UNIVERSITÉ MOHAMMED V – AGDAL
FACULTÉ DES SCIENCES
Rabat
Faculté des Sciences, 4 Avenue Ibn Battouta B.P. 1014 RP, Rabat – Maroc Tel +212 (0) 37 77 18 34/35/38, Fax : +212 (0) 37 77 42 61, http://www.fsr.ac.ma
N° d’ordre………..
THÈSE DE DOCTORAT
Présentée par
BOUKHA Zouhair
Discipline : Chimie
Spécialité : Physico-chimie des Matériaux et Catalyse
PREPARATION, CARACTERISATION ET ETUDE COMPARATIVE DES
PROPRIETES CATALYTIQUES DE M(Pd; Ni)/Ca
10(PO
4)
6(OH)
2ET M(Pd;
Ni)/Ca
10(PO
4)
6F
2DANS LA COMBUSTION ET LE REFORMAGE DU METHANE
Soutenue le samedi 25 novembre 2006 Devant le jury :
Président :
ZIYAD Mahfoud : Professeur à l'université Mohammed V
Examinateurs :
ALBIZANE Abdarrahman : Professeur à l'université Hassan II Mohammadia
KACIMI Mohamed : Professeur à l'université Mohammed V
ROUIMI Mohamed : Professeur à l’Ecole Nationale Supérieure-Rabat
NAJA Jamal : Professeur à l’université Hassan 1
er-Settat
A mes très chers parents, ceux qui ont attendu avec patience le fruit de
leurs sacrifices et leur bonne éducation
A mes frères et sœurs
A ma grande famille
A tous mes amis
Avant propos
Ce travail a été réalisé, sous la direction du professeur M. ZIYAD, au laboratoire de physico-chimie des Matériaux et Catalyse de la Faculté des Sciences de Rabat.
Je suis heureux de pouvoir exprimer ma respectueuse gratitude à Monsieur le Professeur M. ZIYAD pour m’avoir soigneusement initié à la recherche et pour les conseils et les encouragements constants qu’il n’a cessé de me prodiguer tout au long de ce travail. Aussi, c’est un grand honneur qu’il me fait en acceptant de présider le jury de cette thèse.
J’exprime ma profonde reconnaissance à Monsieur M. KACIMI, professeur à la faculté des sciences de Rabat, qui a dirigé mon travail, pour les conseils qu’il m’a prodigués, pour l’expérience dont il a su me faire bénéficier et pour ses qualités humaines.
J’adresse mes vifs remerciements à Monsieur F. BOZON-VERDURAZ, professeur à l’Université Paris 7- Denis Diderot (France), pour l’occasion qui m’a offerte pour effectuer des séjours de recherche au sein du laboratoire de Chimie des Matériaux Divisés et Catalyse, et pour l’intérêt qui a montré à mon sujet développé.
J’associe également à ce travail, Monsieur J. L. FIGUEIREDO, professeur à la Faculté d'Ingénierie de l'Université de PORTO (Portugal), qui a codirigé une partie de ce travail avec beaucoup de soin et d’intérêt. Qu’il soit assuré de ma profonde gratitude pour sa grande disponibilité et ses conseils scientifiques. Je le remercie aussi d’avoir effectuer le déplacement pour être parmi les membres du jury.
Sincères remerciements à Monsieur M. ROUIMI, Professeur à l’Ecole Nationale Supérieure-Rabat, qui a accepté d’être rapporteur de cette thèse. Je le remercie aussi pour ses suggestions et conseils.
Je tiens aussi à remercier, de façon particulière, monsieur A. ALBIZANE, Professeur à l'université Hassan II-Mohammedia qui a accepté d’examiner ce travail et faire partie du Jury.
Je suis également reconnaissant à Monsieur J. Naja, Professeur à l’université Hassan II-Settat d’avoir été examinateur et de faire partie du jury de la thèse.
Une partie de cette thèse s’est déroulée au Laboratoire de Chimie des Matériaux Divisés et Catalyse (ITODYS) (université Paris 7). Je remercie l’ensemble du personnel du laboratoire et notamment Messieurs les professeurs J. Y. PIQUEMAL et A. ENSUQUE pour leurs fructueuses remarques et encouragements.
De même, je n’oublierai jamais mes séjours de recherches au Laboratoire de Catalyse et Matériaux de la Faculté d'Ingénierie de l'Université de Porto. Je suis, de même, reconnaissant aux Messieurs les professeurs M. F. R. PEREIRA et J. L. FARIA pour leur aide précieuse et leurs conseils et suggestions.
Enfin, un grand merci à mes proches qui ont su, tout au long de ces dernières années, m'encourager et, surtout, être là lorsque j’en avais besoin. Particulièrement à ma famille qui m’avait présenté toute sorte de soutien pour mener à bien ce projet. En espérant avoir donné envie à d’autres, et surtout à ma petite sœur, de suivre le chemin passionnant de la recherche scientifique.
SOMMAIRE
INTRODUCTION GENERALE……….. 1
CHAPITRE I : SYNTHESE BIBLIOGRAPHIQUE I- RAPPELS SUR LES APATITES………. 5
I-1- Introduction………... 5
I-2- Structure des apatites………... 5
I-3- Substitutions principales……….. 8
I-3-1- Rappels……… 8
I-3-2- Echanges cationiques et anioniques….………. 9
I-3-3- Substitution des ions XO43-……… 9
I-4- Différentes méthodes de synthèse des apatites………... 10
I-4-1- Réactions en phase aqueuse………... 10
I-4-2- Réactions en phase solide………... 10
I-4-3- Réactions en sels fondus………. 11
I-4-4- Réactions sol-gel………. 11
II- CARACTERISTIQUES DU GAZ NATUREL……….. 11
II-1- Introduction………... 11
II-2- Composition du gaz naturel………. 12
II-3- Propriétés physiques et thermiques du méthane………... 12
III- COMBUSTION DU METHANE……….. 13
III-2- Exemples des systèmes catalytiques testés dans la réaction de
combustion du méthane………... 14
III-3- Conversion du méthane en présence des catalyseurs à base de phosphate……….. 15
IV- REFORMAGE DES GAZ NATURELS………... 16
IV-1- Introduction……….. 16
IV-2- Les types de reformage du méthane………... 18
IV-3- Reformage catalytique du méthane au dioxyde du carbone……… 18
IV-4- Cinétique et schéma réactionnel de la réaction de reformage………. 20
V- CONCLUSION………. 22 Références bibliographiques………. 24 CHAPITRE II : TECHNIQUES EXPERIMENTALES 1- Introduction……… 31
2- Mesures de surface spécifique (BET)……… 31
3- Analyses chimiques……….. 31
4- Diffraction des rayons X (DRX)………. 31
5- Microscopie électronique à balayage (MEB)……… 32
6- Microscopie électronique à transmission (MET)……….. 32
7- Spectroscopie d’absorption Infrarouge……… 32
8- Spectroscopie UV-visible-PIR……… 33
9- Spectroscopie photoélectronique de rayons X (XPS)………... 34
10- Test de décomposition du butanol-2……….. 34
10-1- Conditions opératoires……….. 35
10-2- Bilan réactionnel……… 36
11-1- Combustion du méthane sur les catalyseurs à base du palladium... 37
11-2- Reformage du méthane au dioxyde de carbone sur les catalyseurs dopés au nickel………... 38 12- Thermo réduction programmée et la thermo oxydation programmée…….. 38
12-1- Dispositif expérimental………. 39
12-2- Conditions opératoires de la Thermo réduction programmée (TPR)………... 40 12-3- Conditions opératoires de la Thermo oxydation programmée (TPO)………... 40 13- Analyse thermogravimétrique et thermodifférentielle (ATD-ATG)………... 40
13-1- Rappels……… 40
13-2- Appareillage……… 40
Références bibliographiques………. 42
