Université de Sherbrooke
PROGRAMMATION MÉTABOLIQUE FŒTALE :
ÉTUDE DE L’IMPACT DE L’EXPOSITION AU DIABÈTE GESTATIONNEL SUR LE MÉTHYLOME DU NOUVEAU-NÉ
Par
Andrée-Anne Houde Programme de Biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de Philosophiae Doctor (Ph.D.) en biochimie
Sherbrooke, Québec, Canada Mai, 2015
Membres du jury d’évaluation Guylain Boissonneault – Président de jury
Luigi Bouchard – Directeur de recherche Aris Aziz – Évaluateur externe au programme Jacquetta Trasler – Évaluatrice externe à l’Université
À Emma, Victor, Samuel et à mes futurs enfants, Que vos passions vous poussent à dépasser vos limites et
Programmation métabolique fœtale : étude de l’impact de l’exposition au diabète gestationnel sur le méthylome du nouveau-né
Par
Andrée-Anne Houde Programme de Biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de philosophiae doctor (Ph.D.) en biochimie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 L’obésité est un enjeu de société de première importance; elle est un facteur de risque de plusieurs maladies et engendre d’importantes dépenses en santé. Outre l’alimentation, la sédentarité et les prédispositions génétiques, il semble que l’environnement fœtal soit un facteur déterminant dans le développement de l’obésité. En effet, il a été démontré que les nouveau-nés exposés à un environnement intra-utérin défavorable ont un risque accru de développer, à l’adolescence et à l’âge adulte, l’obésité ainsi que les désordres métaboliques qui y sont associés. Le diabète gestationnel (DG) est l’une des complications de santé maternelle les plus fréquentes et est associé à un risque accru à long terme pour la santé métabolique de l’enfant. Malgré les nombreuses données probantes épidémiologiques concernant le phénomène de la programmation fœtale associée au DG, les mécanismes moléculaires impliqués ont été très peu étudiés. Il est cependant de plus en plus évident que l’épigénétique soit l’un de ces mécanismes. Cette thèse a pour objectif d’identifier les changements de méthylation de l’ADN, la modification épigénétique la plus stable et la plus connue, chez les nouveau-nés exposés in utero au DG. Dans un premier temps, la méthylation de l’ADN de 44 échantillons de placenta et de sang de cordon a été analysée à l’échelle du génome. Cette approche a permis de démontrer que les gènes épigénétiquement modifiés suite à une exposition au DG sont majoritairement retrouvés dans les voies biologiques associées aux maladies métaboliques. Des analyses dans une cohorte indépendante (n=80) ont confirmé l’effet de la glycémie maternelle sur la méthylation de l’ADN des gènes BRD2, LRP1B et CACNA1D impliqués dans la régulation du métabolisme des lipides et du glucose et du système rénine-angiotensine respectivement. Dans un second temps, l’approche par gènes candidats a démontré que l’exposition au DG est associée à la méthylation de l’ADN de gènes du métabolisme des lipides (LPL et ABCA1) du placenta. L’analyse de la méthylation de la LEP et de l’ADIPOQ dans le sang et les tissus adipeux de sujets sévèrement obèses a permis d’identifier des sites de méthylation pouvant potentiellement être utilisés dans le sang comme marqueur de susceptibilité à l’obésité. L’ensemble des résultats de cette thèse démontrent que le DG modifie le profil épigénétique de gènes impliqués dans les voies biologiques des maladies métaboliques (métabolisme énergétique et des lipides) et supportent l’importance de la méthylation de l’ADN dans la programmation de la santé métabolique du nouveau-né ayant été exposé in utero au DG.
Mots clés : méthylation de l’ADN, placenta, sang de cordon, épigénétique, leptine, adiponectine, tissus adipeux, sang
Fetal metabolic programming: the impact of gestational diabetes mellitus exposure on newborn’s epigenetic signature
By
Andrée-Anne Houde Biochemistry Program
Thesis presented at the Faculty of medicine and health sciences for the obtention of Doctor degree diploma Philosophiae Doctor (Ph.D.) in biochemistry, Faculty of medicine and
health sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 Obesity has reached epidemic proportions worldwide in both adult and childhood populations and is now recognized as a major public health issue. Obesity is associated with higher incidence of cardiometabolic complications including type 2 diabetes (T2D), dyslipidemia and hypertension as well as with increased health care costs. The fetal environment now appears, with genetics and the environment, as one cause of the obesity epidemic. Indeed, according to the fetal programming hypothesis, newborns exposed to a detrimental fetal environment are more susceptible to develop obesity, T2D and other related chronic disorders when they become teenagers or adults. Many studies have associated gestational diabetes mellitus (GDM) exposure with these long-term metabolic health risks for the newborn. Although, numerous studies show epidemiological evidence to support the fetal programming hypothesis, only a few studies have been undertaken to understand the underlying molecular mechanisms. However, several studies now suggest that epigenetics may be involved. The objective of this thesis is to study changes in DNA methylation, the more stable and studied epigenetic system, in newborns that have been exposed to GDM in utero. First, a genome-wide DNA methylation analysis (BeadChip) was performed in a sample set of 44 placenta and cord blood samples to identify genes and metabolic pathways dysregulated by GDM. This approach showed that genes epigenetically affected by GDM are predominantly involved in metabolic diseases. The associations between maternal glycemia and DNA methylation levels were confirmed, in an independent birth cohort, for BRD2, LRP1B and CACNA1D gene loci involved in the regulation of lipid and glucose metabolism and the renin-angiotensin system respectively. Then, using a candidate gene approach we reported that DNA methylation levels at gene loci involved in lipid metabolism (LPL and ABCA1) are modified in the placenta following exposure to GDM. Furthermore, analyses of LEP and ADIPOQ DNA methylation levels in blood and adipose tissues of severely obese men and women allowed the identification of CpG sites that might be used in blood as a marker of obesity susceptibility. Altogether the results of this thesis show that GDM affects the epigenetic signature of genes involved in metabolic disease pathways (energy and lipid metabolism) and support the role of DNA methylation in metabolic health programming of the newborn exposed to GDM.
Keywords : DNA methylation, placenta, cord blood, epigenetic, leptin, adiponectin, adipose tissue, blood
Résumé ... iii
Summary ... iv
Table des matières ... v
Liste des figures ... viii
Liste des tableaux ... xi
Liste des abréviations ... xiv
Chapitre 1 - Introduction ... 1
1.1 L’obésité ... 1
1.1.1 Portrait de l’obésité au Canada ... 2
1.1.2 Les principales causes de l’obésité ... 3
1.1.2.1 Les facteurs génétiques ... 3
1.1.2.2 Les facteurs environnementaux ... 4
1.2 La programmation fœtale : de Barker à aujourd’hui ... 6
1.3 Le diabète gestationnel ... 11
1.3.1 La prévalence et les facteurs de risque du diabète gestationnel ... 11
1.3.2 Le dépistage et les critères diagnostiques du diabète gestationnel ... 11
1.3.3 La physiopathologie du diabète gestationnel ... 14
1.3.4 Les conséquences du diabète gestationnel pour la santé de la mère ... 17
1.3.5 Le diabète gestationnel et la programmation fœtale ... 18
1.4 L’épigénétique ... 24
1.4.1 La définition de l’épigénétique ... 24
1.4.2 Les principaux mécanismes épigénétiques... 26
1.4.2.1 Les modifications des histones ... 26
1.4.2.2 Les ARN non codants ... 27
1.4.2.3 La méthylation de l’ADN ... 29
1.4.2.3.1 Mécanismes de méthylation de l’ADN ... 29
1.4.2.3.2 Mécanismes de déméthylation de l’ADN ... 31
1.4.2.3.3 Le méthylome et son rôle dans la régulation de l’expression génique ... 32
1.5 La méthylation de l’ADN pendant le développement embryonnaire ... 35
1.5.1 Différentiation cellulaire et reprogrammation du méthylome chez le zygote et dans les cellules germinales primordiales ... 35
