© Benjamin Duchêne, 2019
Utilisation des technologies CRISPR/Cas9 pour le
développement d'approches thérapeutiques pour le
traitement de la dystrophie musculaire de Duchenne
Thèse
Benjamin Duchêne
Doctorat en médecine moléculaire
Philosophiæ doctor (Ph. D.)
Utilisation des technologies CRISPR/Cas9 pour le
développement d’approches thérapeutiques pour le
traitement de la dystrophie musculaire de Duchenne.
Thèse
Benjamin Duchêne
Sous la direction de :
iii
Résumé
La dystrophie musculaire de Duchenne, est une maladie qui résulte d’une mutation dans le gène codant pour la dystrophine. Cette mutation entraine l'absence de la protéine dystrophine dans les fibres musculaires et mène à une dégénérescence des différents muscles ce qui engendre une défaillance cardiorespiratoire suivie d’un décès prématuré.
La récente découverte du système CRISPR/Cas9 ouvre de nouvelles perspectives pour le développement d’un traitement curatif pour la DMD. A l’aide d’un ARNg, reconnaissant une séquence cible protospacer localisée à proximité d’un PAM (protospacer adjacent motif), l’endonucléase Cas9 génère une coupure double brin dans l’ADN. Il a été démontré que l’utilisation d’une paire d’ARNgs ciblant des introns permettait de générer de larges délétions et de restaurer un cadre de lecture propice à l’expression d’une dystrophine tronquée dans des cellules de patients DMD. Cependant, cette approche ne prend pas en considération la structure de la dystrophine qui résulte de cette délétion. Il a été suggéré que chez les patients atteints de la dystrophie musculaire de Becker, produisant une dystrophine tronquée mais fonctionnelle, la sévérité de la maladie serait reliée au type de délétion et à la structure de la dystrophine qui en résulte. Il semble donc pertinent de travailler au développement d’une approche qui prend aussi en considération la structure des répétitions de type spectrine. D’autre part, le système CRISPR/Cas9 envahit progressivement toutes les sphères des sciences de la vie et soulève par la même occasion des questions de sécurité pour les patients. En effet, la possibilité de mutations hors-cible ou d’une réponse immunitaire dirigée contre ces endonucléases pourrait freiner l’application clinique de ces outils. Ainsi nous avons envisagé différentes approches qui contribueraient à limiter des tels effets pouvant s’avérer néfastes pour les patients.
Nos résultats montrent qu’il est possible d’utiliser la Cas9 de S. aureus ainsi qu’une paire d’ARNgs ciblant des exons pour induire une délétion dans l’ADN génomique. Cette délétion permet la formation d’un exon hybride qui restaure non seulement le cadre de lecture du gène de la dystrophine [1], mais contribue aussi à la formation d’une répétition de type spectrine hybride correctement phasée. Lors de nos expérimentations, nous avons été capables d’induire la production de dystrophine in
iv souris dystrophique.
Ensuite, avec la technologie du Feldan Shuttle nous avons montré qu’il était possible d’induire l’édition du gène de la dystrophine (gène humain ou murin) en livrant directement des complexes ribonucléoprotéiques dans le muscle d’une souris dystrophique. Cette édition a permis d’induire l’expression de protéine dystrophine dans les fibres musculaires, mais cette approche reste pour le moment réduite à des applications localisées.
Enfin, nous avons démontré que l’inactivation de l’activité autocatalytique du ribozyme N79 serait une stratégie envisageable pour contrôler l’expression d’une endonucléase. Présentement, ce système n’a fait ses preuves que lors d’expérimentations in vitro, mais il ouvre la porte au développement de nouveaux moyens de contrôler pharmacologiquement l’édition du génome par le système CRISPR/Cas9.
Finalement, l’ensemble de ces travaux contribuent à une meilleure compréhension des défis à relever pour mettre au point un traitement curatif pour la dystrophie musculaire de Duchenne, de façon plus efficace et sécuritaire.
v
Abstract
Duchenne Muscular Dystrophy is one of the most severe genetic disease. It is caused by a mutation in the dystrophin gene. Such mutation is responsible for the absence of the dystrophin protein in the muscles thus leading to muscle wasting and to a premature death following cardiorespiratory failure.
The discovery of the CRISPR/Cas9 systems opened the path for the establishment of curative treatments for genetic diseases, such as DMD. A Cas9 endonuclease can generate a double strand break in the DNA at a targeted locus through a guide RNA that specifically recognize a DNA protospacer sequence located closed to a protospacer adjacent motif (PAM). Recent work published by others demonstrated that the use of a pair of sgRNAs targeting introns permitted to create a genomic deletion that restores the DMD gene reading frame thus leading to de novo synthesis of a truncated dystrophin protein. However, such deletion does not consider the resulting structure of the central part of the dystrophin. In Becker muscular dystrophic patients, a truncated dystrophin protein is synthesized but the severity of the disease could be related to the structure of this protein. Consequently, it seems relevant to develop a therapeutic approach that considers the structure of the spectrin-like repeat that forms the central rod-domain of the dystrophin protein. Furthermore, while CRISPR/Cas9 is on the rise it also raises safety issues for patients. Indeed, off-target mutations and immune response directed against such endonuclease can occur thus preventing the possibility of starting clinical trials. Consequently, there is an increasing need to develop safer approaches that may counter such undesirable effects.
Our results demonstrated the feasibility of inducing a large genomic deletion with the Cas9 from S. aureus with a pair of sgRNAs targeting exons. Such deletion allows the formation of a hybrid exon that could, in addition to restoring the expression of the dystrophin protein, restore the correct structure of the spectrin-like repeat in its central rod-domain. We have been able to demonstrate such dystrophin expression in
vitro and in vivo in four different DMD patient cell lines and in a dystrophic mouse
model, respectively.
Next, we envisioned the delivery of Cas9/sgRNA ribonucleoprotein complexes using the Feldan Shuttle technology. We provided proof-of-principle that such delivery permits the editing of the dystrophin gene in the TA of mouse models. Following the
vi
editing, dystrophin protein expression was restored in the treated muscles of a dystrophic mouse model. Since this approach remains restricted to in situ treatments, further development should be addressed to allow systemic delivery of Cas9/sgRNA. Finally, we provided evidence that the self-catalytic activity of the ribozyme N79 can be controlled using toyocamycin. Even if it only demonstrated its efficacy in vitro, this system opens the path to the development of a different tool for the pharmacological induction of endonuclease protein expression.
Finally, this work contributes to the improvement of our understanding for the establishment of a potent and safe therapy to find a cure for DMD.
vii
Table des matières
Résumé...iii
Abstract ... v
Listes des figures ...xi
Liste des tables/tableaux ...xiv
Liste des abréviations ...xv
Remerciements ...xvi
Avant-propos ...xx