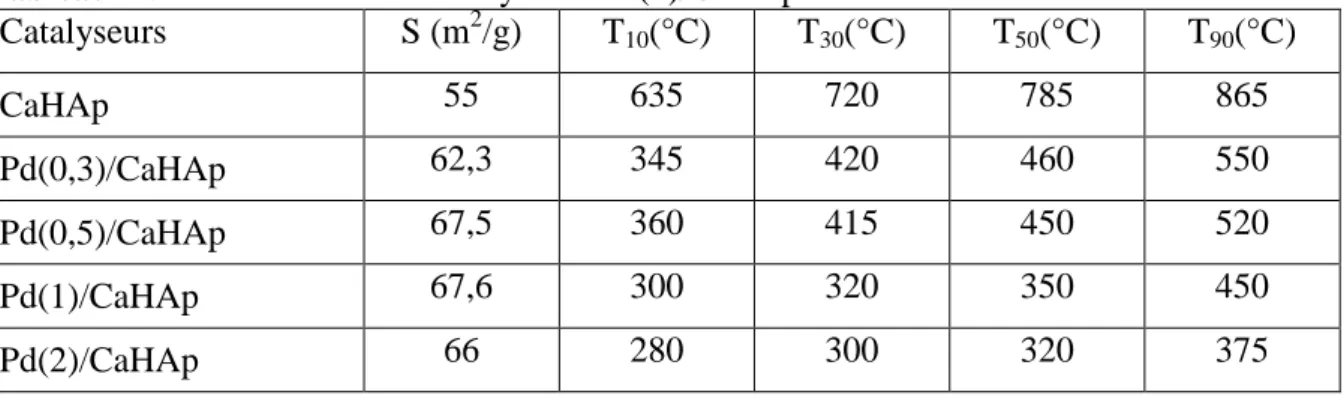
CHAPITRE III : PREPARATION ET CARACTERISATION DES CATALYSEURS Pd(x)/CaHAp et Pd(x)/CaFAp I- PREPARATION DES ECHANTILLONS……….……. 44
I-1- Préparation des supports………. 44
I-2- Préparation des catalyseurs Pd/hydroxyapatite et Pd/fluorapatite…...…….. 44
II- CARACTERISATION DES CATALYSEURS Pd(x)/CaHAp ET Pd(x)/CaFAp.. 45
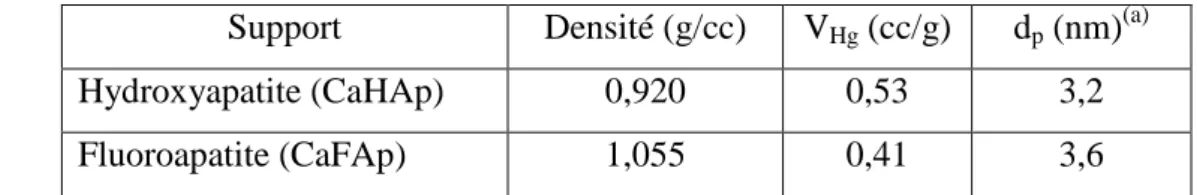
II-1- Isothermes d’adsorption de l’azote………. 45
II-2- Mesure des surfaces spécifiques et analyses chimiques……… 46
II-3- Diffraction des rayons X……….. 47
II-4- Microscopie électronique à transmission (MET)……….. 49
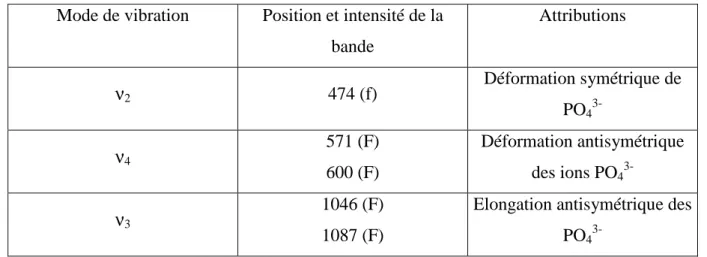
II-5- Spectroscopie d'absorption IR……… 51
II-7-1- Spectre UV-VIS-PIR du précurseur Pd(NO3)2(NH3)2………... 57
II-7-2- Spectres UV-VIS-PIR des catalyseurs Pd(x)/CaHAp et Pd(x)/CaFAp... 58
III- CONCLUSION……… 61
Références bibliographiques………. 63
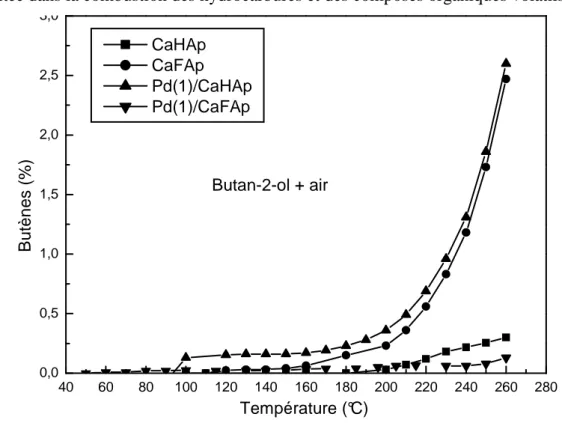
CHAPITRE IV : ETUDE COMPARATIVE DE LA REACTIVITE DES CATALYSEURS Pd(x)/CaHAp ET Pd(x)/CaFAp DANS LA DECOMPOSITION DU BUTANOL-2 ET LA REACTION DU COMBUSTION DU METHANE I- ACTIVITE DE Pd(x)/CaHAp ET Pd(x)/CaFAp DANS LA REACTION DE DECOMPOSITION DU BUTANOL-2……….………... 66
I-1- Activité catalytique sous atmosphère inerte………... 66
I-2- Activité catalytique en présence d’oxygène……… 68
II- ACTIVITE DES CATALYSEURS Pd(x)/CaHAp, Pd(x)/CaFAp DANS LA REACTION DE COMBUSTION DU METHANE……… 70
II-1- Activité des supports………. 70
II-2- Activités des catalyseurs Pd(x)/CaHAp et Pd(x)/CaFAp………... 71
II-2-1- Activité des catalyseurs Pd(x)/CaHAp………. 71
II-2-1-1- Stabilité du catalyseur Pd(1)/CaHAp………... 72
II-2-1-2- Activité du catalyseur Pd(1)/CaHAp en régime isotherme……….... 73
II-2-1-3- Influence de la masse du catalyseur……….. 74
II-2-1-4- Influence de la composition du mélange réactionnel.. 75
II-2-2- Activité catalytique du système Pd(x)/CaFAp………. 76
III- DISCUSSIONS ET COMPARAISON DES ECHANTILLONS……… 77
IV- CONCLUSION……… 80
Références bibliographiques………. 82
CHAPITRE V : PREPARATION ET CARACTERISATION DES CATALYSEURS A BASE DE NICKEL/APATITES A/- PREPARATION ET CARACTERISATION DES CATALYSEURS Ni(x)/CaHAp et Ni(x)/CaFAp I- PREPARATION DES CATALYSEURS………. 85
II- CARACTERISATION DES CATALYSEURS………. 85
II-1- Isothermes d’adsorption de l’azote et surfaces spécifiques BET………….. 85
II-2- Microscopie électronique à balayage………... 87
II-3- Diffraction des rayons X………... 90
II-4- Spectroscopie d’absorption Infrarouge………... 92
II-5- Thermo réduction programmée (TPR)………... 93
II-6- Thermo oxydation programmée à la température (TPO)………. 97
II-7- Spectroscopie de photoélectrons X (XPS)………... 99
II-8- Spectroscopie UV-visible-PIR………. 105
II-9- Décomposition du méthane en programmation de température (CH4 -TPR)... 107
III- CONCLUSION……….. 109
Ni(4)/Cu(x)CaHAp
I- PREPARATIONS DES CATALYSEURS Ni(4)/Cu(x)CaHAp …….………... 110
II- CARACTERISATION DES CATALYSEURS Ni(4)/Cu(x)CaHAp………... 110
II-1- Isothermes d’adsorption de l’azote et surfaces spécifiques BET………….. 110 II-2- Microscopie électronique à balayage (MEB)…….………. 111 II-3- Thermo-réduction et thermo-oxydation programmées (TPR/TPO)…….... 113 II-4- Décomposition du méthane en programmation de température (CH4
-TPR)…... 115 II-5- Spectroscopie de photoélectron X (XPS)………...………….. 116
III- CONCLUSION……….. 118
Références bibliographiques………. 119
CHAPITRE VI :
ACTIVITE CATALYTIQUE DE Ni(x)/CaHAp, Ni(x)/CaFAp ET Ni(4)/Cu(x)CaHAp DANS LA REACTION DE REFORMAGE DU METHANE AU DIOXYDE DE
CARBONE
I-ETUDE DE L’ACTIVITE CATALYTIQUE DES CATALYSEURS
Ni(x)/CaHAp………... 122
I-1- Activité en fonction de la température de réaction (light off)………... 122 I-2- Activité en régime stationnaire……… 129
II-ACTIVITE CATALYTIQUE DES CATALYSEURS
Ni(x)/CaFAp ……….. 132
II-2- Activité en régime stationnaire……… 138 II-3- Analyse de dépôt de carbone sur les catalyseurs Ni(x)/CaHAp et
Ni(x)/CaFAp……….……….. 140
III- ACTIVITE CATALYTIQUE DES CATALYSEURS Ni(4)/Cu(x)CaHAp……... 141
III-1- Activité en fonction de la température de réaction……… 141 III-2- Activité en isotherme……….……… 145
IV- CONCLUSION……… 148
Références bibliographiques………. 150
2
L’ensemble des travaux qui sont présentés dans ce mémoire s’inscrit dans un programme plus large développé dans notre laboratoire depuis sa création. Il concerne l’élaboration de matériaux phosphatés et l’examen de la possibilité de leur utilisation dans diverses réactions catalytiques. Parmi ces phosphates, ceux de structure apatitique présentent des propriétés structurales et acido-basiques qui en font un matériau de choix dans le type d’investigations que nous menons. Ils admettent d’innombrables substitutions et échanges, et leurs propriétés acido-basiques sont modulables. Dans le présent travail nous allons utiliser ces phosphates comme support de phases actives dans l’oxydation du méthane.
Le gaz naturel est très riche en méthane. Compte tenu des problèmes énergétiques que le monde vit actuellement, il sera de plus en plus utilisé grâce à ses nombreux avantages (émissions réduites et bon rendement). Sa combustion comparée à celle d’autres produits fossiles, dégage moins de dioxyde de carbone (CO2), d’oxydes d’azote (NOx), et quasiment
pas de dioxyde de soufre (SO2). Les centrales thermiques modernes au gaz naturel ont des
rendements importants (supérieurs à 50%); en plus elles émettent peu de polluants dans l’atmosphère. Le gaz naturel est aussi utilisé par d’autres filières industrielles qui le transforme en ammoniac, propanal, méthanol, ou diméthyl éther via le gaz de synthèse (H2/CO) et qui sont opérationnelles depuis des années. Il peut également servir à la production
d’hydrogène par vaporeformage par exemple; mais ce procédé présente l’inconvénient de rejeter du gaz carbonique dans l’atmosphère. En revanche, le reformage du méthane au CO2
(reformage à sec) est plus avantageux puisqu’il consomme du gaz carbonique.
Le phosphore est couramment ajouté aux catalyseurs industriels sous forme de phosphate. Ce groupement a pour effet de stabiliser les formulations catalytiques et d’apporter au système des propriétés acido-basiques modulables. C’est dans cette perspective qu’une attention particulière est portée à l’utilisation des apatites comme supports catalytiques dans des réactions de valorisation des gaz naturels. Elles peuvent constituer une excellente alternative aux supports classiques grâce à (i) leur stabilité structurale, (ii) leurs propriétés acido-basiques et (iii) leur capacité d’échanger divers ions de leur charpente.