1.5.2 L’empreinte parentale ... 38
1.5.3 L’inactivation du chromosome X ... 39
1.6 La méthylation de l’ADN dans la programmation métabolique fœtale ... 40
1.6.1 Les modèles animaux ... 40
1.6.2 Les études épidémiologiques ... 42
1.6.2.1 Approche à l’échelle du génome ... 42
1.6.2.2 Approche par gènes candidats ... 46
1.7 Les facteurs contribuant à la variabilité des niveaux de méthylation ... 49
1.7.1 L’environnement post-natal ... 49
1.7.2 La génétique ... 50
1.8 La sélection du tissu pour l’analyse de la méthylation dans le contexte de la programmation fœtale ... 53
1.9 Problématique ... 56
1.9.1 Hypothèse ... 56
1.9.2 Objectifs ... 57
Chapitre 2 - Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic disease ... 58
Chapitre 3 - LRP1B, BRD2 and CACNA1D: New candidate genes in fetal metabolic programming of newborns exposed to maternal hyperglycemia ... 87
Chapitre 4 - Placental lipoprotein lipase DNA methylation levels are associated with gestational diabetes mellitus and maternal and cord blood lipid profiles ... 118
Chapitre 5 - Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status ... 147
Chapitre 6 - Fetal epigenetic programming of adipokines ... 184
Chapitre 7 - Cross-tissue comparisons of leptin and adiponectin DNA methylation profiles ... 202
Chapitre 8 - Leptin and adiponectin DNA methylation levels in adipose tissues and blood cells are associated with BMI, waist girth and LDL-cholesterol levels in severely obese men and women ... 232
Chapitre 9 - Discussion ... 266
9.1 Analyse du méthylome du placenta et du sang de cordon suite à l’exposition au diabète gestationnel ... 267
9.1.2 La validation des résultats de notre étude épigénomique ... 271
9.1.3 Les limites de notre étude épigénomique ... 274
9.1.3.1 L’hétérogénéité cellulaire... 274
9.1.3.2 Les variables biologiques ... 276
9.1.3.2 Les variants génétiques ... 278
9.2 Modifications épigénétiques des gènes du métabolisme des lipides suite à l’exposition au diabète gestationnel ... 280
9.3 Associations entre les modifications épigénétiques observées chez les nouveau-nés exposés au diabète gestationnel et les indicateurs de santé et de croissance fœtale ... 287
9.4 État actuel des connaissances concernant l’identification des gènes épigénétiquement impliqués dans la programmation métabolique fœtale associée au diabète gestationnel ... 290
9.4.1 La spécificité cellulaire de la méthylation de l’ADN des gènes LEP et ADIPOQ .. 293
9.4.2 Les associations entre la méthylation des gènes LEP et ADIPOQ et des facteurs de risques cardiométaboliques chez l’adulte ... 296
Chapitre 10 - Conclusion ... 300
Remerciements ... 301
Liste des références ... 303
Annexe 1 ... 345 Annexe 2 ... 349 Annexe 3 ... 352 Annexe 4 ... 353 Annexe 5 ... 354 Annexe 6 ... 359 Annexe 7 ... 361 Annexe 8 ... 362
Figure 1.1- Réponse adaptative du fœtus et les risques de maladies à long-terme ... 7
Figure 1.2- Risque relatif d’être atteint d’une maladie métabolique en fonction du poids à la naissance ... 9
Figure 1.3- Adaptations physiques et mécanismes physiopathologiques associés à une grossesse normoglycémique et au diabète gestationnel ... 16
Figure 1.4- Le cycle intergénérationnel de l’obésité perpétué par le diabète gestationnel ... 21
Figure 1.5- La méthylation de l'ADN ... 30
Figure 1.6- La déméthylation de l'ADN ... 32
Figure 1.7- La méthylation de l'ADN et la régulation de l'expression des gènes ... 34
Figure 1.8- Reprogrammation du méthylome dans le cycle de vie des mammifères ... 37
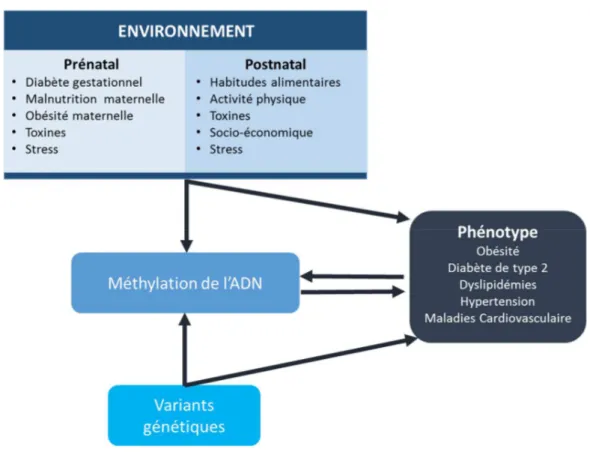
Figure 1.9- Représentation des interactions entre les facteurs environnementaux, la génétique et la méthylation de l’ADN entraînant le phénotype de l’obésité et des complications cardiométaboliques ... 52
Figure 2.1- Overview of the analytical strategy used to identify genes and metabolic pathways showing epigenetic dysregulation in response to GDM exposure ... 66
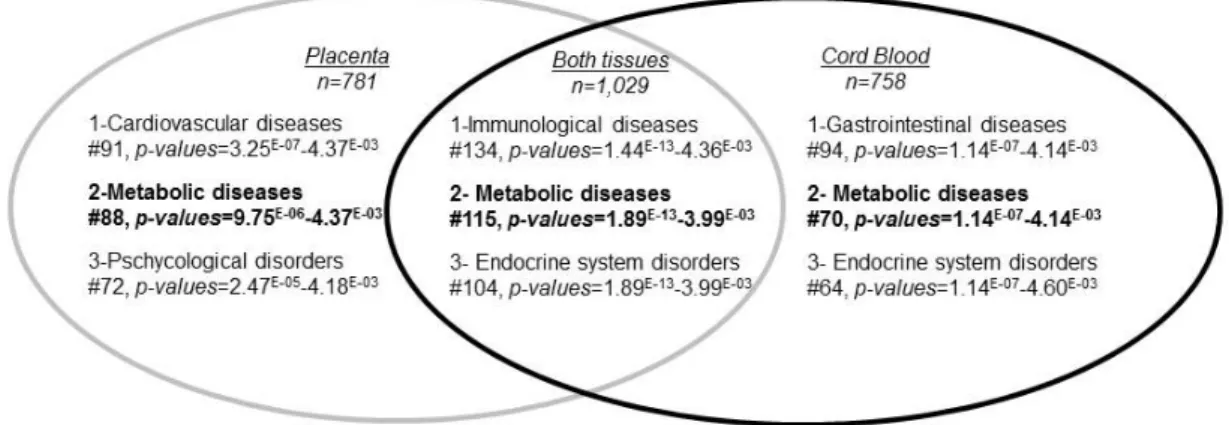
Figure 2.2- Ingenuity Pathway Analysis: Top-ranked common disease and disorder pathways that were epigenetically affected by GDM ... 67
Figure 2.3- Ingenuity Pathway Analysis: Top-ranked diseases and disorders associated to each of the common disease and disorder pathways epigenetically affected by GDM ... 68
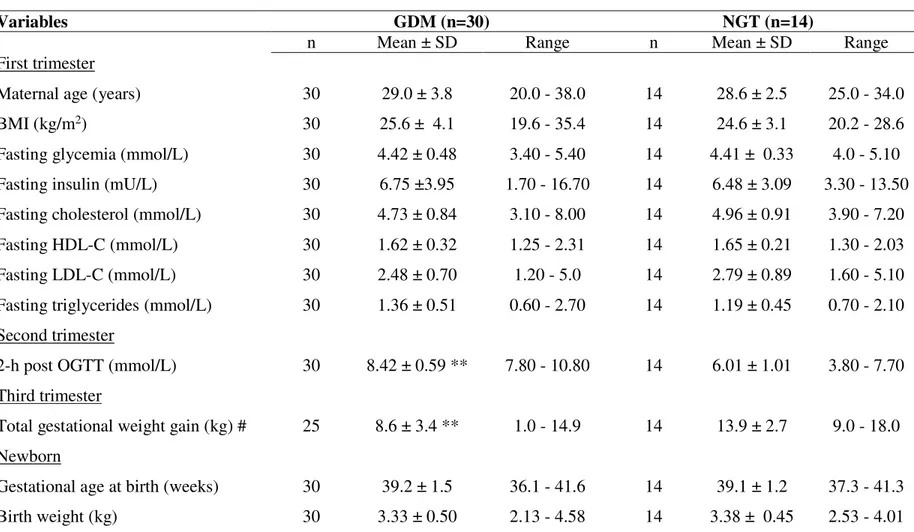
Figure 3.1- Association plots between maternal glucose levels 2h post-OGTT and DNA methylation levels in placenta of NGT (black circle) and GDM (black cross) women ... 100
Figure 3.2- Association plots between maternal glucose levels 2h post-OGTT and DNA methylation levels in cord blood of NGT (black circle) and GDM (black cross) women ... 101
Figure 3.3- Location of CpGs, transcription factor binding sites and highly conserved regions within BRD2 gene locus, adapted from UCSC genome browser tracks ... 104
Figure 4.1- Schematic representation of the LPL gene locus CpG island and localization of the 3 loci epigenotyped ... 131 Figure 4.2- DNA methylation levels at placental LPL loci according to maternal glucose tolerance status ... 132 Figure 4.3- Pearson correlations between LPL gene CpG2 (A) and CpG3 (B) DNA methylation and mRNA levels in placenta (n=126) ... 135 Figure 5.1- ABCA1 CpG island proximal promoter region and localization of the 4 loci epigenotyped in maternal and fetal placenta, as well as in maternal and cord blood samples ... 155 Figure 5.2- Pearson correlation coefficients between CpGs at ABCA1-A locus in maternal (A) and cord (B) blood ... 156 Figure 5.3- Placental ABCA1-CpG5 and -CpG6 DNA methylation levels according to maternal and newborn metabolic profile ... 158 Figure 5.4- DNA methylation levels at ABCA1-CpG6 on the maternal side of the placenta according to glucose tolerance status and HDL-C levels ... 159 Figure 5.5- Cord blood ABCA1-A locus DNA methylation levels according to maternal and newborn characteristics ... 162 Figure 5.6- Functional impact of the ABCA1 DNA methylation level variability ... 163 Figure 5.7- Maternal and fetal metabolic variables associated with placental and cord blood ABCA1 DNA methylation variations ... 167 Figure 6.1- Correlation between leptin gene promoter (A) or adiponectin CpG island E2 (B) DNA methylation levels and 2 h post-OGTT glycemia ... 190 Figure 6.2- Average methylation levels of CpG sites within leptin and adiponectin gene loci in the placenta and cord blood ... 193 Figure 7.1- CpG island within LEP gene proximal promoter region ... 209 Figure 7.2- Pearson correlation coefficients between CpGs at LEP proximal promoter locus in blood (A), subcutaneous (B) and visceral (C) adipose tissues ... 210 Figure 7.3- DNA methylation levels at LEP gene promoter (A) and ADIPOQ CpG island E (B) CpG sites in blood (white), subcutaneous (black) and visceral (grey) adipose tissues (n=73) ... 211
Figure 7.4- Location of transcription factor binding sites and highly conserved regions within the CpG island of the LEP gene proximal promoter region in placental mammals, adapted from UCSC genome browser tracks ... 214 Figure 7.5- CpG islands analysed within the ADIPOQ gene locus ... 218 Figure 7.6- Spearman correlation coefficients between CpGs at ADIPOQ locus in blood (A), subcutaneous (B) and visceral (C) adipose tissues ... 219 Figure 7.7- Average DNA methylation profile by CpG site for LEP gene promoter locus (A) and ADIPOQ CpG island E gene locus (B) in blood (white circles), subcutaneous (black circles) and visceral (grey circles) adipose tissues (n=73) ... 221 Figure 8.1- LEP and ADIPOQ DNA methylation levels according to body mass index (BMI) in severely obese patients ... 247 Figure 8.2- ADIPOQ DNA methylation levels according to waist circumference in severely obese patients ... 247 Figure 9.1- Effet du traitement du DG sur le méthylome du placenta et du sang de cordon
... 273 Figure 9.2- Fonction des protéines des gènes candidats de la programmation métabolique fœtale impliqués dans le transport materno-fœtal des lipides ... 286 Figure 9.3- Interrogations et plan de travail visant l’identification de marqueurs épigénétiques de la susceptibilité à l’obésité et aux complications cardiométaboliques ... 292 Figure 9.4- Proposition des facteurs modulant la méthylation de LEP et ADIPOQ in utero et à l’âge adulte et leur possible contribution au développement de l’obésité et des complications cardiométaboliques ... 299