INTRODUCTION ... 1
Chapitre 1 : Introduction – Généralités sur le muscle squelettique ... 1
1.1. Organisation du muscle squelettique... 1
1.2. La myogenèse ... 5
1.3. Régénération du tissu musculaire et cellules satellites... 6
Chapitre 2 : La dystrophie musculaire de Duchenne ... 8
2.1. Découverte de la dystrophie musculaire de Duchenne ... 8
2.2. Diagnostic de la DMD et signes cliniques ... 9
2.3. La dystrophine et les dystrophinopathies ... 11
2.3.1. La dystrophine : le gène et ses mutations ...11
2.3.2. La protéine dystrophine et protéines associées ...12
2.4. Modèles animaux dystrophiques ... 17
2.4.1. Modèles de souris dystrophiques ...17
2.4.2. Modèles de rats dystrophiques ...19
2.4.3. Modèles de chiens dystrophiques ...20
2.4.4. Modèles de porcs dystrophiques ...21
2.5. Médications et approches thérapeutiques actuelles ... 21
2.5.1. Les corticostéroïdes ...22
2.5.2. Le saut d’exon par l’utilisation d’oligonucléotides antisens...23
2.5.3. Thérapie cellulaire ...25
2.5.4. Thérapie génique par transfert de gène ...26
Chapitre 3 : Une brève histoire de l’édition génomique ; d’hier à CRISPR ...31
6.1. Les endonucléases programmables ... 31
3.1.1. Protéines de liaison à l’ADN fusionnées à Fok1 ...31
3.1.2. Le système CRISPR/Cas9. ...34
6.2. Méthode de livraison pour les endonucléases programmables ... 41
6.2.1. Méthodes physiques ...41
6.2.2. Les vecteurs viraux ...42
viii
6.3. Potentiel d’applications thérapeutiques. ... 49
6.3.1. Rôle des mécanismes de réparation de l’ADN dans l’édition génomique .49 6.3.2. Cibles thérapeutiques...51
Chapitre 4 : Projet de doctorat ...54
Problématique ... 54
Objectifs ... 55
RESULTATS ...56
Chapitre 5: CRISPR-induced deletion with SaCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. ...56
5.1. Résumé ... 57
5.2. Abstract ... 58
5.3. Introduction. ... 59
5.4. Results ... 61
5.4.1. Identification and activity assays of individual sgRNAs in 293T cells ...61
5.4.2. Test of sgRNA pairs in 293T cells ...62
5.4.3. Analysis of hybrid exons...62
5.4.4. 3D spectrin-like models resulting from the formation of hybrid exons 3– 47/16–58 and 5–47/18–58. ...63
5.4.5. Formation of the hybrid exons 47-58 and restoration of dystrophin protein expression in DMD patient cells. ...64
5.4.6. Off-target analysis ...65
5.4.7. Systemic delivery of AAV9 partially restores dystrophin expression in the heart of del52hDMD/mdx mice. ...66
5.5. Discussion ... 67
5.6. Materials & Methods ... 70
5.6.1. Identifications of DNA targets and sgRNA cloning ...70
5.6.2. Expression vector and cloning of sgRNAs ...71
5.6.3. Transfection procedure ...71
5.6.4. Genomic DNA extraction and analysis. ...71
5.6.5. Assessment of the formation by sgRNAs of INDELs on on-target and off-target. 72 5.6.6. Assessment of the formation of hybrid exons ...72
5.6.7. Lentivirus production ...72
5.6.8. Myoblast transduction with recombinant lentiviral vector...73
5.6.9. Western blot analysis ...73
5.6.10. Production of AAV9 viral vectors ...74
5.6.11. In vivo experiments. ...74
ix 5.6.13. Molecular modelling ...75 5.6.14. HDMD mouse models ...75 5.6.15. Immunohistochemistry ...75 5.7. Acknowledgments ... 75 5.8. Author Contributions ... 76
Chapitre 6: Feldan shuttle-based delivery of Cas9/sgRNAs ribonucleoprotein edits the dystrophin gene. ...93
6.1. Résumé ... 94
6.2. Abstract ... 95
6.3. Introduction ... 96
6.4. Results ... 97
6.4.1. Analysis of the in vitro cleavage by the Cas9/sgRNA complex ...97
6.4.2. Surveyor test on the exon 54 of the DMD gene, following the transduction of the CRISPR / Cas9 system with the Feldan Shuttle in Hela cells. ...97
6.4.3. Delivery of the SpCas9: sgRNA complex in the hDMD mouse model. ...98
6.4.4. Screening of sgRNAs for the deletion of the exon 23 of the mdx gene. ....99
6.4.5. Intramuscular injection of RNP-Feldan Shuttle complexes restores dystrophin expression. ...100
6.4. Discussion ... 102
6.5. Materials and methods ... 104
6.5.1. SgRNA in vitro transcription ...104
6.5.2. Cell culture ...104
6.5.3. In vitro activity analysis of crRNA:tracrRNA:SpCas9 ribonucleic complex 104 6.5.4. Protein delivery in HeLa cells ...105
6.5.5. Identification of sgRNAs targeting the mdx gene and their cloning ...105
6.5.6. In vivo protein delivery...106
6.5.7. Genomic DNA extraction and purification ...107
6.5.8. Hybrid exon analysis by PCR ...107
6.5.9. Cloning and sequencing for the hybrid exon 50-54 ...107
6.5.10. Immunohistochemistry ...108
6.1.1. Animal models ...108
Chapitre 7 : Exploration d’approches pour limiter les mutations hors-cible des endonucléases SpCas9 et SaCas9. ...118
7.1. Objectifs ... 118
7.1. Matériel et méthode ... 120
7.1.1. Culture cellulaire ...120
x
7.1.3. Analyses par Western-Blot ...121
7.1.4. Modèles animaux ...122
7.1.5. Vecteurs viraux ...122
7.2. Résultats ... 122
7.2.1. Utilisation de la eSaCas9 pour réduire la fréquence des mutations hors-cible ...122
7.2.2. Utilisation d’un SCR pour le contrôle de l’expression de SaCas9 ou de SpCas9 ...125
7.3. Discussion ... 132
CONCLUSION ...135
Discussion, défis à relever et conclusion ...135
Bilan des résultats obtenus ... 135
La pertinence de développer l’édition génomique par le système CRISPR/Cas9 pour le traitement de la DMD. ...139
De l’importance du design de la dystrophine : ...141
Nouvelles générations d’outils pour l’édition du génome ... 144
Niveau de dystrophine requis ... 146
Défis à relever ou comment limiter les mutations hors-cibles et une réponse immune ... 147
Questions d’éthique ... 148
Perspectives ... 150
Références ...152
Annexe 1: From gRNA Identification to the Restoration of Dystrophin Expression: A Dystrophin Gene Correction Strategy for Duchenne Muscular Dystrophy Mutations Using the CRISPR-Induced Deletion Method...171
xi
Listes des figures
Figure 1. Structure du muscle squelettique ... 2
Figure 2. Organisation des sarcomères, unités structurantes de myofibrilles ... 4
Figure 3. Réparation du tissu musculaire ... 7
Figure 4. Premières observations de patients DMD ... 9
Figure 5. Manoeuvre de Gowers ... 10
Figure 6. Représentation schématique de la dystrophine et des complexes de glycoprotéines associées ... 13
Figure 7. Conséquence de la délétion d'exon(s) sur la structure de la dystrophine .. 16
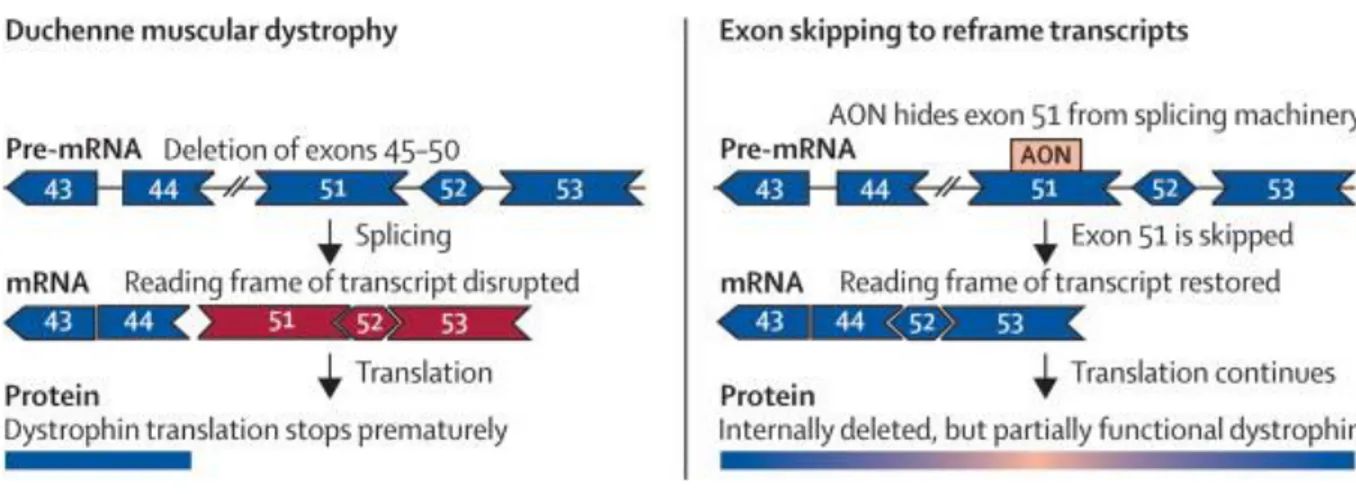
Figure 8. Saut d'exon à l'aide d'AON pour restaurer le cadre de lecture dans l'ARNm de la dystrophine ... 23
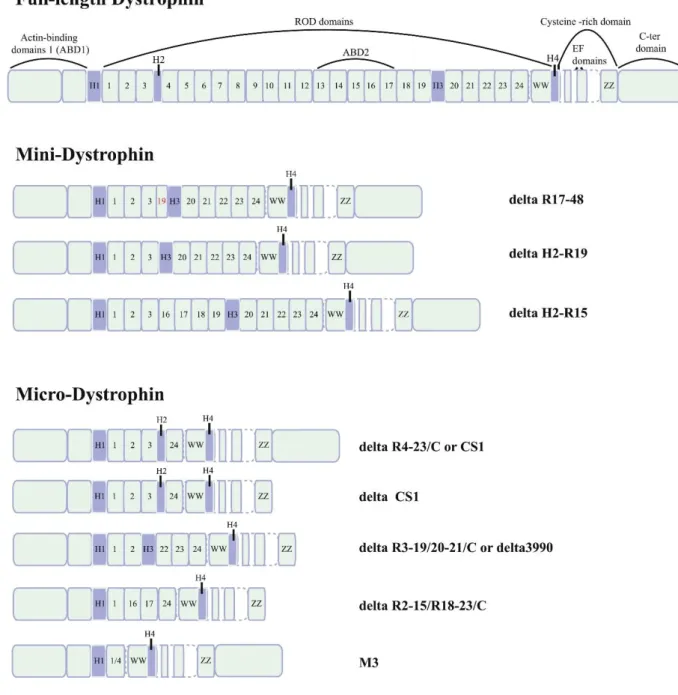
Figure 9. Exemples de mini- et micro-dystrophines ... 28
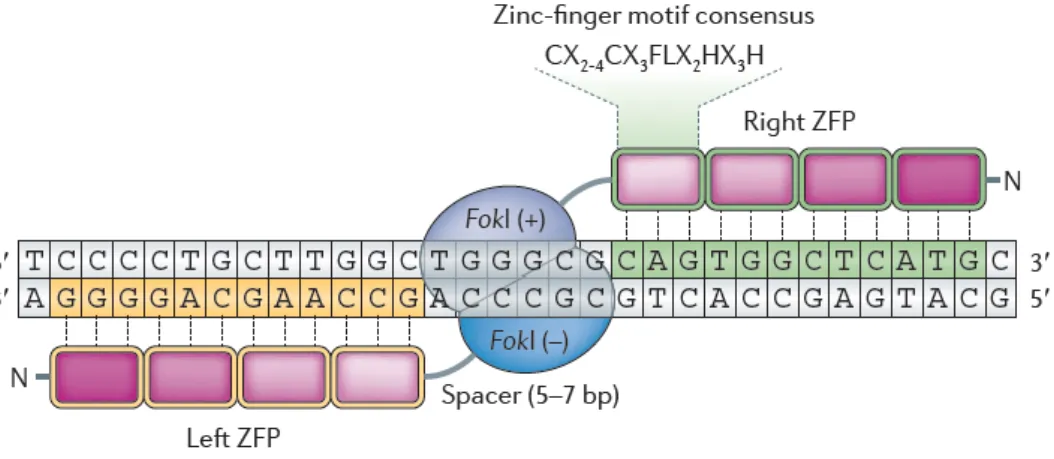
Figure 10. Structure des nucléases à doigt de zinc (ZFN) ... 32
Figure 11. Structure des nucléases effectrices de type activateur de transcription (TALENs) ... 34
Figure 12. Mécanisme de défense immunitaire avec le système CRISPR de type II 36 Figure 13. Représentation schématique de CRISPR/SpCas9 ... 37
Figure 14. Représentation schématique du CRISPR/SaCas9 ... 38
Figure 15. Plusieurs sérotypes de AAV permettent de cibler précisément différents types de tissus. ... 45
Figure 16. Principe de fonctionnement du Feldan Shuttle pour la livraison de protéines et/ou d’acides nucléiques. ... 48
Figure 17. Voies de réparation de l’ADN induite par une coupure double brin. ... 49
Figure 18. Generation of several hybrid exons by genomic edition using pairs of sgRNAs ... 77
Figure 19. Structural representations of SLR18 and two hybrid SLRs ... 78