Notre travail est une application de ces supports apatitiques dans deux types de valorisation du gaz naturel: (i) la combustion et (ii) le reformage au dioxyde de carbone. Des catalyseurs à base du palladium Pd(x)/Ca10(PO4)6(OH)2 et Pd(x)/Ca10(PO4)6F2 seront testés
dans la réaction de combustion du méthane, et d’autres à base de nickel Ni(x)/Ca10(PO4)6(OH)2 et Ni(x)/Ca10(PO4)6F2 seront examinés dans le reformage à sec du
de dégager des corrélations entre les propriétés de ces catalyseurs et leurs performances. Le plan qui sera adopté pour la présentation de cette étude est le suivant :
Le premier chapitre est consacré à une revue bibliographique mettant l’accent sur les propriétés physico-chimiques des apatites et les voies de valorisation des gaz naturels.
Le deuxième chapitre présente les différentes techniques expérimentales utilisées. Le chapitre III décrit l’élaboration des catalyseurs Pd(x)/Ca10(PO4)6(OH)2 et
Pd(x)/Ca10(PO4)6F2 et leur caractérisation par différentes techniques physico-chimiques (BET,
MET, Spectroscopie IR, DRX, XPS, UV-Visible-PIR).
Le chapitre IV présente une étude comparative de la réactivité des catalyseurs Pd(x)/Ca10(PO4)6(OH)2 et Pd(x)/Ca10(PO4)6F2 dans la décomposition du butanol-2 et la
réaction du combustion du méthane.
La synthèse des catalyseurs Ni(x)/Ca10(PO4)6(OH)2, Ni(x)/Ca10(PO4)6F2 et
Ni(4)/Cu(x)CaHAp, ainsi que leur caractérisation par différentes techniques physico-chimiques (BET, MEB, Spectroscopie IR, DRX, XPS, UV-Visible-PIR, TPR/TPO), seront décrites dans le chapitre V.
Le chapitre VI étudie l’activité catalytique de Ni(x)/CaHAp et Ni(x)/CaFAp dans la réaction de reformage du méthane au dioxyde de carbone, ainsi que l’influence de l’ajout d’un deuxième métal (cuivre), par échange, sur l’activité des catalyseurs Ni(4)/Cu(x)CaHAp.
CHAPITRE I
I- RAPPELS SUR LES APATITES
I-1-Introduction
Le nom d’apatite, signifiant « trompeur », a été donné il y a environ un siècle à un groupe de minéraux qui étaient souvent confondus avec d’autres, tels que l’aigue-marine, l’améthyste et l’olivine. De tous les composés phosphatés, l’apatite est la plus importante. Ce sont des composés qui sont de plus en plus étudiés et utilisés dans les domaines de la chimie des matériaux et en catalyse. Leur structure est stable et tolère de grands écarts à la stoechiométrie.
Plusieurs travaux ont montré que les composés phosphatés sont prometteurs et jouent un rôle considérable dans le développement de plusieurs branches de la science des matériaux. Ceci est dû aux propriétés physico-chimiques observées dans le cas des phosphates de métaux alcalins et de terres rares, telles que les propriétés catalytiques, réfractaires, ferroélectriques, de luminescence et la possibilité d'obtention de verres spéciaux [1-2]. Ainsi, la chimie des phosphates est devenue une science à part entière, qui trouve des applications industrielles importantes. Par exemple, plusieurs catalyseurs à base de phosphates ont une application industrielle [3] : Citons à titre d’exemple :
- Les phosphates de silice utilisés pour la polymérisation des hydrocarbures.
- Un catalyseur à base de phosphates de calcium et de nickel pour la déshydrogénation des butènes.
- Les phosphates de vanadium utilisés dans l’oxydation du butane en anhydride malaique - Les phosphates de fer dans l’oxydation de l’acide iso butyrique.
I-2-Structure des apatites
Les apatites de formule générale [Me10(XO4)6Y2] cristallisent dans le système hexagonal et
appartiennent au groupe spatial P63/m (où Me représente un cation bivalent, XO4 un anion
trivalent et Y un anion monovalent) [4-6].
La maille élémentaire du réseau de l’apatite est un prisme droit à base losange (Fig. 1). Le réseau cristallin présente deux types de tunnels dans lesquels sont localisés les ions Ca2+ (Fig. 2). Il apparaît ainsi dans le réseau de l'apatite deux catégories d'ions (CaI et CaII). Les ions CaI,
qui occupent le tunnel de diamètre égal à environ 2,5 Å, sont placés sur les axes ternaires. Ils sont engagés dans des colonnes verticales CaI-O, qui sont reliées entre elles par des ions P
6
qui forment des tétraèdres PO4 avec 3 oxygènes d'une colonne et 1 oxygène de la colonne
suivante (Fig. 3). Les cations de la deuxième catégorie (Ca2+) sont situés à la périphérie du deuxième tunnel (3 à 4,5 Å), et placés à proximité des axes sénaires hélicoïdaux des tunnels en formant des triangles équilatéraux alternés autour de ces axes. Les anions OH- ou F- sont à l'intersection de l'axe sénaire et des plans de symétrie placés aux côtes z=1/4 et z=3/4 (Fig. 4) [7].
Figure 1 : Projection de la structure de l'apatite sur le plan 001
Figure 2 : Caractéristique du système hexagonal avec les nœuds de réseau à gauche et
schéma de l’hydroxyapatite à droite avec en noir les calcium en site I et en blanc les calciums en site II
Figure 3 : Représentation des colonnes CaI-O reliés par des ions P
8
J. C. Elliott a montré que l’hydroxyapatite peut aussi cristalliser dans le système monoclinique P21/b. L’apatite monoclinique n’a pas connu assez d’intérêts, sa transformation en structure
hexagonale s’effectue à des températures comprises entre 200 et 210°C [8, 9].
I-3- Substitutions principales
I-3-1- Rappels
L'hydroxyapatite, Ca10(PO4)6(OH)2, est le principal constituant minéral des os. Elle est stable
jusqu'à 1000°C [10]. Certaines de ses compositions fluorées comportant des ions silicates peuvent conserver leur structure jusqu'à 1500°C [11].
Le produit de solubilité des apatites obtenues par précipitation est très faible [12, 13] (tableau 1). Il diminue quand la température augmente [14] : on parle alors d'une solubilité rétrograde. Cette solubilité dépend également du pH. En effet, l'apatite phosphocalcique est plus soluble en milieu acide qu'en milieu neutre ou basique [12-14]. De même, l’expérience a montré que les hydroxyapatites précipitées rapidement sont non-stœchiométriques. La maturation en milieu aqueux d’hydroxyapatites non-stœchiométriques conduit, par un phénomène de surface, à une évolution vers une phase de plus en plus stœchiométrique et de moins en moins soluble.
Tableau 1 : Produits de solubilité de la fluoroapatite et de l'hydroxyapatite (T = 37°C) Apatite (formule chimique) Produits de solubilité
Ca5(PO4)3F 10-61
Ca5(PO4)3(OH) 10-60
L’une des propriétés particulières de la structure des apatites, est sa capacité à incorporer et à échanger des anions et des cations [15, 16]. Rappelons que ces substitutions, font intervenir des facteurs, tels que la charge et la taille des ions échangés [10, 17-19], et elles s’accompagnent sur le plan structural de légères déformations de la maille.
Dans le tableau 2, on a regroupé quelques exemples de substitutions possibles dans la structure de l’apatite, en considérant la formule générale suivante : Me10(XO4)6(Y)2.
Tableau 2 : Quelques exemples de substitutions dans la structure apatitique : Me2+ XO43- Y -Ca2+ Sr2+ Pb2+ Mg2+ Ba2+ Zn2+ REE3+ □ Na+ ... SiO4 4-... CO3F 3-AsO4 3-PO4 3-VO4 3-... SO4 2-HPO4 2-CO3 2-... CO3 2-S2 2-O 2-... OH -F -Cl -I -Br -O2 -H2O N2 □ ...
I-3-2- Echange cationique et anionique
Les ions Ca2+ peuvent être échangés par des ions divalents tels que Sr2+, Cd2+, Ba2+, Cu2+, Ni2+, Co2+….Ces substitutions ont fait l’objet de plusieurs travaux de recherches dans le domaine de la catalyse hétérogène et le traitement des eaux usées. Ils ont essentiellement pour but la détermination de la capacité d’échange de l’apatite et les sites dans lesquels ils s’effectuent [20-22]. Il a été, par exemple, démontré que les terres rares préfèrent se substituer aux cations Ca2+ qui sont dans les plans de symétrie z=1/4 et z=3/4 [23-25]. Il est aussi possible de remplacer partiellement les ions Ca2+ par des cations monovalents (Na+, K+…), trivalents (La3+, Al3+, Eu3+…) ou des lacunes. Dans les apatites naturelles, la substitution de Ca2+ par Na+ et Mg2+ a été fréquemment signalée [26]. Lehr et coll, ont montré que ces substitutions sont en relation directe avec celle de PO43- par CO32-. Ils ont constaté que le taux
de substitution de Ca2+ par Na+ et Mg2+ augmente avec celui de la substitution des PO43- par
CO32- [27].
Les tunnels, dans lesquels sont situés les ions Y-, confèrent aux apatites des propriétés d'échangeur d'anions (hydroxyde, fluorure, chlorure...) [16, 28]. La substitution de F- par OH -est assez fréquente dans les apatites naturelles. Des études récentes, ont montré qu’avec un traitement hydrothermique par KOH, on peut convertir une fluoroapatite ou chloroapatite en hydroxyapatite [29, 30]. De même, il a été reporté que les ions OH- peuvent être substitués par des ions bivalents (CO32-, S2-, O2-…) ou des lacunes [31].
