encore plus surprenante, Ku est retrouvé à la surface externe de la membrane plasmique de nombreuses cellules normales ou tumorales (Ginis et al. 1995; Ginis and Faller 2000; Lucero et al. 2003; Monferran et al. 2004). En effet, au sein du laboratoire, il a été montré que Ku est sécrété de façon non classique. Une fois réassocié à la membrane, il interagit avec la MMP 9 afin de favoriser le processus d’invasion au travers de matrice de collagène IV (Monferran et al. 2004; Paupert et al. 2007).
2. La DNA-PKcs
La DNA-PKcs est une protéine de 465 kDa qui fait partie de la famille des analogues des Phosphatidylinositol 3-Kinase related Kinase (PI3KK), tout comme ATM et ATR. Cette famille présente un domaine kinase proche de celui des PI3-Kinases mais contrairement aux PI3K, les PI3KK ne peuvent phosphoryler que les protéines et pas les lipides (Abraham 2004). La
PKcs est recrutée par Ku au niveau de la CDB afin de constituer un complexe nommé DNA-PK possédant une activité sérine-thréonine kinase portée par la DNA-PKcs et activée spécifiquement par les CDBs de l’ADN. Plusieurs cibles de l’activité kinase ont été retrouvées in vitro, comme Artémis, Ku, XRCC4, p53 et la DNA-PKcs elle-même (Anderson 1993; Meek et al. 2004).
La DNA-PKcs a deux rôles distincts dans le processus de la NHEJ. Dans un premier temps, elle permet l’alignement des extrémités libres et participe ainsi à la formation de la synapse. Puis elle va coordonner l’accès à la CDB des différents facteurs, elle peut être considérée comme l’activateur de la NHEJ (DeFazio et al. 2002; Meek et al. 2004; Weterings and Chen 2007). Son activation par son autophosphorylation est nécessaire à son action dans la NHEJ. La DNA-PKcs présente 16 sites d’autophosphorylation répartie en plusieurs clusters (Figure 9). Par exemple, l’autophosphorylation en trans de la DNA-PKcs au niveau du cluster 2609-2647 pendant la synapse va permettre de libérer les extrémités libre de l’ADN et ainsi permettre l’accès aux enzymes de maturation et de ligation (Meek et al. 2004; Goodarzi et al. 2006). La régulation de la dissociation du complexe une fois la réparation finie passe encore par la phosphorylation et l’autophosphorylation de la kinase (Costantini et al. 2007). Même si l’autophosphorylation semble être la voie d’activation de la DNA-PKcs, il apparaît que certains sites, comme la thréonine 2609 et la thréonine 2647, sont phosphorylés même si la DNA-PKcs n’a plus son activité kinase. Pour expliquer cela, l’hypothèse selon laquelle ATM pourrait phosphoryler la DNA-PKcs est avancée (Chen et al. 2007; Uematsu et al. 2007). Plusieurs études ont mis en évidence que cette autophosphorylation sur différents sites notamment au niveau de la sérine 2056, et du cluster 2609-2647, est nécessaire à la NHEJ (Meek et al. 2004; Uematsu et al. 2007). Cette autophosphorylation est responsable du changement de conformation de la DNA-PKcs, si elle n’a pas lieu alors la protéine clé de la NHEJ est incapable de remplir ses rôles : la DNA-PKcs va rester bloquée au niveau de la cassure et empêcher la réparation (Figure 10)
Comme Ku, la DNA-PKcs est présente en dehors du noyau. Elle serait présente au niveau de la membrane plasmique où elle pourrait avoir un rôle dans la survie cellulaire ((Lucero et al. 2003); données personnelles). En effet, plusieurs études ont montré que la DNA-PKcs peut se comporter comme un récepteur TLR9 (Toll Like Recepteur 9), en reconnaissant certaines séquences d’ADN bactérien à la surface de la cellule et ainsi activer la réponse immunitaire (Chu et al. 2000; Dragoi et al. 2005). De même, une étude a observé que l’internalisation du récepteur EGFR (Epithélial Growth Factor Receptor), due à son activation après irradiation, induisait la translocation de la DNA-PKcs du noyau au cytoplasme afin d’interagir avec la vésicule d’internalisation du récepteur. Ainsi, on peut penser que si la DNA-PKcs n’est plus au noyau afin de réparer les cassures, cela va rendre les cellules radiosensibles (Bandyopadhyay et al. 1998).
Toutes les données sur les rôles nucléaires ou non de la DNA-PKcs montrent que cette protéine joue un rôle majeur dans la réponse au stress cellulaire.
3. Artémis
Artémis a été découvert en 2001 chez des enfants présentant une immunodéficience et une radiosensibilité (Moshous et al. 2001). Les cellules de ces patients ont un défaut de NHEJ et de recombinaison V(D)J. Le gène Artémis code pour une protéine de 77 kDa qui fait partie de la famille des métallo-B-lactamase. Artémis est une nucléase possédant les deux activités : endonucléase et exonucléase, qui sont requises pour cliver les structures en Hairpins. Ainsi Artémis joue un rôle important dans la recombinaison V(D)J où il va être recruter pour cliver les structures en hairpin afin de permettre la ligation entre deux segments (Ma et al. 2002). Dans le mécanisme de la NHEJ, Artémis interviendrait seulement dans 10 % des cas, spécialement pour les cassures avec des extrémités sous forme de hairpins ou lorsqu’elles possèdent un groupement 3’ phosphoglycolate. Pour cela, Artémis est recrutée par la DNA-PKcs et les deux protéines vont former un complexe permettant la maturation de la cassure. L’activation d’Artémis dépend donc de l’activation de la DNA-PKcs que se soit de façon directe (phosphorylation d’Artémis par DNA-PKcs) ou indirecte (autophosphorylation de la DNA-PKcs qui libère les extrémités) (Meek et al. 2004; Riballo et al. 2004; Weterings and Chen 2008).
4. Le complexe XRCC4/Ligase IV
Ce complexe est composé de 2 protéines, la ligase IV qui porte l’activité de ligation et XRCC4 qui permet la stabilité de la ligase ainsi que la liaison à l’ADN. XRCC4 est une protéine de petite taille, 34 kDa, qui est présente sous forme de tétramère ou de dimère lorsqu’elle est liée avec la ligase IV. La fonction principale de XRCC4 est de stabiliser la ligase IV, en effet les cellules XRCC4 déficientes ont un défaut de NHEJ car la ligase IV est dégradée. L’activité ligase du complexe est donné par la ligase IV qui a la capacité de religuer deux extrémités même si elles ne sont pas compatibles (Meek et al. 2004; Weterings and Chen 2008).
Au site de la cassure, le complexe XRCC4/Ligase IV est recruté par Ku grâce à son interaction avec la ligase IV. Le rôle de la DNA-PKcs dans ce recrutement semble controversé : l’équipe de B. Salles, ont montré que la DNA-PKcs était nécessaire au recrutement de XRCC IV (Drouet et al. 2005), par contre l’étude de Constantini et ses collaborateurs montre qu’elle ne serait pas nécessaire pour ce recrutement, mais que sa présence faciliterait ce recrutement (Costantini et al. 2007). On peut en conclure que même si la DNA-PKcs n’est pas nécessaire à