Figure 20. Lentiviral delivery of the SaCas9 gene and pairs of sgRNAs restored the expression of the dystrophin in DMD patient cells ... 80
Figure 21. Restoration of dystrophin expression in edited myotubes. ... 81
Figure 22. Formation of the hybrid exons 3-47/16-58 and 5-47/18-58 restored the expression of dystrophin protein in vivo in cardiomyocytes of the del52hDMD/mdx mouse model ... 83
Figure 23. Analysis of the activity of the ribonucleoprotein complex CRISPR/SpCas9 in vitro and in vivo ... 110
Figure 24. Intramuscular injection of RNP with Feldan Shuttle B forms a hybrid exon 50-54. (A) Expected and obtained sequence of the hybrid exon 50-54 formed by genome editing using two RNP complexes sgRNA:SpCas9 in hDMD mouse model. (B) Sequence alignment of the expected sequence (Query) of the hybrid exon 50-545 and the sequence experimentally obtained (Sbjct). Analysis show a perfect homology between the two sequences, thus demonstrating the effective formation of the hybrid exon following the transduction of the two RNP complexes targeting exons 50 and 54 of the DMD gene. ... 111
Figure 25. Screening of the activity of sgRNAs in C2C12 cells ... 112
Figure 26. Single intramuscular injection of Cas9/sgRNAs/FS restores dystrophin expression ... 115
Figure 27. Analyse de l'activité de la eSaCas9 pour l'édition du gène DMD humain ... 124
Figure 28. Test d'inhibition de l'activité du ribozyme N79 pour le contrôle de l'expression de SpCas9-T2A-GFP ... 127 Figure 29. L'inhibition du ribozyme N79 permet le contrôle de l'expression de SpCas9
xii
et de SaCas9 et de leur activité d'édition génomique ... 129 Figure 30. Évaluation de la toxicité de la toyocamycine in vivo ... 131
xiii
Figures supplémentaires
Figure S1. In silico identification of sgRNAs for the SaCas9 suitable for the formation hybrid spectrin-like repeats normally phased. ... 87 Figure S2. TIDE profiles of INDEL formation generated by individual sgRNA tested in 293T. ... 89 Figure S3. Nucleotide sequences of the hybrid exons generated by SaCas9 and a pair of sgRNAs ... 90 Figure S4. Analysis of the off-targets in the IL-17A and TRIM67 genes. ... 91 Figure S5. Poison primer-based PCR for the detection of the deletion of exon 23. 116 Figure S6. La livraison de AAV9 codant l’expression de la protéine SaCas9 induit une réponse humorale dans la souris del52hDMD/mdx. ... 119
xiv
Liste des tables/tableaux
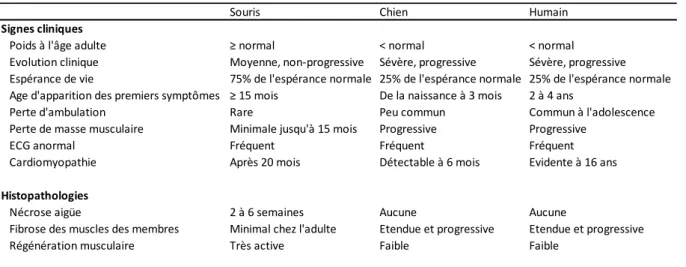
Tableau 1. Comparaison de la sévérité de la dystrophie entre les souris, les chiens et
les patients DMD ... 21
Tableau 2. Essais cliniques en cours pour le traitement de la DMD ... 22
Table 3. List of sgRNA Target Sites in Exons 46 to 58 ... 92
Table 4. Analysis of Hybrid Exon Amplicons by TIDE ... 92
Table 5. Primers used for the amplification of the in vitro transcription templates .. 117
xv
Liste des abréviations
AAV : adeno-associated virus, virus adéno-associé
ABD : Actin Binding Domaine, domaine de liaison à l’actine ADN : acide désoxyribonucléique
ADP : Adénosine biphosphate ATP : Adénosine triphosphate ARN : acide ribonucléique
BMD : Becker Muscular Dystrophy, dystrophie musculaire de Becker Cas9 : CRISPR-associated protein
CK : créatine kinase CMV : cytomegalovirus
CRISPR : Clustered Regularly Interspaced Short Palindromic Repeat DMEM : Dulbecco′s Modified Eagle′s Medium
DMD : Dystrophie Musculaire de Duchenne
Dmd : gène de la dystrophine humain
FS : Feldan Shuttle
gRNA (ARNg): guide RNA (ARN guide) IL : interleukin
kDa: kilo Dalton LB : Luria-Bertani
mdx : gène de la dystrophine de souris MHC : Myosin Heavy Chain
MyoD : Myogenic differentiation factor NaCl : Chlorure de sodium
nNOS : neuronal nitric oxid synthase PAX3 : paired-box 3
PAX7: paired-box 7 pb : paire de base
PBS : Phosphate-buffered saline
PCR : polymerisation chain reaction, réaction de polymérisation en chaine Pi : Phosphate inorganique
RNP : ribonucleoprotein, ribonucléoprotéine SaCas9 : Cas9 de Staphylococcus aureus
SDS-PAGE : sodium dodecyl sulfate polyacrylamide gel electrophoresis SpCas9 : Cas9 de Streptococcus pyogenes
SLR : spectrin-like repeat, repetition de type spectrine TNFα : Tumor necrosis factor alpha
xvi
Remerciements
Tout d’abord je tiens à remercier le Dr Jacques P. Tremblay pour m’avoir accepté dans son équipe de recherche. Initialement je devais travailler sur un projet de recherche rassemblant l’édition génomique et la maladie d’Alzheimer. Finalement il semble que j’ai oublié le projet en question... Le hasard a donc fait que j’ai travaillé sur une pathologie dont mon nom de famille en est l’homonyme. Je lui suis profondément reconnaissant de toujours avoir laissé sa porte ouverte et d’avoir été à l’écoute de mes idées parfois un peu trop farfelue, il faut l’admettre. Ces années dans son équipe auront été particulièrement enrichissantes et m’auront permis de m’épanouir aussi bien au laboratoire, qu’en dehors dans mes activités extra-académiques. Tout simplement, un grand merci.
Je tiens aussi à remercier les membres de l’équipe du Dr Gosselin, Manon, Alexandre, Carine et Anne-Julie. Je n’oublierai pas que vous avez été mes premiers collègues québécois.
La vie au laboratoire n’aurait pas été la même sans eux et c’est pourquoi je tiens à remercier l’ensemble des membres de l’équipe du Dr. Tremblay que j’ai pu côtoyer durant ces quelques années ;
Pierre Chapdelaine, passionné de pêche à la truite, mais pas que. Un grand passionné de sciences aussi, dévoué à son travail et dont la retraite est bien méritée.
Dominique, ta détermination et ton sens de l’organisation m’auront toujours impressionné. Je te souhaite une belle carrière chez CRISPR Therapeutics.
Cathy, je souhaite sincèrement qu’un jour tu trouves une solution à ta guerre de voisinage avec les écureuils. J’espère aussi avoir la chance de rencontrer Jean-Chri de mes propres yeux, de préférence pas de dos.
Jouelle, le coach. Je ne pas encore perdu l’espoir qu’un jour on fera un 5 à 7. Merci pour ton aide précieuse au laboratoire. J’ai appris bien plus que tu le ne penses à tes cotés.
Khadija, avec qui il aura toujours été un plaisir de débattre de géopolitique. J’espère que dans quelques années nous aurons l’opportunité de travailler à nouveau ensemble. Mabrouk.
xvii
Chantale, malgré ton départ nous avons réussi, jusqu’à présent, à survivre dans le labo et aucun n’accident grave n’est à déplorer. Cela pourrait presque relever du miracle... !
Nathalie, l’aventurière, même si on se connait depuis moins longtemps j’apprécie particulièrement nos conversations sur la science et le reste aussi.
Mes potos de labo, partis trop tôt : Will, Justin Bridou, Eric Jalpin, Simon Boulette, Arnaud et son acolyte le Paralysieux. A jamais nous sommes liés par le tragique évènement de la tomate séchée Qui sait ce que la jarre à souhait exaucera de tous ces vœux que nous lui avons confiés ?
Sans oublier tous mes autres collègues pour leur bonne humeur quotidienne. Kekeb le roi du Western-Blot. Jean-Paul et sa technique dite de la « Jean-Paulette » qui aura révolutionné le monde de la transfection, la 2è révolution en sciences de la vie après la découverte du système CRISPR. Anteneh, Antoine et aussi Antoine (parce qu’on ne partage pas seulement un bureau, mais aussi notre passion pour les mélanges de noix), Daniel et Daniel (chacun de vous saura se reconnaitre), Pouiré, Marlyne, Vanessa, Noelia.