I-3-3- Substitution des ions XO4
3-Les ions PO43- peuvent être remplacés par des anions porteurs de même charge tels que AsO43,
10
du cristal est préservée par l'introduction d'ions F- supplémentaires. Les ions PO43- peuvent
aussi être partiellement remplacés par des groupements bivalents (SO2-, SeO42-, HPO42-…) ou
tétravalents (GeO44-, SiO44-…). Riveira et coll, ont étudié la décontamination des eaux
polluées par le sélénium, en utilisant la substitution des ions PO43- par les ions SeO32- [32].
I-4- Différentes méthodes de synthèse des apatites
Parmi ces méthodes, on peut citer la précipitation en solution aqueuse, la réaction solide-solide (voie sèche), la réaction en sels fondus et le procédé sol-gel.
I-4-1- Réactions en phase aqueuse
Les synthèses en phase aqueuse se font selon deux procédés différents : la méthode par double décomposition et la méthode par neutralisation. Ces procédés sont actuellement utilisés pour la production industrielle d’apatite.
La méthode par double décomposition [10, 33, 34] consiste à ajouter de façon contrôlée une solution du sel de cation Me dans une solution du sel de l’anion XO4. Le précipité est ensuite
lavé et séché. Cette technique permet également d’obtenir des apatites mixtes (contenant deux cations différents M1 et M2) avec une maîtrise du rapport Me1/Me2. Les cations sont mélangés
simultanément afin d’éviter une ségrégation lors de la précipitation. La seconde méthode consiste à neutraliser une solution de lait de chaux par une solution d’acide phosphorique. Cette réaction permet d’obtenir rapidement de grandes quantités d’hydroxyapatite phosphocalcique [10, 35]. Il est également possible de synthétiser des fluorapatites en utilisant le même protocole expérimental.
I-4-2- Réactions en phase solide
La synthèse par réaction solide-solide consiste à chauffer un mélange réactionnel, constitué de divers sels de cations et d’anions, dans un rapport Me/XO4 égal à 1,67. Ce mélange doit être
parfaitement homogène pour permettre une réaction totale à une température de 900ºC. La synthèse d’une fluorapatite phosphocalcique peut être effectuée, par exemple, à partir du phosphate tricalcique et du fluorure de calcium selon la réaction suivante [36] :
I-4-3- Réactions en sels fondus
Cette méthode permet d’approcher les conditions de synthèse dans lesquelles on été obtenues certaines apatites naturelles. On a pu ainsi préparer des cristaux de phosphates en utilisant des réactifs de départ en excès.
I-4-4- Réactions sol-gel
Le procédé sol-gel est basé sur la polymérisation de précurseurs organométalliques de type alcoxydes M(OR)n [37]. Après une hydrolyse contrôlée de l’alcoxyde en solution, la
condensation des monomères conduit à des ponts oxo puis à un oxyde organique. La polymérisation progressive de ces précurseurs forme des oligomères puis des polymères en augmentant ainsi la viscosité. Ces solutions polymériques conduisent à des gels qui permettent une mise en forme aisée des matériaux (films denses et transparents, poudres ultra-fines, céramiques, ...) avec de nombreuses applications technologiques [38, 39].
II- CARACTERISTIQUES DU GAZ NATUREL
II-1- Introduction
Pendant longtemps, le gaz naturel a été considéré comme un sous-produit de l’exploitation du pétrole. Il était condamné à être brûlé à la torche, faute de moyens techniques pour l’acheminer vers des points de consommation à des conditions économiquement acceptables. Il ne fut valorisé qu’à partir des années trente, grâce aux progrès des techniques sidérurgiques. Le gaz naturel était généralement présenté comme une source d’énergie « propre ». Sa combustion dégage en effet peu d’émanations polluantes comme les oxydes de soufre ou les oxydes d’azote (NOx). La consommation mondiale de gaz naturel a connu une progression
moyenne de 3,5 % par an entre 1965 et 2000, alors que la demande globale en énergie primaire augmentait en moyenne de 2,4 % par an. Rappelons qu’un quart des besoins énergétiques du monde est aujourd’hui satisfait par le gaz naturel, et que la consommation mondiale devrait encore augmenter dans les décennies à venir. Les réserves de gaz, connues pourraient couvrir au moins soixante années de consommation au rythme actuel.
12
II-2- Composition du gaz naturel
Le gaz naturel est un mélange d'hydrocarbures saturés dans lequel domine le méthane (tableau 3). Après épuration, le gaz naturel contient entre 81 et 97 % de méthane. Il contient aussi des alcanes, des composés oxygénés, azotés ou sulfurés en proportions variables.
Tableau 3 : Composition type de quelques gaz naturels (en pourcentages volumiques)
Origine Maroc (Korimat)
Maroc
(Ndarq) Algérie Pays bas
Mer du nord Russie France (Lacq) Méthane 79,670 47,730 88,500 82,400 85,700 97,000 97,000 Ethane 13,010 6,050 9,100 3,300 9,000 1,100 2,190 Propane 2,540 1,080 1,500 0,570 2,400 0,400 0,163 i-butane 0,734 - 0,180 0,090 0,250 0,100 0,042 n-butane 0,557 - 0,240 0,100 0,480 0,100 0,080 Néo-pentane - - 0,020 0,004 - - - i-pentane - - 0,011 0,023 0,050 0,030 0,014 n-pentane - - 0,002 0,027 0,050 0,030 0,019 Hexane - - - 0,072 0,020 0,020 0,100 Helium - - - 0,047 - 0,020 0,007 Azote 3,490 45,140 0,450 12,324 0,600 1 0,390 CO2 - - - 1,031 1,400 0,200 - P.C.S (kwh/m3) 12,57 6,78 12,2 10,1 12,2 11,2 11,3
II-3- Propriétés physiques et thermiques du méthane
Dans les conditions normales de température et de pression, le méthane est un gaz. Lorsqu'il est pur, il est incolore. Il est peu soluble dans l'eau: on ne peut dissoudre que 40 cm3 de ce gaz dans un litre d'eau dans les conditions normales de température et de pression. Des mesures ont permis de déterminer la longueur des liaisons C – H : 1,09.10-10 mètres.
Le tableau 4 regroupe les principales propriétés physiques et thermiques du méthane. Lors de la réaction de combustion, l'énergie dégagée peut être exprimée par unités de quantité de combustible. Ainsi, la combustion de 1 kg de combustible, solide ou liquide, ou de 1 m3 de gaz (1 m3 pris dans les conditions normales de pression, 1 atm, et de température 25 °C) fournit une quantité d'énergie appelée «pouvoir calorifique» et notée «PC». Dans le cas du
méthane son pouvoir calorifique est de l’ordre de 35,87-39,89 MJ/m3. On distingue le PCS
(supérieur) et le PCI (inférieur), selon que l'eau formée est à l'état liquide ou à l'état vapeur.
Tableau 4 : Propriétés physiques et thermiques du méthane
Symbole chimique CH4
Masse molaire (g.mol-1) 16,043
Masse volumique (kg.m-3) 0,715 (273 k, 1atm) Température critique (°C) -85,5
Volume critique (cm3.mol-1) 99,03
Pression critique (atm) 45,8
Enthalpie de formation (kcal.mol-1) -17,889 (25°C) Température de liquéfaction (°C) -161,4 (1 atm) Température de solidification (°C) -182,5 (1atm) Viscosité cinématique (kg.m-1.s-1) 1,013.10-5 (0°C)
Energie de liaison C-H 420 kj/mole
Conductivité thermique (w.m-1.k-1) 219,63.10-4 (0°C) Capacité calorifique (J.mol-1.k-1) 34,45 (0°C)
III- COMBUSTION DU METHANE
III-1-Combustion catalytique du méthane
La combustion catalytique du méthane, qui peut avoir lieu à basse température, présente beaucoup d'avantages par rapport à la combustion vive traditionnelle. De ce fait, elle a fait l’objet de plusieurs études [40-53]. Elle est la pierre angulaire des nouvelles technologies gazières [54-57]. Généralement, les systèmes de combustion catalytique du gaz naturel doivent satisfaire les exigences suivantes [40]:
- Abaisser la température de la réaction de combustion.
- Récupération d’une énergie élevée avec le minimum d’excès de l’air. - Minimiser les émissions en CO et en NOx,gaz nuisibles à l’environnement.
14
III-2- Exemples des systèmes catalytiques testés dans la réaction de combustion du méthane
Dans la combustion catalytique du méthane, les oxydes sont généralement moins actifs que les métaux de transitions supportés, mais ils présentent l’avantage d’être plus résistants et moins volatiles [58]. Les supports les plus utilisés sont les fibres d’alumine gamma qui ont une surface spécifique de l’ordre de 150 m2/g [59]. Les promoteurs qui sont inclus dans la préparation des catalyseurs supportés sont utilisés pour minimiser les effets des poisons, du frittage thermique et pour stimuler l’activité du catalyseur.
En général, l’activité des catalyseurs supportés dépend de la structure cristalline de l’élément actif, du promoteur, de la nature du support et de la méthode d’imprégnation utilisée [60-62]. Une étude comparative de 28 catalyseurs supportés et non supportés utilisés dans la combustion du méthane a montré que les catalyseurs à base de palladium sont les plus actifs, suivis par les catalyseurs à base de platine [63]. L’activité catalytique des métaux et des oxydes correspondants décroît dans l’ordre suivant : Pd, Pt, Cr, Mn, Cu, Ce, Co, Fe, Ni et Ag [64]. Plusieurs chercheurs qui ont étudié les catalyseurs à base de Rh, Pt ,Ni, Mn, Fe et Co, ont conclu que leurs performances sont affectées par la nature du support (Al2O3, MnO2,
MgO, SiO2, MoO3…) [65-68].
Au cours de ces dernières années une attention particulière a été portée à la synthèse de matériaux performants et stables, car dans ce type de réactions, les catalyseurs sont utilisés dans des conditions sévères. Actuellement, les recherches sont essentiellement axées vers la modification de l’alumine et l’élaboration d’autres supports moins sensibles au frittage et possédant des propriétés acido-basiques convenables.