Tableau 1.1- Classes d’obésité et le risque de comorbidités associé à l’IMC selon les critères
de l’OMS. ... 2
Tableau 1.2- Stratégie de dépistage du diabète gestationnel selon les associations ... 13
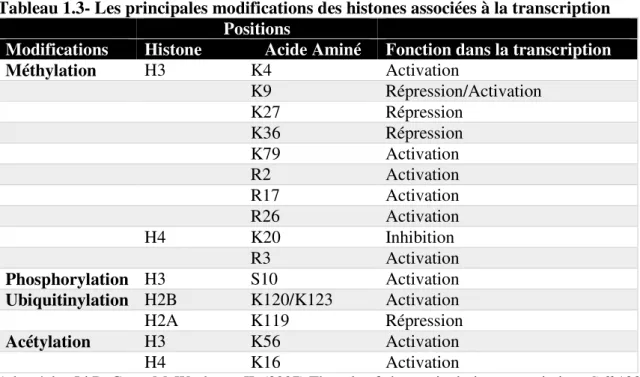
Tableau 1.3- Les principales modifications des histones associées à la transcription ... 27
Tableau 2.1- Maternal and newborn characteristics ... 65
Tableau 2.2- Ingenuity Pathway Analysis. Top-ranked common disease and disorder pathways that were epigenetically affected by GDM ... 69
Tableau 3.1- PCR and pyrosequencing primers for amplification and pyrosequencing of the 15 CpGs analysed in the Gen3G birth cohort. ... 97
Tableau 3.2- Comparison of the characteristics of mothers and newborns according to glucose tolerance status ... 99
Tableau 3.3- Mann-Whitney U comparisons of DNA methylation levels (%) in placenta and cord blood samples not exposed to GDM (NGT, n=60) or exposed to GDM (n=20) in the Gen3G birth cohort ... 102
Tableau 4.1- Characteristics of women and newborn according to glucose tolerance status ... 129
Tableau 4.2- Pearson correlation coefficient between placental LPL DNA methylation and maternal metabolic profile ... 134
Tableau 5.1- Maternal and newborn characteristics according to glucose tolerance status ... 154
Tableau 5.2- Multivariable linear regression analyses of ABCA1 DNA methylation levels predictors in placenta and cord blood ... 160
Tableau 5.3- PCR and pyrosequencing primers for ABCA1 gene CpG island locus amplification ... 173
Tableau 6.1- Spearman correlation coefficient between CpG sites methylation of leptin gene promoter and adiponectin gene loci C1 and E2 in maternal and fetal sides of placenta ... 194 Tableau 6.2- Spearman correlation coefficient between cord blood CpG sites methylation for adiponectin gene loci C1 and E2 and maternal and fetal sides of placenta ... 195 Tableau 7.1- Characteristics of subjects according to sex ... 208 Tableau 7.2- Pearson correlation coefficients between DNA methylation levels at LEP gene promoter CpG sites in blood, subcutaneous (SAT) and visceral adipose (VAT) tissue ... 213 Tableau 7.3- Pearson correlation coefficients between DNA methylation and mRNA levels at LEP gene promoter CpG sites in subcutaneous (SAT) and visceral adipose (VAT) tissues according to rs2167270 genotype and adjusted for sex ... 215 Tableau 7.4- Comparison of DNA methylation levels (%) at LEP proximal promoter CpG sites and LEP mRNA levels (AU) in SAT and VAT according to rs2167270 genotype ... 216 Tableau 7.5- Spearman rank correlation coefficients between DNA methylation levels at ADIPOQ CpG island E loci in blood, subcutaneous (SAT) and visceral adipose (VAT) tissues ... 219 Tableau 7.6- Pearson correlation coefficients between DNA methylation and mRNA levels at ADIPOQ CpG island E in subcutaneous (SAT) and visceral adipose (VAT) tissues ... 220 Tableau 7.7- PCR and pyrosequencing primers for LEP gene proximal promoter CpG island amplification and pyrosequencing ... 223 Tableau 8.1- PCR and pyrosequencing primers for LEP gene proximal promoter CpG island amplification and pyrosequencing ... 242 Tableau 8.2- PCR and pyrosequencing primers for ADIPOQ gene CpG islands amplification and pyrosequencing ... 243 Tableau 8.3- Characteristics of the subjects studied (n=73) ... 245
Tableau 8.4- Pearson correlation coefficients between LEP and ADIPOQ DNA methylation and mRNA levels in blood, subcutaneous (SAT) and visceral adipose tissue (VAT) and anthropometric variables (n=73) ... 248 Tableau 8.5- Pearson correlation coefficients between fasting LDL-C levels and both LEP and ADIPOQ DNA methylation and mRNA levels in subcutaneous (SAT) and visceral adipose tissues (VAT) (n=73) ... 251 Tableau 8.6- Pearson correlation coefficients between LEP and ADIPOQ DNA methylation and mRNA levels in blood, subcutaneous (SAT) and visceral adipose tissue (VAT) and cardiometabolic risk factors (n=73) ... 252 Tableau 8.7- Pearson correlation coefficients between LDL-C levels, LEP mRNA and LEP gene CpG17 DNA methylation levels in subcutaneous adipose tissue (SAT) according to the rs2167270 genotype ... 254 Tableau 9.1- Les 5 gènes démontrant les différences de méthylation les plus significatives entre les échantillons de placenta ou de sang de cordon exposés au DG et à un environnement in utero normoglycémique dans la cohorte E-21... 270 Tableau 9.2- Nombre de sites CpG affectés par l’exposition au DG dans le placenta et le sang de cordon avant et après la correction pour le décompte cellulaire... 276 Tableau 9.3- Comparaison des facteurs pouvant influencer le méthylome du nouveau-né chez les mères avec un diabète gestationnel (DG) et les mères avec une tolérance normale au glucose (TNG) de la cohorte E-21 ... 277
5-hmC 5-hydroxyméthylcytosine
ABCA1 ATP-binding cassette transporte A1 ABCG1 ATP-binding cassette transporte G1 ADA American diabetes association
ADIPOQ Adiponectine
AGL Acide gras libre
AOCG American college of obstetricians and gynecologists
ARNi ARN interférents
ARNnc ARN non codant
ARNpi ARN interagissant avec Piwi Avy Agouti viable yellow gene
BER Base excision repair
BRD2 Bromodomain-Containing 2
CACNA1D Calcium Channel, Voltage-Dependent, L Type, Alpha 1D Subunit
CCM Complication cardiométabolique
CDA Canadian diabetes association
CGI Ilôt CpG
c-HDL Cholestérol des lipoprotéines de haute densité c-LDL Cholestérol des lipoprotéines de basse densité CpG
CT/c-HDL
Dinucléotide CpG
Ratio de cholestérol total sur cholestérol des lipoprotéines de haute densité
DG Diabète gestationnel
DMR Differentially methylated region
DNMT Méthyltransférase de l’ADN
DT2 Diabète de type 2
FABP Protéine de liaison des acides gras FATP Protéine de transport des acides gras
GLUT Transporteur de glucose
GWAS Genome wide association studies
HAPO Hyperglycemia and adverse pregnancy outcome HGPO Hyperglycémie provoquée par voie orale
IADPSG International association of diabetes and pregnancy study groups ICE Imprinting control element
IGF Insulin-like growth factor
IMC Indice de masse corporelle
LDLR Récepteur des lipoprotéines de faible densité
LEP Leptine
LEPR Gène codant pour le récepteur de la leptine LIPG Gène codant pour la lipase endothéliale
LPL Lipoprotéine lipase
LRP1B Low-Density Lipoprotein Receptor-Related 1B LRP1P Low-Density Lipoprotein Receptor-Related MC4R Récepteur de type 4 de la mélanocortine
MCV Maladie cardiovasculaire
meQTL DNA methylation quantitative trait loci
miARN microARN
NDDG National diabetes data group
NPY Neuropeptide Y
OMS Organisation mondiale de la santé PLTP Protéine de transfert des phospholipides
POMC Pro-opiomélanocortine
SAM S-adénosyl-L-méthionine
SNP Single-nucleotide polymorphism SRB1 Scavenger receptor class B, type 1
STZ Streptozotocine
TET Ten eleven translocation
TG Triglycéride
tsDMR VLDL
Tissue-specific differentially methylated regions Lipoprotéines de très faible densité
1.1 L’obésité
L’obésité est actuellement l’enjeu de santé mondial le plus important, devançant maintenant la dénutrition (James, 2008). Le nombre de femmes et d’hommes adultes en surpoids ou obèses est aujourd’hui estimé au double de ce qu’il était dans les 1980 et ce autant dans les pays industrialisés quand dans ceux en voie de développement (Ahima, 2011). Cette fulgurante augmentation de surpoids et d’obésité est responsable de l’accroissement de la prévalence de nombreuses comorbidités incluant le diabète de type 2 (DT2), les dyslipidémies, les maladies cardiovasculaires (MCV) et certains type de cancers (Must et al., 1999; Calle et Kaaks, 2004; Poirier et al., 2006; Bays et al., 2013). Par conséquent, l’obésité représente un lourd fardeau économique pour les systèmes de santé et est aujourd'hui reconnue comme l’un des premiers facteurs de risque de décès au monde (Swinburn, 2011). Elle est d’ailleurs qualifiée d’épidémie par l’organisation mondiale de la santé (OMS) depuis 1997 (James, 2008).