Parce que faire un doctorat, ce n’est pas une sinécure, merci à mes amis d’être là : Mes coloc’ de cette exceptionnelle, désormais célèbre, Cagette Espagnole : Camille, Sam, Marie, Patrick et aussi Krokie, toujours prêts pour l’apéro et prendre juste « un » verre quand l’opportunité se présente. Et aussi ceux qui ont pu nous y rendre visite ou pas : Marine & Vincent, Emilie, Vincent, Edwige, Maëlle & Jérem’, Mérilie, Anne-Elodie (et Sofia), Marie & Valentin, Julien, Mathieu, Bertrand, David & Mélissa.
Que de bons moments passés avec vous tous en chalets, randonnées, barathon, à l’escalade, au ski, à manger des galettes saucisses, barbecues et autres.
Une pensé pour mes ami(e)s de France : mes ami(e)s du lycée : Gaël & Audrey ( et Raphaël) - Clémentine & Simon – Marine – Marion - Pierre – Yves - Romain, mes potes de prépa : Adri, Quentin, Tudu, Mr Roncier, Baptiste, les puercos et autres ami(e)s de Grenoble : Max, Pauline, Rémi, Julie, Xav, Julio, sans oublier ceux de la fac de Rennes : Lisa, Laurent, Gé, Edouardo, Sophie.
« Eventuellement » (dans le sens acadien), je voudrais remercier ma Cathou pour tous les bons moments passés ensemble, pour ton soutien inconditionnel durant ces dernières années et pour m’avoir enduré durant ma rédaction. « Tu es une belle
xviii
personne ». J’ai hâte de voir à quoi ressemblera notre vie avec des poules hollandaises, une chèvre et surtout Marius.
Finalement, je souhaiterai remercier toute ma famille. Particulièrement mes parents pour m’avoir accompagné pendant toute ma vie et soutenus durant mes études. Sans vous tout cela n’aurait pas été possible et je ne saurais suffisamment vous en remercier.
xix
« Soyez curieux en toute circonstance,
il y a toujours quelque chose à faire et à réussir, n’abandonnez jamais. »
xx
Avant-propos
Le chapitre 5 est un article intitulé « CRISPR-Induced Deletion with SaCas9 Restores Dystrophin Expression in Dystrophic Models In Vitro and In Vivo » qui a été publié dans la revue Molecular Therapy en 2018. Des modifications mineures ont été apportées pour son insertion dans cette thèse. En particulier, les numéros des figures ont été ajustés afin de suivre la succession de toutes les figures présentes dans ce manuscrit de thèse. Je suis le premier auteur de cette étude. A ce titre, j’ai élaboré et réalisé la plupart des expériences. J’ai rédigé le manuscrit, et celui-ci a été relu et corrigé par le Dr. Jacques P. Tremblay et Dominique Ouellet. Khadija Chérif m’a assisté dans la réalisation de certains Western-Blot. Antoine Guyon m’a initié aux joies de la bio-informatique pour les analyses des fichiers de deep-sequencing. Xavier Barbeau et Patrick Lague ont réalisé la modélisation de la structure des répétitions de type spectrine.
Le chapitre 6 est un article qui sera prochainement soumis dans la revue Molecular Therapy, et dont le titre sera « Feldan shuttle-based delivery of Cas9/sgRNAs ribonucleoprotein edits the dystrophin gene. ». Je suis le premier auteur de cette étude. Daniel Agudelo et Jean-Paul Iyombe-Engembe ont réalisé les expériences portant sur la formation de l’exon hybride 50-54. Pour ma part, j’ai réalisé et élaboré toutes les autres expériences, et rédigé le manuscrit. Le Dr. Jacques P. Tremblay a relu puis corrigé le manuscrit.
L’annexe 1 est un chapitre livre de méthodologie qui a été publié dans Methods in Molecular Biology - Duchenne Muscular Dystrophy. Il présente de façon détaillée certains aspects techniques utilisés au cours des différentes expériences de ce projet de recherche. Je sus le premier auteur de ce chapitre. A ce titre, j’ai rédigé l’intégralité de son contenu. Le Dr Dominique Ouellet et le Dr Jacques P. Tremblay ont relu et corrigé le manuscrit.
1
INTRODUCTION
Chapitre 1 : Introduction
– Généralités sur le muscle
squelettique
Le corps humain comprend plus de 600 muscles qui se décomposent en trois grands groupes : les muscles lisses viscéraux, les muscles striés cardiaques, et majoritairement les muscles striés squelettiques. Au total, l’ensemble des tissus musculaires représente environ 40% de notre masse corporelle. Plus particulièrement, les muscles squelettiques nous permettent de maintenir notre posture, d’effectuer de simples mouvements, de nous déplacer, etc... Étant donné la multitude de tâches qu’ils nous permettent d’accomplir, si l’intégrité de ces muscles est affectée, c’est notre qualité de vie et notre autonomie quotidienne qui s’en retrouve menacée
1.1. Organisation du muscle squelettique
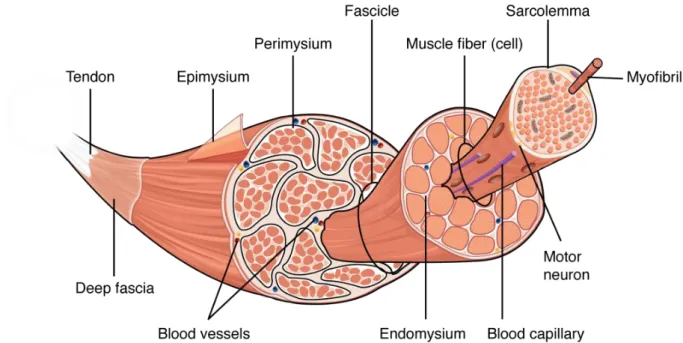
Les muscles squelettiques permettent à l’organisme de générer une force qui est responsable des mouvements. A leurs extrémités, ils sont ancrés à des os par l’intermédiaire de tendons qui sont constitués de faisceaux de fibres de collagène. A leur surface, les muscles sont entièrement recouverts par du tissu conjonctif dense appelé épimysium. Sous cette structure, des faisceaux de 10 à 100 fibres musculaires sont enveloppés par le périmysium. Au sein de ces faisceaux, chaque fibre musculaire est entourée par l’endomysium (Figure 1).
Les fibres musculaires, aussi appelées myotubes, sont les unités cellulaires essentielles des muscles qui possèdent des propriétés contractiles. Dans le corps humain, ces unités sont les seules à être multinucléés et peuvent contenir 50 à 70 noyaux par millimètre. Ces fibres peuvent atteindre jusqu’à 30 cm de longueur avec un diamètre qui varie de 10 à 100 µm. Elles sont issues de la fusion de plusieurs cellules musculaires, appelées myoblastes, qui dérivent de la division cellulaire asymétrique de cellules satellites qui constituent les cellules souches musculaires [2, 3]. Dans les fibres musculaires normales, les noyaux se localisent en périphérie, directement sous le sarcolemme, leur membrane plasmique. Cette localisation particulière libère donc le centre du sarcoplasme (cytoplasme des fibres musculaires), pour laisser place à l’organisation des myofibrilles qui permettent notamment la
2 contraction.
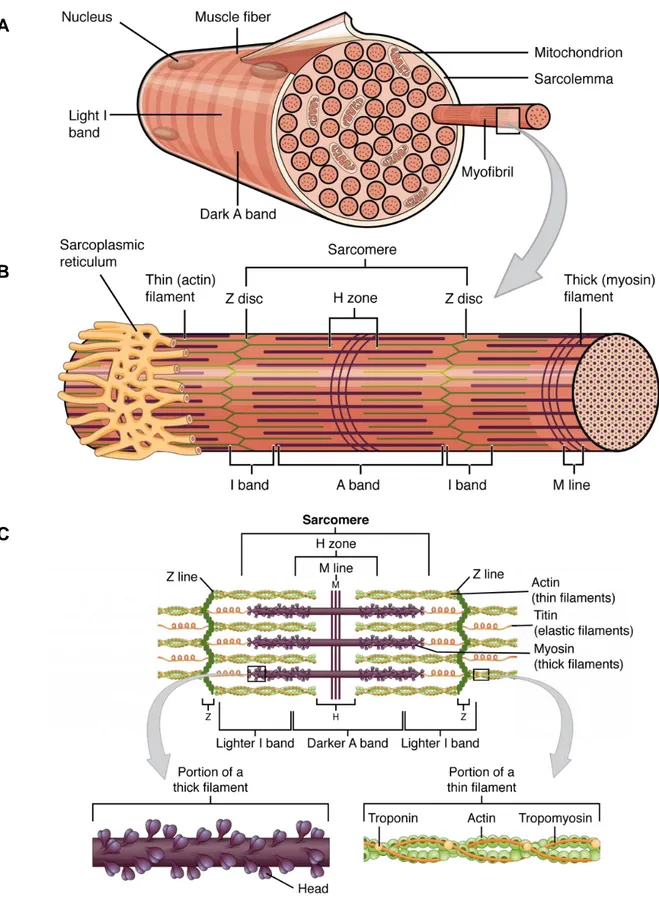
Les myofibrilles sont essentiellement constituées d’actine et de myosine. Des molécules d’actine polymérisées interagissent avec la tropomyosine et la troponine pour former de fins filaments qui s’étendent sur toute la longueur du sarcomère, l’unité fonctionnelle contractile des myofibrilles. Dans ces structures, des dimères de molécules d’actine forment la base d’un filament avec lequel interagissent des molécules de tropomyosine ce qui forme un complexe interagissant avec des molécules de troponine. Ces filaments s’étirent depuis les disques Z jusqu’aux stries M, respectivement placés aux extrémités et au centre du sarcomère.
Depuis les stries M, des filaments de myosine s’étirent vers les disques Z, parallèlement aux filaments d’actine avec lesquels les têtes de myosine vont interagir et permettre la fonction de contraction. Cette organisation laisse ainsi apparaitre des bandes I de faible densité optique, essentiellement formées par les filaments d’actines, et des bandes A de plus forte densité optique où interagissent filaments d’actine et de myosine, facilement observables en microscopie (Figure 2).