Par ailleurs, il a été rapporté que le palladium est actif lorsqu’il est supporté sur un catalyseur présentant des propriétés faiblement acides. En revanche, le platine est actif en présence d’un support ayant une forte acidité [69].
L’ajout de métaux alcalins et alcalino-terreux a un effet bénéfique et contribue à diminuer le frittage du palladium. Le recouvrement de l’alumine par quelques couches de silice améliore aussi ses performances en diminuant sa tendance au frittage. En revanche, Cullis et Willatt, ont montré que les halogènes ont un effet inhibiteur sur l’activité des catalyseurs à base de palladium et de platine [70].
Bien que les catalyseurs les plus performants, soient ceux à base de palladium supporté, plusieurs difficultés limitent leur utilisation [71-78]. Ces difficultés ont été énumérées par Lyubovsky et Pfefferle [79].
L’état d’oxydation du palladium semble jouer un rôle déterminant dans son activité. Il est presque admis que c’est PdO qui constitue la phase active dans la réaction de combustion du méthane [80-82]. Choudhary et coll, ont étudié l’effet de cycles d’oxydo-réduction du palladium sur l’évolution de l’activité de Pd/Al2O3. Ils ont trouvé que le palladium est plus
actif quand il est sous forme oxydé (PdO) [83]. Cependant, il n’est pas exclu que ce soit la présence simultanée d’une forme réduite et oxydée qui soit responsable de l’activité. D’autres auteurs, préconisent que c’est la formation de grandes cristallites de Pd métallique obtenues par réduction partielle de PdO en présence du mélange réactionnel qui conduit à une augmentation de l’activité catalytique à cause de la présence simultanée des deux formes oxydées et réduites [84].
En utilisant les oxydes Ta2O3, TiO2, CeO2 et ZrO2 comme supports de PdO, Farrauto et coll
[85] ont pu étudier l’influence de la nature du support sur la stabilité de l’oxyde de palladium déposé. Ils ont conclu que la température de décomposition de PdO varie d’un support à l’autre. Par exemple, sur ZrO2, PdO se décompose à des températures plus basses par rapport
à celles mesurées sur les autres supports.
Fujimoto et coll ayant étudié le système PdOx/ZrO2 ont montré que la combustion du
méthane à basse température est liée à la présence d’atomes et de lacunes d’oxygène sur la surface du catalyseur [84]. Un mécanisme de Mars et Van Krevelen est préconisé par ces auteurs.
Ciuparu et coll ont étudié l’échange d’oxygène entre le palladium et les supports oxydes [73]. Ils ont montré qu’aux basses températures, l’oxygène provenant du support participe au mécanisme de combustion du méthane.
Carstens et coll ont remarqué aussi, qu’en présence du mélange méthane/air, il y a formation de larges cristallites de PdO. En présence d’oxygène seul, PdO existe sous deux formes : amorphe et cristallisée [82]. Des hystérèses en températures de réaction ont été observées lorsqu’on applique des cycles de températures. Ces hystérèses sont liées à l’oxydoréduction du palladium et conduisent à des processus oscillants [86-88].
III-3- Conversion du méthane en présence des catalyseurs à base de phosphate
Malgré les nombreuses applications de l’apatite, son étude en catalyse reste relativement limitée. Rappelons que ce type de phosphate est connu pour ses propriétés basiques. En effet, Hall et coll. ont montré que l’hydroxyapatite stœchiométrique (Ca/P = 1,66) catalyse aussi bien la déshydrogénation et la déshydratation du butan-2-ol alors que la non stœchiométrique
16
(Ca/P = 1,58) conduit uniquement à la déshydratation [89]. L’activité déshydratante est liée à l’augmentation de l’acidité du catalyseur et à sa déficience en calcium. Il est à remarquer que l’écart à la stoechiométrie semble entraîner la disparition de la basicité du catalyseur. Dans la structure, ils existent donc des ions Ca2+ responsables de la basicité. La substitution des ions Ca2+ par des ions Cu2+ ou Ni2+ confère au matériau des propriétés déshydrogénantes et oxydo-réductrices remarquables [90]. Il a été montré aussi que les propriétés acido-basiques des surfaces phosphatés, ainsi que leur activité catalytique, peuvent être modelées par les échanges cationiques [91 ; 92].
Dans une étude réalisée sur l’oxydation du méthane en présence de l’hydroxyapatite, il a été conclu que les sélectivités des produits de la réaction dépendent du rapport Ca/P de l'hydroxyapatite. A 600°C une sélectivité importante en CO2 est obtenue pour un rapport de
de Ca /P égale à 1,65 [93].
La substitution des ions de calcium (35 %), dans le réseau de l’apatite, par le plomb améliore nettement la sélectivité en dioxyde de carbone [94]. De même, des phosphates d’alcalins et d’alcalino-terreux (Li, Na, K, Rb, Ca, Cs, Be, Mg, Sr, Ba) ont été testés dans l'oxydation du méthane. Les résultats obtenus ont montré que le phosphate de sodium présente la meilleure sélectivité en CO2 [95]. Ces résultats rendent bien compte de l’importance des propriétés
basiques du support sur l’évolution de l’activité catalytique.
IV- REFORMAGE DU GAZ NATUREL
IV-1- Introduction
L’élément hydrogène est très abondant sur Terre, dans l’eau des lacs, des rivières et des océans ainsi que dans les combustibles fossiles. Il est le constituant essentiel de notre Univers, mais il n’existe pas à l’état libre. Le tableau 5 regroupe les sources principales de sa production.
Tableau 5 : Sources de production de l’hydrogène
Source Capacité mondiale (%)
Gaz naturels 48
Pétrole 30
Charbon (Coal) 18
C’est un combustible à pouvoir énergétique élevé, non polluant, non toxique et sa combustion ne produit que de l’eau. Son énergie massique, délivrée lors de sa combustion avec l’oxygène, est élevée (120 MJ.kg-1), comparée à celle de l’essence (45 MJ.kg-1) ou du méthane (50 MJ.kg-1) (tableau 6).
Tableau 6 : Propriétés comparées de l’hydrogène et du méthane
Propriétés Dihydrogène Méthane
Masse molaire 2,0158 g.mol-1 16,043 g.mol-1
Température d’ébullition (à 1013 hPa) 20,27 K 109,15 K Température de solidification 14,01 K 90,67 K Masse volumique gazeuse à 273K 0,08988 kg.m-3 0,6512 kg.m-3 Masse volumique liquide à 20,3K 70,79 kg.m-3
Masse volumique gazeuse à 20,3K 1,34 kg.m-3
PCI 119930 kJ.kg -1 50020 kJ.kg-1
PCS 141860 kJ.kg-1
Conductivité thermique du gaz 0,1897 W.m-1.K-1 Température critique (température au dessus de
laquelle la liquéfaction est impossible) 33,30K
Température d’auto inflammation dans l’air 858 K 813 K Température de flamme dans l’air à 300K 2318 K 2148 K Limites d’inflammabilité dans l’air (vol%) 4 - 75 5,3 -15 Limites de détonation dans l’air (vol%) 13 - 65 6,3 -13,5 Energie explosive (kg de TNT.m-3 de gaz) 2,02 7,03 Vitesse de flamme dans l’air 260 cm.s-1 37 cm.s-1 Vitesse de détonation dans l’air 2,0 km.s -1 1,8 km.s-1 Mélange stoechiométrique dans l’air (vol) 29,53% 9,48%
L’hydrogène est appelé à jouer un rôle essentiel dans les systèmes énergétiques du futur. Le développement de son utilisation comme carburant du XXIe siècle est devenu indispensable, face au problème préoccupant de l’augmentation de l’effet de serre (dû principalement au CO2) et à la diminution des ressources d’énergies fossiles.
Ainsi, du fait des contraintes très évolutives en matière d'environnement et de la nécessaire diversification des sources d'énergie, les réactions d'activation catalytique du gaz naturel (essentiellement le méthane) en gaz de synthèse (mélange hydrogène et monoxyde de carbone) sont aujourd'hui des objectifs industriels hautement stratégiques[96 ;97]. On peut
18
citer à titre d'exemple son utilisation pour la production de l'ammoniac et l’utilisation du mélange H2/CO pour la production du méthanol et les synthèses de Fischer-Tropsch [98].
IV-2- Les types de reformage du méthane
Le reformage du méthane regroupe les réactions ayant lieu entre le méthane et un oxydant (O2, CO2 ou H2O) pour former du gaz de synthèse, qui est un mélange de CO et H2 [99]. Afin
d’être réalisable dans des domaines de température et de pression pas trop élevés, ces réactions sont effectuées en présence d’un catalyseur [100-102].
Oxydation partielle : CH4 + 1/2 O2 2 H2 + CO -36 kJ.mol-1
Reformage à sec : CH4 + CO2 2 CO + 2 H2 247 kJ.mol-1
Vapo-reformage : CH4 + H2O CO + 3 H2 206 kJ.mol-1
En dépit de sa faible réactivité, le méthane reste la matière première la plus économique pour la production d'hydrogène et du monoxyde de carbone [103]. Bien que, le reformage du méthane en présence de la vapeur d’eau soit le processus qui a prévalu pendant 70 ans [104], il présente l'inconvénient de sa grande endothermicité. De ce fait, les réactions d'oxydation partielle et de reformage " à sec " par le dioxyde de carbone en mélange CO/H2 constituent
une alternative intéressante au reformage classique "à l'eau". En effet, la réaction de vapo-reformage présente les inconvénients d'être endothermique et de produire des gaz nuisibles et toxiques comme NOx, SOx, COx. En revanche, l'oxydation partielle, offre l'avantage de
conserver un rapport H2/CO élevé, proche de 2, tout en étant quasi athermique [105].