L’obésité est définie comme une accumulation anormale ou excessive de tissu adipeux pouvant nuire à la santé (Persson et Bondke Persson, 2013). Plusieurs mesures anthropométriques sont utilisées pour évaluer le degré d’obésité : la circonférence de la taille, l’indice de masse corporelle (IMC) (la masse (en kg) divisé par la taille (en m) au carré (kg/m2)), le rapport entre la circonférence de la taille sur celle des hanches et le pourcentage
de gras corporel. La circonférence de la taille, marqueur de l’adiposité viscérale, est la variable qui prédit le mieux les risques de complications cardiométaboliques (CCM) associées à l’obésité (Poirier et Després, 2003). Par contre, l’IMC, l’unité de mesure la plus facilement accessible, est celle qui est utilisée internationalement par l’OMS afin de déterminer le degré d’obésité et les risques associés pour la santé (WHO, 2000). Selon les recommandations de l’OMS, un IMC entre 25,0 et 29,9 kg/m2 est associé au surpoids.
L’obésité, quant à elle, est caractérisée par une IMC de plus de 30 kg/m2. Le tableau 1.1
présente les différentes classes d’obésité et le risque de comorbidité associé à chacune d’entre elles.
Tableau 1.1- Classes d’obésité et le risque de comorbidités associé à l’IMC selon les critères de l’OMS Classes d’obésité Poids insuffisant Poids Normal Surpoids Obèse Classe I Obèse Classe II Obèse Classe III IMC (kg/m2) ≤ 18,5 18,5-24,9 25,0-29,9 30,0-34,9 35,0-39,9 ≥40,0 Risque de
comorbidité Faible Moyen Accru Modéré Élevé Très élevé IMC, indice de masse corporelle
Tiré et modifié de World Health Organization, WHO (2000) dans Obesity: preventing and managing
the global epidemic. Report of a WHO consultation. World Health Organization Technical Report Series 894 : page 10.
1.1.1 Portrait de l’obésité au Canada
La population canadienne est aussi largement touchée par l’épidémie d’obésité. Depuis les trois dernières décennies, la prévalence de l’obésité a triplé chez les adultes canadiens passant de 6,1 à 18,0% (Twells et al., 2014). Selon les prédictions, le nombre d’adultes en surpoids ou obèses augmentera encore au cours des 5 prochaines années et ce dans l’ensemble des provinces du Canada (Twells et al., 2014). Les coûts de santé engendrés par le surpoids, l’obésité et ses comorbidités sont présentement estimés à près 4,3 milliards de dollars par année et une augmentation est anticipée dans les années à venir (Katzmarzyk et Janssen, 2004).
Les statistiques sur l’obésité infantile canadienne sont également très inquiétantes. Dans une enquête menée récemment par l’UNICEF auprès de 29 pays industrialisés, le Canada se situe au 3e rang des pays avec les taux d’obésités infantiles les plus élevés derrière
les États-Unis et la Grèce (UNICEF, 2013). Le nombre de canadiens âgés de 2 à 17 ans souffrant d’un excès poids ou d’obésité est maintenant évalué à 29%, soit le triple de ce qui avait été rapporté en 1975 (Shields et Tjepkema, 2006). Cet accroissement d’obésité est la cause de l’apparition, chez les enfants, de désordres métaboliques (DT2, dyslipidémie, hypertension artérielle) qui étaient auparavant restreints à la population adulte (Deckelbaum et Williams, 2001; Nathan et Moran, 2008). Par ailleurs, il est reconnu qu’environ 2/3 des enfants et adolescents obèses le resteront toute leur vie (Singh et al., 2008). De ce fait, si la tendance se maintient, il est anticipé que près de 70% de la population canadienne âgée de
40 ans sera obèse en 2040 (Le Petit et Berthelot, 2006). Devant cette situation alarmante et afin de freiner l’augmentation de l’obésité, il est impératif de mieux comprendre ses causes. Les facteurs génétiques et environnementaux de même que l’interaction entre ces deux facteurs constituent les principales causes de l’épidémie d’obésité.
1.1.2 Les principales causes de l’obésité 1.1.2.1 Les facteurs génétiques
Des études familiales et sur des jumeaux ont permis de mettre en évidence qu’il existe une composante génétique à l’obésité. L’héritabilité de l’obésité, tel qu’estimée par ces études, varie de 6 à 85% selon les variables anthropométriques analysées (Comuzzie et allisson, 1998; Barsh et al., 2000; Yang et al., 2007). Les variants génétiques qui sont associés au phénotype de l’obésité peuvent être divisés en deux catégories : 1) les variants rares associés aux formes sévères et monogéniques de l’obésité et 2) les variants communs associés à la forme fréquente et polygénique de l’obésité.
Les formes monogéniques de l’obésité, causées par des variants génétiques rares, sont très sévères et font leur apparition à un âge très précoce, soit entre un et deux ans. Ce type d’obésité représente moins de 5% des cas d’obésité (Ranadive et Vaisse, 2008). Plus d’une vingtaine de mutations causales de la forme monogénique de l’obésité ont jusqu’à présent été identifiées. La plupart de ces mutations modifies la séquence de gènes codant pour des protéines impliquées dans la régulation hypothalamique du métabolisme énergétique (Farooqi et O’Rahilly, 2004; Hochberg et Hochberg, 2010). Parmi celles-ci, des mutations des gènes de la leptine (LEP), du récepteur de la leptine (LEPR), du récepteur de type 4 de la mélanocortine (MC4R), et de la pro-opiomélanocortine (POMC), impliqués dans le contrôle de la satiété et de la prise alimentaire, ont notamment été associées à l’hyperphagie et à des formes très sévères de l’obésité dès la petite enfance (Ranadive et Vaisse, 2008; Dubern et Clément, 2012; Valette et al., 2013).
Les variants génétiques communs associés à la forme polygénique de l’obésité ont pour la majorité été identifiés par des études d’association pangénomique (genome wide
association studies (GWAS)). Les GWAS, en analysant jusqu’à un million de SNP (single nucleotide polymorphism) communs couvrant l’ensemble du génome, ont permis l’identification de plus de 50 variants associés à différentes mesures anthropométriques de l’obésité, soit : l’IMC, la circonférence de la taille, le ratio de la circonférence de la taille sur celle des hanches et le pourcentage de gras corporel (Heid et al., 2010; Day et Loos, 2011; Herrera et al., 2011). Par contre, ces SNP expliquent moins de 2% de la variabilité phénotypique de l’obésité (Waalen, 2014). Ainsi, il semble que d’autres facteurs contribuent à l’héritabilité de l’obésité. Des études ont suggéré que certains variants rares ainsi que l’interaction entre plusieurs variants génétiques rares ou communs, permettraient d’expliquer une fraction du phénotype de l’obésité (Yang et al., 2007; McCarthy et al., 2008; Manolio et al., 2009). En outre, l’interaction entre les gènes et les facteurs environnementaux obésogènes permettrait d’expliquer une partie importante de l’héritabilité manquante de l’obésité (Agus-Collins et Bouchard, 2008; Ahmad et al., 2013; Chaput et al., 2014).
1.1.2.2 Les facteurs environnementaux
Les facteurs environnementaux contribuant à l’augmentation de la prévalence de l’obésité sont nombreux. Plusieurs s’entendent pour dire que le mode vie de notre société occidentale, caractérisé par une alimentation riche en gras et en sucre ainsi que par la sédentarité, est en partie responsable de l’épidémie d’obésité (WHO, 2000). Plus récemment, il a été démontré que des composés organiques dérivés des plastiques et des pesticides (bisphénol-A et tributylétain) peuvent modifier les mécanismes moléculaires impliqués dans la différenciation adipocytaire (Grun, 2010; Holtcamp, 2012). Ces toxines environnementales pourraient donc également jouer un rôle important dans le développement de l’obésité.
Au-delà des facteurs obésogènes précédemment énumérés, l’environnement auquel le fœtus est exposé pendant son développement apparaît maintenant comme un facteur déterminant dans le développement de l’obésité. En effet, de nombreuses études ont démontré que les nouveau-nés ayant été exposés à un environnement fœtal défavorable présentent un risque accru de développer, à l’adolescence et à l’âge adulte, l’obésité et les désordres métaboliques qui y sont associés. Ce phénomène appelé programmation métabolique fœtale est le thème central de cette thèse qui a pour objectif d’identifier des
modifications épigénétiques impliquées dans la programmation métabolique des nouveau-nés exposés in utero au diabète gestationnel (DG). Les deux prochaines sections de ce chapitre introduiront la programmation métabolique fœtale et plus spécifiquement, celle associée à l’exposition au DG. Par la suite, les mécanismes épigénétiques de même que les évidences permettant d’associer la méthylation de l’ADN (la modification épigénétique la plus étudiée) à la programmation métabolique fœtale seront abordés. Les chapitres 2 à 5, constitués de manuscrits publiés ou soumis, présenteront l’impact du DG sur la méthylation de l’ADN du nouveau-né (placenta et sang de cordon) selon deux approches, soit l’approche à l’échelle du génome et celle par gènes candidats. Les chapitres 6 à 8 présenteront les résultats d’une étude exploratoire qui visait à mieux comprendre le profil de la méthylation de deux gènes candidats de la programation métabolique foteale, les gènes LEP et de l’adiponectine (ADIPOQ), dans différents tissus et d’évaluer le potentiel de ces deux gènes comme marqueurs épigénétiques de la susceptibilité à l’obésité en clinique. Le chapitre 9 conclura cette thèse en discutant des résultats obtenus ainsi que des enjeux et perspectives associés à l’étude des mécanismes épigénétiques impliqués dans la programmation métabolique fœtale.