Figure 1. Structure du muscle squelettique
Le muscle squelettique est structuré par plusieurs niveaux de tissus conjonctifs : l’épimysium, le périmysium et l’endomysium. Dans chaque faisceau se trouve plusieurs fibres musculaires qui contiennent de nombreuses myofibrilles jouant un rôle fondamental dans la contraction musculaire. Image tirée de [4].
L’ensemble de cette structure est également soutenue par des molécules de titines qui permettent de stabiliser le sarcomère par leur propriété d’élasticité [5].
3
Dans les myofibrilles, le glissement des filaments d’actine par rapport aux filaments de myosine permet de raccourcir la longueur du sarcomère et donc un raccourcissement du muscle ce qui permet de réaliser une contraction musculaire. Durant cette contraction, les têtes de myosine qui sont fixées sur les filaments d’actine, hydrolysent de l’ATP en ADP + Pi. Cela entraine un changement d’orientation des têtes de myosine, ce qui fait glisser les filaments d’actine vers le centre sur sarcomère. Ainsi, cette réaction permet de transformer une énergie chimique en une force mécanique capable de réaliser un mouvement.
Bien que les muscles squelettiques partagent une organisation structurale commune, les fibres musculaires qui les composent présentent une certaine hétérogénéité qui permet de les classifier en trois grandes catégories distinctes : les fibres rouges de type I et les fibres blanches de type IIa et IIb [6].
Les fibres de type I, sont des fibres musculaires à contraction lente. Elles sont particulièrement bien vascularisées permettant ainsi un bon apport en oxygène qui favorise un taux de myoglobine élevé (d’où leur nom de fibres rouges), et un métabolisme oxydatif. Cela corrèle donc avec une quantité importante de mitochondries dont l’une des propriétés est la production de l’ATP par un métabolisme aérobique. Dans ces fibres, l’ATP est lentement hydrolysée ce qui est directement en lien avec leur vitesse de contraction. Par ailleurs, elles ont une grande capacité de résistance à la fatigue et soutiennent des efforts physiques prolongés. Ainsi, elles sont davantage présentes dans les muscles du maintien de la posture et sont aussi particulièrement sollicitées dans les muscles des membres inférieurs chez les sportifs d’endurance.
Les fibres de type Ila, sont des fibres à contraction rapide. Comme les fibres de types I, elles sont bien vascularisées avec une teneur élevée en myoglobine et mitochondries qui favorisent le métabolisme oxydatif. Cependant, dans les fibres de type IIa des contractions plus rapides sont favorisées par l’hydrolyse de l’ATP plus active mais ces fibres ne sont pas aussi résistantes que les fibres de type I.
Comme les fibres de type IIa, les fibres de type IIb sont à contractions rapides. Moins bien vascularisées que les autres, ces fibres pauvres en myoglobine et mitochondries produisent leur énergie par un métabolisme glycolytique anaérobique. De par une activité d’hydrolyse d’ATP supérieure à celle des autres fibres, elles peuvent se contracter plus rapidement et déployer une plus grande force bien qu’elles ne sont pas résistantes à la fatigue.
4
Figure 2. Organisation des sarcomères, unités structurantes de myofibrilles
(A) Chaque fibre musculaire est constituée de plusieurs myofibrilles distribuées les unes parallèles aux autres. (B) Ces myofibrilles sont formées par une multitude de sarcomères dont les filaments d’actine et de myosine sont les constituants principaux. (C) Les unités de sarcomère se caractérisent par l’alternance de bandes claires I et de bandes foncées A. Chaque unité est délimitée à ces extrémités par les lignes Z. Cette architecture confère ainsi un aspect strié qui est caractéristique du muscle squelettique. Image tirée de [4].
A
B
5
Basées sur ces trois grandes catégories de fibres, certaines équipes de recherche ont établi d’autres niveaux de classification de fibres musculaire. Ils prennent en considération les profils d’expression de MYH, le type d’isoforme de MYH, ou encore le profil d’un marquage histochimique de l’ATPase, mais ces catégories de seront pas discutées [7-9].
1.2. La myogenèse
La myogenèse est le processus par lequel le tissu musculaire est mis en place.
Cela débute durant le processus de la gastrulation où l’embryon se retrouve formé par trois feuillets embryonnaires : l’ectoderme, l’endoderme et le mésoderme. C’est à partir de ce dernier feuillet embryonnaire que les vaisseaux sanguins, les os, mais aussi et surtout les muscles vont se développer.
Les cellules du mésoderme paraxial, au travers des voies de signalisation de Wnt [10] et du FGF (fibroblast growth factor) [11], se différentient en somites où la voie de signalisation de l’acide rétinoïque est très active. Les somites se divisent en deux sous-ensembles : le sclérotome et le dermomyotome. Les somites qui composent le dermomyotome expriment particulièrement le facteur de transcription PAX3, un marqueur de précurseur des cellules myogéniques, impliqué dans la migration des cellules du dermomyotome vers le bourgeonnement des membres en cours de formation [12, 13]. L’expression de PAX7, combinée à celle de PAX3, joue un rôle prédominant dans l’entrée dans la voie de la différentiation des somites en cellules myogéniques qui ont des propriétés de prolifération [14]. Ces cellules myogéniques Pax3+/Pax7+, forment ainsi le myotome à partir duquel les premières fibres musculaires seront formées. Cependant, la voie de signalisation de Notch maintient ces cellules myogéniques dans un état de prolifération qui bloque leur différentiation terminale. Lorsque cette voie est inactivée, la répression de l’expression plusieurs facteurs de régulation myogénique (MRF) tels que Myf5 ou encore MyoD, est interrompue [15, 16]. Ces cellules qui expriment MyoD et Myf5, sont appelées myoblastes [17]. Les myoblastes, marquées par l’expression de Pax7, se multiplient et vont progressivement exprimer des MRFs tels que MyoG et Mrf4. L’expression de ces MRFs tardifs permet la différentiation des myoblastes en myocytes qui n’expriment alors plus Myf5 ni Pax7 (donc incapable de prolifération) mais synthétisent la créatine kinase musculaire [18] et surtout la chaine lourde de myosine (MHC), que l’on retrouve
6
dans la structure des myofibrilles. Ces myocytes ont la propriété unique de fusionner entre eux afin de former un myotube (ou fibre musculaire).
Il est important de noter que dans cette phase finale de développement du muscle, lorsque les myoblastes quittent le cycle de prolifération, un réservoir de cellules dîtes « satellites » exprimant Pax7 persistent et demeurent proches des fibres musculaires au niveau de la lame basale [3, 19]. Chez les souris, ces cellules sont généralement très abondantes dans les premières semaines de vie (~30% des cellules) mais leur nombre chute rapidement (~5%) [20]. Elles jouent ensuite un rôle essentiel dans le maintien de l’intégrité des muscles, qui sera évoqué dans la section suivante.
1.3. Régénération du tissu musculaire et cellules satellites
Dans des conditions physiologiques normales, le taux de renouvellement des noyaux des fibres musculaires est de l’ordre 1 à 2% par semaine [21]. Mais un effort physique intense (traumatisme mécanique), des agents myotoxiques, une ischémie, ou encore des myopathies, peuvent entrainer des lésions musculaires qui nécessitent de réparer le tissu endommagé. La régénération du muscle va alors se faire en deux phases successives ; une phase de nécrose/dégénérescence et d’inflammation suivie d’une phase de réparation du tissu [22].
À la suite de la lésion du sarcolemme, du Ca2+ extracellulaire va s’infiltrer dans les fibres musculaires et activer des protéases endogènes entrainant leur nécrose [23, 24]. Parallèlement à cette perméabilisation du sarcolemme, des composants intracellulaires vont diffuser à l’extérieur des fibres. C’est le cas de la créatine kinase (CK), dont la concentration sérique augmente, et qui est un indicateur de lésion musculaire mais aussi un marqueur dans le diagnostic de myopathies.
Une réponse inflammatoire est médiée par l’infiltration, à partir des vaisseaux sanguins, de neutrophiles, macrophages et lymphocytes T [25]. Ces cellules inflammatoires vont libérer des cytokines (IL-1, IL-6, IL-8, TNF-α) qui amplifient la réponse inflammatoire, mais aussi et surtout des facteurs de croissance (IGF-1, TGF-α, TGF-β, PDGF-AA, PDGF-BB) qui sont impliqués dans l’activation des cellules satellites quiescentes présentes à proximités [26, 27].
7
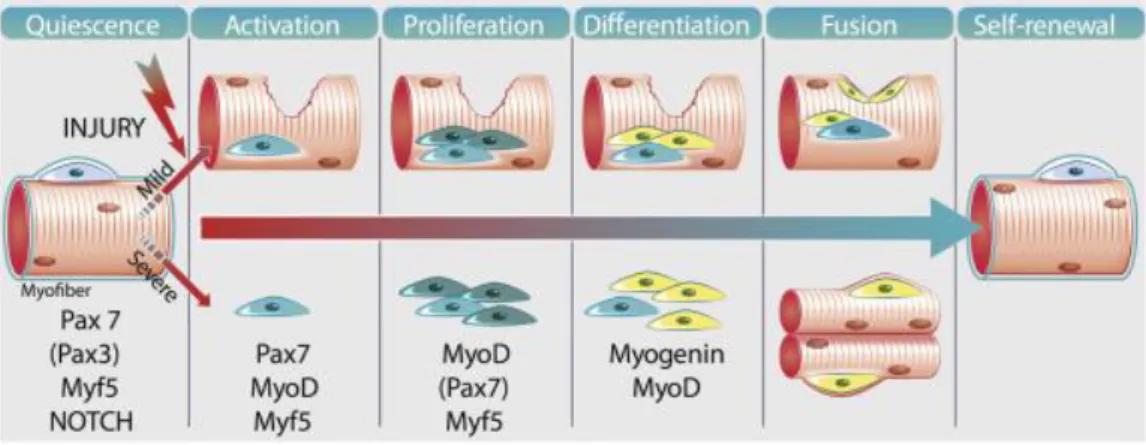
Figure 3. Réparation du tissu musculaire
Sous l’effet d’une lésion musculaire, plusieurs facteurs vont induire l’activation des cellules satellites et leur prolifération. Progressivement, ces cellules vont se différentier en myoblastes, dont les propriétés de fusion vont permettre la réparation des fibres endommagées ou la formation de nouvelles fibres musculaires. Image tirée de[28].