IV-3- Reformage catalytique du méthane au dioxyde du carbone
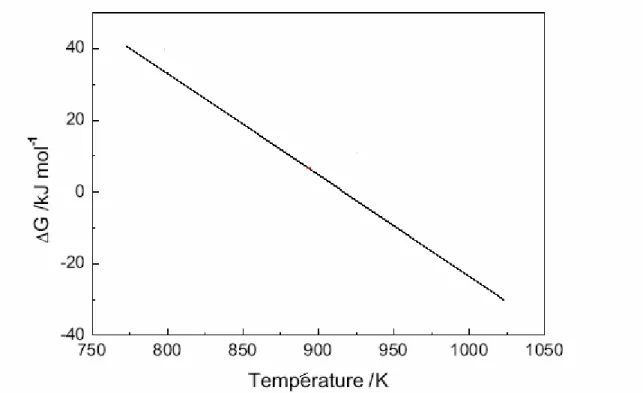
Malgré que la réaction de reformage du méthane au CO2 (à sec) soit fortement endothermique
(Fig. 5), elle présente l'intérêt de consommer du gaz carbonique qui est souvent présent dans le gaz naturel et qui est un agent important de l'effet de serre.
De même, cette réaction produit du monoxyde de carbone avec une grande sélectivité. Le problème majeur de cette voie est la désactivation rapide des catalyseurs par dépôt de carbone [104-110]. La figure 6 montre l’influence du rapport CO2/CH4 sur la formation de carbone en
Figure 5 : Thermodynamique de la réaction de reformage du méthane au dioxyde du carbone
Figure 6 : Formation du carbone en fonction de la température et du rapport CO2/CH4 de la réaction
du reformage du méthane au CO2
Les catalyseurs classiques des réactions de reformage sont généralement à base de métaux nobles (Ru, Pd, Pt…) déposés sur des supports oxydes (SiO2, Al2O3, ZrO2 ...) par
imprégnation ou co-précipitation [112-113]. Cependant, de grands espoirs sont placés dans les métaux de transition Ni, Co… [114-122], qui sont actifs et moins coûteux que les métaux nobles. Ils présentent l’inconvénient majeur de se désactiver suite aux faibles interactions métal-support. Les supports non classiques suscitent beaucoup d’intérêt, car ils permettent
20
d’élaborer des catalyseurs qui présentent de fortes interactions entre le métal et son support [123, 124].
Bradford et coll., ont étudié les performances des catalyseurs à base de Ni et de métaux nobles supportés [125]. Pour des raisons économiques, le nickel supporté sur alumine est employé en industrie comme catalyseur de la réaction de reformage du méthane au dioxyde carbone, pour produire les gaz de synthèse [126].
Récemment, une attention particulière est portée à l’utilisation des composés phosphatés comme supports. Les catalyseurs phosphate de Nickel-calcium/hydroxyapatite présentent une grande activité et une bonne sélectivité [127]. Sur ces catalyseurs, trois espèces de nickel ont été dénombrées. Dans une autre étude, le catalyseur Ni-Sr/phosphate a été testé. Il a été montré que le nickel métallique est la phase active et elle résulte de la réduction de trois espèces différentes de nickel [128].
La modification des propriétés acido-basiques pourrait aussi avoir une influence sur le taux de dépôt de carbone. Ainsi, plusieurs chercheurs ont proposé des additifs et des promoteurs pour minimiser ces dépôts et stabiliser la réactivité des catalyseurs [129-132].
Bouarab et coll, ont étudié l’effet de l’ajout de MgO sur les propriétés catalytiques de Co/SiO2. Ils ont constaté que MgO limite la formation du coke [133]. L’addition de MgO a
été également étudiée sur Rh/SiO2 [134]. Il a été constaté que le MgO améliore les
performances catalytiques en activant la dissociation de CO2. L’effet de cuivre sur le
comportement de Ni/SiO2 a été également étudié [135]. Cette addition stabilise le catalyseur
pendant 120 heures de réaction et favorise le nettoyage des particules de Ni, du carbone accumulé.
De même, la résistance au cockage a été étudiée en utilisant des additifs tels que CeO2 [136]
et La2O3 [137]. Osaki et coll, ont examiné le rôle du potassium comme agent promoteur des
catalyseurs Ni/Al2O3. Ils ont conclu que cette addition améliore nettement l’activité et réduit
le dépôt de coke [138]. L’ensemble de ces travaux met en évidence le rôle important que joue la basicité de surface dans l’amélioration des performances catalytiques.
IV-4- Cinétique et schéma réactionnel du reformage
Bien que la réaction de reformage du méthane au dioxyde de carbone ait été largement étudiée, sa cinétique n’est pas encore clairement établie. Les étapes élémentaires du mécanisme restent mal définies. Wei et coll. ont montré que la vitesse de la réaction de reformage du méthane, est seulement limitée par l’étape d’activation de la liaison C-H. Elle
n’est pas affectée par la nature ou la concentration des co-réactifs (CO2 ou H2O) [139]. Le
tableau 7 présente quelques modèles cinétiques proposés dans la littérature.
Tableau 7 : Quelques modèles cinétiques de la réaction de reformage du méthane :
Modèle Catalyseurs Référence
r = k PCH4 (PCO2+PH2O). [1+24(PCO2+PH2O) +8PH2]-2 Cu/SiO2 [140]
r = k PCH4 . [1+a PH2O / PH2 + b PCO] -1
Ni/Al2O3 [141]
r = kR.kCO2 .kCH4 .PCO2 . PCH4 [1+ kCO2 PCO2 + kCH4 PCH4] -2 Rh/ Al2O3 [142] r = a PCH4.P 2 CO2 .(PCO2 + bP 2 CO2 + cPCH4) -2 Ni/CaO- Al2O3 [143] r = a PCH4.PCO2 /.[b.PCO.PH2(4-x)/2 + PCO2 (1+ cPCH4)] Ni/TiO2 Ni/MgO Ni/SiO2 [144]
r = k PCH4.PCO2 /.[1+kCH4.PCH4][1+ kCO2 .PCO2] Ni/CeO2-Al2O3 [145]
D’autres études ont permis de montrer que dans la réaction de reformage du méthane au dioxyde de carbone, celui ci devait d'abord se décomposer en hydrogène et en carbone à la surface du métal [146 ; 147]. Les différences apparaissent dans les étapes ultérieures destinées à débarrasser le métal du carbone déposé. Les métaux, comme le nickel, stockent l’oxygène issu du CO2 qui se recombine lentement avec le carbone déposé pour former du CO, suivant
le schéma réactionnel [146]:
CH4 C* + 2H2
CO2 CO + O*
O* + C* CO
En revanche, pour les métaux nobles comme le ruthénium, aucune accumulation d’oxygène n'a lieu. La réaction directe du gaz carbonique avec le carbone du méthane, devient alors plus rapide [148].
L’adsorption du méthane et sa dissociation en fragments CHx, ont fait l’objet de plusieurs
études menées sur des catalyseurs à base de nickel. La stabilité des fragments CHx semble être influencée par la température de réaction et la nature du support utilisé [149,150]. Dans le cas d’une surface riche en hydroxyles, les fragments CHx se transforment en CHxO avant de
se convertir en CO [151, 152].
Par ailleurs, en plus de la réaction principale, qui est le reformage du méthane au dioxyde de carbone (H2/CO<1), une réaction simultanée peut avoir lieu entre le CO2 et l’hydrogène
22
CO2 + H2 CO + H2O
Wei et coll. ont proposé un schéma des étapes élémentaires mises en jeu, dans ces réactions, sur des catalyseurs à base de nickel [139] (Fig. 7).
Figure 7 : Etapes élémentaires de reformage du méthane en présence d’une réaction
simultanée
V- CONCLUSION
Les catalyseurs au palladium supporté sont les matériaux les plus actifs dans la réaction de combustion du méthane. Les études menées sur ces catalyseurs ont été orientées vers la détermination de la nature des sites actifs, et leur évolution au cours des tests. Une attention particulière est accordée aux cycles rédox du palladium, car son état d’oxydation joue un rôle important dans les performances catalytiques. Actuellement, les recherches sont essentiellement focalisées sur la modification de l’alumine et l’élaboration de nouveaux supports moins sensibles au frittage et présentant des propriétés acido-basiques adéquates.
Les catalyseurs classiques des réactions de reformage du méthane au dioxyde de carbone, sont les métaux nobles (Ru, Rh, Ir, Pt et Pd) ou de transition (Ni, Co, Fe) déposés sur des supports oxydes (SiO2, Al2O3, ZrO2, TiO2, MgO...). Ceux à base de nickel présentent l’avantage d’être
actifs et moins coûteux.
La cinétique de la réaction de reformage du méthane n’est pas complètement établie. Il n'existe aucun mécanisme général de la réaction. Des travaux ont cependant montré que la
vitesse de la réaction est uniquement limitée par l’étape d’activation de la liaison C-H. Elle n’est pas affectée par la concentration de CO2. Les mécanismes déjà élaborés, passent par, soit
la décomposition du méthane en carbone à la surface du métal, soit par l’adsorption du méthane et sa dissociation en fragments CHx. Dans le cas d’une surface riche en hydroxyles,
ces derniers se transforment en CHxO avant de se convertir en CO. Notons que, pour des
rapports H2/CO < 1, une réaction simultanée entre le CO2 et l’hydrogène peut conduire à la
formation de CO et de H2O.
Les phosphates minéraux en général et ceux de structure apatitique en particulier constituent une importante alternative aux supports habituellement utilisés en catalyse. Ce point de vue se justifie par la richesse structurale de ces composés et leurs propriétés d’échanges d’ions. Ils figurent parmi les rares solides à posséder des propriétés basiques modelables.