1.2 La programmation fœtale : de Barker à aujourd’hui
Les études à l’origine de la théorie de la programmation fœtale ont été conduites par le Professeur David J.P. Barker à la fin des années 1980. Le Pr Barker et son équipe se sont intéressés à la santé métabolique et aux causes de décès de plus de 16 000 adultes britanniques nés entre les années 1911 et 1930. Les premiers résultats publiés par ces derniers démontrent que les sujets avec un faible poids à la naissance ont un taux de décès de causes cardiovasculaires beaucoup plus élevé que ceux avec un poids plus élevé (Barker et al., 1989; Barker et al., 1993a; Osmond et al., 1993). Dans une seconde série de publications, l’équipe du Pr Barker rapporte de fortes corrélations négatives entre le poids à la naissance et le risque d’être atteint d’hypertension, DT2 et de MCV à l’âge adulte (Barker et al., 1993b; Law et al., 1993; Phipps et al., 1993). Suite à ces travaux, le Pr Barker propose que la diminution de la vitesse de croissance fœtale, marquée entre autre par un faible poids à la naissance, augmente le risque d’être atteint d’obésité ou d’une maladie métabolique plus tard dans la vie (Barker, 1995). En 1995, le British Medical Journal nomme cette hypothèse l’ « hypothèse de Barker » en l’honneur de son instigateur (Paneth et Susser, 1995).
Se basant sur cette hypothèse, plusieurs équipes de recherche se sont penchées sur l’étude de la croissance fœtale et du poids à la naissance comme facteur prédicatif de la susceptibilité aux maladies métaboliques à l’âge adulte. Les premiers résultats du Pr Barker furent validés par de nombreuses études épidémiologiques, dont celles de la Dutch Hunger Famine. La Dutch Hunger Famine a eu lieu pendant la seconde guerre mondiale alors que l’occupation du territoire néerlandais par les allemands entraîne une grève des réseaux ferroviaires et la mise en place d’un système rationnement alimentaire. En raison de ce rationnement, tous les habitants de la région nord des Pays-Bas, incluant les femmes enceintes, ont vu leur apport calorique quotidien limité de 1800 calories à moins de 1000 calories à partir de décembre 1944 jusqu’à aussi peu que 580 kcal par jour en février 1945 (Roseboom et al., 2006). Ce tragique évènement a permis d’étudier les conséquences à long-terme de la restriction alimentaire maternelle à différents stades du développement fœtale sur plus de 2400 nouveau-nés. Cette étude démontre que l’exposition in utero à la famine pendant la seconde moitié de la grossesse est associée à un poids plus faible à la naissance et à une diminution de la tolérance au glucose à l’âge adulte, et ce indépendamment du poids à
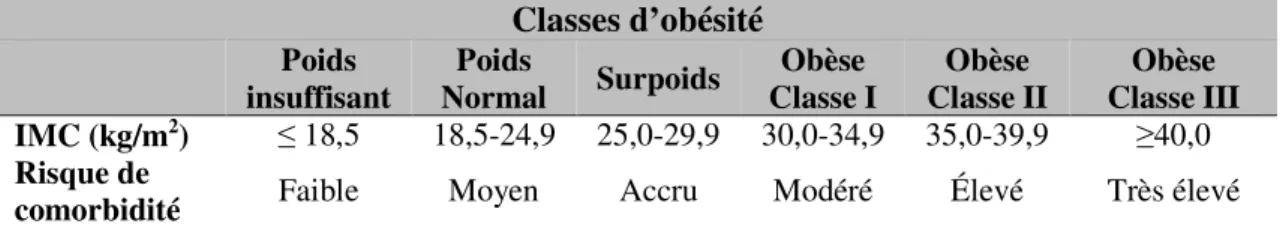
la naissance (Ravelli et al., 1998). De leur côté, les sujets exposés à la famine au début du développement embryonnaire ont un poids à la naissance normal, mais sont plus à risque d’être obèse, d’avoir un profil lipidique athérogénique et de souffrir de MCV à l’âge adulte (Ravelli et al., 1999; Roseboom et al., 2000a; Roseboom et al., 2000b). Les études sur la Dutch Hunger Famine ont grandement contribué à l’avancement des connaissances sur la programmation fœtale en démontrant que la période du développement durant laquelle le fœtus est exposé à un environnement défavorable a un impact sur sa santé à long-terme. De plus, ces études sont les premières à prouver que l’exposition à un environnement fœtal sous-optimal peut programmer la santé métabolique indépendamment du poids à la naissance. Suite à ces résultats, le Pr Barker a proposé l’hypothèse du phénotype d’épargne (Thrifty Phenotype) selon laquelle le fœtus modifie son métabolisme pour s’adapter aux stimuli environnementaux in utero. Ces modifications assureraient un développement fœtal optimal lorsque les ressources alimentaires sont insuffisantes, tout en permettant une programmation métabolique favorisant la survie en situation de dénutrition. Cette programmation serait par contre mal-adaptée à un environnement post-natal où les ressources alimentaires sont abondantes et augmenterait donc la susceptibilité du nouveau-né de développer une maladie chronique (Figure 1.1) (Hales et Barker, 2001).
Figure 1.1- Réponse adaptative du fœtus et les risques de maladies à long-terme
La combinaison de l’environnement in utero et post-natal peut être adaptative ou maladaptative et augmenter ou diminuer le risque d’être atteint de maladies chroniques. Pendant le développement, le fœtus s’adapte aux signaux de la mère afin d’augmenter ses chances de survie. Toutefois, si les signaux post-nataux diffèrent de ceux auxquels il a été exposé in utero, cette adaptation devient désavantageuse et le rend plus susceptible de souffrir d’obésité et de complications cardiométaboliques (CCM).
Adaptée de : Boekelheide K et al. (2012) Predicting Later-Life Outcomes of Early-Life Exposures.
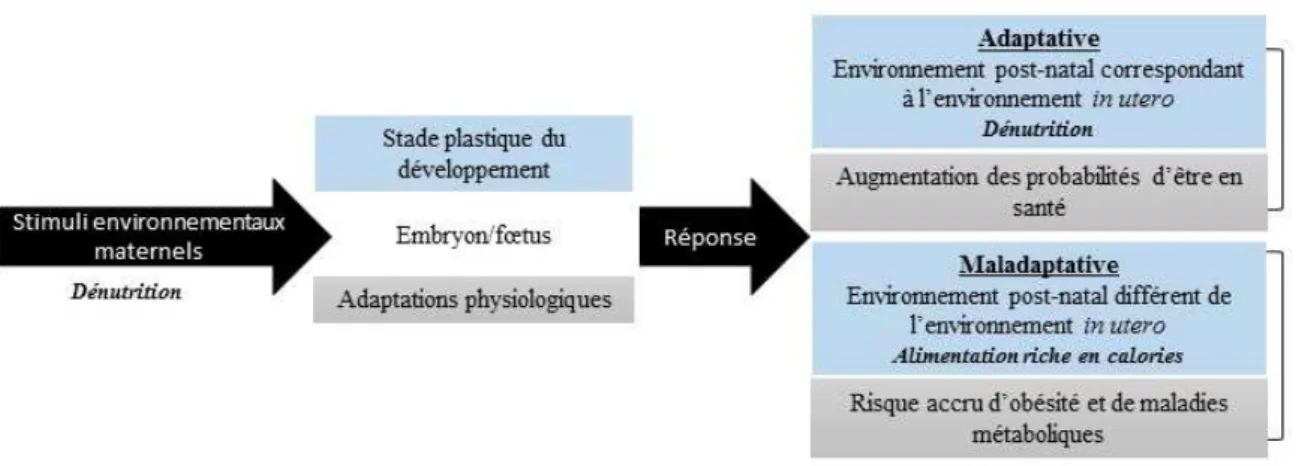
Dans notre société où le nombre de femmes obèses en âge de procréer est en constante augmentation, un grand intérêt s’est développé pour l’étude de la programmation fœtale dans un contexte d’obésité maternelle, de gain de poids excessif pendant la grossesse et de DG. Ces trois conditions maternelles sont associées à un transport excessif de nutriments vers le fœtus, à l’augmentation du poids à la naissance et également à un risque accru d’être atteint d’obésité et de CCM à l’âge adulte (Boney et al., 2005; Kim et al., 2011; Gallino et Bellver, 2013; O’Reilly et Reynolds, 2013). Un poids élevé à la naissance, tout comme un faible poids, est associé associé au risque d’être atteint d’obésité et de CCM dans le futur. Par conséquent, la relation entre le poids à la naissance et le risque relatif de développer l’obésité ou une CCM à l’âge adulte est représentée par une courbe en U (Figure 1.2) (Baker et al., 2008; Whincup et al., 2008).