L’activation des cellules satellites (cellules myogéniques) est le point de départ de la régénération des fibres musculaires (Figure 4) qui se réalise par un processus semblable à la myogenèse [3]. L’inhibition de la voie de signalisation Notch entraine l’expression de Myf5 et MyoD qui permet la prolifération asymétrique des cellules satellites activées, formant ainsi des myoblastes. Ces myoblastes maintiennent des propriétés de prolifération jusqu’à l’expression de MyoG, qui induit leur différenciation puis leur fusion avec les fibres endommagées, ou entre myoblastes pour former de nouvelles fibres musculaires intègres. Les fibres en régénération ont la caractéristique d’avoir une localisation centrale des noyaux qui les composent. Ces noyaux retrouvent leur position périphérique lorsque la régénération se finalise et les cellules satellites actives retournent à un état de quiescence ce qui achève la phase de réparation du tissu et permet un retour à l’homéostasie du muscle.
8
Chapitre 2 : La dystrophie musculaire de Duchenne
Les myopathies (myo-, du grec mus signifiant muscle ; et -pathie, du grec pathos qui signifie souffrance), aussi connues sous le nom de maladies neuromusculaires, regroupent l’ensemble des pathologies qui affectent l’intégrité d’un ou plusieurs muscles. Il en existe plus de 200 et leur degré d’atteinte musculaire et de sévérité sont variables. Ces maladies peuvent être d’origines inflammatoires auto-immunes (polymyosites, dermatomyosites, myosites à inclusions) mais elles sont essentiellement d’origine génétique. Parmi ces dernières, la dystrophie musculaire de Duchenne [29] en est la forme la plus répandue et sera la seule discutée dans ce chapitre.2.1. Découverte de la dystrophie musculaire de Duchenne
En 1852, Edward Meryon a caractérisé pour la première fois plusieurs patients atteints de désordres musculaires. Ces huit patients exclusivement masculins issus de trois familles différentes présentaient notamment des difficultés de locomotion dans leurs jeunes années, suivi d’une perte complète de locomotion et d’un décès précoce durant l’adolescence [30, 31]. Au cours de l’autopsie, Meryon a décrit le corps d’un patient qui était très amaigri, surtout au niveau des extrémités inférieures, et qui présentait une importante courbure latérale de la colonne vertébrale. Par l’analyse microscopique de tissus musculaires récoltés post-mortem (Figure 4), Meryon identifiait l’absence de fibres musculaires qui étaient remplacées par du tissu adipeux et granuleux [30, 32]. Une dizaine d’année plus tard Guillaume-Benjamin Amand Duchenne de Boulogne, médecin neurologue français, caractérisait la maladie comme une paralysie musculaire pseudo-hypertrophique ou paralysie myosclérosique [33, 34]. Ce nom mettait l’emphase sur le fait que les faiblesses et changement musculaires atteignent l’ensemble du corps ; les jambes d’abord, puis le dos et les bras. Progressivement, cette pathologie fut davantage connue sous l’appellation de dystrophie musculaire de Duchenne.
9
Figure 4. Premières observations de patients DMD
(A) Premières observations microscopiques de la biopsie musculaire du cas de DMD observé par
Meryon. (B) Photographie de Joseph Sarrazin, du premier cas de DMD caractérisé par Duchenne. Images tirées de [32].
2.2. Diagnostic de la DMD et signes cliniques
La DMD est une maladie qui se manifeste très tôt chez les patients. Les premiers signent peuvent apparaitre dès l’âge de 2 ans lorsque l’enfant présente un retard dans l’acquisition de la marche, chute fréquemment et présente des difficultés à courir ou monter des escaliers [35]. L’un des signes cliniques le plus caractéristique de la DMD est le recours à la manœuvre de Gowers, effectuée par l’enfant lorsqu’il se relève (Figure 6) [36]. L’observation d’un tel phénotype va diriger vers des analyses sanguines afin de doser la créatine kinase (CK) présente dans le sérum, qui est un marqueur de lésions musculaires. Une quantité sérique élevée de CK soulève alors la possibilité d’un diagnostic de DMD. Ce n’est que lorsque l’absence de la protéine dystrophine est observée sur des biopsies de muscles (généralement prélevées dans le quadriceps ou le deltoïde), que le diagnostic de la DMD sera confirmé. Des tests génétiques seront ensuite réalisés pour confirmer celui-ci et pour identifier le type de mutations dans le gène codant pour la dystrophine.
10
Figure 5. Manoeuvre de Gowers
Dans l’exécution de ce mouvement particulier, l’enfant se positionne d’abord à genoux. Il utilise ensuite ses bras pour redresser ses jambes et poser ses mains sur ses genoux. En s’appuyant sur ces derniers avec ces bras, il parvient à relever le haut de son corps. Images tirées de [32].
Durant la vie du patient, l’ensemble de ses muscles squelettiques et cardiaques se détériorent. Cette dégradation est notamment liée à une incapacité de régénération efficace. En effet, l’absence de dystrophine fragilise le sarcolemme des fibres musculaires et expose davantage les muscles qui vont subir de nombreux cycles de nécrose/régénération. Sur-sollicitées, le réservoir de cellules satellites s’épuise peu à peu et ces cellules deviennent peu nombreuses. De plus, l’absence de dystrophine dans les cellules satellites activités empêche leur polarisation et altère leur division asymétrique [37]. Ainsi, les cellules satellites ne peuvent plus générer de précurseurs myogéniques qui contribuent normalement à la formation de nouvelles fibres musculaires ou à leur régénération.
Ne pouvant retourner dans un état d’homéostasie, le tissu musculaire demeure dans un état pro-inflammatoire favorisant le développement de fibrose ainsi que la formation de tissus adipeux qui remplacent physiquement les fibres musculaires [38]. Cette présence de fibrose et d’adipose s’observe également sur la biopsie musculaire des patients. C’est ainsi qu’après les premières faiblesses musculaires, observées à un jeune âge, le patient perd graduellement son autonomie d’ambulation et doit se déplacer en fauteuil roulant dans le début de son adolescence.
11
avoir un impact significatif sur le système respiratoire du patient [39]. En effet, sous l’effet de l’augmentation de courbure de la colonne vertébrale, le volume pulmonaire se retrouve réduit et facilite la survenue de maladies pulmonaires restrictives. De plus, l’affaiblissement des muscles du diaphragme empêche le patient de dégager convenablement ces voies respiratoires ce qui l’expose davantage à de sévères infections respiratoires [40]. L’insuffisance respiratoire est ainsi l’une des premières causes de mortalité chez les patients DMD [41].
Les atteintes cardiaques sont celles qui apparaissent le plus tardivement chez les patients, soit vers l’âge de 15 ans. Les cardiomyocytes (cellules cardiaques) sont progressivement détruits ce qui provoque une atrophie du myocarde conduisant à une diminution de la fonction systolique, de la tachycardie, ou encore une anomalie ventriculaire [40, 42]. L’insuffisance cardiaque est ainsi la deuxième cause de mort prématurée chez les patients DMD, mais il est à noter que sa prévalence (20% des décès) est en partie due à une amélioration dans la prise en charge du système respiratoire [42].
2.3. La dystrophine et les dystrophinopathies 2.3.1. La dystrophine : le gène et ses mutations
C’est un peu plus d’un siècle après la découverte de la DMD que le gène de la dystrophine [1] est découvert dans le locus Xp21 et est associé à la dystrophie musculaire de Duchenne [43]. Avec 2.4 Mb (incluant les introns et les exons), le gène
Dmd est l’un des plus grands parmi les gènes mammifères et représente près de 1%
du génome humain. Ce gène est constitué de 79 exons qui correspondent à un ARNm d’environ 14 kb, soit 0.6% seulement du gène, et codant pour une protéine de 3685 acides aminés pour un total de 427 kDa [44].
A la naissance, la prévalence d’une DMD chez les garçons est de 1/3500 à 1/5000 [45, 46]. Dans 1/3 des cas de DMD, la mutation se fait de novo, tandis que les 2/3 restants sont dus à l’héritage d’un chromosome X muté provenant de la mère du patient. Les dystrophinopathies sont des maladies monosomiques récessives, ce qui explique pourquoi les garçons sont presque exclusivement les seuls atteints. Les mères des patients sont des porteuses saines car la seconde copie du chromosome X, n’ayant pas de mutation dans le gène DMD, permet de compenser la mutation présente sur l’autre. Généralement asymptomatiques, chez 2.5 % à 10 % des
12
porteuses saines (prévalence de 1 femme sur 100000) des faiblesses musculaires ou de légères cardiomyopathies peuvent malgré tout se manifester [47, 48]. Chez les jeunes filles, la prévalence de la DMD est de seulement 1 sur 50 millions [49, 50]. Chez ces patientes, appelées porteuses symptomatiques de DMD, les signes cliniques et l’évolution de la maladie sont cependant très variables.