Dans la suite de ce travail, on étudiera les propriétés physico-chimiques des catalyseurs à base de palladium et de nickel dispersés sur des apatites (hydroxyapatite et fluorapatite). On examinera également leurs performances catalytiques dans les réactions de combustion et de reformage du méthane.
24
Références bibliographiques
1- H. G. Danielmeyer, H. P. Weber, J. Quantum Electr, 8, 805 (1972) 2- P. Maestro, P. Dougier, L'actualité chimique, (june-july) 15 (1982)
3- E. Weisang, P. A. Engeihad, "Colloque international sur les phosphates minéraux" Société chimique de France, ed., Paris, 165 16-20 Mai (1968)
4- A. S. Posner, A. Perloff and A. F. Diorio, Acta Cristallogr. 11, 308 (1958) 5- C. B. Beevers, D. B. Mc Intyre, Miner. Mag., 27, 254 (1946)
6- K. Sudarsnan, P. E. Mackie, and R. A. Young, Mater. Res. Bull. 7, 1331 (1972) 7- M. I. Kay, R. A. Young and A. S. Posner, Nature 204, 1050 (1964)
8- H. Suda, M. Kakihana, and Yoshimura, J. Phys. Chem. 99, 6752 (1995) 9- J. C. Elliott, Nature, 230, 72 (1971)
10- J. C. Trombe, Contribution à l’étude de la décomposition et de la réactivité de certaines apatites, Thèse, Université Paul-Sabatier Toulouse, (1972)
11- E. D. Dzyuba, M.T. Sokolov, L. P. Valyukevich, Inorganic materials, 18, 89-92 (1982) 12- E. C. Moreno, M. Kresak, R. T. Zahradnik, Nature, 247, 64-65 (1974)
13- E. C. Moreno, M. Kresak, R.T. Zahradnik, Caries Research, 11 (1), 142-160 (1977) 14- V. M. Valyashko, L.N. Korgardo, I.L. Khodakpovskyi, Geokhimiya, 1, 26-36 (1968) 15- T. S. B. Nasaraju and D. E. Phebe, J. Mater. Sci. 31, 1 (1996)
16- J. Brenan, Chem. Geol. 110, 195 (1994)
17- Mc Connel, Applied Mineralogy, New York, 5 (1973) 18- B. O. Fowler, Inorg. Chem. 13, 194 (1974)
19- A. Nounah, Thèse de Doctorat, Toulouse (1992)
20- M. E. Fleet and Y. Pan, J. Solid state Chem, 111, 78 (1994) 21- M. E. Fleet and Y. Pan, Am. Miner 80, 329 (1995)
22- M. E. Fleet and Y. Pan, Am. Miner 82, 870 (1997)
23- J. M. Hughes, M. Cameron, and A. N. Mariano, Am. Miner. 76, 1165 (1991) 24- P. E. Mackie and R. A. Young, J. Appl. Crystallogr. 6, 26 (1973)
25- R. P. Gunawardane, R. A. Howie, and F. P. Glasser, Acta Crystallogr. Sect. B 38, 1564 (1982) 26- M. Slansky, Mémoire du BRGM, 114 (1980)
27- J. R. Lehr, G. H. Mc Clellan, J. P. Smith, A. W. Frazier, Caracterization of apatites in commercial phosphate rocks, Colloque international sur les phosphates minéraux solides, Toulouse, 29-44 (1967)
28- J. C. Elliott, R. A. Young, Nature, 214, 904 (1967)
29- J. C. Rendon-Angeles, K. Yanakisawa, N. Ishizawa, and S. Oishi, J. Solid State. Chemistry, 151, 65-72 (2000)
30- J. C. Rendon-Angeles, K. Yanakisawa, N. Ishizawa, and S. Oishi, Chem. Mater. 12, 2143-2150 (2000)
31- M. Mikou, A. Taitai, J. L. Lacout, Ann. Chim. Fr., 10, 645 (1985) 32- F. M. Riveira et al, J. Sol. State. Chem. 221, 291-300 (2000)
33- E. Hayek, H. Newesely, Pentacalcium monohydroxyorthophosphate-hydroxyapatite. Inorg. Syntheses, 7, 63-65 (1963)
34- J. Arends, J. Christoffersen, M. R. Christoffersen, H. Eckert, O. Fowler, J. C. Heughebaert, G. H. Nancollas, J. P. Yesinowski, S. J. Zawacki, a calcium hydroxyapatite precipitated from aqueous solution-an international multimethod analysis. J.Crystal Growth, 84, 515-532 (1987)
35- A. Osaka, Y. Miura, K. Takeuchi, M. Asada et K. Takahashi, « Calcium apatite prepared from calcium hydroxyde and orthophosphoric acid », J.Mater. Sci.Mat.Med., 2, 51-55 (1991)
36- R. Wallaeys, Ann. Chim. 7, 808-848 (1952)
37- D. M. Liu, T. Troczynski, W. J. Tseng, Biomaterials 22, 1721-1730 (2001) 38- L. Sanchez, journal of non-crystalline solides, 145, 11-19 (1992)
39- Y. H. Chin, and D. Resasco, « Catalysis-Specialists Periodical Reports », Royal Society of Chemistry, Cambridge, 14, 1 (1999)
40- J. M. Beer, Progress in Energy and Combustion Science 26, 301-327 (2000) 41- O. Deutschmann, F. Behrendt, J. Warnatz, catal. Today. 46, 155-163 (1998)
42- Y. Tan, P. Dagaut, M. Cathonnet, J. C. Boettner, « Pyrolysis, oxidation and ignition of C1 and C2 hydrocarbons: Experiments and modelling », J. Chim. Phys, 72, 726-746 (1995)
43- J. R. Creighton, J. Phys. Chem. 81, 2520 (1977)
44- M. Niwa, K. Awano, Y. Murakami, Appl. Catal. 7, 317 (1983) 45- K. Muto, N. Katada, M. Niwa, Appl. Catal. A 134, 203 (1996) 46- L. Ma, D. L. Trimm, C. Jiang, Appl. Catal. A 138, 275 (1996)
47- J. C. van Giezen, F. R. van den Berg, J. L. Kleinen, A. J. van Dillen, J. W. Geus, Catal. Today 47, 287 (1999)
48- D. L. Trium, Appl. Catal. 7, 249 (1983)
49- J. Revel, «Modélisation de la combustion du méthane : validation d’un mécanisme détaillé, Construction d’un modèle cinétique global équivalent », Thèse de Doctorat de l’Université Pierre et Marie Curie, Orléans (1991)
50- T. P. Coffee, A. J. Kotlar and M. S. Miller, Combust. Flame, 54, 155-169 (1983)
51- F. L. Dryer and I. Glassman, “High temperature oxidation of CO and CH4,” Fourteenth Symp. Int. on Combustion, The Combustion Institute. Pittsburg, pp. 987-1003 (1973)
52- W. P. Jones and R. P. Lindstedt, Combustion and Flame, 73, 233-249 (1988) 53- C. K. Westbrook and F. L. Dryer, Combust. Sci. and Tech. 27, 31-43 (1981) 54- R.Prasard, L.A. Kennedy, E. Ruckenstein, Catal. Rev. Sci. Eng. 26, 1 (1984)
26
55- L. D. Pfefferle, W.C. Pfefferle, Catal.Rev. Sci. Eng. 29, 219 (1987) 56- Z. R. Ismagilov, M. A. Kerzhenzev, Catal. Rev. Sci. Eng.32, 51 (1990)
57- M. F. M. Zwinkels, S.G. Jaras, P.G. Menon, T.A Griffin, Catal. Rev. Sci. Eng. 35, 319 (1993) 58- P. O. Thevenin, A. G. Etsson, H. M. J. Kusar, P. G. Menon, S. G. Jaras, Appl. Catal. A : Gen
212, 189-197 (2001)
59- S. Siacardi, V. Specchia and F. Ferrero, kinetics of combustion of methane with different catalysts. Chimica industriale, 3, 217 (1982)
60- P. Gélin, M. Primet, Appl. Catal B : Env 39, 1-37 (2002)
61- R. J. Farrauto, M. C. Hobson, T. Kennelly, E. M. Waterman, Appl. Catal. A 81, 227 (1992) 62- Nan Yi, Yong Cao, Yang Su, Wei-Lin Dai, He-Yong He and Kang-Nian Fan, J. Catal., 230, 249
(2005)
63- H. Wise and M. Quinlan, Elementary processes in the catalytic combustion of methane. Gas research Institute report, Chicago, II (1986)
64- D. L. Trim and C. LAM, the combustion of methane on platinium-alumina. I kinetics and mechanism. Chem. Eng. Sci., 35, 1405 (1980)
65- D. A. Hickman and L. D. Scmidt, science. 259, 343 (1993)
66- D. Dissanayake, M. P. Rosynek, K. C. C. Kharas and J. H. Lunsford, J. Catal. 132, 117 (1991) 67- A. Slagtern, and U. Olsbye, Appl.catal.A.Gen. 110, 99 (1994)
68- E. Ruckenstein, and H. Y. Wang, J.Catal. 177, 386 (1998)
69- Y. Yazwa, H; Yoshida, N. Takagi, S. Komai, A. Satsuma and T. Hattori, J. Catal. 187, 15 (1999) 70- C. F. Cullis, B. M. Willatt, J. Catal. 86, 187 (1984)