L’avantage évolutif de l’adaptation de la croissance fœtale en situation de suralimentation est moins bien compris que celui lié à la dénutrition. Ma et ses collaborateurs (2013) ont récemment proposé que la programmation métabolique fœtale observée chez les nouveau-nés exposés in utero à l’obésité maternelle ou au DG soit plutôt contre-évolutive et attribuable à une mal-adaptation du placenta face à des concentrations de nutriments trop élevées. Au cours de l’évolution, les mécanismes de transport des nutriments du placenta se seraient adaptés à la restriction alimentaire, mais en situation de suralimentation ils ne réussiraient pas à limiter l’apport nutritionnel au fœtus. Cette théorie est appuyée par l’hypothèse proposée par Pedersen en 1954. Selon Pedersen, le glucose, présent en concentration excessive chez les mères avec un diabète, est directement transféré vers le fœtus entraînant l’hyperglycémie et l’augmentation de sa production d’insuline (Pedersen, 1954). L’insuline augmente la croissance du fœtus notamment en stimulant l’expression des facteurs de croissance ressemblant à l’insuline (IGFs), l’adipogénèse et la déposition de tissu adipeux (Catalano et Hauguel-De Mouzon, 2011; Negrato et al., 2012). Les modèles animaux ont aussi démontré que le transfert excessif de nutriments favorise la production d’insuline, la croissance, la prolifération et l’hypertrophie des adipocytes ainsi que la résistance à la leptine et à l’insuline chez le fœtus (Rkhzay-Jaf et al., 2012).
Figure 1.2- Risque relatif d’être atteint d’une maladie métabolique en fonction du poids à la naissance
Un poids trop faible ou trop élevé à la naissance est associé avec un risque accru de développer de l’obésité, un diabète de type 2 (DT2) ou une maladie cardiovasculaire (MCV). Les facteurs associés à la diminution et à l’augmentation du poids à la naissance sont énumérés dans les parties inférieures gauche et droite de la figure respectivement.
Tirée et modifiée de : Isganaitis E, Patti ME (2011). Adipocyte Development and Experimental Obesity, page 332. Dans: RH Lustig (ed.), Obesity Before Birth: Maternal and Prenatal Influences
De nombreuses conditions maternelles et environnementales pouvant perturber le développement du fœtus sont aujourd’hui associées à la programmation de la santé métabolique du fœtus: l’alcool, l’altitude, l’anémie, la cigarette, le DG, les drogues, le gain de poids excessif pendant la grossesse, la malnutrition, l’obésité maternelle, les polluants organiques persistants et le stress (Figure 1.2) (Boney et al., 2005; Meyer et Lubo, 2007; Fei et al., 2007; Kim et al., 2011; Minnes et al., 2011; Hamlin, 2012; Galliano et Bellver, 2013; O’Reilly et Reynolds, 2013; Soto et Bahado-Singh, 2013). Il a également été mis en évidence que des facteurs environnementaux périnataux tels que l’allaitement, l’alimentation et le stress joueraient un rôle considérable dans la programmation de la santé métabolique du nouveau-né (Tamashiro et Moran, 2010; Moore et al., 2011; Koletzko et al., 2012). Dans le cadre de cette thèse, seule la programmation fœtale associée au DG sera abordée en détail.
1.3 Le diabète gestationnel
1.3.1 La prévalence et les facteurs de risque du diabète gestationnel
Le DG, défini comme une hyperglycémie diagnostiquée pour la première fois lors de la grossesse, est l’une des complications de santé maternelles les plus fréquentes (Thompson et al., 2013). Puisqu’il n’existe pas de consensus international sur les critères diagnostiques à utiliser pour dépister le DG, les statistiques sur la prévalence du DG sont très variables. Néanmoins, selon la majorité des études, la prévalence du DG serait de 2 à 6 % et elle pourrait atteindre de 10 à 20% dans certaines populations à haut risque, notamment chez les autochtones, les asiatiques et les hispaniques (Galtier, 2010). Outre l’origine ethnique, l’âge maternel, les antécédents familiaux de DT2, les antécédents de DG et de macrosomie ainsi que l’obésité sont des facteurs de risque reconnus du DG (Ben-Haroush et al., 2004; Galtier, 2010). L’obésité maternelle, un des facteurs de risque le plus important, contribuerait à plus de 20% des cas de DG (Kim et al., 2010; Kim et al., 2013). De ce fait, et en raison de l’épidémie d’obésité qui touche les femmes de plus en plus jeunes, la prévalence du DG sera vraisemblablement appelée à augmenter dans les prochaines années. Les nouveaux critères diagnostiques récemment adoptés par l’International Association of Diabetes and Pregnancy Study Groups (IADPSG), qui seront discutés dans la prochaine section, pourraient également contribuer à améliorer le dépistage du DG et à augmenter le nombre de femmes diagnostiquées et traitées pour le DG.
1.3.2 Le dépistage et les critères diagnostiques du diabète gestationnel
La méthode de dépistage et les critères utilisés pour diagnostiquer le DG sèment, depuis longtemps, la controverse dans la communauté médicale internationale. La méthode la plus couramment utilisée pour dépister le DG est le test d’hyperglycémie provoquée par voie orale (HGPO) entre la 24e et la 28e semaine de grossesse (Coustan, 2013). Cette méthode consiste
à mesurer la variation de la glycémie des mères suite à l’ingestion d’une boisson sucrée. Les quantités de glucose ingérées par les mères et les temps auxquels les prélèvements sanguins sont faits pour doser la glycémie dépendent de la stratégie de dépistage utilisée (Tableau 1.2) (Cosson, 2010). Avant l’étude HAPO (Hyperglycemia and Adverse Pregnancy Outcome) en 2008, les bénéfices, pour la mère et le nouveau-né, associés au dépistage du DG étaient très
variables d’une étude à l’autre et en fonction des protocoles et des critères diagnostiques utilisés (Cosson, 2010). Par conséquent, les endocrinologues se sont beaucoup questionnés sur les seuils diagnostiques de glycémie à utiliser de même que sur la nécessité de dépister et de traiter le DG (Dornhorst et Chan, 1998; Buchanan et Kjos, 1999; Khandelwal et al., 1999).
L’étude HAPO réalisée dans 9 pays sur plus de 23 000 femmes enceintes, avait parmi ses objectifs la standardisation de la méthode de dépistage et des critères diagnostiques du DG. Cette étude a démontré que la glycémie maternelle (à jeun, 1h et 2h post-HGPO entre la 24e et la 28e semaine de grossesse) est associée, de façon linéaire, à des complications de
grossesse pour la mère et le nouveau-né (Metzger et al., 2008; HAPO, 2009). Afin de diminuer ces complications, l’IADPSG a recommandé que toutes les femmes enceintes soit dépistées pour le DG et que trois seuils glycémiques consensus (un à jeun, un 1h et un 2h post-HGPO) soient utilisés pour le diagnostic du DG (Tableau 1.2) (Metzger et al., 2010). Ces recommandations, si elles sont adoptées internationalement, permettront de cibler une plus grande proportion des mères qui sont à risque d’avoir des complications et des issues de grossesse indésirables. Ceci entrainera une augmentation significative du nombre de femmes diagnostiquées avec un DG et devant être traitées. De récentes études ont d’ailleurs déjà rapporté que le nombre de femmes diagnostiquées avec un DG selon les critères de l’IADPSG était deux fois plus élevé que lorsque les critères de l’American Diabetes Association (ADA), de l’Association Canadienne du Diabète (CDA) ou de l’American College of Obstetricians and Gynecologists (AOCG) étaient utilisés (Shang et Lin, 2014; Oriot et al., 2014; Mayo et al., 2014; Shang et al., 2014). En 2013, l’ADA a adopté les critères diagnostiques de l’IADPSG. Par contre, ceux-ci ne font pas encore l’unanimité puisque les critères de la CDA, de l’AOCG, et de l’OMS, plus laxistes que ceux de l’IADPSG, sont encore utilisés (Tableau 1.2) (WHO, 1999; ADA, 2013; Thompson et al., 2013; AOCG, 2013). Dans le cadre de ce projet de recherche, les femmes de la cohorte E-21 de Chicoutimi ont été diagnostiquées selon les critères de l’OMS tandis que la cohorte de validation, Gen3G de Sherbrooke, selon ceux de l’IADSPG.
Tableau 1.2- Stratégie de dépistage du diabète gestationnel selon les associations Étape Test* Seuils glycémiques pour le diagnostic du DG ADA
(2013) 1 HGPO : 75g de glucose À jeun : ≥ 5,1 mmol/L 1h post-HGPO : ≥ 10,0 mmol/L 2h post-HGPO : ≥ 8,5 mmol/L
*Une valeur anormale pour le diagnostic du DG
AOCG (2013)
2 Étape 1 HGPO : 50g de glucose
Les seuils du NDDG ou de Carpenter et Coustan peuvent être utilisés NDDG (1979) Carpenter et Coustan (1982) 1h post-HGPO ≥ 7,8 mmol/L ≥ 7,2 mmol/L
*Si la glycémie est plus élevée que 7,8 ou 7,2 mmol/L, passer à l’étape 2
Étape 2 HGPO : 100g de glucose
Les seuils du NDDG ou de Carpenter et Coustan peuvent être utilisés NDDG (1989) Carpenter et Coustan (1982)
À jeun ≥ 5,8 mmol/L ≥ 5,3 mmol/L
1h post-HGPO ≥ 10,6 mmol/L ≥ 10,0 mmol/L 2h post-HGPO ≥ 9,2 mmol/L ≥ 8,6 mmol/L 3h post-HGPO ≥ 8,0 mmol/L ≥ 7,8 mmol/L
*Deux valeurs anormales du NDDG ou de Carpenter et Coustan pour le diagnostic du DG
IADPSG (2010)
1 HGPO : 75g de glucose À jeun : ≥ 5,1 mmol/L
1h post-HGPO : ≥ 10,0 mmol/L 2h post-HGPO : ≥ 8,5 mmol/L
*Une valeur anormale pour le diagnostic du DG
CDA (2013) 1 TTG : 50g de glucose ou TTG: ≥ 11.1 mmol/L ou
HGPO : 75g de glucose À jeun : ≥ 5.3 mmol/L
1h post-HGPO : ≥ 10.6 mmol/L 2h post-HGPO : ≥ 9.0 mmol/L
*Une valeur anormale pour le diagnostic du DG
OMS
(1999) 1 HGPO : 75g de glucose À jeun : ≥ 7.0 mmol/L 2h post-HGPO : ≥ 7.8 mmol/L
*Une valeur anormale pour le diagnostic du DG
ADA, American Diabetes Association; AOCG, American College of Obstetricians and
Gynecologists; CDA, Canadian Diabetes Association; DG, diabète gestationnel; HGPO; hyperglycémie provoquée par voie orale, IADPSG, l’International Association of Diabetes and
Pregnancy Study Groups; NDDG; National Diabetes Data Group; OMS, Organisation mondiale de la santé; TTG; test de tolérance au glucose.