Ce sont près de 7000 mutations différentes qui sont référencées dans le gène DMD (http://www.dmd.nl). Celles-ci peuvent être des mutations ponctuelles, des duplications d’exon(s), ou encore des délétions. Les larges délétions d’un ou plusieurs exons représentent près de 70% de toutes ces mutations [51]. Il faut distinguer les mutations qui maintiennent un cadre de lecture de celles qui forment un codon STOP prématuré ; elles sont respectivement responsables d’une dystrophie musculaire de Becker (DMB) ou d’une dystrophie musculaire de Duchenne, groupe de maladies nommées dystrophinopathies. De façon intéressante, l’essentielle des mutations sont concentrées des exons 45 à 55, région du gène Dmd aussi connue sous le nom de « hotspot » [52, 53]. Mais l’observation de cette distribution asymétrique peut suggérer que des individus ayant une délétion qui maintient le cadre de lecture en dehors de cette région sont asymptomatiques ou ne montrent pas un phénotype dystrophique [54].
Dans les cas des DMBs, la mutation permet la synthèse d’une protéine dystrophine tronquée d’une partie centrale mais possiblement fonctionnelle [55]. A l’opposé, pour les patients DMD, la présence d’un codon STOP prématuré, conséquent à la mutation, empêche la synthèse d’une protéine dystrophine complète et fonctionnelle qui résulte en une pathologie particulièrement sévère.
2.3.2. La protéine dystrophine et protéines associées 2.3.2.1. Structure de la dystrophine
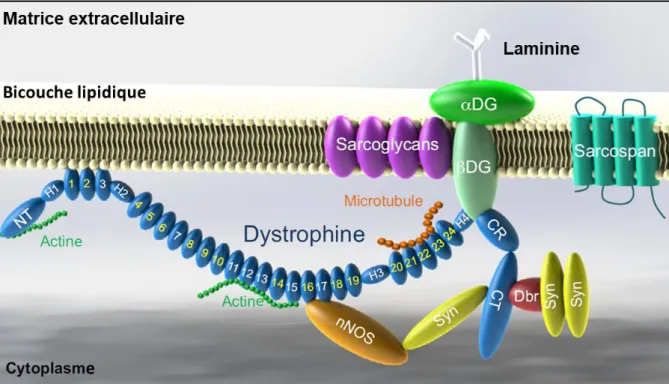
La dystrophine est une protéine de 427 kDa qui participe à la structuration des fibres musculaires et au maintien de l’intégrité du sarcolemme par des interactions entre le cytosquelette, la membrane et la matrice extracellulaire. Ces propriétés lui sont conférées par quatre domaines (Figure 6) [56] :
13
• Le domaine central Rod, qui représente près de 75% de la protéine dystrophine. Il est formé par 24 répétitions de type spectrine (SLR). Ce domaine inclus aussi 4 « Hinges », des charnières qui sont riches en proline et qui confèrent une certaine flexibilité à la dystrophine.
• Une région riche en cystéines (CR) reconnait la sous-unité β du dystroglycan qui est indirectement reliée à la matrice extracellulaire via la laminine.
• Un domaine C-terminal (CT) qui se lie à la synthrophine et à la dystrobrevine.
Figure 6. Représentation schématique de la dystrophine et des complexes de glycoprotéines associées
La dystrophine se localise sous le sarcolemme (membrane des cellules musculaires) et sert de lien entre le cytosquelette de la cellule, par des interactions avec des filaments d’actine ou les microtubules, et la matrice extracellulaire, par des interactions avec la laminine au travers des protéines associées à la dystrophine. Figure adaptée de [56].
2.3.2.2. Rôle des protéines associées à la dystrophine
Actine & Microtubule
Au travers du domaine NT, principalement formé par deux domaines « Calponin Homology », et des motifs SLRs 11-15, la dystrophine interagit avec des filaments d’actine [57, 58]. Ces interactions contribuent ainsi à la formation d’un lien indirect entre le cytosquelette des fibres musculaires la matrice extracellulaire.
Oxyde nitrique synthase neuronal (nNOS)
Ce facteur joue un rôle important dans la physiologie du muscle. Il est colocalisé avec la dystrophine par une liaison avec les SLR16/17 de celle-ci [59-61]. Par sa présence
14
sous-membranaire, nNOS facilite la vasodilatation des vaisseaux environnant sous l’effet de l’oxyde nitrique (NO) qui est produit par l’hydrolyse de arginine en L-Citruline + NO. Ainsi la libération de NO permettrait de limiter la survenue d’ischémie fonctionnelle et de réduire les dommages musculaires lors d’un effort [62].
Syntrophine
Il existe 3 isoformes de la syntrophine (α, β1 et β2). Parmi ces isoformes, seule l’α-syntrophine participerait au recrutement de nNOS au niveau de la dystrophine et donc à sa localisation sous membranaire. Les isoformes β ne sont présents que dans les fibres de type II ou proches des jonctions neuromusculaires [63, 64].
Dystrobrevine
La dystrobrevine (isoformes α ou β) se lie à la dystrophine dans son domaine CT par l’intermédiaire d’un domaine qui est similaire à celui-ci. Cette protéine joue un rôle d’adaptateur qui stabilise l’interaction entre la dystrophine et la syntrophine [65].
β-Dystroglycan
Dans la région CR, un domaine de doigt de zinc (ZZ), un domaine formé de 2 tryptophanes (WW) sont à l’origine d’une liaison forte avec le β-dystroglycan. Cette protéine ancrée dans le sarcolemme est impliquée dans une interaction impérative pour la localisation de la dystrophine sous la membrane [66, 67]. Malgré la production de dystrophine, l’absence d’une telle liaison entraine un phénotype similaire à la DMD et est associée à un retard mental important [68].
La dystrophine, en plus d’être un lien entre la matrice extracellulaire et le cytosquelette, contribue à la localisation sous le sarcolemme de certains facteurs ayant un impact sur la physiologie du muscle. La dystrophine est le cœur d’un grand interactôme qui forme le complexe de protéines associées à la dystrophine (DAPC, dystrophin associated protein complex)
2.3.2.3. De l’importance des répétitions de type spectrine, motif conservé du domaine rod.
Structure des SLRs du domaine rod
15
de ces SLRs est formée par trois hélices alpha successives (A, B et C) alternées de façon antiparallèle. La jonction entre deux SLRs se fait généralement par l’alignement direct de l’hélice C d’une première SLR avec l’hélice A de la SLR qui la suit. Ces éléments sont structurés par des motifs de 7 acides aminés (a, b, c, d, e, f, g) formant des heptades, dans lesquels des acides aminés hydrophobes sont dans les positions « a » et « d » et permettent d’obtenir une structure en hélice alpha.
Relation entre le domaine rod et les propriétés fonctionnelles de la dystrophine
De plus en plus de travaux tendent à démontrer que la dystrophine n’est pas qu’un simple filament linéaire. En effet, il semblerait que la structure de celle-ci joue un rôle important dans sa fonctionnalité. Ces informations proviennent essentiellement de ce que nous pouvons apprendre des patients BMD chez qui les mutations du gène Dmd maintiennent un cadre de lecture.
Par exemple, la mutation ponctuelle T1280C dans l’exon 11 du gène Dmd entraine une modification conformationnelle de la structure de la SLR 1. Cette mutation a des répercussions directes sur la fonction de liaison à l’actine du domaine CR adjacent et est responsable d’une des formes de dystrophie les plus sévères pour les patients BMD [69].
Comme décrit précédemment, les hélices B et C de la SLR 16 et l’hélice A de la SLR 17 sont responsables de la liaison de nNOS. Sans la totalité de ces régions codées par les exons 42 à 45, ce facteur ne peut se localiser sous le sarcolemme. Chez les patients BMD pour lesquels cette localisation ne se fait pas, malgré la production d’une dystrophine tronquée, le phénotype dystrophique peut être tout aussi sévère que celle d’un patient DMD. Cela s’observera également au niveau histologique avec la présence de fibrose et d’adipocytes, que l’on retrouve chez les patients DMD [70]. Au-delà des conséquences qu’une mutation de la dystrophine peut avoir sur un domaine de liaison à une protéine du DAPC, la structure même de la dystrophine et des SLRs peuvent être à mettre en lien avec la sévérité du phénotype dystrophique qui se développe. Les travaux du Dr Elisabeth Le Rumeur montrent des faits intéressants pour certains patients BMD qui ont été l’objet d’une étude [71]. Notamment, les patients BMD Δ45-47 et Δ45-48 ne sont qu’environ 30% à devoir utiliser un fauteuil roulant et 35% à développer une cardiomyopathie tandis que les patients BMD Δ45-49 sont respectivement 60% et 55%. Parmi ces patients, les BMD Δ45-47 et Δ45-48 sont seulement 5 à 6% à développer une cardiomyopathie dilatée
16
et à nécessiter une chaise roulante de façon permanente. A l’opposé, 23% des patients BMD Δ45-49 sont concernés par ces 2 aspects.
Au niveau structurale (Figure 7), les délétions d’exons entrainent la formation d’une répétition de type spectrine hybride. Cependant, ces nouvelles structures ne sont pas toujours correctement phasées. En d’autres mots, la succession des hélices A, B puis C n’est pas toujours préservée. Ainsi, pour la délétion 45-47 la topologie filamenteuse du domaine rod est conservée alors que dans le cas de la délétion 45-49 cette topologie est profondément altérée. Le fait de ne pas maintenir le bon phasage des SLRs, et donc la structure filamenteuse de la dystrophine, compromet par effet boule de neige le bon fonctionnement de la dystrophine.
Figure 7. Conséquence de la délétion d'exon(s) sur la structure de la dystrophine
Figure tirée de [71].
Il est donc fondamental de comprendre que chez les patients BMDs, ce n’est pas la taille de la délétion qui impacterait la sévérité de la maladie, mais la structure de la protéine dystrophine qui en résulte [72].
C’est pourquoi, il apparait déterminant d’étudier les patients DMB et surtout d’en extraire les informations clés sur la relation entre la structure de la dystrophine et sa fonctionnalité dépendamment des délétions. Cela contribuerait à mieux anticiper les bénéfices cliniques qu’un nouveau traitement pourrait apporter aux patients souffrant de DMD.