71- C. A. Muller, M. Maciejewski, R. A. Koeppel, and A. Baiker, J. Catal. 166, 36 (1997) 72- M. Lyubovsky, L. Pfefferle, A. Datye, J. Bravo and T. Nelson, J. Catal. 187, 275 (1999) 73- D. Ciuparu, F. Bozon-Verduraz and L. Pfefferle, J. Phys. Chem. B 106, 3434-3442 (2002) 74- K. Eguchi and H. Arai, Applied catalysis A: 222, 359 (2001)
75- H. Widjaja, K. Seikizawa, K. Eguchi and H. Arai, Catalysis Today, 35, 197 (1997) 76- R. Spinicci and A. Tofanari, Appl. Catal A: 227, 159 (2002)
77- R. F. Hicks, H. M. L. Young and R. G. Lee, J. Catal. 122, 280 (1990) 78- P. Briot and M.Primet, Appl. Catal. 68, 308 (1990)
79- M. Lyubovsky and L. Pfefferle, Catalysis Today 47, 29 (1999) 80- R. Burch, F. J. Urbano, Appl. Catal. A 124, 121 (1995) 81- R. Burch, Catal. Today 35, 27 (1997)
82- J. N. Carstens, S. C. Su, A. T. Bell, J. Catal. 176, 136 (1998)
83- T. V. Choudhary, S. Banerjee, V. R. Choudhary, Cat. Communications, 6, 97-100 (2005) 84- K. Fujimoto, F. H. Ribeiro, M. Avalos-Borja and E. Iglesia, J. Catal. 179, 431 (1998) 85- R. J. Farrauto, J. K. Lampert, M. C. Hobson, E. M. Waterman, Appl. Catal. B 6, 263 (1995)
86- D. Ciuparu, L. Pfefferle, appl. Catal. A : Gen. 218, 197-209 (2001) 87- Y. Deng and T. G. Nevell, J. Mol. Catal. 142, 51 (1999)
88- T. R. Baldwin and R. Burch, Appl. Catal. 66, 337 (1990) 89- J. A. S. Bett and W. K. Hall, J. Catal., 10, 105 (1968) 90- C. L. Kibby and W. K. Hall, J. Catal., 29, 144 (1973) 91- J. B. Moffat, Catal. Rev. -Sci. Eng. 18, 199 (1978)
92- J. B. Moffat, M. Grayson and G. J. Griffiths, Topics in phosphorus Chemistry, Wiley Interscience, New York. 10, 285 (1980)
93- Y. Matsumura and J. B. Moffat, J. Catal. 148, 323-333 (1994) 94- S. Sugiyama et al, J. Sol. Stat. Chem. 135, 86-95 (1998) 95- T. Ohno and J. B. Moffat, catal. Lett. 9, 23-24 (1991)
96- M. Prigent. Les piles à combustible; état du développement et des recherches en cours, Editions Technip, 1997
97- A. SCHWARZENEGGER "Hydrogen fueling stations every 20 miles", Election Speech of September 21, Fox News, Califórnia (2003)
98- A. T. Ascroft, A.K. Cheetman, M. L. Green, P. D. F. Vernon, Nature 352, 225 (1991) 99- J. Zhu, D. Zhang, K. D. King, Fuel 80, 899-905 (2001)
100- J. R. Rostrup-Nielsen, J. Catal, 85, 31 (1984)
101- H. C. Dibbern, P. Olesen, J. R. Rostrup-Nielsen, P. B. Tottrup and N. R. Udengaard, Hydrocarbon Process, 65, 3 (1986)
102- N. R. Udengaard, J. –H. B. Hansen and D. C. Hanson, Oil Gas J, 90, 62 (1992) 103- T. S. Pugsley, S. Malcus, Ind. Engng. Chem. Res 36, 4567-4571 (1997) 104- A. M. Adris, et al, Can. J. Chem. Engng., 74, 177-186 (1996)
105- N. Dave, G. A. Folds, Ind. Engng. Chem. Res, 34, 1037-1043 (1995)
106- G. Q. Lu, S. Wang, J. Dev. Chem. Engng. Miner. Process 7(5/6), 443-462 (1999) 107- J. H. Edwards, Catal. Today 23, 29 (1995)
108- V. C. H. Kroll, H. M. Swaan, C. Miradotos, J. Catal. 161, 409 (1996)
109- D. Halliche, R. Bouarab, O. Cherifi, M. M. Bettahar, Stud. Surf. Sci. Catal. 119, 705 (1998) 110- A. Guerrero-Ruiz, A. Sepulveda-Escribano, I. Rodriguez-Ramos, Catal. Today. 21, 545 (1994) 111- J. N. Armor, Appl. Catal A: Gen. 176, 159 (1999)
112- P. D. F. Vernon, M. L. Green, A. K. Cheethman, A. T. Ashcroft, Catal. Lett. 6 (2), 181 (1990) 113- A. M. O’Connor, J. R. H. Ross, Catal. Today. 46, 203 (1998)
114- M. A. Ermakova, D. Yu. Ermakov, G. Kuvshinov, Appl. Catal. A : General., 201, 61-70 (2000) 115- Katsoumi Takehira, Tetsuya Shishido, Peng Wang, Tokuhisa KKosaka, and Ken Takaki, J.
Catal., 221, 43-54 (2004)
272-28
275 (2004)
117- M. C. J. Bradford and M. A. Vannice, Appl. Catal. A 142, 73 (1996)
118- D. Dissanayake, M. P. Rosynek, K. C. C. Kharas and J. H. Lunsford, J. Catal. 132, 117 (1991) 119- J. N. Armor, Appl. Catal. A 176, 159 (1999)
120- M. A. Pena, J. P. Gomez, J. L. G. Fierro, Appl. Catal. A 144, 7 (1996) 121- V. R. Choudhary, A. M. Rajput, B. Prabhaker, Catal. Lett. 32, 391 (1995) 122- V. R. Choudhary, A. S. Mamman, Appl. Energy 66, 161 (2000)
123- K. Elkabouss, M. Kacimi, S. Amar, F. Bozon-Verduraz and M. Ziyad, J. Catal. 226, 16-24 (2004)
124- J. El-Edrissi, M. Kacimi,M. Loukah, M. Ziyad, J. Chim. Phys, 94, 1984 (1997) 125- M. C. J. Bradford, M. A. Vannice, Catal. Rev. Sci. Eng. 41, 1 (1999)
126- J. R. Rostrup-Nielsen, in : J. R. Anderson, M. Boudart (Eds), Catalysis, Science and Technology, Springer, Berlin, 5 (1999), 1 (1999)
127- J. H. Jun, T. Lee, T. H. Lim, S. Nam, S. Hong, K. J. Yoon, J. Catal. 221, 178-190 (2004)
128- S. J. Lee, J. H. Jun, S. Lee, K. J. Yoon, T. H. Lim, S. Nam, S. Hong, Appl. Catal. A :Gen. 230, 61-71 (2002)
129- A. M. Gadallah and M. E. Sommer, J. Am. Ceram. Soc, 72, 683 (1989) 130- A. M. Gadallah and M. E. Sommer, Chem. Eng. Sci, 44, 2825 (1989)
131- K. S. M. Bhatta and G. M. Bixon, Ind. Eng. Chem. Prod. Res. Dev. 8, 324 (1969)
132- T. Horiuchi, K. Sakuma, T. Fukui, Y. Kubo, T. Osaki and T. Mori, Appl. Catal. A: Gen. 144, 111 (1996)
133- R. Bouarab, O. Akdim, A. Auroux, O. Cherifi, C. Mirodatos., Appl. Catal. A : General 264, 161-168 (2004)
134- J. Nakamura, K. Aikawa, K. Sato, T. Uchijima, Catal. Lett. 25, 265 (1994)
135- H. W. Chen, C. Y. Wang, C. H. Yu, L. T. Tseng and P. H. Liao, Catal. Today 97, 173-180 (2004)
136- Q. Zhuang, Y. Qin, L. Chang, Appl. Catal. 70, 1 (1991)
137- K. B. Mok, J. R. H. Ross, R. M. Sambrook, Preparation of catalysts, Elseveier, Amsterdam, III , 291 (1983)
138- T. Osaki and T. Mori, J. Catal. 204, 89-97 (2001) 139- J. Wei and E. Iglesia, J. Catal. 224, 370-383 (2004)
140- W. K. Lewis, E. R. Gilliland and W. A. Reed, Ind. Eng. Chem 41, 1227 (1949) 141- I. M. Bordov and L. O Appel’baum, Kinet. Catal., 8, 326 (1967)
142- J. T. Richardson and S. A. Paripatyadar, Appl. Catal., 61, 293 (1990) 143- Z. L. Zhang and X. E. Verykios, Catal. Today, 21, 589 (1994)
145- S. Wang, G. Q. Lu, React. Eng. Pollut. Prev. 75 (2000)
146- V. C. H. Kroll, H. M. Swaan, S. Lacombe and C. J. Mirodatos, J. Catal. 167, 387 (1997) 147- Y. Schuurman and C. Mirodatos, Appl. Catal. 151 (1), 305 (1997)
148- K. Walter, O. V. Buyevskaya, D. Wolf and M. Baems, Catal. Lett. 29, 261 (1994)
149- S. T. Seyer, Q. Y. Yang, M. B. Lee and A. D. Johnson, Methane conversion, Elsevier, Amsterdam, 51 (1993)
150- T. Osaki, H. Masuda and T. Mori, Catal. Lett. 29, 33 (1994) 151- C. Li, W. Yan and Q. Xin, Catal. Lett. 24, 249 (1994) 152- F. Solymosi and J. Cserenyi, Catal. Today, 21, 561 (1994) 153- A. M. Gadalla and B. Bower, Chem. Eng. Sci., 43, 3049 (1988) 154- J. R. R. Nielsen and J. H. B. Hansen, J. Catal., 144, 38 (1993)
155- H. M. Swaan, V. C. H. Kroll, G. A. Martin and C. Mirodatos, Catal. Today, 21, 571 (1994) 156- H. D. Giesser, N. R. Hunter, A. N. Shigapov and V. Januati, Energy and Fuels, 8, 1123 (1994)


![Tableau 7 : Attributions des bandes des groupements OH dans le domaine du PIR [25]](https://thumb-eu.123doks.com/thumbv2/123doknet/2196272.12039/69.892.98.799.383.556/tableau-attributions-bandes-groupements-domaine-pir.webp)