*Les tests de dépistage sont faits entre la 24e et la 28e semaine de la grossesse. ou
1.3.3 La physiopathologie du diabète gestationnel
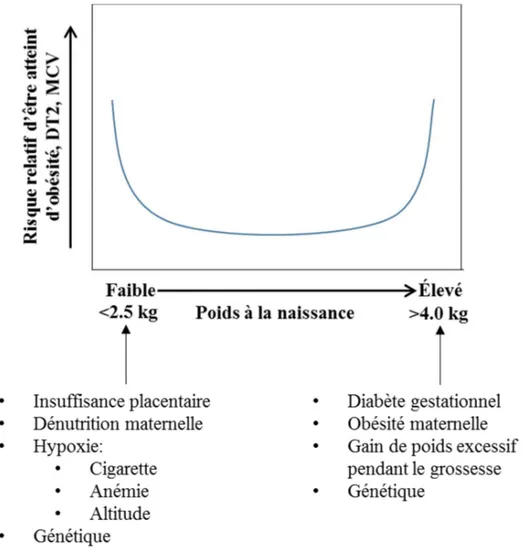
La grossesse est marquée par de nombreux changements anatomiques, cardiovasculaires, respiratoires et endocriniens chez la mère. Ces adaptations physiologiques, non-pathologiques, sont essentielles au développement du fœtus et pour préparer la mère à l’accouchement et à l’allaitement (Tan et Tan, 2013). Au début de la seconde moitié de la gestation, une résistance des tissus périphériques à l’insuline s’installe lentement et progresse jusqu’à la fin du troisième trimestre pour atteindre des niveaux similaires à ceux observés chez les patients souffrant de DT2 (Lain et Catalano, 2007). L’insulino-résistance musculaire, hépatique et adipocytaire permet d’augmenter les quantités de glucose, d’acides gras libres et d’acides aminés en circulation afin d’assurer un apport suffisant de ces nutriments pour le fœtus qui entre dans sa phase de croissance la plus importante (Boden, 1996). Bien que les mécanismes soient encore mal compris, il semble que certaines hormones placentaires et maternelles, soient impliquées dans le développement de cet état de résistance à l’insuline (Figure 1.3) (Barbour et al., 2007; Zavalza-Gomez et al., 2008; D’ippolito et al., 2012). Chez la majorité des femmes enceintes, la production d’insuline est augmentée de 200% à 250% pour maintenir l’euglycémie (Catalano et al., 1999). Par contre, chez certaines femmes, la production d’insuline est insuffisante pour contrer la résistance à l’insuline, entraînant ainsi une hyperglycémie, puis un DG (Figure 1.3).
Deux mécanismes sont impliqués dans la physiopathologie du DG. Dans un premier temps, il est reconnu que la réponse insulinique suite à un stimulus nutritionnel est beaucoup plus faible chez les femmes avec DG que chez les femmes avec une grossesse normoglycémique. Pour un degré d’insulino-résistance similaire, la sécrétion d’insuline des mères avec DG est jusqu’à 50% plus faible que celle des mères normoglycémiques et ce, pendant et après la grossesse (Buchanan, 2001; Homko et al., 2001; Moleda et al., 2013). La défaillance des cellules ß-pancréatiques serait responsable de la réduction de la production d’insuline observée chez les femmes avec un DG (Figure 1.3). Les causes exactes de la perte de fonction des cellules ß-pancréatiques demeurent, à ce jour, inconnues. Il est cependant suggéré qu’une exposition prolongée à l’hyperglycémie désensibilise les cellules ß-pancréatiques, induit leur apoptose ainsi que la diminution de la production d’insuline. Ensemble, ces mécanismes alimenteraient le cycle de la résistance à l’insuline (Cerf, 2013).
Le deuxième mécanisme pathologique responsable du DG est la résistance à l’insuline. L’aggravation de la résistance à l’insuline chez les femmes avec un DG fut démontrée pour la première fois par en 1985 par Ryan et son équipe avec la méthode du clamp euglycémique hyperinsulinémique (Ryan et al., 1985). Cette méthode de référence permet de quantifier l’action de l’insuline sur les flux de glucose dans l’ensemble de l’organisme (DeFronzo et al, 1979). En perfusant des concentrations stables d’insuline, ces derniers ont observé que la quantité de glucose absorbée par les tissus des femmes avec DG était plus basse que chez celles avec une glycémie normale au 3e trimestre de la grossesse
(Ryan et al., 1985). Les résultats de plusieurs équipes ont ensuite confirmé que les femmes avec un DG étaient plus résistantes à l’insuline en fin de grossesse que les femmes normoglycémiques (Catalano et al., 1993; Kautzky-Willer et al., 1997; Catalano et al., 1999; Homko et al., 2001). Lorsqu’elles ne sont pas enceintes, les femmes avec un antécédent de DG ont également une résistance à l’insuline plus grande que celles qui n’ont jamais développer cette pathologie (Kautzky-Willer et al., 1997; Catalano et al., 1999; Verma et al., 2002). Ces résultats suggèrent que les femmes qui développent un DG ont une résistance à l’insuline chronique, ce qui pourrait expliquer, du moins en partie, la raison pour laquelle elles sont plus susceptibles d’être atteintes d’un DT2 plus tard dans leur vie. L’altération de la voie de signalisation de l’insuline dans les tissus périphériques (muscle et tissu adipeux) contribuerait également à l’augmentation de la résistance à l’insuline (Figure 1.3) (Friedman et al., 1999; Shao et al., 2002; Barbour et al., 2011). De sucroît, les adipokines, reconnus pour leur rôle dans la résistance à l’insuline induite par l’obésité, seraient également impliqués dans l’aggravation de l’insulino-résistance observée chez les mères avec un DG (Figure 1.3). Les concentrations plasmatiques d’adiponectine, de leptine, de facteur de nécrose tumoral alpha (TNFα) et de plusieurs autres adipokines ont d’ailleurs été associées au DG et à des index de résistance à l’insuline pendant la grossesse (Lacroix et al., 2013a; Lacroix et al., 2013b.; Guillemette et al., 2014; Fasshauer et al., 2014).
En somme, le DG est caractérisé par une hyperglycémie persistante causée par une résistance à l’insuline trop grande pour être contrebalancée par la production d’insuline des cellules ß-pancréatiques dysfonctionnelles (Figure 1.3). Les femmes diagnostiquées avec un DG entre la 24e et la 28e semaine de grossesse sont traitées et prises en charge par leur
médecin jusqu’à la fin de leur grossesse afin de diminuer les complications à l’accouchement et les effets indésirables sur la santé à court et à long-terme du nouveau-né (présentés dans les deux prochaines sections). La première prescription pour diminuer la glycémie des mères avec un DG consiste à apporter des modifications au régime alimentaire et à intégrer la pratique régulière d’activité physique (Ruchat et Mottola, 2013; Hernandez et al., 2013). Lorsque ces deux éléments sont insuffisants pour rétablir la glycémie sous le seuil recommandé en deux semaines, le traitement par l’insuline ou par d’autres agents hypoglycémiants ne traversant pas la barrière placentaire (glyburide ou metformine) est recommandé (Thompson et al., 2013; Durnwald, 2013).
Figure 1.3- Adaptations physiques et mécanismes physiopathologiques associés à une grossesse normoglycémique et au diabète gestationnel
Au début du 2e trimestre de la grossesse, une résistance périphérique à l’insuline s’installe chez la mère. Chez la majorité des mères, les cellules ß-pancréatiques produisent assez d’insuline pour maintenir l’euglycémie. Chez les mères qui développent un diabète gestationnel, les cellules ß-pancréatiques sont défaillantes et la résistance à l’insuline est plus importante que chez les mères normoglycémiques. Par conséquent, les cellules ß-pancréatiques ne réussissent pas à produire assez d’insuline pour contrer la résistance à l’insuline et une hyperglycémie s’ensuit.
1.3.4 Les conséquences du diabète gestationnel pour la santé de la mère
Chez la majorité des femmes diagnostiquées avec un DG, la glycémie redevient normale suite à la naissance du bébé. Toutefois, ces femmes ont un risque accru de développer des complications métaboliques dans les années suivant l’accouchement (Kitzmiller et al., 2007).
Le DT2 est la complication métabolique la plus étudiée et celle qui est la plus fréquente chez les femmes présentant des antécédents de DG. Chez ces femmes, le risque d’être atteint d’un DT2 est jusqu’à 7 fois plus grand que chez les femmes qui ont eu une grossesse normoglycémique (Bellamy et al., 2009; Malcolm, 2012). Une étude ontarienne sur le suivi post-partum de plus de 660 000 femmes a estimé que près de 20% des femmes avec des antécédents de DG reçoivent un diagnostic de DT2 dans les 9 années suivant leur accouchement (Feig et al., 2008).