17 2.4. Modèles animaux dystrophiques
Afin de valider les approches expérimentales envisagées pour le traitement de la DMD, des modèles animaux dystrophiques ont été mis au point. Les modèles mammifères sont les plus couramment utilisés en recherche. Les modèles de souris, de chiens et de porcs sont présentés ici.
2.4.1. Modèles de souris dystrophiques
2.4.1.1. Modèles de souris dystrophiques mdx
Les modèles de souris mdx sont les plus largement utilisés pour l’étude de la DMD. Parmi ces modèles on retrouve les souris mdx, mdx2cv, mdx3cv, mdx4cv, mdx5cv, mdx52 qui présentent chacun des mutations différentes dans leur gène codant pour la dystrophine [73] (http://www.dmd.nl/DMD_animal_mut.html) . Le premier modèle de souris dystrophique mdx fut découvert au début des années 1980 [74]. Celui-ci se caractérise par une mutation dans l’exon 23 du gène mdx qui est responsable la formation d’un codon STOP (mutation non-sens C→T) qui empêche la synthèse d’une protéine dystrophine complète et fonctionnelle et donc son absence de localisation sous la membrane du sarcolemme [43].
Les modèles créés en laboratoire mdx2cv (mutation du site d’épissage 3’ dans l’intron 42), mdx3cv (mutation du site d’épissage 3’ dans l’intron 65), mdx4cv (mutation non-sens dans l’exon 53), mdx5cv (mutation formant un nouveau site d’épissage dans l’exon 10) et mdx52 (délétion de l’exon 52) ne présentent pas de grandes différences de phénotype avec le modèle mdx. Néanmoins, les modèles mdx4cv et mdx5cv possèdent significativement moins de fibres musculaires révertantes que les autres modèles mdx [75]. Bien que moins utilisés, les modèles de souris mdx4cv et mdx52 demeurent intéressants car les mutations dans leur gène mdx, sont localisées dans une région génomique qui est fréquemment muté chez les patients DMD, c’est-à-dire dans le hot-stop 45-55.
Bien que ces différents modèles permettent de valider les preuves de principe de diverses approches thérapeutiques, comme le saut d’exon ou encore la thérapie génique pour la livraison de mini- ou de micro-dystrophine, ces modèles ne portent pas un gène de dystrophine humain qu’il serait pertinent d’avoir pour l’étude d’approches par édition du génome.
18
2.4.1.2. Modèles de souris humanisées hDMD
Les séquences en acides aminés de la dystrophine humaine et de souris sont similaires à 91%. Cette différence significative ne permet donc pas d’étudier in vivo dans les modèles murins mdx les approches thérapeutiques dépendantes des séquences du gène Dmd ou encore du transcrit qui lui correspond. Afin de combler le besoin d’un modèle humanisé, le modèle hDMD/mdx a été développé [76] [77]. Ce modèle est muté pour le gène de la dystrophine de souris (mutation non-sens dans l’exon 23) mais il possède aussi le gène complet (introns et exons) de la dystrophine humaine, inséré dans le chromosome 5 à partir d’un chromosome artificiel de levure contenant le gène Dmd, humain [78]. Ce modèle présente donc la particularité de ne produire que la protéine dystrophine humaine. Il est à noter que dans ce modèle, la protéine dystrophine humaine permet de compenser l’absence de la dystrophine de souris tel qu’indiqué par le profil histologique comparable à celui une souris de type sauvage [76].
Cependant, ce modèle hDMD/mdx ne permet pas d’étudier les approches thérapeutiques visant à corriger directement le gène Dmd ou étudier les effets du saut d’exons qui permettraient de restaurer l’expression d’une protéine dystrophine tronquée.
Mais plus récemment, le modèle de souris hDMD52/mdx a été mis au point et permet à présent d’avoir un modèle murin plus adapté au besoin de la recherche sur la DMD [79]. Ce nouveau modèle a été développé à partir du modèle hDMD/mdx dans lequel la délétion de l’exon 52 du gène Dmd a été induite à l’aide de TALENs, qui seront décrits dans une section de chapitre suivante. La délétion de l’exon 52, qui ne contient pas un multiple de 3 nucléotides, est alors responsable d’un décalage du cadre de lecture du gène Dmd qui entraine l’absence de la production de la protéine dystrophine humaine complète et qui est donc absente sous le sarcolemme.
2.4.1.3. Phénotype des souris mdx et del52hDMD/mdx
Au niveau moléculaire, sur des coupes histologiques de muscles squelettique de souris mdx, on relève l’absence de la localisation sous le sarcolemme des protéines dystroglycan, synthrophine, dystrobrevine ou encore nNOS. Ces modèles se caractérisent aussi par un niveau de créatine kinase (CK) élevé dans le sérum, l’infiltration de macrophages et neutrophiles dans le tissu musculaire, ou encore une
19
localisation centrale des noyaux dans les fibres musculaires. Cependant les phénotypes dystrophiques plus sévères tels que la dégradation des fibres musculaires, les atteintes cardiaques et respiratoires ne surviennent qu’à un âge très avancé de la souris. D’autres part, leur espérance de vie n’est que faiblement affectée.
L’apparition tardive de ces phénotypes pourraient en partie s’expliquer par une surexpression de l’utrophine qui compense l’absence de dystrophine dans les modèles murins [80] [81]. Ainsi, afin d’éviter l’effet compensatoire de l’utrophine, des modèles double-ko utrophine/mdx ont été mis au point de sorte à reproduire davantage le phénotype observé chez les patients DMD. Ce type de modèle permet d’observer des pertes de mobilités, d’augmenter fortement les atteintes cardiaques ou encore de réduire l’espérance de vie des souris [82, 83]. Cependant, il est a noté qu’un tel génotype n’a pas été observé chez les patients DMD et le maintien de telles colonies de souris est particulièrement compliqué. D’autre part, le développement limité d’un phénotype dystrophique chez les souris mdx peut s’expliquer par une meilleure conservation de la longueur des télomères [84]. Cela favorise un maintien des capacités des cellules satellites à participer très activement à la régénération musculaire et de compenser les lésions dues à l’absence de la protéine dystrophine qui engendrent de nombreux cycles de lésion/régénération sous l’effet de contraintes mécaniques. Ainsi, le phénotype dystrophique des souris mdx n’apparait qu’après plusieurs générations au cours desquelles la longueur des télomères réduits progressivement.
Certains paramètres fonctionnels peuvent cependant être utilisés pour caractériser le phénotype des souris mdx. Par exemple, les muscles Tibialis anterior (TA) de souris mdx, soumis à des étirements ex vivo, démontrent un déficit de force 4 à 7 fois supérieur à celui d’une souris normale [85]. Il est aussi possible d’évaluer l’intégrité des fibres musculaires avec le colorant bleu d’Evans [86]. Ce marqueur, injecté par voie intraveineuse avant l’essai, peut diffuser dans les fibres musculaires endommagées où il émettra une fluorescence.
2.4.2. Modèles de rats dystrophiques
Les modèles de rats ont l’avantage d’être beaucoup plus gros que les modèles de souris mais ils restent facilement utilisables en laboratoire, contrairement à de plus
20
gros mammifères (comme les chiens ou les porcs) dont les coûts d’hébergement en animalerie sont beaucoup plus élevés. Les modèles de rats pourraient donc être un bon intermédiaire entre les petits et grands modèles animaux pour l’étude de la DMD. Différents modèles de rats dystrophiques ont été générés à l’aide de TALENs (délétion de 11 nucléotides dans l’exon 23 du gène de la dystrophine) [87] ou avec la technologie CRISPR/Cas9 (formation de INDELs dans l’exon 3 ou dans l’exon 16 du gène de la dystrophine) [88]. Ces modèles présentent de nombreuses similitudes avec les modèles murins telles que : la présence de noyaux en position centrale dans les fibres, des infiltrations de cellules inflammatoires, un niveau de CK sérique élevé. Cependant certaines différences qui rapprochent ces modèles de rats du phénotype des patients DMD sont à noter. Dès trois mois, ces modèles présentent des calcifications mais aussi des fibres nécrotiques par la suite remplacées par de la fibrose et du tissus adipeux qui entrainent une dégradation progressive du tissu musculaire, comme observé chez les patients DMD. Au niveau cardiaque les atteintes sont plus sévères que dans les modèles murins avec notamment des nécroses et fibroses importantes qui s’accompagnent de distension du cœur et d’une altération de la fonction diastolique.
Les modèles de rats dystrophiques présentent donc des intérêts particuliers pour l’étude de la DMD ainsi que pour tester certaines approches thérapeutiques. Il serait donc intéressant d’avoir accès à des modèles qui soient porteurs d’un gène Dmd humain muté de la même façon que dans le modèle murin del52hDMD/mdx.
2.4.3. Modèles de chiens dystrophiques
Bien qu’il existe plusieurs races de chiens pour lesquelles différentes mutations ont été identifiées dans le gène de la dystrophine [89-93], le modèle dystrophique de Golden Retriever (GRDM) est le plus utilisé.
Le chien GRMD est un modèle naturel qui présente une mutation dans l’intron 6 du gène de la dystrophine [78]. Sa taille et le phénotype dystrophique très sévère qu’il présente en font un modèle beaucoup plus proche des patients Duchenne que des modèles murins, voir tableau 1. Plus particulièrement, le modèle GRDM va présenter une faiblesse de ses membres dès son jeune âge, et l’espérance de vie est d’environ 3 ans.
![Figure 7. Conséquence de la délétion d'exon(s) sur la structure de la dystrophine Figure tirée de [71]](https://thumb-eu.123doks.com/thumbv2/123doknet/3269708.93777/37.892.119.819.446.788/figure-conséquence-délétion-exon-structure-dystrophine-figure-tirée.webp)