Les femmes ayant eu une grossesse compliquée par un DG sont également plus susceptibles de développer le syndrome métabolique après l’accouchement. La définition du syndrome métabolique ainsi que les critères diagnostiques sont variables d’un organisme à un autre (Grundy et al., 2004; Alberti et al., 2005; Huang, 2009). Cependant, ils s’entendent tous sur le fait que l’obésité, l’hypertension, la résistance à l’insuline et les dyslipidémies sont les principales composantes du syndrome métabolique. Toutes ces complications métaboliques sont plus fréquentes chez les femmes avec des antécédents de DG que chez les femmes ayant eu une grossesse normoglycémique (Roca-Rodriguez et al., 2012). Conséquemment, le risque de développer un syndrome métabolique est de 1,3 à 4 fois plus élevé chez les mères avec un antécédent de DG que chez celles qui n’en ont pas (Xu et al., 2014; Noctor et al, 2014; Lauenborg et al., 2005).
Les composantes du syndrome métabolique sont également des facteurs de risques de la MCV. Ainsi, les femmes avec des antécédents de DG ont donc aussi un risque accru d’avoir une MCV post-partum. Des études d’observations rétrospectives, conduites chez différents groupes ethniques, ont démontré que les femmes avec des antécédents de DG étaient 1,7 à 2,6 fois plus à risque de subir un évènement cardiovasculaire que celles ayant eu une grossesse normoglycémique (Carr et al., 2006; Shah et al., 2008; Kessous et al., 2013;
Archambault et al., 2014). Le risque de développer des complications cardiovasculaires serait principalement attribuable au DT2. Par conséquent, le risque cardiovasculaires associé au DG chez les femmes qui ne développent pas de DT2 post-partum est, jusqu’à présent, considéré équivalent à celui des femmes qui n’ont pas d’antécédent de DG (Kim, 2010; Archambault et al., 2014).
Au-delà des facteurs de risques reconnus du DG et du DT2 (l’origine ethnique, l’obésité, l’âge, les antécédents familiaux de DT2 ou de DG, les variants génétiques), l’ampleur de la résistance à l’insuline pendant la grossesse ainsi que la prise de poids post-partum sont associées à une augmentation des risques de complications métaboliques suite à l’accouchement (Kim et al., 2002; Ben-Haroush et al., 2004; Kwak et al., 2013). Des études d’interventions randomisées ont démontré que l’adoption de saines habitudes de vie, la médication et l’allaitement pouvaient améliorer la santé métabolique des femmes tout en prévenant et retardant l’apparition du DT2 et des autres CCM (Ratner et al., 2008; Feig, 2012; Gunderson et al., 2012; Chasan-Tabler, 2014; Much et al., 2014). De ce fait, le suivi médical post-partum des femmes avec un DG apparaît comme étant tout aussi primordial que celui qui est fait pendant la grossesse.
1.3.5 Le diabète gestationnel et la programmation fœtale
Pour le nouveau-né, l’exposition à l’environnement in utero hyperglycémique est associée à l’augmentation de sa croissance fœtale et à un risque accru pour sa santé métabolique à long terme. Pendant la grossesse, le transport du glucose maternel à travers la barrière placentaire se fait selon le gradient de concentration materno-fœtal et via deux transporteurs de glucose (transporteur de glucose de type 1 et 3 (GLUT1) et (GLUT3)) qui sont présents en quantité suffisante pour ne pas limiter le transfert du glucose vers le fœtus (Hay, 2006; Gauster et al., 2012). Chez les mères avec DG, le fœtus est donc exposé à des concentrations élevées de glucose qu’il doit lui-même métaboliser puisque le placenta est imperméable à l’insuline (Challier et al., 1986). Afin de métaboliser le glucose et de prévenir l’hyperglycémie, les cellules ß-pancréatiques de fœtus augmentent leur production d’insuline. L’insuline stimule la production de facteurs de croissance IGFs et contribue donc à l’augmentation du dépôt de tissus adipeux chez le fœtus (Pedersen, 1954; Sacks, 2007). Les mécanismes de transport
placentaire des lipides et des acides aminés sont également modifiés dans les grossesses compliquées par un DG favorisant le transfert des nutriments vers la circulation fœtale et l’augmentation de la réponse insulinémique du foetus (Jansson et al, 2002; Cetin et al., 2005; Jansson et al., 2006; Radaelli et al., 2009). La corrélation positive entre la glycémie maternelle au 2e trimestre de la grossesse et les niveaux d’insuline du fœtus est supportée par
plusieurs études dont l’étude HAPO (Metzger et al., 2008; HAPO, 2009; Luo et al., 2010; Gesteiro et al., 2011 ). Des associations linéaires entre la glycémie maternelle 2h post-HGPO au 2e trimestre, le poids et l’adiposité du bébé à la naissance ont aussi été rapportées à maintes
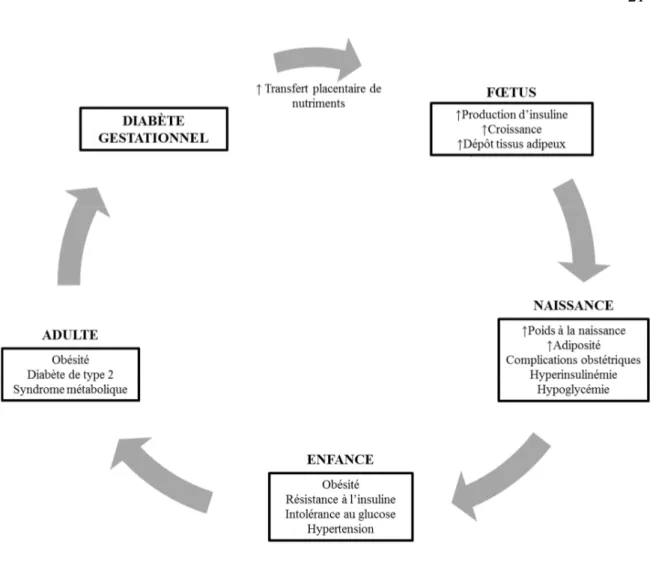
reprises (Catalano et al., 2003; Hill et al., 2005; Metzger et al., 2008; HAPO, 2009; Farah et al., 2011). Le DG est donc associé à une augmentation du risque de complications néonatales (macrosomie (poids ≥ 4 kg), hypoglycémie, détresse respiratoire, dystocie, hyperbilirubinémie) et obstétricales lors de l’accouchement (pré-éclampsie, césarienne, accouchement prématuré) (Jensen et al., 2000; Fadl et al., 2010; Catalano et al., 2012; Ovesen et al., 2014;) (Figure 1.4).
L’effet de l’exposition au DG sur la croissance et les indicateurs de santé métabolique des enfants semble être différent selon l’âge des enfants. En effet, chez les enfants de moins de 2 ans, les indicateurs de santé métaboliques des enfants nés de mères avec ou sans DG sont très similaires (Knight et al., 2007;Pettitt et al., 2010; Chatzi et al., 2011; Retnakaran et al., 2013). Ceci pourrait notamment être expliqué par un ralentissement de la croissance post-natal (catch down) des enfants ayant été exposés au DG (Stenhouse et al., 2006; Crume et al., 2011). Chez les enfants exposés au DG in utero, la croissance serait ralentie pendant les premiers 24 mois de vie et suivi d’une période de gain de poids accéléré. De nombreuses études démontrent d’ailleurs qu’à partir de l’âge de 3 ans et jusqu’à l’âge adulte, les enfants ayant été exposés au DG sont plus susceptibles d’être obèse, intolérant au glucose, résistant à l’insuline et hypertendue que les enfants ayant été exposés à un environnement normoglycémique (Tam et al, 2008; Wright et al., 2009; Vaarasmaki et al., 2009; Patel et al, 2012; Nehring et al., 2013; Page et al., 2014) (Figure 1.4). Il est intéressant de noter que les associations entre l’hyperglycémie maternelle et les CCM sont également observées chez les enfants et adolescents avec un poids normal à la naissance (≥ 2,0 kg et ≤ 4,0 kg) (Hillier et al., 2007). Le DG peut donc programmer la santé métabolique du
nouveau-né indépendamment de son poids à la naissance. En outre, même si l’exposition au DG soit suffisante pour programmer la santé métabolique du nouveau-né, il est suggéré que l’exposition à l’obésité maternelle en combinaison avec le DG potentialise les risques de complications métaboliques chez les enfants (Gillman et al., 2003; Boerschman et al., 2010;Pirkola et al., 2010; Catalano et al., 2012; Kubo et al., 2014).
Chez les adultes, des études de cohortes rétrospectives démontrent que ceux qui ont été exposés in utero au DG sont deux fois plus à risque d’être obèse et jusqu’à 7,8 fois plus susceptible de développer un pré-diabètes ou un DT2 que ceux qui ont été exposé à un environnement in utero normoglycémique (Clausen et al., 2008; Clausen et al., 2009; Kelstrup et al., 2013). De plus, le risque d’être atteint d’un syndrome métabolique serait jusqu’à 4 fois plus élevé chez les adultes nés de mères avec un DG que chez ceux nés de mère avec une glycémie normale (Clausen et al., 2009) (Figure 1.4). Tel que mentionné précédemment, l’obésité et le DT2 sont des facteurs de risque du DG. Conséquemment, les femmes en âge de procréer qui ont été exposées in utero au DG sont plus susceptibles d’avoir une grossesse compliquée par un DG et d’engendrer des enfants présentant un risque accru de développer de l’obésité et des CCM; perpétuant ainsi le cycle intergénérationnel de l’obésité (Figure 1.4) (Dabelea et Crume, 2011).